 Heta Huttunen1
Heta Huttunen1 Matti Hero2
Matti Hero2 Mitja Lääperi1
Mitja Lääperi1 Johanna Känsäkoski1Heikki Swan3Joel A. Hirsch4Päivi J. Miettinen2Taneli Raivio1,2*
Johanna Känsäkoski1Heikki Swan3Joel A. Hirsch4Päivi J. Miettinen2Taneli Raivio1,2*
- 1Department of Physiology, Faculty of Medicine, University of Helsinki, Helsinki, Finland
- 2Children’s Hospital, Pediatric Research Center, University of Helsinki, Helsinki University Hospital, Helsinki, Finland
- 3Heart and Lung Center, Helsinki University Hospital, Helsinki, Finland
- 4Department of Biochemistry and Molecular Biology, George S. Wise Faculty of Life Sciences, Institute of Structural Biology, Tel Aviv University, Ramat Aviv, Israel
Objective: Two missense mutations in KCNQ1, an imprinted gene that encodes the alpha subunit of the voltage-gated potassium channel Kv7.1, cause autosomal dominant growth hormone deficiency and maternally inherited gingival fibromatosis. We evaluated endocrine features, birth size, and subsequent somatic growth of patients with long QT syndrome 1 (LQT1) due to loss-of-function mutations in KCNQ1.
Design: Medical records of 104 patients with LQT1 in a single tertiary care center between 1995 and 2015 were retrospectively reviewed.
Methods: Clinical and endocrine data of the LQT1 patients were included in the analyses.
Results: At birth, patients with a maternally inherited mutation (n = 52) were shorter than those with paternal inheritance of the mutation (n = 29) (birth length, −0.70 ± 1.1 SDS vs. −0.2 ± 1.0 SDS, P < 0.05). Further analyses showed, however, that only newborns (n = 19) of mothers who had received beta blockers during pregnancy were shorter and lighter at birth than those with paternal inheritance of the mutation (n = 29) (−0.89 ± 1.0 SDS vs. −0.20 ± 1.0 SDS, P < 0.05; and 3,173 ± 469 vs. 3,515 ± 466 g, P < 0.05). Maternal beta blocker treatment during the pregnancy was also associated with lower cord blood TSH levels (P = 0.011) and significant catch-up growth during the first year of life (Δ0.08 SDS/month, P = 0.004). Later, childhood growth of the patients was unremarkable.
Conclusion: Loss-of-function mutations in KCNQ1 are not associated with abnormalities in growth, whereas maternal beta blocker use during pregnancy seems to modify prenatal growth of LQT1 patients—a phenomenon followed by catch-up growth after birth.
Introduction
Defects in ion channels are increasingly implicated in endocrine diseases. For example, dysfunctional or inactive K-ATP channel function due to mutations in KIR6.2/SUR1 genes leads to congenital hyperinsulinism and/or neonatal diabetes, and defects in potassium channel KCNJ18 underlie periodic hypokalemic paralysis (1–3). We recently reported that two specific mutations in KCNQ1, a gene encoding the alpha subunit of a voltage-gated K+ channel (Kv7.1), result in a gain-of-function in patch clamp analyses and cause pituitary hormone deficiency and maternally inherited gingival fibromatosis (4). This finding was unexpected, since KCNQ1 is traditionally considered as an important regulator of cardiac repolarization rather than one of the key regulators of human growth (5). A prelude for this possibility was achieved already in 2015, however, when Zoledziewska et al. showed that maternally inherited variation in KCNQ1 was associated with shorter adult height in the Sardinian population, although the mechanism of this finding has not been established (6). In other words, there may be growth-regulating mechanisms unrelated to growth hormone secretion by which KCNQ1 affects height. Indeed, KCNQ1 is located on chromosome 11p15.5 in a cluster of imprinted growth-regulating genes. The KCNQ1 locus contains imprinting control region 2, which regulates the imprinting of nearby genes, such as CDKN1C (7). Mutations in this imprinted region cause the growth disorders Beckwith–Wiedemann syndrome (BWS) and Silver–Russell syndrome (8). BWS, which is characterized by placental and pre- and postnatal overgrowth, is occasionally caused by maternally inherited mutations which also disrupt KCNQ1 (9).
According to the parental conflict theory, imprinting has evolved to serve the conflicting interests of the father and the mother; paternally expressed genes promote fetal growth to ensure the viability of the present offspring, whereas maternally expressed genes restrict fetal growth to preserve the mother’s resources to her future offspring (10). Herein, we investigated the growth and endocrine features of patient with long QT syndrome (LQTS) due to loss-of-function mutations in KCNQ1. We paid special attention to the relationship between early growth and parent-of-origin of the mutation.
Subjects and Methods
Patients
In this retrospective study, the patients diagnosed with LQTS in Helsinki University Central Hospital between 1997 and 2015 were first identified from the electronic health records by using the ICD-10 code I49.8 (other cardiac arrhythmia) which has been used for all LQTS patients. From this population, we excluded those with an erroneous diagnosis (other cardiac arrhythmia than LQTS), other LQTS than LQT1, and those with missing medical or growth data (Figure 1). Overall, 104 patients with genetically diagnosed LQT1 and representative growth data were followed up in the Children’s Hospital from 1997 to 2015, comprising the population of this study. Data collected from all LQT1-patients included gender, specification of the KCNQ1 gene mutation and its inheritance, treatment modalities, and growth data including measurements from the birth to the adulthood and parental heights. Additionally, all available endocrine data, including the laboratory test results and diagnoses were recorded.
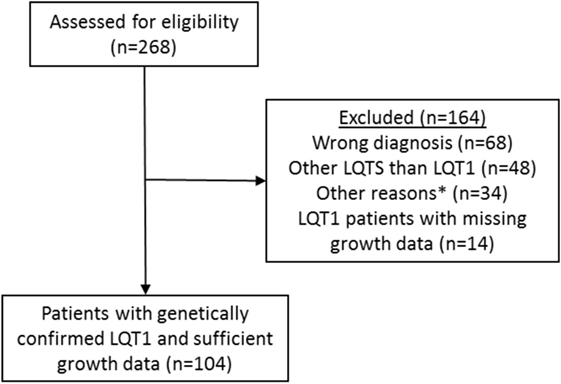
Figure 1. Identification and verification process of the patients with long QT syndrome 1 (LQT1) due to KCNQ1 mutations at the Children’s Hospital, Helsinki University Hospital. Medical records of 104 patients with genetically verified LQT1 and growth data were identified. See main text for details. *Miscellaneous reasons such as patient’s medical records were not found.
Out of the 104 LQT1 patients, 53 (51%) were males and 7 (6.7%) had been born prematurely. Birth length and weight data were available from 81 patients who were born full term; of them, 29 had inherited the mutation from the father and 52 from the mother. Infancy growth data (at least one length measurement was available from 82 patients, altogether 287 measurements). There were 94 (90%) patients who were recorded to receive beta blocker therapy, 8 symptom-free patients had stopped the medication. The inheritance of KCNQ1 mutation was documented in all except 2 patients; 67 patients had inherited the mutation from the mother, 33 from the father, and 2 from both parents. The most common mutation was c.1766G > A p.(Gly589Asp) (63%). The molecular genetic findings are described in Table 1.
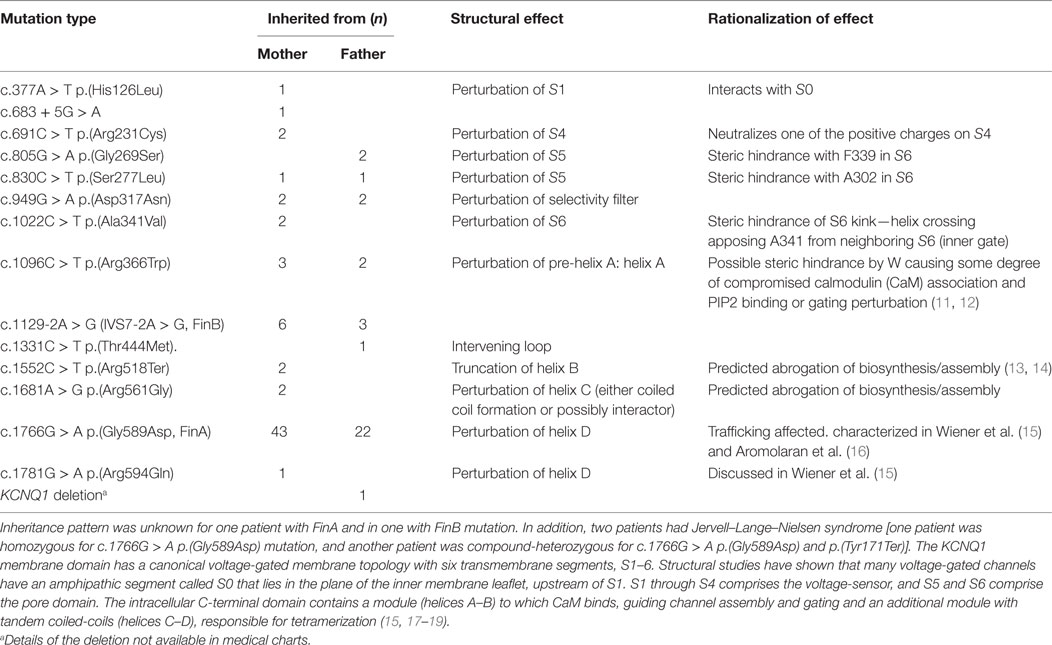
Table 1. Molecular genetic diagnoses of long QT syndrome 1 (LQT1) patients and the structural and rationalized effects of the mutant proteins.
Statistical Analyses
Values are mean (SD) unless otherwise stated. The analyses were carried out with the SPSS statistical software for Windows, release 22.0 (SPSS, Chicago, IL, USA) and with R® using packages lme4 and lmerTest (20–22). Premature infants were excluded. The one sample t-test was used for comparing the height SDS at different ages with the general population. The independent samples t-test was employed in analyzing the effect of paternal/maternal inheritance on growth. The linear mixed-model was used for analyzing the associations between birth length, first year growth, beta blocker use during pregnancy and genetic features, and for comparing cord blood TSH values between the subgroups. To account for the sibship between the patients and the differences between the growth of the individuals in the first year model, random intercepts, and slopes were fitted for both the family and the individual. The effects of the beta blocker use during pregnancy combined with the inheritance of the KCNQ1 mutation were estimated both on birth and per month during the first year. The p-values were calculated using Satterthwaite’s degrees of freedom approximation. The statistically significant level was set to P < 0.05.
Informed Consent and Ethical Approval
Since this study is entirely based on health record data, no ethical permission was required according to the Finnish Medical Research Act. The Helsinki University Central Hospital approved the study.
Results
Birth Size
Birth length and weight data were available for 81 patients with known maternal or paternal inheritance of the mutation. Those with maternal inheritance of the KCNQ1 mutation (n = 52) were born significantly shorter than those (n = 29) with paternal inheritance (−0.70 ± 1.1 SDS vs. −0.20 ± 1.0 SDS, respectively; P = 0.046). There was no significant difference in birth weight (3,371 ± 519 vs. 3,515 ± 466 g, respectively; P = NS). When the respective comparison was carried out after adjusting for sibship in the linear mixed model, the difference in birth length between the groups remained significant (data not shown). However, some of the patients with maternal inheritance of the KCNQ1 mutation (and none of those with paternal inheritance) had been exposed to beta blocker treatment prenatally. We, therefore, divided the patients into three groups: those with paternal inheritance of the mutation (PI, n = 29), maternal inheritance of the mutation without mother’s beta blocker treatment (MI/BB−, n = 33), and those with beta blocker exposure during pregnancy (MI/BB+, n = 19). The patients born in the MI/BB+ group were significantly shorter at birth as compared to those with PI, but the birth lengths of MI/BB− and PI groups did not differ (Table 2). The placental weights (n = 64) did not differ between the MI/BB+, MI/BB−, and PI groups (512 ± 85 vs. 577 ± 114 g vs. 575 ± 127 g, respectively; P = NS).

Table 2. Distributions (mean ± SD) of birth length [in standard deviation scores (SDS)] and birth weight in patients with long QT syndrome 1 due to paternally or maternally inherited KCNQ1 mutations.
Postnatal Growth
Growth data during the first year of life was available for 82 patients and included 287 measurements. During this period, patients with maternal inheritance and intrauterine beta blocker exposure showed significant catch-up growth (Δ0.08 SDS/month, P = 0.004), whereas patients with maternal inheritance and no such exposure grew steadily (Δ0.01 SDS/month, P = NS) (Figure 2). Similarly, length SDS in patients with paternal inheritance did not change significantly during the same period (Δ−0.04 SDS/month, P = NS). After the first 2 years of life, the patients’ mean height SDS did not significantly differ from the population mean (Figure 3). The two patients with biallelic loss-of-function mutations of the KCNQ1 had normal birth lengths (−0.7 and 1.5 SDS). Postnatal growth data were available only for one of them and this patient showed normal first year growth (length −0.2 SDS at 1 year of age).
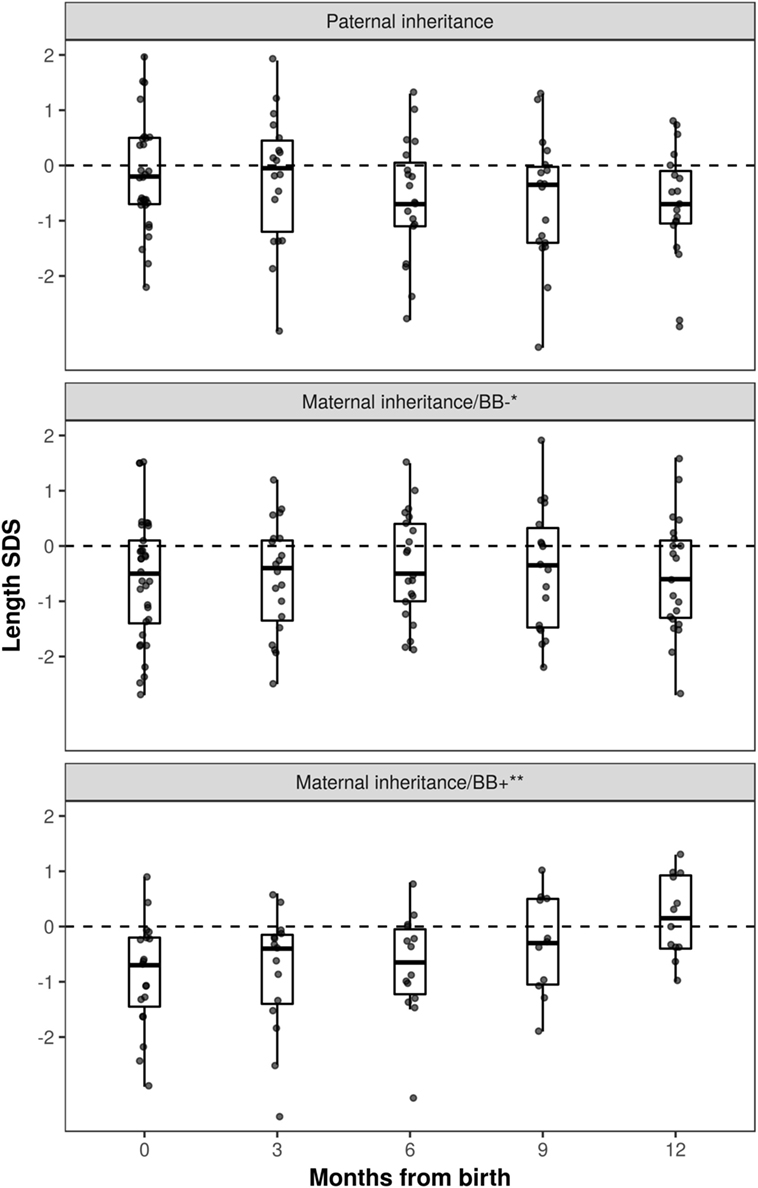
Figure 2. Length standard deviation score (SDS) during the first year of life in long QT syndrome 1 patients. Patients with maternal inheritance of KCNQ1 mutation and prenatal beta blocker exposure were shorter than those with paternal inheritance at birth (P < 0.05), and displayed significant catch-up growth during the first year of life (Δ0.08 SDS/month, P = 0.004). *No beta blocker exposure during pregnancy; **prenatal beta blocker exposure.
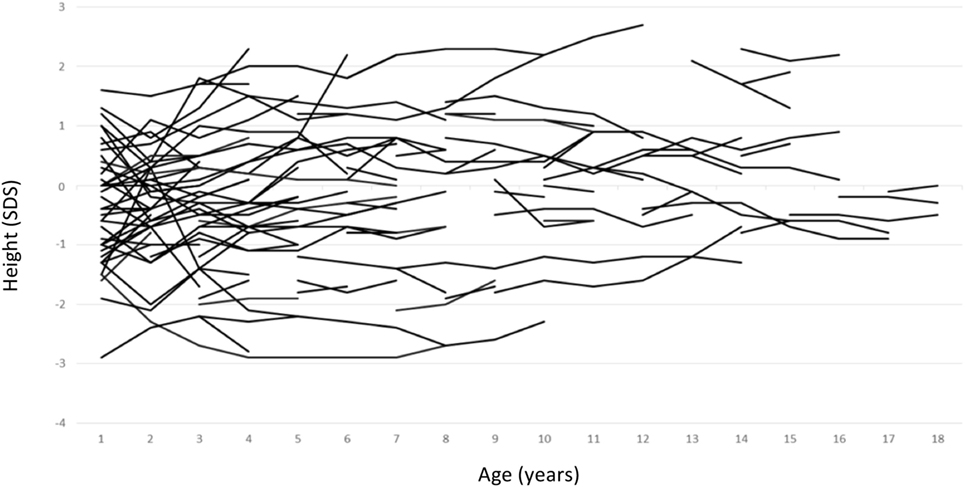
Figure 3. Individual childhood height standard deviation score curves of patients with long QT syndrome 1 due to loss-of-function mutations in KCNQ1.
Other Endocrine Features
As beta blocker treatment appeared to modulate intrauterine growth environment, we compared cord blood TSH levels (n = 41) between MI/BB+, MI/BB−, and PI groups. The mean cord blood TSH level in MI/BB− group did not differ from the PI group [6.1 ± 2.3 vs. 8.3 ± 4.7 mU/L, P = NS (0.07)]. The lowest mean level was found in the MI/BB+ and the highest mean level in the PI group (4.8 ± 1.6 vs. 8.3 ± 4.7 mU/L, P = 0.011). TSH and free T4 levels were measured in 33 (32%) and 24 (23%) LQT1 patients during pediatric follow-up, respectively, and none of the patients were diagnosed with hypo- or hyperthyroidism (data not shown). Insulin-like growth factor I levels were available only from four patients (3.8%) and they were within age- or bone age adjusted reference range. None of the patients received any treatment for endocrine diseases during childhood.
Discussion
The current work was sparked by our recent finding which showed that two specific mutations in KCNQ1, p.(Arg116Leu) and p.(Pro369Leu), underlie growth hormone deficiency and maternally inherited gingival fibromatosis (4). KCNQ1 is expressed in mouse hypothalamic GHRH neurons, and also in pituitary somatotropes (4), lending credence to the hypothesis that KCNQ1 may act at different levels of the hypothalamic–pituitary axis. KCNQ1 can associate with one of the five KCNE beta subunits that are known to modulate the potassium channel function. KCNE1 is important in the heart, whereas KCNE2 is ubiquitously expressed in different tissues (23). Recently, KCNQ1–KCNE2 complexes were suggested to participate in the regulation of myoinositol transport in the choroid plexus, secretion of insulin, and recently we found that KCNQ1 is implicated in cellular processes, such as spliceosomal snRNP assembly, nuclear import, and intracellular vesicle trafficking (4, 24, 25). The concept of KCNQ1 having a role beyond the regulation of cardiac repolarization is further enforced by the finding that KCNQ1 is implicated in the regulation of beta-catenin signaling in colon cancer (26). We examined endocrine phenotypic features in a large cohort of pediatric patients with loss-of-function mutations in this gene. Our results, however, do not demonstrate clinically relevant associations between the loss-of-function KCNQ1 mutations and early growth or the endocrine system. These findings suggest that the role of KCNQ1 in the maintenance of the hypothalamic–pituitary axis integrity is not necessarily associated with the electrical activity of the KCNQ1–KCNE channels, and underscore the fact that to date only the codons 116 and 369 link KCNQ1 to pituitary hormone deficiency (4). To our knowledge, mutations in these codons have not been reported in patients with LQT1, and rare amino acid-changing variants are not reported in codons 116 or 369 of KCNQ1 in Genome Aggregation Database (gnomAD r2.0.2)1 or Exome Aggregation Consortium database (ExAC vs. 0.3.1).2 Similarly, aberrations in growth or pituitary dysfunction have not been described in patients with the extremely rare short QT syndrome (27, 28), or familial atrial fibrilliation (29) due to activating KCNQ1 mutations, which also corroborates that the electrical activity of KCNQ1–KCNE channels may not be the primary mechanism underlying the KCNQ1-related pituitary dysfynction (4). A clear limitation of our study is, however, paucity of IGF-I levels and other endocrine data.
We found that LQT1 patients exposed to maternal beta blocker medication during pregnancy were smaller at birth than those with paternally inherited mutations, and showed subsequent catch-up growth during the first year of life. Although impaired potassium channel function has been proposed to cause intrauterine growth restriction (30), loss-of-function mutations in KCNQ1 may not account for this phenotype. Instead, beta blocker use has been previously associated to reduced birth weight in preeclampsia (31–33). Interestingly, ablation of Kcne2 causes hypothyroidism in pregnant and lactating mice and in their pups (34), yet we did not find any enrichment of thyroid problems in our pediatric LQT1 patient series. Maternal beta blocker use, rather than the parental origin of the KCNQ1 mutation, was also associated with lower cord TSH levels. To our knowledge, however, beta blocker use during pregnancy has not been reported to modulate offspring thyroid function. On the other hand, cord blood TSH levels are known to be affected by stress-related factors (35), and it is tempting to hypothesize that the effects of such factors on cord blood TSH levels are modified by maternal beta blocker use during the pregnancy. We did not investigate the incidence of disturbances in the glucose homeostasis in our patient cohort, since the frequency of hypoglycemia in Finnish children with LQT1 was very recently reported (36).
In conclusion, loss-of-function mutations in KCNQ1 have no apparent effects on growth, suggesting that impaired electrical activity of KCNQ1-KCNE channels may not alter growth hormone secretion in a clinically significant manner. Our analyses show, however, that maternal beta blocker use during pregnancy seems to restrict prenatal growth with subsequent and rapid catch-up during the first year of life.
Ethics Statement
Since this study is entirely based on health record data, no ethical permission was required according to the Finnish Medical Research Act. The Helsinki University Central Hospital approved the study.
Author Contributions
All authors contributed substantially to manuscript drafting. MH and TR designed the study. HH collected the data. HH, MH, JK, ML, PM, and TR analyzed and interpreted the data and wrote the first version of the manuscript. JH interpreted the predicted effects of KCNQ1 mutations. All authors approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Dr. Jari Petäjä is thanked for commenting the manuscript.
Funding
This work was supported by Sigrid Juselius Foundation, the Academy of Finland, Foundation for Pediatric Research, University of Helsinki, and Helsinki Central Hospital research funds.
Footnotes
- ^http://gnomad.broadinstitute.org/ (Accessed: April 14, 2018).
- ^http://exac.broadinstitute.org/ (Accessed: April 14, 2018).
References
1. Edghill EL, Flanagan SE, Ellard S. Permanent neonatal diabetes due to activating mutations in ABCC8 and KCNJ11. Rev Endocr Metab Disord (2010) 11:193–8. doi:10.1007/s11154-010-9149-x
2. Lord K, Dzata E, Snider KE, Gallagher PR, De Leon DD. Clinical presentation and management of children with diffuse and focal hyperinsulinism: a review of 223 cases. J Clin Endocrinol Metab (2013) 98:E1786–9. doi:10.1210/jc.2013-2094
3. Zheng J, Liang Z, Hou Y, Liu F, Hu Y, Lin P, et al. A novel Kir2.6 mutation associated with hypokalemic periodic paralysis. Neurophysiol Clin (2016) 127:2503–8. doi:10.1016/j.clinph.2016.03.008
4. Tommiska J, Känsäkoski J, Skibsbye L, Vaaralahti K, Liu X, Lodge EJ, et al. Two missense mutations in KCNQ1 cause pituitary hormone deficiency and maternally inherited gingival fibromatosis. Nat Commun (2017) 8:1289. doi:10.1038/s41467-017-01429-z
5. Wu J, Ding WG, Horie M. Molecular pathogenesis of long QT syndrome type 1. J Arrhythm (2016) 32:381–8. doi:10.1016/j.joa.2015.12.006
6. Zoledziewska M, Sidore C, Chiang CWK, Sanna S, Mulas A, Steri M, et al. Height-reducing variants and selection for short stature in Sardinia. Nat Genet (2015) 47:1352–6. doi:10.1038/ng.3403
7. Shmela ME, Gicquel CF. Human diseases versus mouse models: insights into the regulation of genomic imprinting at the human 11p15/mouse distal chromosome 7 region. J Med Genet (2013) 50:11–20. doi:10.1136/jmedgenet-2012-101321
8. Demars J, Gicquel C. Epigenetic and genetic disturbance of the imprinted 11p15 region in Beckwith-Wiedemann and Silver-Russell syndromes. Clin Genet (2012) 81:350–61. doi:10.1111/j.1399-0004.2011.01822.x
9. Beygo J, Joksic I, Strom TM, Lüdecke HJ, Kolarova J, Siebert R, et al. A maternal deletion upstream of the imprint control region 2 in 11p15 causes loss of methylation and familial Beckwith-Wiedemann syndrome. Eur J Hum Genet (2016) 24:1280–6. doi:10.1038/ejhg.2016.3
10. Jurkowska RZ, Jeltsch A. Genomic imprinting – the struggle of the genders at the molecular level. Angewandte Chem Int Ed (2013) 52:13524–36. doi:10.1002/anie.201307005
11. Shamgar L, Ma L, Schmitt N, Haitin Y, Peretz A, Wiener R, et al. Calmodulin is essential for cardiac IKS channel gating and assembly: impaired function in long-QT mutations. Circ Res (2006) 98:1055–63. doi:10.1161/01.RES.0000218979.40770.69
12. Matavel A, Medei E, Lopes CM. PKA and PKC partially rescue long QT type 1 phenotype by restoring channel-PIP2 interactions. Channels (Austin) (2010) 4:3–11. doi:10.4161/chan.4.1.10227
13. Schmitt N, Schwarz M, Peretz A, Abitbol I, Attali B, Pongs O. A recessive C-terminal Jervell and Lange-Nielsen mutation of the KCNQ1 channel impairs subunit assembly. EMBO J (2000) 19:332–40. doi:10.1093/emboj/19.3.332
14. Sachyani D, Dvir M, Strulovich R, Tria G, Tobelaim W, Peretz A, et al. Structural basis of a Kv7.1 potassium channel gating module: studies of the intracellular c-terminal domain in complex with calmodulin. Structure (2014) 22:1582–94. doi:10.1016/j.str.2014.07.016
15. Wiener R, Haitin Y, Shamgar L, Fernández-Alonso MC, Martos A, Chomsky-Hecht O, et al. The KCNQ1 (Kv7.1) COOH terminus, a multitiered scaffold for subunit assembly and protein interaction. J Biol Chem (2008) 283:5815–30. doi:10.1074/jbc.M707541200
16. Aromolaran AS, Subramanyam P, Chang DD, Kobertz WR, Colecraft HM. LQT1 mutations in KCNQ1 C-terminus assembly domain suppress IKs using different mechanisms. Cardiovasc Res (2014) 104:501–11. doi:10.1093/cvr/cvu231
17. Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature (1996) 384:80–3. doi:10.1038/384080a0
18. Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature (1996) 384:78–80. doi:10.1038/384078a0
19. Sun J, MacKinnon R. Cryo-EM structure of a KCNQ1/CaM complex reveals insights into congenital long QT syndrome. Cell (2017) 169:1042–50.e9. doi:10.1016/j.cell.2017.05.019
20. Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Software (2015) 67:1–48. doi:10.18637/jss.v067.i01
21. Kuznetsova A, Brockhoff PB, Christensen RHB. lmerTest package: tests in linear mixed effects models. J Stat Softw (2017) 82(13):1–26. doi:10.18637/jss.v082.i13
22. R Core Team. R: A Language and Environment for Statistical Computing Vienna, Austria: R Foundation for Statistical Computing (2017). Available from: https://www.R-project.org/ (Accessed: August 15, 2017).
23. Abbott GW. The KCNE2 K(+) channel regulatory subunit: ubiquitous influence, complex pathobiology. Gene (2015) 569:162–72. doi:10.1016/j.gene.2015.06.061
24. Lee SM, Baik J, Nguyen D, Nguyen V, Liu S, Hu Z, et al. Kcne2 deletion impairs insulin secretion and causes type 2 diabetes mellitus. FASEB J (2017) 31:2674–85. doi:10.1096/fj.201601347
25. Abbott GW, Tai KK, Neverisky DL, Hansler A, Hu Z, Roepke TK, et al. KCNQ1, KCNE2, and Na+-coupled solute transporters form reciprocally regulating complexes that affect neuronal excitability. Sci Signal (2014) 7:ra22. doi:10.1126/scisignal.2005025
26. Rapetti-Mauss R, Bustos V, Thomas W, McBryan J, Harvey H, Lajczak N, et al. Bidirectional KCNQ1:Beta-catenin interaction drives colorectal cancer cell differentiation. Proc Natl Acad Sci U S A (2017) 114:4159–64. doi:10.1073/pnas.1702913114
27. Hedley PL, Jørgensen P, Schlamowitz S, Wangari R, Moolman-Smook J, Brink PA, et al. The genetic basis of long QT and short QT syndromes: a mutation update. Hum Mutat (2009) 30:1486–511. doi:10.1002/humu.21106
28. Bellocq C, van Ginneken AC, Bezzina CR, Alders M, Escande D, Mannens MM, et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation (2004) 109:2394–7. doi:10.1161/01.CIR.0000130409.72142.FE
29. Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science (2003) 299:251–4. doi:10.1126/science.1077771
30. Luo Y, Kumar P, Mendelson CR. Estrogen-related receptor gamma (ERRgamma) regulates oxygen-dependent expression of voltage-gated potassium (K+) channels and tissue kallikrein during human trophoblast differentiation. Mol Endocrinol (2013) 27:940–52. doi:10.1210/me.2013-1038
31. Fuerst M. Beta-blockers may have role in preeclampsia. JAMA (1982) 248:516–8. doi:10.1001/jama.1982.03330050006004
32. Heida KY, Zeeman GG, Van Veen TR, Hulzebos CV. Neonatal side effects of maternal labetalol treatment in severe preeclampsia. Early Hum Dev (2012) 88:503–7. doi:10.1016/j.earlhumdev.2011.12.012
33. Xie RH, Guo Y, Krewski D, Mattison D, Walker MC, Nerenberg K, et al. Beta-blockers increase the risk of being born small for gestational age or of being institutionalised during infancy. BJOG (2014) 121:1090–6. doi:10.1111/1471-0528.12678
34. Roepke TK, King EC, Reyna-Neyra A, Paroder M, Purtell K, Koba W, et al. Kcne2 deletion uncovers its crucial role in thyroid hormone biosynthesis. Nat Med (2009) 15:1186–94. doi:10.1038/nm.2029
35. Korevaar TI, Chaker L, Jaddoe VW, Visser TJ, Medici M, Peeters RP. Maternal and birth characteristics are determinants of offspring thyroid function. J Clin Endocrinol Metab (2016) 101:206–13. doi:10.1210/jc.2015-3559
Keywords: growth, ion channels, KCNQ1, long QT syndrome 1, beta blocker
Citation: Huttunen H, Hero M, Lääperi M, Känsäkoski J, Swan H, Hirsch JA, Miettinen PJ and Raivio T (2018) The Role of KCNQ1 Mutations and Maternal Beta Blocker Use During Pregnancy in the Growth of Children With Long QT Syndrome. Front. Endocrinol. 9:194. doi: 10.3389/fendo.2018.00194
Received: 08 February 2018; Accepted: 09 April 2018;
Published: 24 April 2018
Edited by:
Andrea Enzo Scaramuzza, Istituti Ospitalieri di Cremona, ItalyReviewed by:
Gianpaolo De Filippo, Bicêtre Hospital, FranceEli Hershkovitz, Soroka Medical Center, Israel
Copyright: © 2018 Huttunen, Hero, Lääperi, Känsäkoski, Swan, Hirsch, Miettinen and Raivio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Taneli Raivio, taneli.raivio@helsinki.fi