 Michaela Krupková1František Liška1Ludmila Kazdová2Lucie Šedová1,3Adéla Kábelová1Drahomíra Křenová1Vladimír Křen1
Michaela Krupková1František Liška1Ludmila Kazdová2Lucie Šedová1,3Adéla Kábelová1Drahomíra Křenová1Vladimír Křen1 Ondřej Šeda1*
Ondřej Šeda1*
- 1The First Faculty of Medicine, Institute of Biology and Medical Genetics, Charles University, The General Teaching Hospital, Prague, Czechia
- 2Centre for Experimental Medicine, Institute for Clinical and Experimental Medicine, Prague, Czechia
- 3Laboratory of Transgenic Models of Diseases, Division BIOCEV, Institute of Molecular Genetics of the Czech Academy of Sciences, v.v.i., Vestec, Prague, Czechia
Background: Glucocorticoids (GCs) are potent therapeutic agents frequently used for treatment of number of conditions, including hematologic, inflammatory, and allergic diseases. Both their therapeutic and adverse effects display significant interindividual variation, partially attributable to genetic factors. We have previously isolated a seven-gene region of rat chromosome 8 sensitizing to dexamethasone (DEX)-induced dyslipidemia and insulin resistance (IR) of skeletal muscle. Using two newly derived congenic strains, we aimed to investigate the effect of one of the prime candidates for this pharmacogenetic interaction, the Zbtb16 gene.
Methods: Adult male rats of SHR-Lx.PD5PD-Zbtb16 (n = 9) and SHR-Lx.PD5SHR-Zbtb16 (n = 8) were fed standard diet (STD) and subsequently treated with DEX in drinking water (2.6 µg/ml) for 3 days. The morphometric and metabolic profiles of both strains including oral glucose tolerance test, triacylglycerols (TGs), free fatty acids, insulin, and C-reactive protein levels were assessed before and after the DEX treatment. Insulin sensitivity of skeletal muscle and visceral adipose tissue was determined by incorporation of radioactively labeled glucose.
Results: The differential segment of SHR-Lx.PD5SHR-Zbtb16 rat strain spans 563 kb and contains six genes: Htr3a, Htr3b, Usp28, Zw10, Tmprss5, and part of Drd2. The SHR-Lx.PD5PD-Zbtb16 minimal congenic strain contains only Zbtb16 gene on SHR genomic background and its differential segment spans 254 kb. Total body weight was significantly increased in SHR-Lx.PD5PD-Zbtb16 strain compared with SHR-Lx.PD5SHR-Zbtb16, however, no differences in the weights of adipose tissue depots were observed. While STD-fed rats of both strains did not show major differences in their metabolic profiles, after DEX treatment the SHR-Lx.PD5PD-Zbtb16 congenic strain showed increased levels of TGs, glucose, and blunted inhibition of lipolysis by insulin. Both basal and insulin-stimulated incorporation of radioactively labeled glucose into skeletal muscle glycogen were significantly reduced in SHR-Lx.PD5PD-Zbtb16 strain, but the insulin sensitivity of adipose tissue was comparable between the two strains.
Conclusion: The metabolic disturbances including impaired glucose tolerance, dyslipidemia, and IR of skeletal muscle observed after DEX treatment in the congenic SHR-Lx.PD5PD-Zbtb16 reveal the Zbtb16 locus as a possible sensitizing factor for side effects of GC therapy.
Introduction
Glucocorticoids (GCs) are potent therapeutic agents frequently used for treatment of number of conditions, including hematologic, inflammatory, and allergic diseases (1). The therapeutic effect of GCs administration has been repeatedly shown to vary interindividually due to numerous factors like gender, age, and particularly, genetic constitution of the treated individual. One extreme of the range of varied responses is a near- to complete failure to respond to GC therapy, described as GC insensitivity or resistance (2, 3). Similar variety is observed in unwanted, side effects accompanying GCs therapy—lower bone mineral density and osteoporosis, diabetes, dyslipidemia, obesity, impaired wound healing, or muscle wasting (4). Manifestation of the adverse effects is, to certain extent, dependent on the therapeutic dose and time of treatment; however, pharmacogenetic interactions have been implicated as substantial contributing factors (5, 6). Variation in genes coding for proteins directly involved in GC genomic actions were shown to alter GC sensitivity, namely, the GC receptor (NR3C1), recently summarized by Nicolaides and Charmandari (7). Clearly, other genes can also mediate the clinically relevant adverse effects or changes in response to GC. For instance, genome-wide association study (GWAS) of response to inhaled GCs in asthma patients identified a polymorphism in glucocorticoid-induced transcript 1 gene (GLCCI1) and further replicated the lower response of individuals carrying two copies of the variant allele in four independent cohorts (8). In another GWAS performed in acute lymphoblastic leukemia patients, dexamethasone (DEX)-induced pleiotropic side effects were significantly associated with variation in F2RL1 (F2R like trypsin receptor 1) gene (9). In an experimental, genetically designed model strain set, we showed previously that several genomic loci affect sensitivity toward DEX-induced unfavorable changes in carbohydrate and lipid metabolism (10). Later, we reported that a presence of defective Cd36/Fat-containing genomic segment substantially blunted DEX-induced glucose intolerance and dyslipidemia (11). Also, we have isolated a small region of rat chromosome 8, which sensitized its carrier toward DEX-induced, muscle-specific insulin resistance (IR). In the process of positional cloning of gene(s) connected to several aspects of metabolic syndrome based on results of linkage and association studies in segregating populations and recombinant inbred strains, we derived congenic strain SHR-Lx.PD5 (12). This strain harbors a limited rat chromosome 8 segment of polydactylous rat (PD/Cub) origin on the genomic background of the spontaneously hypertensive rat (SHR). While SHR represents one of the most studied models of essential hypertension (13), it shows also IR, dyslipidemia, and other features of metabolic syndrome (14). By contrast, the PD/Cub strain is a model of metabolic syndrome without elevated blood pressure (15), particularly sensitive to nutritional and pharmacologic challenges (10, 16–18). We have hypothesized that a PD/Cub-derived, GC-responsive gene present within the isolated region, Zbtb16 (zinc finger and BTB domain containing 16), is a prime candidate for the observed pharmacogenetic interaction (12). Our previous sequence analysis revealed a conservative amino acid substitution (T208S) and a 2,964-bp deletion in intron 2 of PD/Cub’s Zbtb16 gene (19). Zbtb16 is a pleiotropic transcription factor involved, among others, in processes underlying pathogenesis of practically all major constituents of metabolic syndrome, as reviewed recently (20). In this study, we created two new models, one of them carrying only the Zbtb16 gene from the original segment to retest its effect on DEX-induced metabolic syndrome features by comparing their metabolic profiles including global and tissue-specific insulin sensitivity.
Materials and Methods
Ethics Statement
All experiments were performed in agreement with the Animal Protection Law of the Czech Republic (311/1997) which is in compliance with the European Community Council recommendations for the use of laboratory animals 86/609/ECC and were evaluated and approved by the Ethical committee of the First Faculty of Medicine and the Ministry of Education, Youth and Sports of the Czech Republic (protocol ID MSMT-1461/2015-17). The health of the rats was examined daily and monitored every 1 h during the experimental procedures. There were no unexpected deaths throughout the experiment. All efforts were made to minimize suffering of the experimental animals.
Derivation of the SHR-Lx.PD5PD-Zbtb16 and SHR-Lx.PD5SHR-Zbtb16 Congenic Strains
The SHR/OlaIpcv [SHR hereafter, RGD (21) ID no. 631848] and SHR-Lx.PD5 (RGD ID no. 1641851) strains were maintained at the Institute of Medical Biology and Genetics, Charles University in Prague. To derive SHR-Lx.PD5PD-Zbtb16 and SHR-Lx.PD5SHR-Zbtb16 congenic strains, a marker-assisted backcross breeding approach was used, as described previously (12, 22, 23). In short, SHR rats were crossed with SHR-Lx.PD5 rats, and the subsequent F1 hybrids were repeatedly backcrossed to SHR. The differential segments in the respective strains were fixed by intercrossing heterozygotes and selecting the progeny with homozygous SHR-Lx.PD5-derived chromosome 8 segments. The congenic status of the new strains was validated with a whole-genome marker scan.
DNA Extraction and Genotyping
Rat DNA was isolated from tail samples by the modified phenol extraction method. Primer nucleotide sequences were obtained from public databases, particularly the Rat Genome Database (RGD, http://rgd.mcw.edu), or designed using Primer3 (24). Polymerase chain reaction (PCR) was used for genotyping markers polymorphic between progenitor strains. PCR was performed in 20 µl reaction volume containing 50–100 ng of genomic DNA, 1 U Taq polymerase (Fermentas), 1× KCl reaction buffer (Fermentas), 1 M betaine (Sigma-Aldrich), 1.5 mM MgCl2, 50 µM of each dNTP, 200 nM of each forward and reverse primers, and 5 mM cresol red (Sigma-Aldrich, for instant gel loading purpose). Cycling conditions were as follows: initial denaturation for 2 min 30 s at 93°C, then 35 cycles of denaturation at 93°C/30 s, annealing at 53°C/40 s and elongation at 72°C/50 s; 72°C/5 min final elongation. Zbtb16 2,964 bp deletion was genotyped using Phusion DNA polymerase (New England Biolabs) according to the manufacturer’s protocol, cycling conditions were as follows: initial denaturation for 30 s at 98°C, then 30 cycles of denaturation at 98°C/10 s, annealing at 63°C/30 s and elongation at 72°C/1 min 30 s; 72°C/5 min final elongation. We tested DNA from the congenic strains (SHR-Lx.PD5PD-Zbtb16 and SHR-Lx.PD5SHR-Zbtb16, n = 10 per strain) and the progenitor strains SHR, SHR-Lx.PD5, as well as the donor of differential segment in SHR-Lx.PD5, the PD/Cub strain. For genotyping the microsatellites, the PCR products were separated on polyacrylamide (7–10%) gels and detected in UV light after ethidium bromide staining using Syngene G:Box (Synoptics Ltd., Cambridge, UK). For genotyping single nucleotide variants, PCR products were sequenced directly using BigDye v1.1 Cycle Sequencing kit and sequencing reactions electrophoresed using ABI PRISM 310 (Applied Biosystems). The complete set of markers used to determine the extents of differential segments is listed in Table S1 in Supplementary Material, and representative gel images are shown in Figure S1 in Supplementary Material.
Experimental Protocol
The selection of experimental protocol and specific ages of rats was made to match our previous study (12) to replicate our findings in the newly derived congenic strains. Adult rat males were held under temperature and humidity controlled conditions on 12 h/12 h light–dark cycle. At all times, the animals had free access to food (except the single overnight fast) and water. Male SHR-Lx.PD5PD-Zbtb16 (n = 9) and SHR-Lx.PD5SHR-Zbtb16 (n = 8) rats were fed standard laboratory chow ad libitum. At the age of 5 months, the oral glucose tolerance test (OGTT) was performed after overnight fasting. Blood for determination of glycemia (Ascensia Elite Blood Glucose Meter; Bayer HealthCare, Mishawaka, IN, USA, validated by Institute of Clinical Biochemistry and Laboratory Diagnostics of the First Faculty of Medicine), insulin (ELISA kit for rat insulin assay, Mercodia, Uppsala, Sweden), free fatty acids (FFAs) (acyl-CoA oxidase-based colorimetric kit, Roche Diagnostics GmbH, Mannheim, Germany), and triacylglycerols (TGs) (standard enzymatic method, Erba-Lachema, Brno, Czech Republic), was drawn from the tail at intervals of 0, 30, 60, 120, and 180 min after the intragastric glucose administration to conscious rats (3 g/kg body weight, 30% aqueous solution). Seven days after the original OGTT, all rats were administered DEX (Dexamed, Medochemie) in drinking water (2.6 µg/ml) for 3 days as described previously (11, 12), and blood was drawn in fed state for determination of TGs, FFAs, insulin, and C-reactive protein (ELISA kit, Alpha Diagnostics International, San Antonio, TX, USA). After an overnight fast, the OGTT was repeated including all the measurements described earlier. Then, the animals were sacrificed, and the total weight and weight of heart, liver, kidneys, adrenals, epididymal, and retroperitoneal fat pads were determined.
For determination of TG content in the liver, tissue was powdered under liquid N2 and extracted over 16 h in chloroform:methanol after which 2% KH2PO4 was added and the solution centrifuged. The organic phase was removed and evaporated under N2. The resulting pellet was dissolved in isopropyl alcohol, and the TG content was determined by enzymatic assay (Erba-Lachema, Brno, Czech Republic).
Basal- and Insulin-Stimulated Glycogen Synthesis in Muscle, and Glucose Utilization in Isolated White Adipose Tissue
Diaphragmatic muscles were immediately after excision incubated in Krebs-Ringer bicarbonate buffer (pH 7.4) containing 0.1 μCi of [U-14C] glucose, 5 mmol/l of unlabeled glucose, and 2.5 mg/ml of bovine serum albumin (Fraction V; Sigma-Aldrich, St. Louis, MO, USA) in the presence (250 μU/ml) or absence of insulin in incubation media. All incubations were performed in a 95% O2 + 5% CO2 atmosphere in sealed vials at 37°C in a shaking water bath. After 2 h incubation tissue glycogen was extracted, and basal and insulin-stimulated incorporation of [U-14C] glucose was determined.
Pieces of epididymal fat were rapidly dissected and incubated for 2 h in Krebs-Ringer bicarbonate buffer with 5 mmol/l glucose, 0.1 μCi [U-14C] glucose/ml (UVVR, Prague, Czech Republic), and 2% bovine serum albumin, gaseous phase 95% O2 + 5% CO2 in the presence (250 μU/ml) or absence of insulin in incubation media. All incubations were performed at 37°C in sealed vials in a shaking water bath.
We estimated incorporation of [U-14C] glucose into neutral lipids. Briefly, fat was removed from incubation medium, rinsed in saline, and immediately placed into chloroform. Pieces of tissue were dissolved using a teflon pestle homogenizer, methanol was added (chloroform:methanol 2:1), and lipids were extracted at 4°C overnight. The remaining tissue was removed, KH2PO4 was added, and the clear extract was taken for further analysis. An aliquot was evaporated, reconstituted in scintillation liquid, and radioactivity measured by scintillation counting. Incremental glucose utilization was calculated as the difference between the insulin-stimulated and basal incorporation of glucose into neutral lipids.
Statistical Analysis
All statistical analyses were performed using STATISTICA 13.2. Unpaired Student’s t-test was used for comparison of morphometric traits and metabolic of the SHR-Lx.PD5PD-Zbtb16 vs. SHR-Lx.PD5SHR-Zbtb16 strains. Repeated measures ANOVA was used for comparison of repeatedly measured parameters. The differences were considered statistically significant when P < 0.05.
Results
Genomic Characterization of the SHR-Lx.PD5 Congenic Substrains
The derivation of the original SHR-Lx.PD5, initial genomic characterization and detailed genetic scan including in silico analysis and manual sequencing of the coding parts of the genes and selected conserved non-coding regions was reported previously (12, 19). We utilized the same extensive set of polymorphic markers to characterize the span of rat chromosome 8 (RNO8) differential segments in the newly derived SHR-Lx.PD5SHR-Zbtb16 and SHR-Lx.PD5PD-Zbtb16 congenic strains. As shown in Figure 1, the SHR-Lx.PD5PD-Zbtb16 harbors a 254 kb segment of the PD/Cub origin including only the Zbtb16 gene on SHR genomic background while the SHR-Lx.PD5SHR-Zbtb16 carries the SHR variant of the Zbtb16 gene together with remaining 563 kb-long part of the original SHR-Lx.PD5 segment. Since we did not identify any additional non-SHR alleles throughout the genomic scan of polymorphic markers in either of the new strains, the congenic status of SHR-Lx.PD5SHR-Zbtb16 and SHR-Lx.PD5PD-Zbtb16 can be considered as validated.
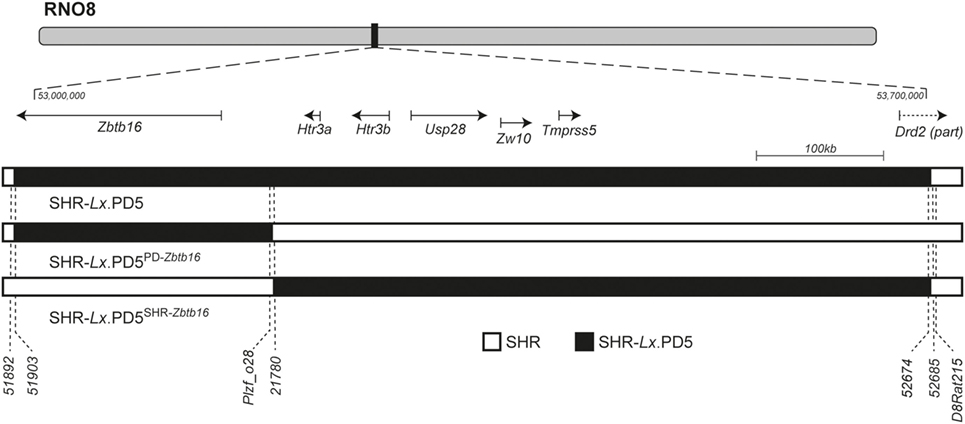
Figure 1. The rat chromosome 8 (RNO8) differential segments in SHR-Lx.PD5 congenic strains. The schematic representation of RNO8 with indication of the relative position and extent of the differential segments in the congenic strains is shown at the top. The portrayed region corresponds to the interval between 53 and 53.7 Mb [based on rat genome assembly: Rnor_6.0 (GCA_000001895.4)]. The gene track shows the RefSeq genes present within the differential segments: Zbtb16 (zinc finger and BTB domain containing 16), Htr3a (5-hydroxytryptamine receptor 3A), Htr3b (5-hydroxytryptamine receptor 3B), Usp28 (ubiquitin specific peptidase 28), Zw10 (zw10 kinetochore protein), Tmprss5 (transmembrane protease, serine 5), and Drd2 (dopamine receptor D2). SHR-Lx.PD5PD-Zbtb16 harbors only 254 kb segment of the PD/Cub origin (black bar) including only the Zbtb16 gene on SHR genomic background (open bar) while the SHR-Lx.PD5SHR-Zbtb16 carries the remaining 563 kb-long part of the original SHR-Lx.PD5 segment (black bar), while the rest of RNO8 and its genome is of the SHR origin (open bar). The position of selected polymorphic markers is shown under the chromosome schemes. The complete annotation of markers used for genotypic characterization of the differential segments is given in Table S1 in Supplementary Material.
Morphometric and Metabolic Profile Comparison
Both before an after DEX administration, the SHR-Lx.PD5PD-Zbtb16 rats were significantly heavier in comparison with SHR-Lx.PD5SHR-Zbtb16 strain. Weights of liver, heart, kidneys, and the visceral and retroperitoneal adipose tissue depots were higher in SHR-Lx.PD5PD-Zbtb16 rats (Table 1). When corrected for body weight, morphometric parameters did not differ between the two strains except for liver, which remained significantly heavier in SHR-Lx.PD5PD-Zbtb16. In fed state, we observed significantly higher TG concentrations in SHR-Lx.PD5PD-Zbtb16 in combination with lower TG content in liver when compared with SHR-Lx.PD5SHR-Zbtb16. The concentrations of FFAs, insulin, TG in muscle, and serum CRP were comparable in the two strains (Table 1).
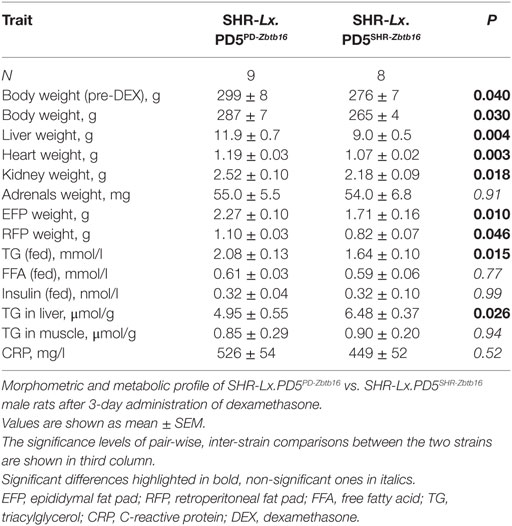
Table 1. Morphometric and metabolic profile of the congenic rat strains.
When fed standard diet (STD), the glycemia time courses during the OGTT were nearly identical (Figure 2A). The levels of insulin also did not differ until the second hour of the test, when the SHR-Lx.PD5PD-Zbtb16 exhibited lower values compared with SHR-Lx.PD5SHR-Zbtb16 (Figure 2C). The fasting concentrations of FFA were significantly higher in SHR-Lx.PD5PD-Zbtb16, yet in the remaining time points of OGTT the two strains did not diverge. No significant differences were observed for TG concentrations. After the DEX challenge (performed in the identical animals assessed under STD conditions), fasting glycemia and glucose concentrations during the first hour of OGTT were significantly higher in SHR-Lx.PD5PD-Zbtb16 in comparison with SHR-Lx.PD5SHR-Zbtb16 with no difference in insulin levels all the way through the test. The suppression of FFA levels by insulin was less effective in SHR-Lx.PD5PD-Zbtb16 as evidenced in Figure 2B, ending in significantly higher FFA concentrations 120 min after the glucose bolus administration. Contrasting with baseline conditions, SHR-Lx.PD5PD-Zbtb16 exhibited elevated TG at fasting state and 30 min into the OGTT, then the values converged (Figure 2D).
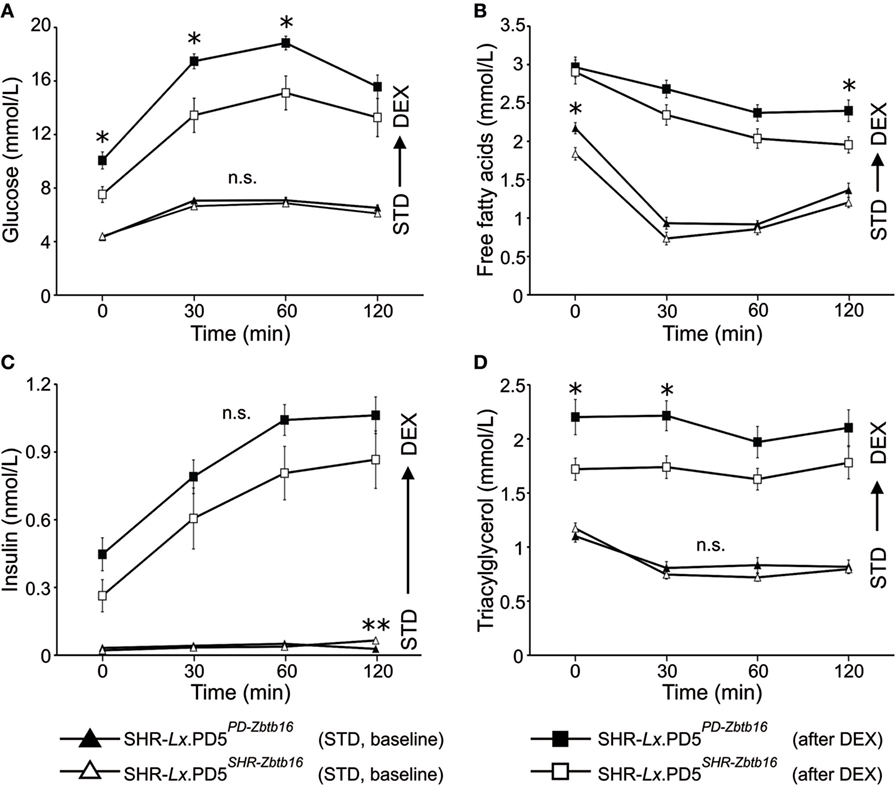
Figure 2. The oral glucose tolerance test (OGTT). Time course of glucose (A), free fatty acids (B), insulin (C), and triacylglycerols (D) during OGTT is shown for standard diet-fed SHR-Lx.PD5PD-Zbtb16 (black triangles, n = 9) and SHR-Lx.PD5SHR-Zbtb16 (open triangles, n = 8) male rats. The procedure was then repeated in the same animals after administration of dexamethasone (values for SHR-Lx.PD5PD-Zbtb16 shown as black squares, SHR-Lx.PD5SHR-Zbtb16 shown in open squares). Adjusted statistical significance levels using the repeated measured ANOVA are indicated for differences between the strains under identical conditions as *P < 0.05.
Insulin Sensitivity of Visceral Adipose Tissue and Skeletal Muscle
While the insulin sensitivity of visceral adipose tissue did not differ between the two congenic strains, both basal and insulin-stimulated incorporation of radioactively labeled glucose into skeletal muscle glycogen were substantially reduced in SHR-Lx.PD5PD-Zbtb16 in comparison with SHR-Lx.PD5SHR-Zbtb16 (Figure 3).
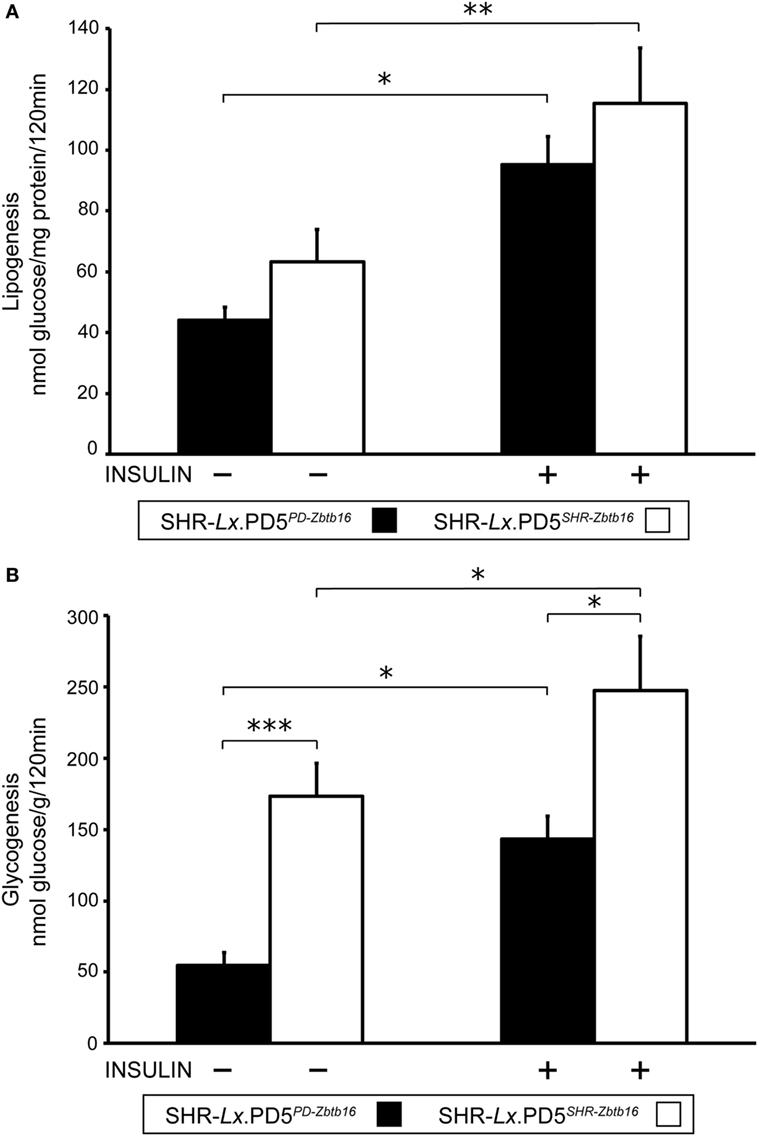
Figure 3. Insulin sensitivity of peripheral tissues in the SHR-Lx.PD5PD-Zbtb16 and SHR-Lx.PD5SHR-Zbtb16 male rats. Basal- and insulin-stimulated [U-14C] glucose incorporation into total lipids of epididymal (visceral) adipose tissue [lipogenesis (A)] and into glycogen of the diaphragm [glycogenesis (B)] in the SHR-Lx.PD5PD-Zbtb16 (full bars) and SHR-Lx.PD5SHR-Zbtb16 (open bars) are shown. Data are expressed as mean ± SEM (n = 8 individual rats/group). Adjusted statistical significance levels using the unpaired t-test are indicated as *P < 0.05; **P < 0.01; and ***P < 0.001.
Discussion
In a follow-up to previous identification of limited genomic region responsible for DEX-induced dyslipidemia and muscle-specific IR, we present in this study further evidence that GC-inducible transcription factor Zbtb16 (25) plays a major role in such sensitization. Zbtb16, also known as Plzf (promyelocytic leukemia zinc finger), belongs to POK (POZ and Krüppel zinc finger) family of proteins and is involved in plethora of physiological processes including maintenance of spermatogenesis (26), stem cell self-renewal (27), hematopoiesis (28), limb development (29, 30) and regulation of metabolism (31, 32). The gene is quite conserved in mammals, the human and rat ZBTB16/Zbtb16 proteins show 96% identity over 673 amino acids constituting the protein. The new, single-gene congenic strain carrying the variant of Zbtb16 gene (3 kb intronic deletion + T208S substitution) showed increased plasma TG and diminished insulin sensitivity of skeletal muscle tissue. Both these observations correspond to the original findings in parental strain SHR-Lx.PD5 (with seven genes in its differential segment, including Zbtb16) when compared with SHR (12). Reduced visceral adiposity observed in the previous study was not confirmed despite the matching difference trend (P = 0.06). The concept of DEX-induced IR of muscle tissue is well known, and several mechanisms have been proposed to convey this effect including direct effects on glucose transporter GLUT4 trafficking (33), increase in glycogen synthase phosphorylation together with decrease in expression and insulin-stimulated phosphorylation of protein kinase B (34) or dysregulation of intramyocellular lipid metabolism (35). To the best of our knowledge, involvement of Zbtb16 in this process has not been reported so far. We have recently reviewed in detail the pleiotropic connections of Zbtb16 to the facets of metabolic syndrome, including IR and dyslipidemia (20). Zbtb16 is strongly induced by GCs (36) and at the same time it negatively regulates the insulin signaling pathway by decreasing the phosphorylation of IRS1, Akt, and FoxO1 in normal mice (32). This presents potential basis for a cross-talk involving DEX, Zbtb16, and insulin sensitivity of muscle, somewhat similar to the one reported for the role of Zbtb16 in regulation of hepatic gluconeogenesis previously. In the suggested model, Zbtb16 acts as a downstream effector of the PGC-1α/GR complex, activated by gluconeogenic signals. Activated PGC-1α/GR then induces Zbtb16 expression, triggering hepatic gluconeogenesis and, at the same time, negatively regulating the insulin signaling pathway (32). The insulin sensitivity of skeletal muscle determined repeatedly in DEX-treated SHR [unpublished data and Ref. (12)] is comparable to that of SHR-Lx.PD5SHR-Zbtb16 strain (SHR basal 121 ± 13; insulin-stimulated 226 ± 25 nmol glucose/g/120 min vs. SHR-Lx.PD5SHR-Zbtb16 basal 173 ± 23; insulin-stimulated 247 ± 38 nmol glucose/g/120 min) contrasting with decreased sensitivity in the single-gene congenic strain with the PD/Cub mutant allele of Zbtb16 gene as well as the original SHR-Lx.PD5 strain (SHR-Lx.PD5 basal 82 ± 6; insulin-stimulated 142 ± 10 nmol glucose/g/120 min vs. SHR-Lx.PD5PD-Zbtb16 basal 55 ± 9; insulin-stimulated 144 ± 16 nmol glucose/g/120 min). In skeletal muscle, GCs were shown to cause IR by reducing transcription of IRS1, while increasing expression of proteins that interfere with insulin action, including protein tyrosine phosphatase type 1B, leukocyte common antigen-related protein (LAR) and p38MAPK (37). However, over 170 potential GC targets have been identified in muscle (38) affecting not only insulin sensitivity but also protein metabolism with effects extending to other organs, as documented in the muscle-specific GR knockout animals (39). While interactions between Zbtb16 and several of the implicated genes have been described, the identification of causal perturbed pathways in the SHR-Lx.PD5PD-Zbtb16 remains to be established. The enhanced TG storage in liver due to GC administration (40) was more prominent in the SHR-Lx.PD5SHR-Zbtb16 strain. In an experimental model with TALEN (transcription activator-like effector nuclease)-mediated Zbtb16 knockout, the heterozygous SHR-Plzf+/− animals showed decreased concentrations of TG and cholesterol in liver and plasma (41). Hepatic Zbtb16 is upregulated in several murine models showing severe hepatic steatosis (32). However, given the higher fasting and satient plasma concentrations of TG in SHR-Lx.PD5PD-Zbtb16, we can speculate that DEX-induced activation of liver Zbtb16 in the single congenic strain could cause a decrease of phosphorylation of FoxO1 (32), which in turn activated both gluconeogenesis (leading to increased glycemia) and increased expression of microsomal triglyceride transfer protein, augmenting very low-density lipoprotein, TG-rich particles assembly and secretion (42). The reduced total body weight and relative weight of liver in the original SHR-Lx.PD5 strain compared with SHR is consistent with the morphometric profile of the strain without the variant Zbtb16 allele, i.e., the SHR-Lx.PD5SHR-Zbtb16 strain. Although at this point it is impossible to pinpoint any of the six genes present in the differential segment as a major contributor, the serotonin receptor Htr3a was recently shown to modulate energy expenditure and weight gain via adipose tissue (43) and can be thus considered as a potential candidate. The limitations of this study include the use of only male rats as sex-specific genetic architecture of the metabolic syndrome and its components was demonstrated (44). Also, as the experimental protocol was set up to mimic the original set of experiments comparing SHR rats to SHR-Lx.PD5 congenic, only one dose of DEX and a short-term administration in 5-month-old animals was used. It is possible that the sensitizing effect would be modified under different conditions, yet the observed robust changes, particularly regarding the blunted insulin sensitivity of the skeletal muscle tissue, would most likely not be affected. Further studies should address in detail the mechanisms and pathways, through which the Zbtb16 mediates the sensitization to DEX-induced IR of skeletal muscle. Also, the relevance of our findings for human condition needs to be validated. So far, there is not any pharmacogenetic study involving variation in human ZBTB16 gene available. In a study of 1517 non-diabetic subjects without hypolipidemic treatment, several single nucleotide polymorphisms in ZBTB16 gene were associated with adiposity measures, LDL, and total cholesterol (45), suggesting the importance of ZBTB16 for metabolic syndrome in humans. The metabolic disturbances including impaired glucose tolerance, dyslipidemia, and IR of skeletal muscle observed after DEX treatment in the congenic SHR-Lx.PD5PD-Zbtb16 reveal the Zbtb16 locus as a possible sensitizing factor for side effects of GC therapy.
Ethics Statement
All experiments were performed in agreement with the Animal Protection Law of the Czech Republic (311/1997) which is in compliance with the European Community Council recommendations for the use of laboratory animals 86/609/ECC and were approved by the Ethical committee of the First Faculty of Medicine and the Ministry of Education, Youth and Sports of the Czech Republic (protocol ID MSMT-1461/2015-17).
Author Contributions
VK and DK derived the new congenic substrains. OS, LS, MK, AK, and LK carried out the experimental components of the study and drafted the manuscript. FL participated in the design of the study and performed the statistical analysis. OS, VK, and LK conceived the study, participated in its design and coordination, and helped to draft the manuscript. All the authors participated in the manuscript preparation and read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by Czech Science Foundation Project 15-04871S and Charles University in Prague [PROGRES-Q25/LF1, SVV 260367].
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fendo.2018.00185/full#supplementary-material.
Figure S1. Representative genotyping results. The results of electrophoretic separation of polymerase chain reaction products of polymorphic markers are shown for individual rat strains (primer sequences and genomic positions of individual markers are shown in Table S1 in Supplementary Material) as follows: lane 1: ladder; lanes 2–4: PD/Cub; lanes 5–7: SHR-Lx.PD5; lanes 8–10: SHR-Lx.PD5PD-Zbtb16; lanes 11–13: SHR-Lx.PD5SHR-Zbtb16; lanes 14–16: SHR; and lane 17: negative control.
References
1. Vandewalle J, Luypaert A, De Bosscher K, Libert C. Therapeutic mechanisms of glucocorticoids. Trends Endocrinol Metab (2018) 29(1):42–54. doi:10.1016/j.tem.2017.10.010
2. Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet (2009) 373(9678):1905–17. doi:10.1016/S0140-6736(09)60326-3
3. Hampl R, Vondra K. Peripheral sensitivity to steroids revisited. Physiol Res (2017) 66(Suppl 3):S295–303.
4. Brotman DJ, Girod JP, Garcia MJ, Patel JV, Gupta M, Posch A, et al. Effects of short-term glucocorticoids on cardiovascular biomarkers. J Clin Endocrinol Metab (2005) 90(6):3202–8. doi:10.1210/jc.2004-2379
5. Cuzzoni E, De Iudicibus S, Franca R, Stocco G, Lucafo M, Pelin M, et al. Glucocorticoid pharmacogenetics in pediatric idiopathic nephrotic syndrome. Pharmacogenomics (2015) 16(14):1631–48. doi:10.2217/pgs.15.101
6. Jackson RK, Irving JA, Veal GJ. Personalization of dexamethasone therapy in childhood acute lymphoblastic leukaemia. Br J Haematol (2016) 173(1):13–24. doi:10.1111/bjh.13924
7. Nicolaides NC, Charmandari E. Novel insights into the molecular mechanisms underlying generalized glucocorticoid resistance and hypersensitivity syndromes. Hormones (Athens) (2017) 16(2):124–38. doi:10.14310/horm.2002.1728
8. Tantisira KG, Lasky-Su J, Harada M, Murphy A, Litonjua AA, Himes BE, et al. Genomewide association between GLCCI1 and response to glucocorticoid therapy in asthma. N Engl J Med (2011) 365(13):1173–83. doi:10.1056/NEJMoa0911353
9. Ramsey LB, Pounds S, Cheng C, Cao X, Yang W, Smith C, et al. Genetics of pleiotropic effects of dexamethasone. Pharmacogenet Genomics (2017) 27(8):294–302. doi:10.1097/FPC.0000000000000293
10. Seda O, Liska F, Krenova D, Kazdova L, Sedova L, Zima T, et al. Dynamic genetic architecture of metabolic syndrome attributes in the rat. Physiol Genomics (2005) 21(2):243–52. doi:10.1152/physiolgenomics.00230.2004
11. Krupkova M, Sedova L, Liska F, Krenova D, Kren V, Seda O. Pharmacogenetic interaction between dexamethasone and Cd36-deficient segment of spontaneously hypertensive rat chromosome 4 affects triacylglycerol and cholesterol distribution into lipoprotein fractions. Lipids Health Dis (2010) 9:38. doi:10.1186/1476-511X-9-38
12. Seda O, Liska F, Sedova L, Kazdova L, Krenova D, Kren V. A 14-gene region of rat chromosome 8 in SHR-derived polydactylous congenic substrain affects muscle-specific insulin resistance, dyslipidaemia and visceral adiposity. Folia Biol (Praha) (2005) 51(3):53–61.
13. Doris PA. Genetics of hypertension: an assessment of progress in the spontaneously hypertensive rat. Physiol Genomics (2017) 49(11):601–17. doi:10.1152/physiolgenomics.00065.2017
14. Pravenec M, Kren V, Landa V, Mlejnek P, Musilova A, Silhavy J, et al. Recent progress in the genetics of spontaneously hypertensive rats. Physiol Res (2014) 63(Suppl 1):S1–8.
15. Sedova L, Kazdova L, Seda O, Krenova D, Kren V. Rat inbred PD/cub strain as a model of dyslipidemia and insulin resistance. Folia Biol (Praha) (2000) 46(3):99–106.
16. Sedova L, Seda O, Krenova D, Kren V, Kazdova L. Isotretinoin and fenofibrate induce adiposity with distinct effect on metabolic profile in a rat model of the insulin resistance syndrome. Int J Obes Relat Metab Disord (2004) 28(5):719–25. doi:10.1038/sj.ijo.0802613
17. Sedova L, Seda O, Kazdova L, Chylikova B, Hamet P, Tremblay J, et al. Sucrose feeding during pregnancy and lactation elicits distinct metabolic response in offspring of an inbred genetic model of metabolic syndrome. Am J Physiol Endocrinol Metab (2007) 292(5):E1318–24. doi:10.1152/ajpendo.00526.2006
18. Seda O, Sedova L, Oliyarnyk O, Kazdova L, Krenova D, Corbeil G, et al. Pharmacogenomics of metabolic effects of rosiglitazone. Pharmacogenomics (2008) 9(2):141–55. doi:10.2217/14622416.9.2.141
19. Liska F, Mancini M, Krupkova M, Chylikova B, Krenova D, Seda O, et al. Plzf as a candidate gene predisposing the spontaneously hypertensive rat to hypertension, left ventricular hypertrophy, and interstitial fibrosis. Am J Hypertens (2014) 27(1):99–106. doi:10.1093/ajh/hpt156
20. Seda O, Sedova L, Vcelak J, Vankova M, Liska F, Bendlova B. ZBTB16 and metabolic syndrome: a network perspective. Physiol Res (2017) 66(Suppl 3):S357–65.
21. Shimoyama M, Smith JR, Bryda E, Kuramoto T, Saba L, Dwinell M. Rat genome and model resources. ILAR J (2017) 58(1):42–58. doi:10.1093/ilar/ilw041
22. Sedova L, Liska F, Krenova D, Kazdova L, Tremblay J, Krupkova M, et al. CD36-deficient congenic strains show improved glucose tolerance and distinct shifts in metabolic and transcriptomic profiles. Heredity (Edinb) (2012) 109(1):63–70. doi:10.1038/hdy.2012.14
23. Sedova L, Pravenec M, Krenova D, Kazdova L, Zidek V, Krupkova M, et al. Isolation of a genomic region affecting most components of metabolic syndrome in a chromosome-16 congenic rat model. PLoS One (2016) 11(3):e0152708. doi:10.1371/journal.pone.0152708
24. Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3 – new capabilities and interfaces. Nucleic Acids Res (2012) 40(15):e115. doi:10.1093/nar/gks596
25. Wasim M, Mansha M, Kofler A, Awan AR, Babar ME, Kofler R. Promyelocytic leukemia zinc finger protein (PLZF) enhances glucocorticoid-induced apoptosis in leukemic cell line NALM6. Pak J Pharm Sci (2012) 25(3):617–21.
26. Costoya JA, Hobbs RM, Barna M, Cattoretti G, Manova K, Sukhwani M, et al. Essential role of Plzf in maintenance of spermatogonial stem cells. Nat Genet (2004) 36(6):653–9. doi:10.1038/ng1367
27. Liu TM, Lee EH, Lim B, Shyh-Chang N. Concise review: balancing stem cell self-renewal and differentiation with PLZF. Stem Cells (2016) 34(2):277–87. doi:10.1002/stem.2270
28. Maeda T. Regulation of hematopoietic development by ZBTB transcription factors. Int J Hematol (2016) 104(3):310–23. doi:10.1007/s12185-016-2035-x
29. Barna M, Hawe N, Niswander L, Pandolfi PP. Plzf regulates limb and axial skeletal patterning. Nat Genet (2000) 25(2):166–72. doi:10.1038/76014
30. Liska F, Snajdr P, Sedova L, Seda O, Chylikova B, Slamova P, et al. Deletion of a conserved noncoding sequence in Plzf intron leads to Plzf down-regulation in limb bud and polydactyly in the rat. Dev Dyn (2009) 238(3):673–84. doi:10.1002/dvdy.21859
31. Plaisier CL, Bennett BJ, He A, Guan B, Lusis AJ, Reue K, et al. Zbtb16 has a role in brown adipocyte bioenergetics. Nutr Diabetes (2012) 2:e46. doi:10.1038/nutd.2012.21
32. Chen S, Qian J, Shi X, Gao T, Liang T, Liu C. Control of hepatic gluconeogenesis by the promyelocytic leukemia zinc finger protein. Mol Endocrinol (2014) 28(12):1987–98. doi:10.1210/me.2014-1164
33. Weinstein SP, Paquin T, Pritsker A, Haber RS. Glucocorticoid-induced insulin resistance: dexamethasone inhibits the activation of glucose transport in rat skeletal muscle by both insulin- and non-insulin-related stimuli. Diabetes (1995) 44(4):441–5. doi:10.2337/diabetes.44.4.441
34. Buren J, Lai YC, Lundgren M, Eriksson JW, Jensen J. Insulin action and signalling in fat and muscle from dexamethasone-treated rats. Arch Biochem Biophys (2008) 474(1):91–101. doi:10.1016/j.abb.2008.02.034
35. Morgan SA, Gathercole LL, Simonet C, Hassan-Smith ZK, Bujalska I, Guest P, et al. Regulation of lipid metabolism by glucocorticoids and 11beta-HSD1 in skeletal muscle. Endocrinology (2013) 154(7):2374–84. doi:10.1210/en.2012-2214
36. Fahnenstich J, Nandy A, Milde-Langosch K, Schneider-Merck T, Walther N, Gellersen B. Promyelocytic leukaemia zinc finger protein (PLZF) is a glucocorticoid- and progesterone-induced transcription factor in human endometrial stromal cells and myometrial smooth muscle cells. Mol Hum Reprod (2003) 9(10):611–23. doi:10.1093/molehr/gag080
37. Almon RR, Dubois DC, Jin JY, Jusko WJ. Temporal profiling of the transcriptional basis for the development of corticosteroid-induced insulin resistance in rat muscle. J Endocrinol (2005) 184(1):219–32. doi:10.1677/joe.1.05953
38. Kuo T, Harris CA, Wang JC. Metabolic functions of glucocorticoid receptor in skeletal muscle. Mol Cell Endocrinol (2013) 380(1–2):79–88. doi:10.1016/j.mce.2013.03.003
39. Shimizu N, Maruyama T, Yoshikawa N, Matsumiya R, Ma Y, Ito N, et al. A muscle-liver-fat signalling axis is essential for central control of adaptive adipose remodelling. Nat Commun (2015) 6:6693. doi:10.1038/ncomms7693
40. John K, Marino JS, Sanchez ER, Hinds TD Jr. The glucocorticoid receptor: cause of or cure for obesity? Am J Physiol Endocrinol Metab (2016) 310(4):E249–57. doi:10.1152/ajpendo.00478.2015
41. Liska F, Landa V, Zidek V, Mlejnek P, Silhavy J, Simakova M, et al. Downregulation of Plzf gene ameliorates metabolic and cardiac traits in the spontaneously hypertensive rat. Hypertension (2017) 69(6):1084–91. doi:10.1161/HYPERTENSIONAHA.116.08798
42. Kamagate A, Dong HH. FoxO1 integrates insulin signaling to VLDL production. Cell Cycle (2008) 7(20):3162–70. doi:10.4161/cc.7.20.6882
43. Oh CM, Namkung J, Go Y, Shong KE, Kim K, Kim H, et al. Regulation of systemic energy homeostasis by serotonin in adipose tissues. Nat Commun (2015) 6:6794. doi:10.1038/ncomms7794
44. Seda O, Tremblay J, Gaudet D, Brunelle PL, Gurau A, Merlo E, et al. Systematic, genome-wide, sex-specific linkage of cardiovascular traits in French Canadians. Hypertension (2008) 51(4):1156–62. doi:10.1161/HYPERTENSIONAHA.107.105247
Keywords: ZBTB16, dexamethasone, rat models, congenic strain, pharmacogenetics and pharmacogenomics, insulin resistance
Citation: Krupková M, Liška F, Kazdová L, Šedová L, Kábelová A, Křenová D, Křen V and Šeda O (2018) Single-Gene Congenic Strain Reveals the Effect of Zbtb16 on Dexamethasone-Induced Insulin Resistance. Front. Endocrinol. 9:185. doi: 10.3389/fendo.2018.00185
Received: 12 December 2017; Accepted: 05 April 2018;
Published: 20 April 2018
Edited by:
Tarunveer Singh Ahluwalia, Steno Diabetes Center Copenhagen (SDCC), DenmarkReviewed by:
Matthew Brook, University of Edinburgh, United KingdomLaura Ellestad, University of Georgia, United States
Copyright: © 2018 Krupková, Liška, Kazdová, Šedová, Kábelová, Křenová, Křen and Šeda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ondřej Šeda, oseda@lf1.cuni.cz