Validation and Development of COI Metabarcoding Primers for Freshwater Macroinvertebrate Bioassessment
 Vasco Elbrecht
Vasco Elbrecht Florian Leese
Florian Leese- 1Aquatic Ecosystem Research, Faculty of Biology, University of Duisburg-Essen, Essen, Germany
- 2Centre for Water and Environmental Research, University of Duisburg-Essen, Essen, Germany
A central challenge in the present era of biodiversity loss is to assess and manage human impacts on freshwater ecosystems. Macroinvertebrates are an important group for such bioassessments as many taxa show specific responses to environmental conditions. However, generating accurate macroinvertebrate inventories based on primarily larval morphology is difficult and error-prone. Here, DNA metabarcoding provides new opportunities. Its potential to accurately identify invertebrates in bulk samples to the species level has been demonstrated in several case studies. However, DNA based identification is often limited by primer bias, potentially leading to taxa in the sample remaining undetected. Thus, the success of DNA metabarcoding as an emerging technique for bioassessment critically relies on carefully evaluated primers. We used the R package PrimerMiner to obtain and process cytochrome c oxidase I (COI) sequence data for the 15 globally most relevant freshwater invertebrate groups for stream assessment. Using these sequence alignments, we developed four primer combinations optimized for freshwater macroinvertebrates. All primers were evaluated by sequencing 10 mock community samples, each consisting of 52 freshwater invertebrate taxa. Additionally, popular metabarcoding primers from the literature and the developed primers were tested in silico against these 15 relevant invertebrate groups. The developed primers varied in amplification efficiency and the number of detected taxa, yet, all detected more taxa than standard “Folmer” barcoding primers. Two new primer combinations showed even more consistent amplification than a previously tested ribosomal marker (16S) and detected all 42 insect taxa present in the mock community samples. In silico evaluation revealed critical design flaws in some commonly used primers from the literature. We demonstrate a reliable strategy to develop optimized primers using the tool PrimerMiner. The developed primers detected almost all taxa present in the mock samples, and we argue that high base degeneracy is necessary to decrease primer bias as confirmed by experimental results and in silico primer evaluation. We further demonstrate that some primers currently used in metabarcoding studies may not be suitable for amplification of freshwater macroinvertebrates. Therefore, careful primer evaluation and more region/ecosystem specific primers are needed before DNA metabarcoding can be used for routine bioassessment of freshwater ecosystems.
Introduction
Freshwater resources worldwide are threatened by anthropogenic activities and the pressure on these sensitive ecosystems will intensify with the exponential increase of the human population (Dudgeon et al., 2005; Vörösmarty et al., 2010). Ambitious aquatic ecosystem assessment and biomonitoring programs have been launched globally in the last decades for Environmental Impact Assessment and to provide solid data in order to protect and restore freshwater ecosystems (EU Water Framework Directive, US Clean Water Act). Macroinvertebrates are often a biological key component (“biological quality element”) for assessing stream health, as many taxa are sensitive to stressors. While many bioassessment protocols only require identification at higher taxonomic level (family, genus), it is highly beneficial to include precise species-level information, as even closely related species can show different tolerances to environmental stressors (Macher et al., 2016). However, accurate species-level identification of freshwater macroinvertebrates can be difficult for larval specimens, often leading to low taxonomic resolution or misidentifications (Haase et al., 2010; Sweeney et al., 2011). This in turn decreases the accuracy of the approach and may result in imprecise bioassessment or even misguided management (Stein et al., 2014). Additionally, identification accuracy is affected by different levels of taxonomic expertise amongst specialists, limiting the comparability of assessments (Haase et al., 2010). With the decline of available taxonomic expertise and much of the world's diversity not being properly described, morphology-based monitoring cannot keep pace with current challenges of sustainable water management.
A promising alternative to morphological identification is DNA based determination of macroinvertebrates, which has been demonstrated in multiple case studies (Hajibabaei et al., 2011; Sweeney et al., 2011; Carew et al., 2013; Stein et al., 2013; Elbrecht and Leese, 2015). Prerequisite of such “DNA barcoding” techniques is an appropriate reference data base. In short, a fragment of a standardized genetic marker sequence is obtained from well-determined invertebrate material (typically male adult specimens, which can be determined to species level often) is obtained and stored in a reference database. This reference database can then be used for the identification of larval specimens. The cytochrome c oxidase I (COI) gene is typically used for this DNA barcoding technique and extensive reference sequences are already available in online databases (Ratnasingham and Hebert, 2007, 2013). However, identifying single specimens using DNA barcoding is still quite expensive because each specimen has to be processed and sequenced individually (Cameron et al., 2006; Stein et al., 2014). Recent advances in high throughput sequencing (HTS) have made it possible to characterize the species composition for complete bulk samples often containing hundreds to thousands of specimens. This technique, coined “DNA metabarcoding,” has already been widely used to generate comprehensive taxa lists for many ecosystems and environments (Taberlet et al., 2012). However, the utility of DNA metabarcoding remains limited due to severe primer bias, which prevents the detection of all taxa present in a sample and hinders precise quantification of taxon biomass and abundances (Piñol et al., 2014; Elbrecht and Leese, 2015).
A barcoding primer pair, which amplifies a marker sequence of suitable length for HTS for ideally all taxa contained in the sample, is therefore the most critical component to assess macroinvertebrate bulk samples with DNA metabarcoding. However, the COI barcoding gene region shows high codon degeneracy throughout its sequence, making the design of such “truly” universal primers difficult (Deagle et al., 2014; Sharma and Kobayashi, 2014). Several COI barcoding primers with different levels of base degeneracy have been developed of which many are now used or could be suitable for metabarcoding studies (Figure 1, e.g., Folmer et al., 1994; Hebert et al., 2004; Meusnier et al., 2008; Van Houdt et al., 2010; Shokralla et al., 2011, 2015; Zeale et al., 2011; Geller et al., 2013; Leray et al., 2013; Gibson et al., 2014; Brandon-Mong et al., 2015). However, often these primers were developed for a specific taxonomic group, purpose or ecosystem, for example the primers by Zeale et al. (2011) which were originally developed for gut content analysis on bats but are now more widely used. Thus, despite including several degenerate bases, metabarcoding primers typically recover only 80–90% or even less of the taxa present in a sample (Leray et al., 2013; Brandon-Mong et al., 2015; Elbrecht and Leese, 2015). Furthermore, many primers have not been thoroughly evaluated for primer bias and the proportion of undetected taxa, making development and testing of universal primers a pressing issue. Additionally, details on criteria for primer design such as the used reference sequence data are often not described extensively (e.g., Hajibabaei et al., 2011; Shokralla et al., 2015). Typically, primers are developed either with aligned reference barcode sequences for the taxonomic target groups available from NCBI or BOLD (Zeale et al., 2011; Leray et al., 2013; Gibson et al., 2014), or alternatively only mitochondrial genomes or a small subset of barcoding sequences are used (Geller et al., 2013; Deagle et al., 2014; Brandon-Mong et al., 2015). These two approaches are typically biased, as sequences for certain taxa are overrepresented in big datasets (e.g., from population genetic studies), while datasets containing only mitochondrial genomes have an underrepresented number of reference sequences which is insufficient to capture sequence variation for primer design.
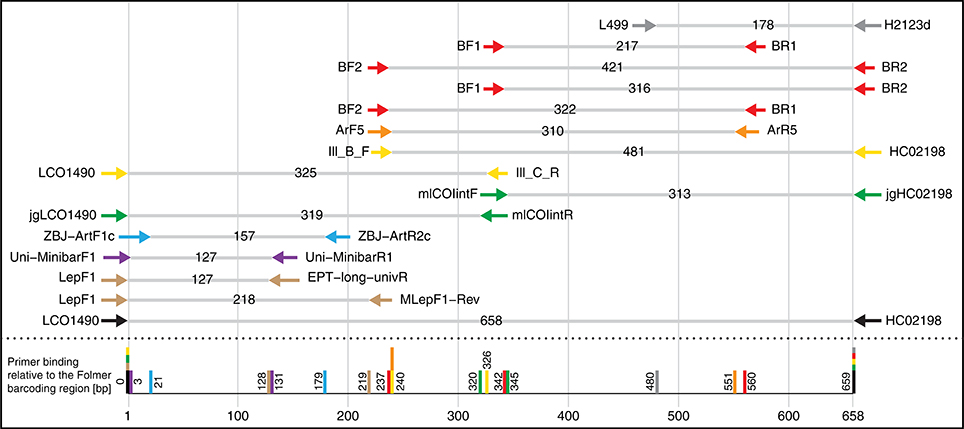
Figure 1. Selection of potential COI primer sets for DNA metabarcoding of insects, targeting the Folmer region. Primer pairs shown are typically used/suggested combinations from the literature. Table S1 gives an overview of the exact primer sequences and references.
In this study, we used the recently developed R package PrimerMiner to explore these two problems for primer development and evaluated the suitability of existing primers for freshwater invertebrate metabarcoding using computational, i.e., in silico analyses. Specifically, we downloaded available sequences from public archives and checked whether published primers showed obvious mismatches to the references which limit their probability of amplification. Furthermore, we experimentally evaluated own optimized primer sets using 10 macroinvertebrate mock communities consisting of 52 freshwater species that were also used for method evaluation in in previous studies (Elbrecht and Leese, 2015; Elbrecht et al., 2016).
Materials and Methods
Primer Development and In Silico Evaluation
The PrimerMiner package v0.7 was used to download and cluster COI sequences for the 15 most relevant freshwater invertebrate groups for bioassessment (accessed September 2016, Table S2, Elbrecht and Leese, 2016). Sequences were aligned with MAFFT v7.017 (Katoh et al., 2002) as implemented in Geneious 8.1.7 (Kearse et al., 2012). PrimerMiner's “selectivetrim” function was used to trim 26 bp in the HCO and 25 bp in the LCO binding sites, and the alignment for each group was visualized with PrimerMiner to manually identify suitable primer binding sites. Two forward (BF1, BF2) and two reverse primers (BR1, BR2) were designed with high base degeneracy. Fusion primers were designed by adding Illumina adapters and inline barcodes, as described by Elbrecht and Leese (2015), to increase per-base pair sequence diversity during sequencing and allow for a one step PCR protocol.
PrimerMiner was also used to evaluate all primers shown in Figure 1 against alignments of the 15 freshwater invertebrate groups, using the default “Position_v1.csv” and “Type_v1.csv” table for mismatch scoring (tables are included in the PrimerMiner example data). Primers that obtained a penalty score of >120 were considered as inappropriate for metabarcoding.
Testing of DNA Metabarcoding Primers on Mock Communities
Amplification success of the BF/BR primers was evaluated using 10 mock communities, each containing a set of 52 different freshwater invertebrates also used in previous studies (Elbrecht and Leese, 2015; Elbrecht et al., 2016). The DNA aliquots and the one-step PCR protocol as in Elbrecht and Leese (2015) was used for all four primer combinations, but the number of PCR cycles was increased from 30 to 35 and the annealing temperature increased to 50°C. As in the previous studies, each sample was uniquely tagged from both sides, but for half of the samples only 25 ng instead of 50 ng DNA was used in PCR (see Figure S1). For each primer combination, all 10 samples were run in the same PCR setup, using one PCR replicate per sample. Ready-to-load products were purified with magnetic beads (left sided, 0.8x SPRIselect, Beckman Coulter, Bread, CA, USA) and quantified using the Qubit HS DNA Kit (Thermofisher Scientific, Carlsbad, CA, USA). For each primer combination, equimolar amounts of amplicons were pooled into one library (amplicon concentrations had to be adjusted due to variation in amplicon length, see Figure S1). The library was sequenced on one lane of a HiSeq 2500 (rapid run, 2 × 250 bp) with 5% PhiX spike-in, carried out by the DNA Sequencing Center of Brigham Young University, USA.
Bioinformatic processing of HTS data was kept as similar as possible to previous studies (Elbrecht and Leese, 2015; Elbrecht et al., 2016). In short, reads were demultiplexed (Script S1 in Supplementary Material) and paired end reads merged using Usearch v8.1.1831 -fastq_mergepairs with -fastq_merge_maxee 1.0 (Edgar and Flyvbjerg, 2015). Where necessary, reads were converted into reverse complement. For each primer combination all 10 replicates were pooled and sequences which were present only one single time in the dataset (singletons) were removed prior to clustering with Usearch (cluster_otus, 97% identity, strand plus, includes chimera removal) (Edgar, 2013). Dereplicated reads for each of the 40 samples (including singletons) were compared against the respective Operational Taxonomic Unit (OTU) dataset, using usearch_global with a minimum match of 97% and strand plus. As in previous studies, low abundance OTUs without at least one sample above 0.003% sequences assigned, were considered unreliable and excluded from the dataset. Taxonomy of the remaining OTUs were identified and manually verified using the BOLD and NCBI databases. To ensure that the same taxonomy was assigned across primer combinations and the reference COI study (Elbrecht and Leese, 2015), the most abundant sequence for each OTU in each sample was extracted using an R script (Script S2 in Supplementary Material) and the haplotype of all individual specimens assembled, if amplified by more than one primer combination.
Results
Developed Primers Using PrimerMiner
We designed four primer pairs (Table 1) using the alignments of 15 major freshwater groups relevant for bioassessment (Figure S2). The two BF and two BR primers show high base degeneracy to amplify as many insect taxa as possible. Amplified regions range from 217 bp for internal barcodes and up to 421 bp for combinations using a degenerated version of the HCO2198 primer (Figure 1). While samples in this study were tagged uniquely from both sides using fusion primers (Figure S3), the inline barcodes allow for tagging of up to 72 samples for each primer combination (see Figure S4 for recommended primer combinations).
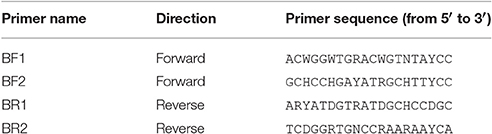
Table 1. Newly developed universal primers targeting freshwater macroinvertebrates relevant for aquatic bioassessment.
All four BF/BR primer combinations were tested on 10 invertebrate mock community samples on an Illumina HiSeq sequencer. PCR efficiency varied across primer combinations, with PCRs involving the BF2 primer showing good amplification whereas those with the BF1 primer always showing decreased yields (Figure S5). Amplification efficiency with fusion primers was always lower than in the positive control (standard COI Folmer primers without Illumina tail, data not shown). Sequencing was successful for all samples, with very similar numbers of sequences obtained for all replicates (on average 1.55 million reads per sample, SD = 0.2, Figure S1A). Cluster density on the lane was low (402 k/mm2) yielding only 48.74% of the expected sequencing output, yet with good sequence quality (Phred Q30 score ≥92.17%, raw data deposited on SRA: SRX1619153). The amplified read lengths had an influence on the number of sequences retained in bioinformatic processing. Longer amplicons showed less overlap when paired-end merged and were thus excluded more often due to expected errors > 1 (Figure S1B). Additionally, for primer combinations that used the P5_BF1_2 primer, more sequences were discarded than with other primer combinations, as ~1/5 of the reads had poor Phred quality scores (see Figure S1B). There were also issues with the BF1 and BF2 primers which showed insertions or deletions on the 3′–end affecting total sequence length by 1–2 bp across all replicates (Figure S6). Some primer combinations also amplified up to 1.35% shorter or longer fragments than expected (Figure S7).
Number of Taxa Recovered
All insect taxa present in the mock samples were detected with each primer combination, with exception of the BF1 + BR1 combination that failed to amplify the Scirtidae (Coleoptera) specimens (Table 2, raw OTU data Table S3, for haplotype sequences see Script S2 in Supplementary Material). All primers failed for some of the other metazoan taxa, with the BF1 + BR2 combination showing the lowest number of undetected taxa. In comparison to the traditional Folmer primers (Folmer et al., 1994), all BF/BR freshwater primers showed a more consistent and equal read abundance across the mock samples (Figure 2). As in Elbrecht et al. (2016), the standard deviation from the expected abundance and precision for the primer pairs was estimated, which summarizes the variance in amplification for each morphotaxon. The primer combination BF1 + BR1 showed the highest inconsistencies in read abundance, while the BF2 + BR1 and BF2 + BR2 combination showed even higher precision than a previously tested 16S marker (Elbrecht et al., 2016). The proportion of detected non-insect metazoan taxa varied between primer combinations, with the combination BF1 + BR2 detecting all but one taxon.
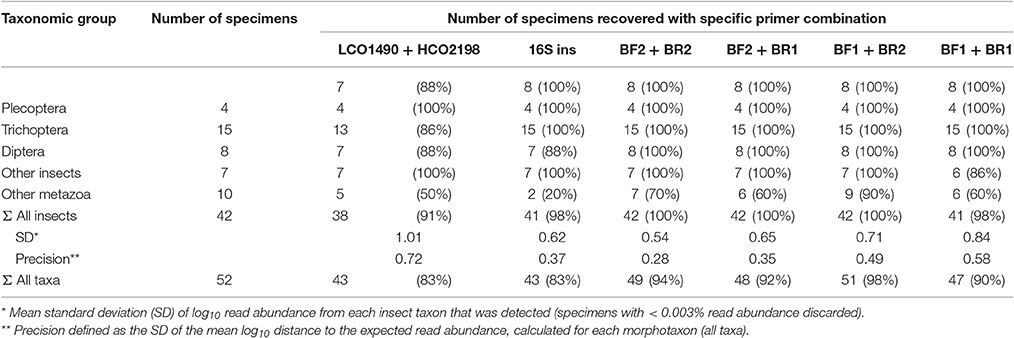
Table 2. Number of species recovered with the newly developed primers and data on 16S and Folmer primers from previous tests (Elbrecht and Leese, 2015; Elbrecht et al., 2016).
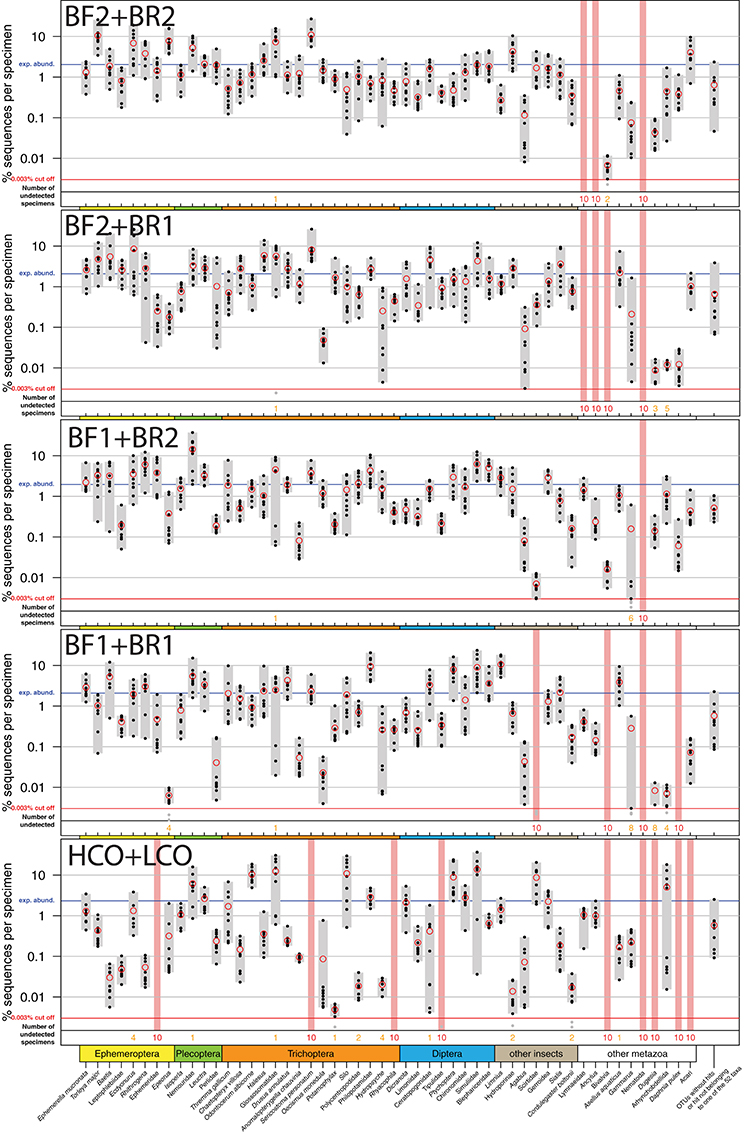
Figure 2. Comparison of the COI Folmer primer performance and the four tested newly developed primer combinations. All primer combinations were tested with the same 10 bulk samples each containing 52 morphologically distinct macroinvertebrate taxa. The 52 taxa are shown on the x-axis with the relative number of reads obtained for each morphotaxon by black dots on the logarithmic y-axis (mean read abundance indicated by red circles), for each respective primer combination. Sequence abundance was normalized across the 10 replicates and the amount of tissue used in each DNA extraction. Only OTUs with a minimum read abundance of 0.003% in at least 1 of the 10 samples were included in analyses. Number of samples for which a morphotaxon was not detected is indicated by orange and red numbers in each plot. A thick vertical line in light red indicates that a morphotaxon was not detected.
In Silico Evaluation of Primers
Performances of the 11 forward and 12 reverse primers were computationally evaluated against OTUs of all insect orders (Figure 3). Reference data for binding sites of the standard Folmer primers HCO and LCO were very limited and Megaloptera and Turbellaria had below 100 OTUs. Primer efficiencies were very similar across orders but varied slightly between primers. However, Bivalvia, Turbellaria, and Hirudinea showed higher penalty scores than other groups, while the high penalty scores for Amphipoda are likely due to the low sequence coverage and one mismatching sequence in the binding region (Figure 3). In silico and PCR (mock community samples) amplification success of BF/BR primer combinations were similar, but not always consistent. For example, while the BR1 primer showed a mean in silico amplification of only 77% (Figure 3), the BF2 + BR1 primer combination performed well with actual samples (Figure 2). In general, primers incorporating wobble bases (jgLCO1490, BF1, BF2, BR1, BR2, jgHCO2198, H2123d) or inosine (Ill_B_F, ArF5, Il_C_R, ArR5) at the 3′-end performed better than primers with no or just few wobble bases (linear regression mean penalty scores against log10 primer degeneracy: p = 0.004, adj. R2 = 0.296).
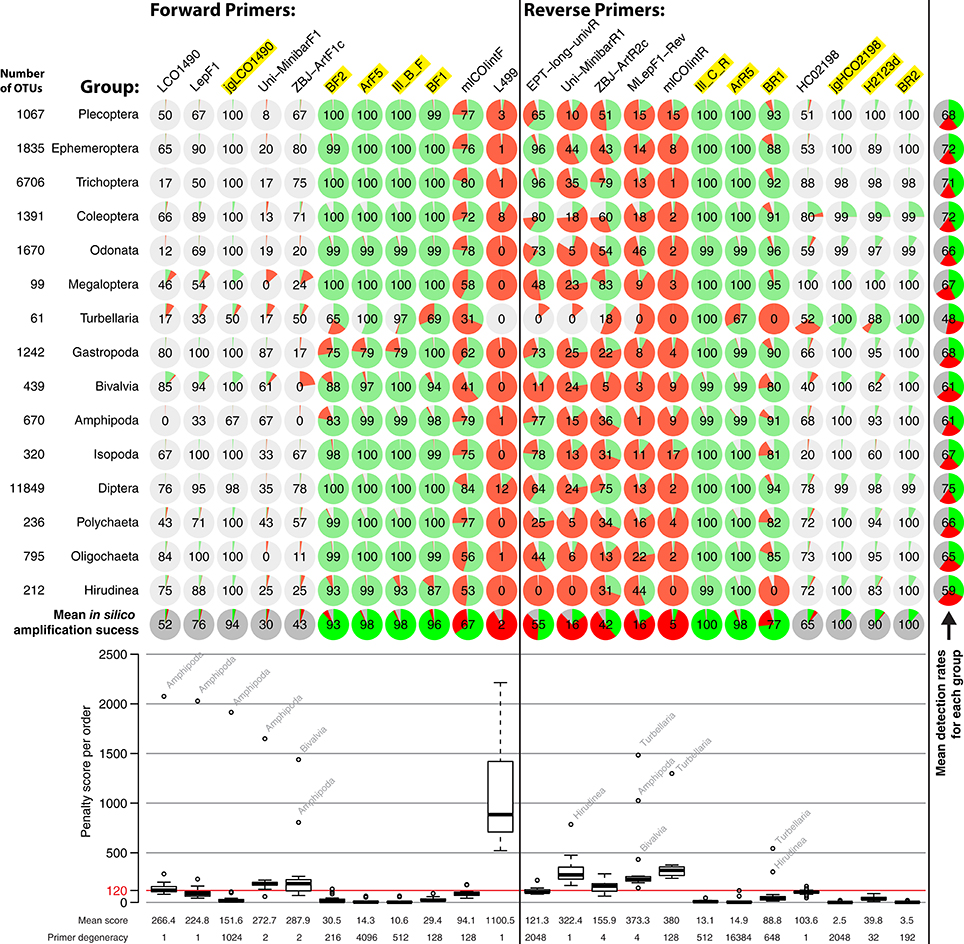
Figure 3. Overview of in silico evaluation of primer performance using PrimerMiner v0.7 with OTU data from 15 freshwater assessment relevant invertebrate groups. Primer performance is shown for each group in pie charts (red = failure, green = working, gray = missing data/gaps). Every primer sequence match with a mismatch penalty score of above 120 is considered a failure, and the amplification success displayed in each circle (excluding missing data). The box plot is based on the mean penalty scores for each group, with the mean penalty score and degeneracy given for each primer. For metabarcoding, potentially suitable primers have a yellow background. For detailed evaluation parameters, see scripts S2 in Supplementary Material. The L499 primer for the Turbellaria group could not be evaluated due to a 3 bp deletion in the reference sequences, but the primer is unlikely to amplify well.
It should be noted that some primers from the literature are not only poorly matching because they lack wobble bases, but are rather affected by additional problems (see Figure S2, “critical mismatches”). For instance, near the 3′-ends, the EPT-long-univR has a completely unnecessary second inosine at a conserved position, while the Uni-MinibarF1 has a “T” at a position where more than half of the reference OTUs have an “A.” Furthermore, the L499 primer targets a highly variable region. The mlCOIintR primer incorporates S (= C or G) leading to many mismatches (Figure S2), while the forward version of the same primer uses W (= A or T) wobble bases which match better. The reverse primers listed in the supplementary information of Gibson et al. (2014) are not written in reverse complement, and will not work if ordered as provided (we evaluated the ArR5 primer in the reverse complement in silico). Finally, certain primers show mismatches to particular groups, e.g., the ZBJ-ArtF1c primer does not match well to sequences of Bivalvia and the BR1 primer shows an unambiguous mismatch to Turbellaria and Hirudinea at the fifth position (Figure S2).
Discussion
Amplification Success of Mock Communities
Aquatic bioassessments require standardized and reliable data on biological quality elements such as macroinvertebrate communities. Metabarcoding holds the potential to assess biodiversity of freshwater ecosystems quickly and more reliably, if suitable primers are available. We used PrimerMiner to obtain freshwater invertebrate specific sequence information based on OTU sequence alignments generated from mitochondrial and COI barcodes obtained from the NCBI and BOLD databases. Using this well-balanced dataset as a reference, we developed and experimentally tested four primer sets targeting freshwater invertebrates. We deliberately decided to not factor-in nucleotide variability present in only a few groups (mostly non-insect Metazoa) to limit the degeneracy of the primers to a reasonable level.
All four BF/BR primer combinations amplified the 10 mock communities successfully, especially well for insect taxa. By factoring-in the different amplicon lengths in library pooling, preparation, we obtained similar numbers of reads for each sample. All degenerated COI primers showed superior detection rates (up to 100% of insects and 98% of all morphotaxa) and more consistent read abundances compared to the standard Folmer barcoding primers that lacked any base degeneracy (Folmer et al., 1994; Elbrecht and Leese, 2015). The primer BF2 in combination with BR1/BR2 even showed better detection rates and higher precision than a previously used primer targeting a more conserved region of the mitochondrial 16S rRNA gene, which was tested on the same communities (Elbrecht et al., 2016). An in silico analysis of the BF/BR primers against 15 freshwater groups obtained from the NCBI and BOLD databases confirmed their good detection rates (especially the BF2 + BR2 combination). However, other primer sets from the literature are also suitable for amplification of insect taxa based on our in silico testing (e.g., the primers by Geller et al., 2013; Gibson et al., 2014; Shokralla et al., 2015). Deagle and co-authors argued strongly against the use of degenerated primers in DNA metabarcoding and instead proposed the use of ribosomal markers with more conserved binding regions (Deagle et al., 2014). However, using a highly standardized approach with 10 independent taxa-rich mock communities, we clearly show that the application of highly degenerated COI primers is not only feasible but even superior to ribosomal metabarcoding of animals with respect to primer performance and available reference databases. Additionally, ribosomal markers often have limited taxonomic resolution, which is less of an issue for the COI barcoding marker (Meusnier et al., 2008; Clarke et al., 2014, 2017).
While our developed primers showed very reliable amplification results, we also identified problems associated with the primers and the metabarcoding protocol. First, while the use of fusion primers potentially decreases the chance of tag switching and reduces the laboratory work needed, it also reduces PCR efficiency substantially (Schnell et al., 2015). Primer combinations involving BF2 primers were less affected by this issue, but it was more pronounced with the BF1 primer (especially in combination with BR1). Concerns have also been raised by amplification biases associated with use of tagged primers (O'Donnell et al., 2016). While we could not directly test for this bias due to the lack of replicates we did not observe any obvious effects in our current dataset (most taxa were detected to equal proportions regardless of primer tag), there was a decrease in sequence quality when using the P5_BF1_2 primer. Whether this was a systematic effect associated with the tag of the P5_BF1_2 primer or a problem in primer synthesis/quality could not be determined from this dataset. Independently of the source of this possible bias, no effects on the number of detected taxa was observed. Further, 17% of reads from the BF2 + BR2 primer combinations were discarded due to low read quality, as the paired end reads have only a small overlap of ~35 bp. Additionally, with highly degenerated primers the specificity of the primers decreases (Deagle et al., 2014), potentially amplifying non-target regions. This effect was often minimal, with few sequences deviating from the expected length (below <0.5 % for most primer sets). These numbers were potentially inflated by PCR/sequencing errors and pseudogenes (Bensasson et al., 2001; Eren et al., 2013). More problematically, the BF1 and BF2 primers were affected by insertion/deletion (“indel”) effects making up to 40% of the sequences 1–2 bp shorter or longer at the primer binding site. The reasons for these effects, which were also observed to a lesser degree in datasets from previous studies (Elbrecht and Leese, 2015; Elbrecht et al., 2016), are unclear. It is possible that the high degeneracy of the forward primers in combination with low diversity nucleotides at the primer's 3′-end (e.g., C[cta]TT[tc]CC in BF2) makes this effect particularly pronounced. Therefore, we recommend designing primers with two unique nucleotides at the 3′-end e.g., CG and additionally considering common primer design guidelines (Kwok et al., 1994; Mülhardt, 2008; Shen et al., 2010). The effect of this minimal shifting, shortens read length by 1–2 bp while having no effect on the detection of taxa (OTUs will still match the same reference taxon, regardless of 1–2 bp being clipped from the sequence). However, when calculating OTU based biodiversity indices, the small shift might lead to a bias in these metrics due to inflated OTU numbers. While this might be solved by aligning OTU sequences and trimming them to the same length, we still advise that OTU-based diversity measures should be taken with caution when using the BF/BR primer set. Finally, we must acknowledge that the BF/BR primer sets showed poor performance on non-insect metazoans like Bivalvia, Turbellaria, Amphipoda, and Hirudinea, which are genetically distant to insects, making the development of a universal primer difficult.
While the primer sets developed and thoroughly evaluated in this study provide enhancements to existing primer resources, they are by no means perfect. While we can recommend using the BF2 + BR2 or BF2 + BR1 primer set for targeting freshwater taxa with DNA metabarcoding, we explicitly express that for routine monitoring further improved primers would be desirable. This can be archived by testing additional degenerated primer pairs or develop multiplex primer sets (targeting the same or similar regions), while the latter have the disadvantage of adding additional laboratory costs (Mülhardt, 2008; Shen et al., 2010; Hajibabaei et al., 2012; Gibson et al., 2014).
Primer Success Is Determined by Base Degeneracy and Reference Data
In silico analysis of 23 potentially suitable primers for COI DNA metabarcoding showed that high primer degeneracy leads to the best amplification of freshwater and insect taxa. We verified this also experimentally with the tested macroinvertebrate mock communities that showed high primer bias with standard Folmer primers (Elbrecht and Leese, 2015) but a very consistent amplification with higher detection rates with the BF/BR primers developed in this study. It is possible that other primers (Gibson et al., 2014; Shokralla et al., 2015) may lead to equally good amplification. However, a lack of degeneracy can lead to substantial bias in many of the other evaluated primers. These biases might not strongly affect PCR for DNA barcoding on single organisms, but they may substantially skew detection rates of complex multispecies bulk samples and lead to taxa remaining undetected (Piñol et al., 2014; Elbrecht and Leese, 2015). For example, the mlCOIint primers which have a maximum degeneracy of two nucleotides at each position (Leray et al., 2013), were previously tested with two mock communities and up to 35% of taxa remained undetected (Leray and Knowlton, 2015). Probably even more problematic are primers that lack base degeneracy. Despite primer bias associated with the high variation of the COI gene having been well-documented (Clarke et al., 2014; Deagle et al., 2014; Piñol et al., 2014; Sharma and Kobayashi, 2014; Elbrecht and Leese, 2015), primers without base degeneracy like ZBJ-Art by Zeale et al. (2011) are widely used e.g., for gut content analysis (153 citations as of March 2017). It is critical therefore that degenerate primers optimized for the ecosystems and organism groups under study are employed. If using primers derived from the literature, these should be tested a priori to investigate if they are suitable for the planned metabarcoding project.
We also demonstrated that several popular primers from the literature contain critical design flaws, possibly introduced by accident (e.g., EPT-long-univR, mlCOIintR, Uni-MinibarF1). It has to be kept in mind that a typographical error, or just one mismatching base at the 3′-end can make or break a primer (Stadhouders et al., 2010; Piñol et al., 2014). Additionally, primers are often developed on a small set of taxa, and thus might not work well for the ecosystem, geographic region or taxa under study. For example, Clarke and co-authors evaluated the L499 + H2123d as a metabarcoding primer (Clarke et al., 2014), but it was originally only developed to target tephritid fruit flies and probably was never intended to be used beyond this dipteran family (Van Houdt et al., 2010). Therefore, careful in silico evaluation and mock community testing of newly developed primers or primers from the literature against the specific taxa of interest is crucial for metabarcoding projects. We highly recommend evaluation primers not only in silico but also using mock communities of known composition, to validate that the primers work well for the targeted groups and purpose. Unfortunately, resources are limited and metabarcoding primers are not always tested and validated before being used in larger scale ecological or monitoring studies.
Recommended Approaches for Freshwater Bio-Assessment Using Macroinvertebrates
The success of DNA metabarcoding for bioassessment and specific environmental impact assessment of freshwater ecosystems depends on well-designed primers that reliably amplify the target communities. The more conserved primer binding regions, the greater the amplification efficiency (Deagle et al., 2014). Therefore, 18S and 16S ribosomal markers have been proposed as suitable alternative markers to the COI gene, despite lacking comprehensive reference databases for animal taxa and potential limitations in taxonomic resolution (Clarke et al., 2014; Deagle et al., 2014; Elbrecht et al., 2016). However, the in silico evaluations and documented good performance of the BF2 + BR1 and BF2 + BR2 primer sets of the COI gene shown in this study suggest clearly that ribosomal markers are not necessary for reliable DNA metabarcoding on animal species tested here (see also Clarke et al., 2017). The COI marker can lead to equally good results or better detection rates (Elbrecht et al., 2016; Clarke et al., 2017), but already has large reference databases available for animals. Therefore, we strongly encourage focusing efforts on developing optimized ecosystem or community-specific COI primers.
When using DNA metabarcoding approaches for bioassessment, protocols from the literature should be critically evaluated as success may be flawed by unsuitable primer design. Additionally, we recommend that replicates are included to reduce the chance of tag switching and exclude false OTUs from the dataset (Lange et al., 2015). While we have previously encouraged the use of fusion primers due to their ease of use (single step PCR, Elbrecht and Leese, 2015), we have to acknowledge that they decrease PCR efficiency (Schnell et al., 2015). Additionally, environmental samples often contain PCR inhibitors, further decreasing amplification efficiency. In these cases, two step PCR which is the recommended approach by Illumina (e.g., Miya et al., 2015; Carvalho et al., 2017) might lead to more reliable amplification results, even though two step PCR can be more prone to tag switching (Esling et al., 2015; Schnell et al., 2015).
Besides metabarcoding, metagenomic approaches using enrichment for mitochondrial genomes may also become suitable for bio-assessment, with potentially less bias as the PCR amplification step can be omitted (Liu et al., 2016). However, as briefly discussed in Elbrecht et al. (2016), metagenomic methods have to be further validated and mitochondrial reference genome libraries ideally need to be completed (Dowle et al., 2015; Papadopoulou et al., 2015).
Thus, DNA metabarcoding using the COI marker for DNA based monitoring of stream ecosystems, is currently the most cost-effective approach for reliable bulk sample assessment. However, primers for DNA metabarcoding of macroinvertebrates ideally need to be further optimized and primers from the literature should be tested more extensively on mock communities.
Conclusions
Reliable and quick bioassessments are of critical importance for biomonitoring and environmental impact assessment of aquatic ecosystems. DNA metabarcoding has the potential to meet this challenge if suitable primers can be obtained. Through computational evaluations as well as experimental data, we showed that almost the entire aquatic macroinvertebrate community can be reliably detected with COI metabarcoding. We provide novel degenerated primer sets with high detection rates and greatly reduced primer bias. As databases are still incomplete, we encourage further such in silico and in vivo evaluations of existing primers as well as the development of improved metabarcoding primers to unlock the full potential of metabarcoding for bioassessment. However, our data already suggests that for freshwater ecosystems, DNA metabarcoding is ready to complement biomonitoring programs on a large scale.
Ethics Statement
DNA from invertebrates from a previous project was used in this study. Permission for collection of invertebrates was granted, see previous project; doi: 10.1371/journal.pone.0130324.
Author Contributions
VE and FL conceived the ideas and designed methodology, VE carried out the laboratory work and analyzed the data, VE led the writing of the manuscript. All authors contributed critically to the drafts and gave final approval for publication.
Funding
FL and VE were supported by a grant of the Kurt Eberhard Bode Foundation to FL.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Bianca Peinert for her help with primer development, and Edward Wilcox and ScienceExchange for HiSeq sequencing. We would like to thank Edith Vamos, Jan Macher, and Vera Zizka, Romana Salis for their helpful suggestions that improved this manuscript. Simon Creer and two additional reviewers provided helpful comments that greatly improved the quality of this manuscript. A preprint of this manuscript is available at PeerJ PrePrints: https://peerj.com/preprints/2044/.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fenvs.2017.00011/full#supplementary-material
References
Bensasson, D., Zhang, D. X., Hartl, D. L., and Hewitt, G. M. (2001). Mitochondrial pseudogenes: evolution's misplaced witnesses. Trends Ecol. Evol. 16, 314–321. doi: 10.1016/S0169-5347(01)02151-6
Brandon-Mong, G. J., Gan, H. M., Sing, K. W., Lee, P. S., Lim, P. E., and Wilson, J. J. (2015). DNA metabarcoding of insects and allies: an evaluation of primers and pipelines. Bull. Entomol. Res. 105, 717–727. doi: 10.1017/S0007485315000681
Cameron, S., Rubinoff, D., and Will, K. (2006). Who will actually use DNA barcoding and what will it cost? Syst. Biol. 55, 844–847. doi: 10.1080/10635150600960079
Carew, M. E., Pettigrove, V. J., Metzeling, L., and Hoffmann, A. A. (2013). Environmental monitoring using next generation sequencing: rapid identification of macroinvertebrate bioindicator species. Front. Zool. 10:45. doi: 10.1186/1742-9994-10-45
Carvalho, G. R., Walsh, K., Seymour, M., Hajibabaei, M., Lallias, D., Christmas, M., et al. (2017). Annual time-series analysis of aqueous eDNA reveals ecologically relevant dynamics of lake ecosystem biodiversity. Nat. Commun. 8, 1–11. doi: 10.1038/ncomms14087
Clarke, L. J., Beard, J. M., Swadling, K. M., and Deagle, B. E. (2017). Effect of marker choice and thermal cycling protocol on zooplankton DNA metabarcoding studies. Ecol. Evol. 7, 873–883. doi: 10.1002/ece3.2667
Clarke, L. J., Soubrier, J., Weyrich, L. S., and Cooper, A. (2014). Environmental metabarcodes for insects: in silicoPCR reveals potential for taxonomic bias. Mol. Ecol. Resour. 14, 1160–1170. doi: 10.1111/1755-0998.12265
Deagle, B. E., Jarman, S. N., Coissac, E., Pompanon, F., and Taberlet, P. (2014). DNA metabarcoding and the cytochrome c oxidase subunit I marker: not a perfect match. Biol. Lett. 10:20140562. doi: 10.1098/rsbl.2014.0562
Dowle, E. J., Pochon, X., and Banks, J. C. (2015). Targeted gene enrichment and high-throughput sequencing for environmental biomonitoring: a case study using freshwater macroinvertebrates. Mol. Ecol. Resour. 16, 1240–1254. doi: 10.1111/1755-0998.12488
Dudgeon, D., Arthrington, A. H., Gessner, M. O., Kawabata, Z.-I., Knowler, D. J., Lévêque, C., et al. (2005). Freshwater biodiversity: importance, threats, status and conservation challenges. Biol. Rev. 81, 163. doi: 10.1017/S1464793105006950
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., and Flyvbjerg, H. (2015). Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 31, 3476–3482. doi: 10.1093/bioinformatics/btv401
Elbrecht, V., and Leese, F. (2015). Can DNA-based ecosystem assessments quantify species abundance? Testing primer bias and biomass—sequence relationships with an innovative metabarcoding protocol. PLoS ONE 10:e0130324. doi: 10.1371/journal.pone.0130324
Elbrecht, V., and Leese, F. (2016). PrimerMiner: an R package for development and in silico validation of DNA metabarcoding primers. PeerJ. doi: 10.7287/peerj.preprints.2352v1. [Epub ahead of print].
Elbrecht, V., Taberlet, P., Dejean, T., Valentini, A., Usseglio-Polatera, P., Beisel, J.-N., et al. (2016). Testing the potential of a ribosomal 16S marker for DNA metabarcoding of insects. PeerJ 4, e1966–e1912. doi: 10.7717/peerj.1966
Eren, A. M., Vineis, J. H., Morrison, H. G., and Sogin, M. L. (2013). A filtering method to generate high quality short reads using illumina paired-end technology. PLoS ONE 8:e66643. doi: 10.1371/journal.pone.0066643.s003
Esling, P., Lejzerowicz, F., and Pawlowski, J. (2015). Accurate multiplexing and filtering for high-throughput amplicon-sequencing. Nucleic Acids Res. 43, 2513–2524. doi: 10.1093/nar/gkv107
Folmer, O., Black, M., Hoeh, W., Lutz, R., and Vrijenhoek, R. (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299.
Geller, J., Meyer, C., Parker, M., and Hawk, H. (2013). Redesign of PCR primers for mitochondrial cytochrome coxidase subunit I for marine invertebrates and application in all-taxa biotic surveys. Mol. Ecol. Resour. 13, 851–861. doi: 10.1111/1755-0998.12138
Gibson, J., Shokralla, S., Porter, T. M., King, I., van Konynenburg, S., Janzen, D. H., et al. (2014). Simultaneous assessment of the macrobiome and microbiome in a bulk sample of tropical arthropods through DNA metasystematics. Proc. Natl. Acad. Sci.U.S.A. 111, 8007–8012. doi: 10.1073/pnas.1406468111
Haase, P., Pauls, S. U., Schindehütte, K., and Sundermann, A. (2010). First audit of macroinvertebrate samples from an EU Water Framework Directive monitoring program: human error greatly lowers precision of assessment results. J. North Am. Benthol. Soc. 29, 1279–1291. doi: 10.1899/09-183.1
Hajibabaei, M., Shokralla, S., Zhou, X., Singer, G., and Baird, D. J. (2011). Environmental barcoding: a next-generation sequencing approach for biomonitoring applications using River Benthos. PLoS ONE 6:e17497. doi: 10.1371/journal.pone.0017497
Hajibabaei, M., Spall, J. L., Shokralla, S., and van Konynenburg, S. (2012). Assessing biodiversity of a freshwater benthic macroinvertebrate community through non-destructive environmental barcoding of DNA from preservative ethanol. BMC Ecol. 12:28. doi: 10.1186/1472-6785-12-28
Hebert, P. D. N., Penton, E. H., Burns, J. M., Janzen, D. H., and Hallwachs, W. (2004). Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. U.S.A. 101, 14812–14817. doi: 10.1073/pnas.0406166101
Katoh, K., Misawa, K., Kuma, K.-I., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kwok, S., Chang, S. Y., Sninsky, J. J., and Wang, A. (1994). A guide to the design and use of mismatched and degenerate primers. PCR Methods Appl. 3, S39–S47.
Lange, A., Jost, S., Heider, D., Bock, C., Budeus, B., Schilling, E., et al. (2015). AmpliconDuo: a split-sample filtering protocol for high-throughput amplicon sequencing of microbial communities. PLoS ONE 10:e0141590. doi: 10.1371/journal.pone.0141590
Leray, M., and Knowlton, N. (2015). DNA barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. Proc. Natl. Acad. Sci. U.S.A. 112, 2076–2081. doi: 10.1073/pnas.1424997112
Leray, M., Yang, J. Y., Meyer, C. P., Mills, S. C., Agudelo, N., Ranwez, V., et al. (2013). A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents. Front. Zool. 10:34. doi: 10.1186/1742-9994-10-34
Liu, S., Wang, X., Xie, L., Tan, M., Li, Z., Su, X., et al. (2016). Mitochondrial capture enriches mito-DNA 100 fold, enabling PCR-free mitogenomics biodiversity analysis. Mol. Ecol. Resour. 16, 470–479. doi: 10.1111/1755-0998.12472
Macher, J. N., Salis, R. K., Blakemore, K. S., and Tollrian, R. (2016). Multiple-stressor effects on stream invertebrates: DNA barcoding reveals contrasting responses of cryptic mayfly species. Ecol. Indic. 61, 159–169. doi: 10.1016/j.ecolind.2015.08.024
Meusnier, I., Singer, G. A., Landry, J.-F., Hickey, D. A., Hebert, P. D., and Hajibabaei, M. (2008). A universal DNA mini-barcode for biodiversity analysis. BMC Genomics 9:214. doi: 10.1186/1471-2164-9-214
Miya, M., Sato, Y., Fukunaga, T., Sado, T., Poulsen, J. Y., Sato, K., et al. (2015). MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: detection of more than 230 subtropical marine species. R. Soc. Open Sci. 2, 150088. doi: 10.1098/rsos.150088
O'Donnell, J. L., Kelly, R. P., Lowell, N. C., and Port, J. A. (2016). Indexed PCR primers induce template-specific bias in large-scale DNA sequencing studies. PLoS ONE 11:e0148698. doi: 10.1371/journal.pone.0148698
Papadopoulou, A., Taberlet, P., and Zinger, L. (2015). Metagenome skimming for phylogenetic community ecology: a new era in biodiversity research. Mol. Ecol. 24, 3515–3517. doi: 10.1093/molbev/msv111
Piñol, J., Mir, G., Gomez-Polo, P., and Agustí, N. (2014). Universal and blocking primer mismatches limit the use of high-throughput DNA sequencing for the quantitative metabarcoding of arthropods. Mol. Ecol. Resour. 15, 1–12. doi: 10.1111/1755-0998.12355
Ratnasingham, S., and Hebert, P. (2007). BOLD: the Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 7, 355–364. doi: 10.1111/j.1471-8286.2006.01678.x
Ratnasingham, S., and Hebert, P. D. N. (2013). A DNA-based registry for all animal species: the Barcode Index Number (BIN) system. PLoS ONE 8:e66213. doi: 10.1371/journal.pone.0066213
Schnell, I. B., Bohmann, K., and Gilbert, M. T. P. (2015). Tag jumps illuminated-reducing sequence-to-sample misidentifications in metabarcoding studies. Mol. Ecol. Resour. 15, 1289–1303. doi: 10.1111/1755-0998.12402
Sharma, P., and Kobayashi, T. (2014). Are “universal” DNA primers really universal? J. Appl. Genet. 55, 485–496. doi: 10.1007/s13353-014-0218-9
Shen, Z., Qu, W., Wang, W., Lu, Y., Wu, Y., Li, Z., et al. (2010). MPprimer: a program for reliable multiplex PCR primer design. BMC Bioinformatics 11:143. doi: 10.1186/1471-2105-11-143
Shokralla, S., Porter, T. M., Gibson, J. F., Dobosz, R., Janzen, D. H., Hallwachs, W., et al. (2015). Massively parallel multiplex DNA sequencing for specimen identification using an Illumina MiSeq platform. Sci. Rep. 5:9687. doi: 10.1038/srep09687
Shokralla, S., Zhou, X., Janzen, D. H., Hallwachs, W., Landry, J.-F., Jacobus, L. M., et al. (2011). Pyrosequencing for mini-barcoding of fresh and old museum specimens. PLoS ONE 6:e21252. doi: 10.1371/journal.pone.0021252.s002
Stadhouders, R., Pas, S. D., Anber, J., Voermans, J., Mes, T. H. M., and Schutten, M. (2010). The effect of primer-template mismatches on the detection and quantification of nucleic acids using the 5′ nuclease assay. J. Mol. Diagn. 12, 109–117. doi: 10.2353/jmoldx.2010.090035
Stein, E. D., Martinez, M. C., Stiles, S., Miller, P. E., and Zakharov, E. V. (2014). Is DNA barcoding actually cheaper and faster than traditional morphological methods: results from a survey of freshwater bioassessment efforts in the United States? PLoS ONE 9:e95525. doi: 10.1371/journal.pone.0095525.t005
Stein, E. D., White, B. P., Mazor, R. D., Jackson, J. K., and Battle, J. M. (2013). Does DNA barcoding improve performance of traditional stream bioassessment metrics? Freshw. Sci. 33, 302–311. doi: 10.1086/674782
Sweeney, B. W., Battle, J. M., Jackson, J. K., and Dapkey, T. (2011). Can DNA barcodes of stream macroinvertebrates improve descriptions of community structure and water quality? J. North Am. Benthol. Soc. 30, 195–216. doi: 10.1899/10-016.1
Taberlet, P., Coissac, E., Hajibabaei, M., and Rieseberg, L. H. (2012). Environmental DNA. Mol. Ecol. 21, 1789–1793. doi: 10.1111/j.1365-294X.2012.05542.x
Van Houdt, J. K. J., Breman, F. C., Virgilio, M., and De Meyer, M. (2010). Recovering full DNA barcodes from natural history collections of Tephritid fruitflies (Tephritidae, Diptera) using mini barcodes. Mol. Ecol. Resour. 10, 459–465. doi: 10.1111/j.1755-0998.2009.02800.x
Vörösmarty, C. J., McIntyre, P. B., Gessner, M. O., Dudgeon, D., Prusevich, A., Green, P., et al. (2010). Global threats to human water security and river biodiversity. Nature 467, 555–561. doi: 10.1038/nature09440
Keywords: DNA barcoding, primer development, primer evaluation, primer bias, ecosystem assessment, in silico PCR, invertebrates
Citation: Elbrecht V and Leese F (2017) Validation and Development of COI Metabarcoding Primers for Freshwater Macroinvertebrate Bioassessment. Front. Environ. Sci. 5:11. doi: 10.3389/fenvs.2017.00011
Received: 15 October 2016; Accepted: 21 March 2017;
Published: 10 April 2017.
Edited by:
Michael M. Douglas, University of Western Australia, AustraliaCopyright © 2017 Elbrecht and Leese. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vasco Elbrecht, Vasco.Elbrecht@uni-due.de