A practical guide to identifying members of the Bemisia tabaci species complex: and other morphologically identical species
 Laura M. Boykin1*
Laura M. Boykin1*  Paul J. De Barro2
Paul J. De Barro2- 1ARC Centre of Excellence in Plant Energy Biology and School of Chemistry and Biochemistry, The University of Western Australia, Crawley, WA, Australia
- 2CSIRO Biosecurity Flagship, Brisbane, QLD, Australia
Members of the Bemisia tabaci species complex (whiteflies) are a considerable threat to a broad range of agriculture and horticulture food and fiber crops. There are hundreds of papers a year published on the members of B. tabaci species complex, many failing to either correctly identify the species involved or confusing identity. Correct identification is a crucial first step in any study, yet all too often, especially in cases where the primary focus of the study is plant pathology, is overlooked. The whitefly research community has struggled for years with common terminology and consistent methods for species identification of various members of the complex due to (1) the lack of a reliable global genetic resource and (2) the complexities of the phylogenetic methods needed to identify unknown individuals correctly. The goal of this paper are to provide a practical guide for identifying unknown whiteflies using a global curated dataset of mitochondrial COI that is freely available at http://dx.doi.org/10.4225/08/50EB54B6F1042. The methods and resources outlined here can be readily extended to other species that are morphologically indistinguishable.
Introduction
Members of the Bemisia tabaci species complex (whiteflies) cause considerable damage to food and fiber crops globally (Pimental et al., 2005) and are named in International Union for the Conservation of Nature and Natural Resources (IUCN) list (http://www.issg.org) of “100 of the World's Worst Invasive Alien Species” [http://www.issg.org/database/species/search.asp?st=100ss]. They are regarded as a regulated species by Australia, Africa, China, the EU, and the USA. There are two main types of damage caused by B. tabaci. The first is caused by the direct feeding of both immature and adult stages. The second is indirect damage through the excretion of honeydew onto the surfaces of leaves and fruit. This acts as a substrate for the growth of sooty mold fungi which interferes with photosynthesis and reduces the quality of fruit and fiber (Byrne and Bellows, 1991). In addition, the species complex contains vectors of hundreds of economically important plant viral-pathogens, most being begomoviruses (Geminiviridae), a group recognized as the most important emerging plant virus group in subtropical and tropical world regions (Maruthi et al., 2002, 2007; Sseruwagi et al., 2006; Navas-Castillo et al., 2011; Liu et al., 2013; Polston et al., 2014).
In 2007, a mitochondrial COI (mtCOI) dataset for B. tabaci was generated from data in GenBank and used to reconstruct the global phylogenetic relationships of the B. tabaci species complex (Boykin et al., 2007). This was the beginning of the curated dataset that has subsequently (Dinsdale et al., 2010; De Barro et al., 2011) been refined to determine B. tabaci is a species complex composed of at least 34 morphologically indistinguishable species (Boykin et al., 2012b, 2013; Boykin, 2014). A critical point here is that validation of the dataset showed that not just one, but five separate delimitation metrics agreed on the presence of species level bounds (Boykin et al., 2012b). Beyond the molecular data, all mating compatibility studies have shown either complete or partial mating isolation, the latter leading to substantial declines in fitness of F1 and F2 (Elbaz et al., 2010; Wang et al., 2010, 2011; Xu et al., 2010; Liu et al., 2011; Sun et al., 2011).
Despite the genomic revolution there still remains a need for single gene based species identification especially for morphologically identical species where rapid identification is required. There are hundreds of scientists working on the various aspects of the B. tabaci species complex (Boykin, 2014), but species identification is slowing down progress on other important research areas. There has been a rapid increase in B. tabaci studies over the last five years, clearly indicating the need to develop a robust protocol for species identification. The reasons proper phylogenetic methods are not utilized include (1) lack of understanding of the complexities of phylogenetic methods, (2) lack of knowledge as to what mtCOI sequences to include or (3) lack of computer resources.
The most common mistakes in the B. tabaci species literature are (1) relying on neighbor-joining trees to infer evolutionary relationships, (2) arbitrary measures of percentage difference between mtCOI sequences to delimit species, (3) limited taxon sampling, (4) distantly related outgroups and (5) lack of quality control of mtCOI data. The negative impact of these on molecular phylogenetic reconstruction have been well addressed (Susko et al., 2004; Heath et al., 2008; Boykin et al., 2010; Yang and Rannala, 2012); instead we focus on providing a way forward.
Here we provide a practical guide for species identification of members in the B. tabaci complex. We have outlined curation of the global mtCOI dataset from 2007-present and also provide a step-by-step guide as to how to identify recently sequenced samples (unknowns) utilizing best practices in phylogenetics and species delimitation. The approaches outlined here will also be applicable to other morphologically indistinguishable species.
Methods for Identifying B. tabaci Species
DNA Extraction and PCR Amplification
The genetic region used to identify species in the B. tabaci species complex is the 3′ end of the mtCOI gene. There are two sets of primers commonly used 1) C1-J-2195 and TL2-N-3014 (Simon et al., 1994) and Btab-Uni primers (Shatters et al., 2009). The PCR conditions are: a 30 μl PCR reactions heated at 94°C for 2 min followed by 35 cycles of 30 s at 94°C denaturation, 30 s at 53°C annealing, 1 min at 72°C extension and a final extension of 72°C for 10 min in a MJ Research PTC-200 Peltier thermal cycler. The PCR reactions are composed of 27 μL Platinum PCR SuperMix (Invitrogen, catalog number 11306-016), 1 μL forward primer (10 ρ mol), 1 μL reverse primer (10 ρ mol), and 1 μL DNA template. Prior to sequencing, the amplified products were cleaned using the montage PCR filter units from Millipore (catalog number UFC7PCR50). It is important for the whitefly community to continue using this region of mtCOI to avoid diversion of resources and to build on the informatics power of the historical data that is already publicly available (Boykin et al., 2012a). Publicly available data has been abstracted, curated, and placed in a database that can be found at: http://dx.doi.org/10.4225/08/50EB54B6F1042 (supported through the Commonwealth Scientific and Industrial Research Organisation, Australia).
Naming Sequences
Prior to conducting phylogenetic analyses, new sequences should be named in a format that will easily integrate into the global dataset. The recommended format is: Species_Country_GenBank Accession number_host. Prior to identification analyses, the species will be unknown, it is recommended to use UNKNOWN_country_GenBank Accession Number_host. After the analyses described below, replace the UNKNOWN with the species identified in the global phylogeny.
Taxon Sampling and the Utility of the Global Dataset
All 34 putative species in the B. tabaci complex and closely related sister taxa must be included in the systematic investigation of the B. tabaci species. B. tabaci has a global distribution and removing the other members (to focus on one geographic region) of the complex will mislead the investigation due to incomplete taxon sampling (Boykin et al., 2007). Taxon sampling is crucial for phylogenetic systematics and much of the early confusion in B. tabaci systematics was due to incomplete taxon sampling. In addition, accuracy of inferences about evolutionary processes obtained from phylogenetic analyses improved significantly through taxon sampling (Heath et al., 2008; Boykin et al., 2013). The global dataset is designed strictly as a repository of unique haplotypes to aid in species identification by having a large proportion of the known diversity available in the one place; it is not designed to track global distribution. All sequences in the dataset are originally lodged in GenBank and have accession numbers. Sequences have been checked for gaps, provenance, pseudogenes and ambiguous bases are <0.8%; as such complies with many of the data quality elements of DNA barcoding. Sequence length is 657 bases. The dataset also contains 21 outgroup species Aleurocanthus camelliae, Aleurocanthus spiniferus, Aleurochiton aceris, Aleurodicus disperses, Aleurodicus dugesii, Aleurotrachelus camelliae, Bemisia afer, Bemisia atriplex, Bemisia berbericola, Bemisia emiliae, Bemisia on Rhagodia parabolic, Bemisia subdecipiens, Bemisia tuberculata, Neomaskellia andropogonis, Tetraleurodes acacia, Trialeurodes abutilonea, Trialeurodes lauri, Trialeurodes ricini, Trialeurodes vaporariorum, Trialeurodes ricini, Vasdavidius concursus. The model of molecular evolution for the global data set is GTR + I + G.
Sequence Analyses- Option 1- Geneious
Figure 1 shows three options for phylogenetically identifying members of the B. tabaci species complex. We recommend that analysis be done using Geneious (http://www.geneious.com/) because it is user friendly and contains all necessary programs for alignment, model selection and phylogenetic analyses. Geneious is available with a 14 day free trial period. It requires a license. Geneious offers student, non-commercial and commercial licenses and special prices are available for academics based in countries with annual GDP <$USD 2000 per capita. Within Geneious the alignment programs MAFFT (Katoh and Toh, 2008), Muscle (Edgar, 2004) and ClustalX (Thompson et al., 1997) are available. In addition, the phylogenetic programs RaxML (Stamatakis, 2006), GARLI (Brauer et al., 2002), PhyML (Guindon et al., 2010), and MrBayes (Ronquist et al., 2012) are implemented in a user friendly format.
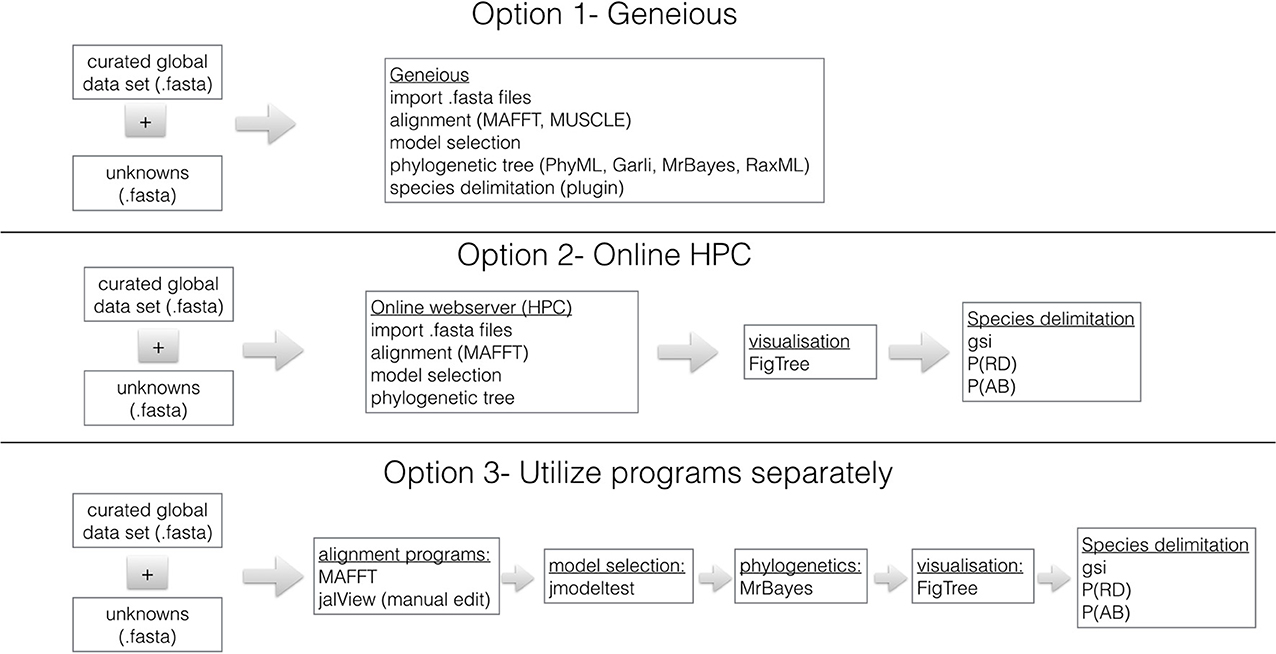
Figure 1. Three work-flows utilizing genomic data and commonly used sequence manipulation software to identify B. tabaci species. The curated mtCOI dataset for B. tabaci species is found: http://dx.doi.org/10.4225/08/50EB54B6F1042. The species delimitation plugin (Masters et al., 2011) for Geneious (Drummond et al., 2010) is used to calculate Rosenberg's reciprocal monophyly, P(AB) (Rosenberg, 2007) and Rodrigo's P(RD) (Rodrigo et al., 2008) measures. The genealogical sorting index (gsi) (Cummings et al., 2008) statistic is calculated in R based on the estimated tree and the assignment file that contains user specified groups (see http://www.genealogicalsorting.org/). High Performance Computing (HPC) resources are available at: http://www.phylo.org/portal2/, http://www.phylogeny.fr/version2_cgi/index.cgi and http://www.ebi.ac.uk/services
Sequence Analyses- Option 2- Online HPC Resources
Depending on the specifications of your computer, it might be necessary to conduct the analyses utilizing online high performance computing (HPC) resources. The Cyberinfrastructure for Phylogenetic Research (CIPRES) Science Gateway (Miller et al., 2010) is a portal to conduct all aspects of phylogenetic analyses using high performance computing (HPC) facilities. Users can create a login at: http://www.phylo.org/portal2/ and all necessary programs for phylogenetic analyses are available. In addition, Phylogeny.fr (http://www.phylogeny.fr/version2_cgi/index.cgi) is available for Robust Phylogenetic Analysis For The Non-Specialist (Dereeper et al., 2008), which contains multiple sequence alignment and phylogenetics programs as well as tree visualization software. Finally, a resource for sequence manipulation is available from the European Bioinformatics Institute, which is part of the European Molecular Biology Laboratory (EMBL-EBI): http://www.ebi.ac.uk/services however, it has limited phylogenetic tree reconstruction resources.
Sequence Analyses - Option 3- Utilize Programs Separately
All the necessary programs needed for a robust phylogenetic analysis of the B. tabaci species and subsequent species delimitation are available freely from independent sources. The first step is to download the global dataset (see above) and align unknowns using MAFFT (Katoh and Toh, 2008). To verify the quality of the data the sequences are translated to make sure there are no stop codons or inadvertent frame shifts using JalView (Waterhouse et al., 2009). The model of molecular evolution should be determined using jModeltest (Posada, 2009) and the phylogenetic method of choice is a Bayesian approach using MrBayes (Ronquist et al., 2012). To assess the convergence of the Bayesian runs utilize Tracer (Rambaut and Drummond, 2010) and visualize the.con.tre file in FigTree (Rambaut, 2012). Further species delimitation analyses maybe necessary and should be done following the methods of Boykin et al. (2012b) references and therein.
Species Identification
The global dataset is curated to include the names of the species as the first part of the sequence title, this facilitates the identification of unknowns when all sequences in the dataset are included in the phylogenetic analyses. Assignment of unknown sequences is based on phylogenetic placement of the unknown in an already defined species, thus, emphasizing the need to include all sequences in the global data set. There is the possibility that the unknown will not be assignable to an already identified species clade i.e., the sequence divergence exceeds the delimitation bounds, when this occurs, species delimitation tests are needed and have been outlined in Boykin et al. (2012b). Researchers need to name species in accordance with established nomenclature. This consistency will ensure ease of communication and help minimize confusion over identity.
Online Phylogenetic Resources
Beyond the phylogenetic programs listed in Figure 1, there are online resources for phylogenetic species delimitation including an online discussion board- phylobabble (http://phylobabble.org) and the evolutionary directory, evoldir, http://evol.mcmaster.ca/evoldir.html. The most comprehensive list of software pertaining to genetic analyses is maintained by Professor Joe Felsenstein and found at: http://evolution.genetics.washington.edu/phylip/software.html
Workshops
There are several workshops devoted to phylogenetics and phylogenomics most notably (1) Woods Hole's Workshop on Molecular Evolution: https://molevol.mbl.edu/ which contains a list of relevant software https://molevol.mbl.edu/index.php/Software and accompanying lectures and (2) Bodega Bay: http://bodegaphylo.wikispot.org/Front_Page. Finally, there is an online seminar series on phylogenetics (http://phyloseminar.org) and past lectures are recorded and available to view.
Concluding Remarks and Future Directions
There are many different options for phylogenetic analyses (Yang and Rannala, 2012) and species delimitation (Rannala and Yang, 2013) beyond the B. tabaci literature. The purpose of this paper is not to review these methods, but to highlight three practical options to assist whitefly researchers in species identification and species delimitation. Program manuals need to be read and understood to carry out the analyses correctly, but we have provided a starting point and a roadmap for future B. tabaci species identification. In addition, we have outlined the curation of the global mtCOI global data set for the B. tabaci species that has been used in previous studies for species identification and delimitation for the complex (De Barro et al., 2011; Boykin et al., 2012b, 2013; Tay et al., 2012). Genomic data will undoubtedly provide insight into the many aspects of B. tabaci, but there will be an ongoing need for rapid identification of these highly invasive species. Current analysis shows that full mitogenome sequence comparison provides no better identification of species than mtCOI alone (Wang et al., 2013).
Also, there is an increasing effort to use the 5′ barcoding region of mtCOI to identify B. tabaci (http://dx.doi.org/10.5883/BOLD:AAM1245). Historically, 5′ DNA barcoding began in 2003, whereas the use of 3′ mtCOI to analyse B. tabaci was first published in 1999 (Frohlich et al., 1999). Based on the analysis of the full mitogenome, the use of the 5′ end of mtCOI provides no better means of identifying species than the 3′ end. It is however less useful than the 3′ end as it prevents comparison against the many 1000s of mtCOI sequences already available. Rapid identification is most easily done using the extensive historical collection of mtCOI data that has been used for the past 25 years to identify species in the B. tabaci species complex. In fact, if you are solely interested in B. tabaci species identification, 657 bases of 3′ end sequence of mtCOI appears to be sufficient.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Laura M. Boykin would like to thank participants of the BMGF Agricultural Research Connection Workshop (2013 and 2014) in Naivasha, Kenya for inspiration in writing this practical guide for species identification and Dr. Ian Small ARC CoE PEB for continued support. Dr. Lava Kumar [International Institute of Tropical Agriculture (IITA), Nigeria] and two anonymous reviewers provided comments that greatly improved the manuscript.
References
Boykin, L. M. (2014). Bemisia tabaci nomenclature: lessons learned. Pest Manag. Sci. doi: 10.1002/ps.3709. [Epub ahead of print].
Boykin, L. M., Armstrong, K., Kubatko, L., and De Barro, P. (2012a). DNA barcoding invasive insects: database roadblocks. Invertebr. Syst. 26, 506–514. doi: 10.1071/IS12025
Boykin, L. M., Armstrong, K. F., Kubatko, L., and De Barro, P. (2012b). Species delimitation and global biosecurity. Evol. Bioinform. 8, 1–37. doi: 10.4137/EBO.S8532
Boykin, L. M., Bell, C. D., Evans, G., Small, I., and De Barro, P. J. (2013). Is agriculture driving the diversification of the Bemisia tabaci species complex (Hemiptera: Sternorrhyncha: Aleyrodidae)?: dating, diversification and biogeographic evidence revealed. BMC Evol. Biol. 13:228. doi: 10.1186/1471-2148-13-228
Boykin, L. M., Kubatko, L. S., and Lowrey, T. K. (2010). Comparison of methods for rooting phylogenetic trees: a case study using Orcuttieae (Poaceae: Chloridoideae). Mol. Phylogenet. Evol. 54, 687–700. doi: 10.1016/j.ympev.2009.11.016
Boykin, L. M., Shatters, R. G. Jr. Rosell, R. C., Mckenzie, C. L., Bagnall, R. A., De Barro, P., et al. (2007). Global relationships of Bemisia tabaci (Hemiptera: Aleyrodidae) revealed using Bayesian analysis of mitochondrial COI DNA sequences. Mol. Phylogenet. Evol. 44, 1306–1319. doi: 10.1016/j.ympev.2007.04.020
Brauer, M. J., Holder, M. T., Dries, L. A., Zwickl, D. J., Lewis, P. O., and Hillis, D. M. (2002). Genetic algorithms and parallel processing in maximum-likelihood phylogeny inference. Mol. Biol. Evol. 19, 1717–1726. doi: 10.1093/oxfordjournals.molbev.a003994
Byrne, D. N., and Bellows, T. S. Jr. (1991). Whitefly biology. Annu. Rev. Entomol. 36, 431–457. doi: 10.1146/annurev.en.36.010191.002243
Cummings, M. P., Neel, M. C., and Shaw, K. L. (2008). A genealogical approach to quantifying lineage divergence. Evolution 62, 2411–2422. doi: 10.1111/j.1558-5646.2008.00442.x
De Barro, P. J., Liu, S. S., Boykin, L. M., and Dinsdale, A. B. (2011). Bemisia tabaci: a statement of species status. Annu. Rev. Entomol. 56, 1–19. doi: 10.1146/annurev-ento-112408-085504
Dereeper, A., Guignon, V., Blanc, G., Audic, S., Buffet, S., Chevenet, F., et al. (2008). Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469. doi: 10.1093/nar/gkn180
Dinsdale, A., Cook, L., Riginos, C., Buckley, Y. M., and De Barro, P. (2010). Refined global analysis of Bemisia tabaci (Gennadius) (Hemiptera: Sternorrhyncha: Aleyroidea) mitochondrial CO1 to identify species level genetic boundries. Ann. Entomol. Soc. Am. 103, 196–208. doi: 10.1603/AN09061
Drummond, A. J., Ashton, B., Buxton, S., Cheung, M., Cooper, A., Heled, J., et al. (2010). Geneious v5.1. Available online at: http://www.geneious.com
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Elbaz, M., Lahav, N., and Morin, S. (2010). Evidence for pre-zygotic reproductive barrier between the B and Q biotypes of Bemisia tabaci (Hemiptera: Aleyrodidae). Bull. Entomol. Res. 100, 581–590. doi: 10.1017/S0007485309990630
Frohlich, D. R., Torres-Jerez, I. I., Bedford, I. D., Markham, P. G., and Brown, J. K. (1999). A phylogeographical analysis of the Bemisia tabaci species complex based on mitochondrial DNA markers. Mol. Ecol. 8, 1683–1691. doi: 10.1046/j.1365-294x.1999.00754.x
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Heath, T. A., Zwickl, D. J., Kim, J., and Hillis, D. M. (2008). Taxon sampling affects inferences of macroevolutionary processes from phylogenetic trees. Syst. Biol. 57, 160–166. doi: 10.1080/10635150701884640
Katoh, K., and Toh, H. (2008). Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinformatics 9, 286–298. doi: 10.1093/bib/bbn013
Liu, B., Preisser, E. L., Chu, D., Pan, H., Xie, W., Wang, S., et al. (2013). Multiple forms of vector manipulation by a plant-infecting virus: Bemisia tabaci and tomato yellow leaf curl virus. J. Virol. 87, 4929–4937. doi: 10.1128/JVI.03571-12
Liu, S. S., Colvin, J., and De Barro, P. (2011). Species concepts as applied to the whitefly Bemisia tabaci systematics: how many species are there? J. Integr. Agric. 11, 176–186. doi: 10.1016/S2095-3119(12)60002-1
Maruthi, M. N., Colvin, J., Seal, S., Gibson, G., and Cooper, J. (2002). Co-adaptation between cassava mosaic geminiviruses and their local vector populations. Virus Res. 86, 71–85. doi: 10.1016/S0168-1702(02)00051-5
Maruthi, M. N., Rekha, A. R., Mirza, S. H., Alam, S. N., and Colvin, J. (2007). PCR-based detection and partial genome sequencing indicate high genetic diversity in Bangladeshi begomoviruses and their whitefly vector, Bemisia tabaci. Virus Genes 34, 373–385. doi: 10.1007/s11262-006-0027-2
Masters, B. C., Fan, V., and Ross, H. A. (2011). Species delimitation–a Geneious plugin for the exploration of species boundaries. Mol. Ecol. Resour. 11, 154–157. doi: 10.1111/j.1755-0998.2010.02896.x
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES science gateway for inference of large phylogenetic trees,” in Proceedings of the Gateway Computing Environments Workshop (GCE) (New Orleans, LA), 1–8. doi: 10.1109/GCE.2010.5676129
Navas-Castillo, J., Fiallo-Olive, E., and Sanchez-Campos, S. (2011). Emerging virus diseases transmitted by whiteflies. Annu. Rev. Phytopathol. 49, 219–248. doi: 10.1146/annurev-phyto-072910-095235
Pimental, D., Zuniga, R., and Morrison, D. (2005). Update of the environmental and economics costs associated with alien-invasive species in the United States. Ecol. Econ. 52, 273–288. doi: 10.1016/j.ecolecon.2004.10.002
Polston, J. E., De Barro, P., and Boykin, L. M. (2014). Transmission specificities of plant viruses with the newly identified species of the Bemisia tabaci species complex. Pest Manag. Sci. doi: 10.1002/ps.3738. [Epub ahead of print].
Posada, D. (2009). Selection of models of DNA evolution with jModelTest. Methods Mol. Biol. 537, 93–112. doi: 10.1007/978-1-59745-251-9_5
Rambaut, A. (2012). FigTree v 1.3.1. [Online]. Available online at: http://tree.bio.ed.ac.uk/software/figtree/
Rambaut, A., and Drummond, A. J. (2010). Tracer v1.5.4 [Online]. Available online at: http://beast.bio.ed.ac.uk/Tracer
Rannala, B., and Yang, Z. (2013). Improved reversible jump algorithms for Bayesian species delimitation. Genetics 194, 245–253. doi: 10.1534/genetics.112.149039
Rodrigo, A., Bertels, F., Heled, J., Noder, R., Shearman, H., and Tsai, P. (2008). The perils of plenty: what are we going to do with all these genes? Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 3893–3902. doi: 10.1098/rstb.2008.0173
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rosenberg, N. A. (2007). Statistical tests for taxonomic distinctiveness from observations of monophyly. Evolution 61, 317–323. doi: 10.1111/j.1558-5646.2007.00023.x
Shatters, R. G. Jr. Powell, C. A., Boykin, L. M., Liansheng, H., and Mckenzie, C. L. (2009). Improved DNA barcoding method for Bemisia tabaci and related Aleyrodidae: development of universal and Bemisia tabaci biotype-specific mitochondrial cytochrome c oxidase I polymerase chain reaction primers. J. Econ. Entomol. 102, 750–758. doi: 10.1603/029.102.0236
Simon, C., Frati, F., Beckenbach, A., Crespi, B., Lui, H., and Flook, P. (1994). Evolution, weighting, and phylogenetic utility of mitochondirial gene sequences and a compilation of conserved polymerase chain reaction “primers.” Ann. Entomol. Soc. Am. 87, 651–701.
Sseruwagi, P., Maruthi, M. N., Colvin, J., Rey, M. E. C., Brown, J. K., and Legg, J. P. (2006). Colonization of non-cassava plant species by whiteflies (Bemisia tabaci) in Uganda. Entomol. Exp. App. 119, 145–153. doi: 10.1111/j.1570-7458.2006.00402.x
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Sun, D. B., Xu, J., Luan, J. B., and Liu, S. S. (2011). Reproductive incompatibility between the B and Q biotypes of the whitefly Bemisia tabaci in China: genetic and behavioural evidence. Bull. Entomol. Res. 101, 211–220. doi: 10.1017/S0007485310000416
Susko, E., Inagaki, Y., and Roger, A. J. (2004). On inconsistency of the neighbor-joining, least squares, and minimum evolution estimation when substitution processes are incorrectly modeled. Mol. Biol. Evol. 21, 1629–1642. doi: 10.1093/molbev/msh159
Tay, W. T., Evans, G. A., Boykin, L. M., and De Barro, P. J. (2012). Will the real Bemisia tabaci please stand up? PLoS ONE 7:e50550. doi: 10.1371/journal.pone.0050550
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 24, 4876–4882. doi: 10.1093/nar/25.24.4876
Wang, H. L., Wang, X. W., and Liu, S. S. (2013). “A global analysis of Bemisia tabaci phylogeny using mitochondrial genomes: a preliminary report,” in 1st International Whitefly Symposium (Kolymbari).
Wang, P., Sun, D. B., Qiu, B. L., and Lui, S. S. (2011). The presence of six cryptic species of the whitefly Bemisia tabaci complex in China as revealed by crossing experiments. Insect Sci. 18, 67–77. doi: 10.1111/j.1744-7917.2010.01381.x
Wang, Z., Yan, H., Yang, Y., and Wu, Y. (2010). Biotype and insecticide resistance status of the whitefly Bemisia tabaci from China. Pest Manag. Sci. 66, 1360–1366. doi: 10.1002/ps.2023
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., and Barton, G. J. (2009). Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi: 10.1093/bioinformatics/btp033
Xu, J., De Barro, P. J., and Liu, S. S. (2010). Reproductive incompatibility among genetic groups of Bemisia tabaci supports the proposition that the whitefly is a cryptic species complex. Bull. Entomol. Res. 100, 359–366. doi: 10.1017/S0007485310000015
Keywords: Bemisia, species identification, species delimitation, whitefly vector, crop protection phylogenetics
Citation: Boykin LM and De Barro PJ (2014) A practical guide to identifying members of the Bemisia tabaci species complex: and other morphologically identical species. Front. Ecol. Evol. 2:45. doi: 10.3389/fevo.2014.00045
Received: 04 June 2014; Accepted: 17 July 2014;
Published online: 05 August 2014.
Edited by:
Mariana Mateos, Texas A&M University, USAReviewed by:
Khidir W. Hilu, Virginia Tech, USALukasz Kajtoch, Institute of Systematics and Evolution of Animals Polish Academy of Science, Poland
Copyright © 2014 Boykin and De Barro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura M. Boykin, ARC Centre of Excellence in Plant Energy Biology and School of Chemistry and Biochemistry, The University of Western Australia, M316, Crawley, Perth, WA 6009, Australia e-mail: lboykin@mac.com