Small genomes and the difficulty to define minimal translation and metabolic machineries
 Rosario Gil1,2*
Rosario Gil1,2*  Juli Peretó1,3*
Juli Peretó1,3*- 1Evolutionary Genetics, Institut Cavanilles de Biodiversitat i Biologia Evolutiva, Universitat de València, València, Spain
- 2Departament de Genètica, Universitat de València, València, Spain
- 3Departament de Bioquímica i Biologia Molecular, Universitat de València, València, Spain
The notion of minimal life has sparked the interest of scientists in different fields, ranging from the origin-of-life research to biotechnology-oriented synthetic biology. Whether the interest is focused on the emergence of protocells out of prebiotic systems or the design of a cell chassis ready to incorporate new devices and functions, proposing minimal combinations of genes for life is not a trivial task. Using comparative genomics and biochemistry of endosymbionts (i.e., intracellular mutualistic symbionts) and intracellular parasites, we proposed a decade ago the core of a minimal gene set for a simple heterotrophic cell adapted to a chemically complex environment. In this work, we discuss the state-of-the-art of the definition of the minimal genome, based on our current knowledge about bacteria with naturally reduced genomes, including both endosymbionts and free-living cells. Any proposed minimal genome would be composed by a set of protein-coding and RNA genes involved in the flux of genetic information (from DNA to functional proteins) and a group of protein-coding sequences embodying a minimalist, stoichiometrically consistent metabolic network. Although the informational portion of the minimal genome is considered quasi-universal, previous proposals have not addressed the need of tRNA post-transcriptional modifications in order to perform their function. For this reason, we have focused on the essentiality of some enzymes involved in such modifications, in order to refine the set of informational genes. As for the metabolic aspect, an obvious difficulty is that there is no one minimal gene set for life but many, depending on the environment. Among cells with reduced genomes, we find a continuum of metabolic modes, from organic matter-dependent heterotrophy to the minimally demanding autotrophy. Both best-known cases of cells with small genomes, the endosymbionts and the ultra-small free-living bacteria, speak in favor of metabolic sharing as a strong force in genome reductive evolution. A hierarchy of minimal cells supported by different metabolic networks with a complexity inversely correlated with the chemical complexity of the environment can be postulated.
The Minimal Genome concept
The chemical complexity of living systems is a major handicap to fully understand life. Biologists have engaged in recent decades in the search of the minimal attributes to consider a system alive, trying to reduce several layers of complexity. It is reasonable to assume that, being less complex, the behavior of simpler cells would be easier to model, predict, and modify.
From a theoretical point of view, Tibor Gánti proposed the chemoton model to reduce cell complexity to the harmonious cooperation of three chemical subsystems based on autocatalytic cycles (Gánti, 1987): (i) an autocatalytic, self-maintained network (metabolism), which provides the molecular building blocks and energy necessary for synthesizing all cellular components; (ii) an informational, self-replicative polymer (genome), which ensures that all genetic instructions pass from generation to generation, and (iii) a self-reproducible boundary (membrane), that encloses the metabolism and the genome, and allows the flow of energy and matter through the system. It is remarkable that the metabolic subsystem uses external sources of energy and matter to provide the building blocks for the envelope and the genome (for original references and a critical review of the chemoton model, see Gánti, 2003). Gánti (1987) discussed the implications of the chemoton model for theoretical and synthetic biology, as well as for the research on the origins of life. Attempts to experimentally reproduce the first steps of life on Earth up to primitive cells (or protocells) follow a research program in systems chemistry for the chemical implementations of Gánti's subsystems (Peretó, 2012; de la Escosura et al., 2015).
In more operational grounds, a minimal (extant, non-primitive) cell has been defined as “a biological system that possesses only the necessary and sufficient attributes to be considered alive. Therefore, it must be able to maintain its own structures (homeostasis), self-reproduce, and evolve in a supportive, protected, and stable environment” (Gil, 2011). Thus, the challenge is to demarcate those “necessary and sufficient attributes” of life, and a functional approach appears adequate for that purpose. The functional elements of a living cell are (lipid) membranes, proteins, and RNA molecules, and the instructions for making these parts, which are encrypted in genes (i.e., DNA) whose information must be “read” by the rest of the molecular machinery. For this reason, a major challenge in biology during the last decades has been to define the minimal number of genes necessary to keep a minimal cell alive, what has been called a minimal genome (Mushegian, 1999). Proposals of a minimal genome have to be tied to a particular level of biological organization, with a defined set of functionalities, as well as to a particular environmental complexity. Most studies have focused on bacteria, due to their apparent simplicity and the amount of information that has already been gathered about them.
Learning from Nature to Define a Minimal Genome
In order to define a set of essential and sufficient genes to keep a bacterial cell alive, it is first necessary to define which are the essential functions that need to be fulfilled. To approach this point, scientists have looked for functions that have been preserved in natural living bacteria with the most reduced genomes, because they must retain all genes involved in informational functions plus a minimal metabolic network for cellular maintenance and reproduction in their given niche. To date, all known cases of reduced bacterial genomes are associated with specific lifestyles linked to stable environments: cosmopolitan oceanic free-living bacteria and obligate symbionts (either parasitic or mutualistic), the latter being the most affected by reductive genome evolution. In other words, natural small genomes have been observed in diverse situations with remarkable dissimilarities, including a dramatic difference in population sizes, from large marine bacterioplankton populations to small populations of endosymbiotic bacteria inside a eukaryotic cell (Batut et al., 2014).
A minimal genome would carry all the information to perform all living functions and, ideally, a microorganism containing it should be able to grow in the laboratory in a defined culture medium and in axenic conditions. Nowadays, the smallest natural genome able to sustain autonomous growth in pure culture and in a defined medium (Yus et al., 2009) belongs to Mycoplasma genitalium, with only 482 annotated protein-coding genes (CDS) in a 580-kb genome (Fraser et al., 1995). Nevertheless, M. genitalium is a human pathogen and, therefore, its genome must also present genes that are not essential for independent life but for its parasitic relationship with its host. Glass et al. (2006) determined experimentally through massive transposon mutagenesis that 100 protein-coding genes were individually dispensable in this bacterium. Therefore, it is clear that, although highly reduced, the M. genitalium genome is far from a minimal genome. Surprisingly, 110 genes encoding for hypothetical proteins and proteins of unknown function appear to be essential, since they were not disrupted in their experiment. This fact can be regarded as a measure of our relative ignorance about even theoretically simple living systems.
To date, all known bacteria with genomes smaller than that of M. genitalium are obligate intracellular mutualistic symbionts (primary or P-endosymbionts) of insects. However, endosymbionts can only survive inside their host cells as they rely on their host (and, in some cases on co-primary endosymbiotic partners) for metabolic and other functions. The conspicuous incompleteness of their metabolic networks supposes an extraordinary obstacle for their cultivation in the laboratory under axenic conditions. All intracellular parasites and endosymbionts described so far exhibit a set of genomic features, namely, genome size reduction, almost total absence of recombination, increased rate of nucleotide substitution, accumulation of deleterious mutations by random genetic drift, loss of codon bias toward A or T, and accelerated sequence evolution. This genome reduction syndrome observed in endosymbiotic bacteria is due to their adaptation to a stable environment, their intracellular isolation from other bacteria and genetic drift, due to the bottlenecks caused by their obligate vertical transmission from mother to offspring (Gil et al., 2010; Batut et al., 2014). The most extreme case corresponds to “Candidatus Nasuia deltocephalinicola,” one of the two partners of the endosymbiotic consortium from the aster leafhopper Macrosteles quadrilineatus, with 137 annotated CDS in a 112-kb genome (Bennett and Moran, 2013). The smallest coding capacity corresponds to “Candidatus Tremblaya princeps,” also one of the two co-primary endosymbions of the citrus mealybug Planococcus citri, with merely 110 predicted CDS in a 139-kb genome (López-Madrigal et al., 2011). But, as stated before, these bacteria are obligate cooperators requiring their corresponding host and accompanying symbiont for survival (López-Madrigal et al., 2011; McCutcheon and von Dohlen, 2011; Bennett and Moran, 2013).
The notion of “symbionelle” has been proposed for these biological entities in which their extreme genomic shrinkage leaves them far beyond the limits to allow them to guarantee essential functions for the making of their own structures or even for replication, transcription and translation (Reyes-Prieto et al., 2014). However, several bacterial endosymbionts of insects have highly reduced genomes while still preserving a fairly complete machinery for DNA replication, transcription, translation and protein folding and, therefore, their informational machinery might be closer to a minimal status. This is the case of Buchnera aphidicola BCc, P-endosymbiont of the aphid Cinara cedri, with a 422-kb genome containing only 362 predicted CDS (Pérez-Brocal et al., 2006). More recently, the genome of “Candidatus Westeberhardia cardiocondylae,” endosymbiont of the invasive ant Cardiocondyla obscurior, has been sequenced (Klein et al., 2015). This streamlined genome (533 kb) contains only 372 predicted CDS and has also retained a simplified but apparently functional informational machinery, similar to the one described for B. aphidicola BCc. The human body louse endosymbiont “Candidatus Riesia pediculicola,” with a 582-kb genome and 528 CDS (Kirkness et al., 2010) has also been used to determine the minimal tRNA modification machinery (see below).
Reductive evolution has not only been observed in host-dependent bacteria, but also in free-living ones. Again, their study faces the difficulty (or impossibility with present cultivation techniques) of growing these bacteria in axenic conditions, in part due to their reduced metabolic networks. However, it is clear that these microorganisms are able to survive with their streamlined genomes, and these species are common in nature (e.g., marine and groundwater bacteria), thus revealing their evolutionary success. In fact, the deep sequencing study of environmental samples by culture-independent methods has revealed that a vast majority of the microbial diversity on Earth cannot be cultured (the so called “dark matter of life”). The metagenomic analysis of environmental samples using deep-sequencing technologies and single-cell sequencing has allowed the assembly of complete genomes from representatives of candidate phyla (i.e., bacterial phyla that lack cultured representatives; CP), even for relatively rare members of the microbial community. Kantor et al. (2013) were able to reconstruct the genomes of single representatives of the four CPs SR1, WWE3, TM7, and OD1 from acetate-stimulated aquifer sediment microbial communities. All four genomes are in the range of 0.7–1.2 Mb (Table 1), while the corresponding cells have a consistently tiny cell size (cell diameter < 0.2 μm and cell volume of around 0.009 μm3) and, for this reason, they are also called ultra-small bacteria. Several additional representatives of phylum TM7 have also been the subject of genomic analyses. Albertsen et al. (2013) sequenced four TM7 genomes from an activated sludge bioreactor, with an average genome size of 1 Mb. Very recently, Brown et al. (2015) were able to sequence 789 partial genomes (more than 50% complete) plus eight more complete genomes from the phylum TM6 and what they call the candidate phyla radiation (CPR, including all previously CP with completely sequenced genomes, except SR1), from ultra-small bacteria collected in an aquifer adjacent to the Colorado River. All complete genomes range from 753 kb to 1 Mb, and most partial ones are estimated to be less than 1 Mb in length. He et al. (2015) were able to cultivate for the first time one member of phylum TM7, the TM7x phylotype found in the human oral cavity, and they also sequenced its complete genome. The study revealed that, with a 705-kb genome containing 699 predicted CDS, oral TM7x is a parasitic epibiont of an oral bacterium (Actinomyces odontolyticus st. XH001), and that its dependence of its host has allowed further reduction of its genome, compared with other TM7 phylotypes.
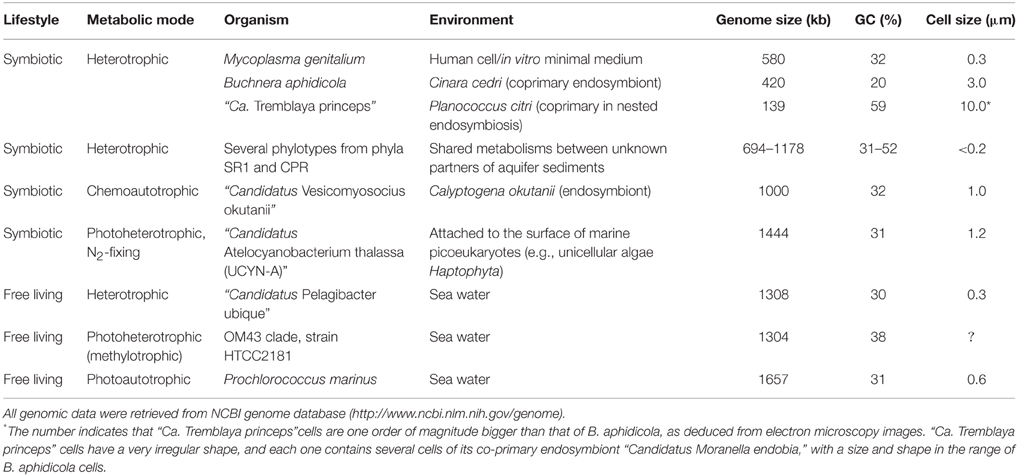
Table 1. Examples of microorganisms with small genomes completely sequenced.
Several hypotheses have been proposed to explain the reductive genomic evolution observed in these host-dependent and free-living bacterial lineages (for reviews see Batut et al., 2014; Giovannoni et al., 2014; Martínez-Cano et al., 2015). According to the streamlining hypothesis, in large populations of prokaryotes living in low-nutrient environments, smaller genomes will be favored by natural selection because they allow cellular minimization (i.e., higher surface-to-volume ratio) and economization, by eliminating superfluous genes and simplifying cell machineries (see further discussions below and Mira et al., 2001; Dufresne et al., 2005). Additionally, in the case of endosymbionts, their isolation inside host cells prevents the acquisition of genes by horizontal gene transfer, while their vertical transmission from mother to offspring causes continuous population bottlenecks over generations, so that genetic drift seems to be the main evolutionary force involved in their reductive syndrome, a hypothesis known as the Muller's ratchet (Moran, 1996). Considering that most bacteria with reduced genomes also live in microbial communities and are very difficult (sometimes impossible, as above stated) to grow in the laboratory in axenic conditions, a third compelling hypothesis, the Black Queen hypothesis, has been proposed (Morris et al., 2012). According to this hypothesis, a vital function can be carried out by a member of the community to benefit the whole community. Therefore, the genes necessary to perform such function become essential for that member but dispensable for the rest of the partners. The loss of a given gene involved in an essential function and the subsequent energy saving would be adaptive at the individual level, as long as the production of public goods is sufficient to support the community. The Black Queen hypothesis was first proposed to explain the reductive genome evolution of bacteria living in environments with limiting resources. However, the same process of sharing public goods could explain why in endosymbiotic consortia one of the partners can end up with a tiny genome of < 200 genes. Nevertheless, in these cases it appears that the reduction has gone too far to consider each one of the partners as a model to approach a minimal genome able to sustain a minimal cell, even for the informational functions.
There is an additional layer of biochemical complexity that has not been explored in the search of minimal genomes, nor is it explicitly considered in the different explanatory models for genome reduction. It must be taken into account that many proteins assemble into protein complexes and networks in order to perform their biological function, and that the combinatorial use of such proteins in different contexts allows them to perform different functions depending on their associated proteins. A proteomic analysis performed on Mycoplasma pneumoniae by Kühner et al. (2009) unveiled an unanticipated proteome complexity for an apparently simple organism (with only 689 CDS annotated in its 816-kb genome; Dandekar et al., 2000). The study revealed that almost 90% of M. pneumoniae soluble proteins are part of at least one protein complex (half of which had not been previously described), and allowed the identification of 156 multifunctional proteins. Many identified proteins are part of molecular machines involved in roles different from that assigned in their functional annotation, while up to 126 proteins that appear within complexes had an unknown or conflicting functional annotation.
Another aspect that must be taken into consideration when searching for minimal gene sets is the possible increase of protein multifunctionality during reductive genome evolution. A general model of metabolic evolution proposes that primitive, short genomes with a limited coding capacity, encoded enzymes with wide specificity (Lazcano et al., 1995), a notion also known as the patchwork model of metabolic pathway evolution based on the pioneering work by Yčas (1974) and Jensen (1976). Gene duplication and divergence could be a mechanism of increasing the specificity of enzymes during the evolutionary expansion of metabolic networks (for critical reviews see Peretó, 2011, 2012). It has been suggested that endosymbiont metabolisms had followed the inverse evolutionary pathway during genome reduction: some enzymes have relaxed specificity to compensate for gene number reduction (Zientz et al., 2004; Peretó, 2011). In this context, the increase in the number of reactions per enzyme that has been observed for the metabolic network of M. pneumoniae is remarkable (Yus et al., 2009). Yet a further feature derived from the intrinsic flexibility of proteins is enzyme promiscuity, defined as the “coincidental catalysis of reactions other than the reaction(s) for which an enzyme evolved” (Khersonsky and Tawfik, 2010). In some circumstances (e.g., during the reduction of the enzyme repertoire by gene loss) those promiscuous activities could take over an essential function, becoming selective and establishing new metabolic connections beyond the canonical pathways. A beautiful experimental test of this concept was performed in Escherichia coli through the random over-expression of enzymes whose promiscuous activities rescued a mutant in the pyridoxal-5′-phosphate biosynthetic pathway (Kim et al., 2010). Those days when it was thought that one gene coded for one protein that performed one function are gone. Current evidence indicates that our knowledge of protein functions, either canonical or promiscuous, is far from complete, and the cell functional messiness (Tawfik, 2010) will become a precious resource for the functional annotation of many genes whose biological roles are elusive.
Finally, in addition to RNA and protein-coding genes, the essential genome of an organism comprises also regulatory (5′-UTRs and non-coding RNAs), and structural elements (Christen et al., 2011). Most of the essentiality studies rely on conventional genome annotations, which are biased against small proteins (less than 100 amino acids; Samayoa et al., 2011) and regulatory elements and, therefore, must be seen as incomplete. In order to define a complete minimal genome with applications in Synthetic Biology, more comprehensive studies, as the one performed by Lluch-Senar et al. (2015) on M. pneumoniae taking also these elements into account, will be highly valuable.
The Minimal Gene Set for Life can be Approached by Comparative Genomics
Comparative genomic approaches have been used since the pioneering work of Mushegian and Koonin (1996). In this work, the reduced genomes of the two first bacterial genomes completely sequenced, from the parasitic bacteria Haemophilus influenzae (Fleischmann et al., 1995), and M. genitalium (Fraser et al., 1995) were compared, in order to identify genes that have been conserved across the large phylogenetic distance that exists between these two species (a gram-negative and a gram-positive bacterium, respectively), assuming that they would be essential. As new reduced genomes were being sequenced, the comparison was extended by the incorporation of the also reduced genomes of bacterial endosymbionts and other parasitic bacteria (Gil et al., 2003; Klasson and Andersson, 2004). In the following years, the identification of essential genes in several model bacteria was also undertaken by experimental approaches (reviewed in Gil, 2014). A decade ago, our group performed a comprehensive analysis of all previously used strategies (both theoretical and experimental) in an attempt to define the core of a minimal genome (CMG; Gil et al., 2004), including only essential genes to sustain a minimal bacterial cell. However, we recognized that the proposed 207 genes are necessary but probably not enough to maintain a cell alive in a realistic environment. We grouped the genes in five main functional categories: (i) information storage and processing; (ii) protein processing, folding and secretion; (iii) cellular processes; (iv) energetic and intermediary metabolism, and (v) poorly characterized genes. The inclusion of the last category indicates that, at that time, there were genes that were present in all reduced genomes analyzed (and, therefore, candidates to be essential), but whose function was still unknown. The main conclusion from that work was that a minimal genome is substantially enriched in genes involved in genetic-information processing, mainly genes of the translational apparatus, which define what has been considered a universal genetic machinery. Additionally, the minimal genome must include the necessary genes to allow the cell to perform all the necessary reactions to maintain a minimal and coherent metabolism able to provide energy and basic components for a minimal living cell. However, from the metabolic point of view, our CMG defined just one of many possible minimal metabolic charts, which would depend on environmental conditions. Gabaldón et al. (2007, 2008) further analyzed this particular minimalist metabolism, using graph-theory based methods and stoichiometric analysis.
Ten years later, Gil (2014) revisited the minimal gene-set by taking into account more recent work on this subject, including both theoretical (comparative genomics, comparative proteomics, and modeling) and experimental approaches. In this revised version, RNA genes that are needed for proper cellular functioning were also included, and some additional genes that might not be strictly essential but could improve cell performance were incorporated. The metabolic chart was maintained essentially as in the former CMG version. Only four out of the five main categories used to classify the minimal gene-set in 2004 were maintained, because only genes with a defined function were included. The translation machinery is, by far, the most complex part of a modern minimal cell, both in its biogenesis and its function. Therefore, it was not surprising that half of the previously classified as poorly characterized genes have been associated with the maturation of the translation apparatus. RsmI (previously called YraL) and MraW are 16S rRNA methyltransferases involved in fine-tuning of the ribosomal decoding center (Kimura and Suzuki, 2010); TilS (formerly MesJ) is the tRNAIle-lysidine synthetase (EC 6.3.4.19) responsible for modifying the wobble base of the CAU anticodon of tRNAIle (Soma et al., 2003), and YbeY has been identified as an endoribonuclease that appears to be involved in the 16S rRNA 3′ terminus maturation (Jacob et al., 2013). Even the former poorly characterized gene yqgF, which was not included in the revised version of the minimal gene-set, seems to participate in the maturation of the ribosome. Recent studies suggest that it is involved in the 5′ processing of pre-16S rRNA, and its absence might affect translational fidelity, as well as reduce the expression of genes containing Shine-Dalgarno-like sequences (Kurata et al., 2015). The fact that several genes of previously unknown function included in the CMG are involved in proper maturation and functioning of the translational apparatus reveals that the detailed repertoire of this complex machinery is far from complete.
Troubles Defining a Minimal Translation Apparatus
Although it must have been highly simple in primitive cells, the translation apparatus has evolved toward its great present complexity ensuring accurate mRNA decoding and optimizing the efficiency of the mRNA-dependent protein synthesis (Petrov et al., 2014). Nevertheless, host-dependent bacteria (whether parasitic or mutualistic) derived from free-living ancestors have lost some of the components of this complex machinery and are good models for identifying the essential components for proper translation. Different studies have been performed since we proposed the CMG in order to get a closer look at this complex translational machinery, using Mollicutes and insect endosymbionts as models (de Crécy-Lagard et al., 2007, 2012; Grosjean et al., 2014). These studies revealed that different minimized but still functional protein-synthetizing machineries have emerged after reductive evolution even inside a given bacterial clade. The recent study of complete and draft genomes from hundreds of CPR bacteria revealed that their ribosomes have an unusual protein composition, and their 16S rRNAs are highly divergent from other bacteria (Brown et al., 2015). Therefore, the definition of a universal minimal gene-set for translation that would be common to very distantly related bacteria seems to be unachievable using comparative analyses. This difficulty is highly relevant for the definition of genes needed for assembly of functional ribosomes and proper maturation of functional tRNAs. Organisms living in widely different environments have developed different rRNA modifications and retained a slightly different set of ribosomal proteins to guarantee translational accuracy and efficiency (Mears et al., 2002). Proteins that have critical roles in the ribosomal structure and function maintain conserved interactions with rRNA and are conserved across the three domains of life. Even though the lack of identification of some ribosomal proteins in some reduced genomes must be regarded with caution, because these are small proteins which are prone to be missed by automatic annotation tools, it is clear that changes in the rRNA sequence would vary the ribosome architecture and the need for some ribosomal proteins because some of the protein-RNA binding sites would also vary.
As for the maturation of tRNAs, tRNA precursors must undergo many post-transcriptional modifications at different positions in order to acquire the proper tRNA tertiary structure and ensure efficiency and fidelity in the decoding process. A detailed study of the essential tRNA modification machinery has been attempted for bacterial endosymbionts, revealing that “Ca. Riesia pediculicola” might possess a close to minimal number of tRNA modification enzymes required for life (de Crécy-Lagard et al., 2012). The absence of all genes involved in modifications of the tRNA body suggested that only modifications at the anticodon loop region are essential for intracellular organisms. We have performed an analysis of the presence of these genes in all currently available endosymbiont genomes within the genome-size range of “Ca. Riesia pediculicola” which also possess a complete set of tRNA genes that allow protein-synthesis with all 20 canonical amino acids (Table 2). As in the set of endosymbionts used by de Crécy-Lagard and coworkers for comparison, all except “Ca. Riesia pediculicola” lack cmoA and B, responsible of the biosynthesis of the 5-oxyacetyl uridine (cmo5U) modification at the U34 of certain tRNAs, which the authors suggest might be dispensable. The translation machinery that relies on the derived simplified set of mature tRNAs might not be very accurate, but it appears to be sufficient to sustain life. Our proposed CMG (Gil et al., 2004; Gil, 2014) included several genes for these tRNA modifications, namely, tilS, mnmA, mnmE, mnmG, trmD, and tsaD. It should be mentioned that in both versions of the CMG, tsaD (formerly gcp) was included in the category of information storage and processing but as a glycoprotein-related chaperon protease (Katz et al., 2010). However, more recent studies have determined that it is involved in the biosynthesis of the threonylcarbamoyl adenosine (t6A) at position 37, adjacent to the anticodon in ANN-decoding tRNAs, an important modification for translational fidelity (Srinivasan et al., 2011; Deutsch et al., 2012). In the light of our recent search, it appears that at least three additional genes should be incorporated to the minimal tRNA modification machinery, i.e., tadA, miaA, and truA. TadA (EC 3.5.4.33) is a tRNA-specific adenosine deaminase that catalyzes deamination of the adenosine at position 34, the wobble base of the tRNAArg2 anticodon, resulting in an inosine at this position. MiaA (EC 2.5.1.75) is the tRNA dimethylallyltransferase that catalyzes the first step in the pathway for hypermodification of the A37. The tRNA pseudouridine synthase A (EC 5.4.99.12), encoded by truA, catalyzes pseudouridine formation in nucleotides 38–40 at the anticodon loop. Nevertheless, it should be taken into account that different modifications would be needed depending on the specific set of tRNAs retained in a given genome, which would vary depending on evolutionary, physiological and environmental factors. Therefore, experimental analysis would be necessary in order to define the proper composition of the minimal gene-set of tRNAs and their modifying enzymes for a given organism.
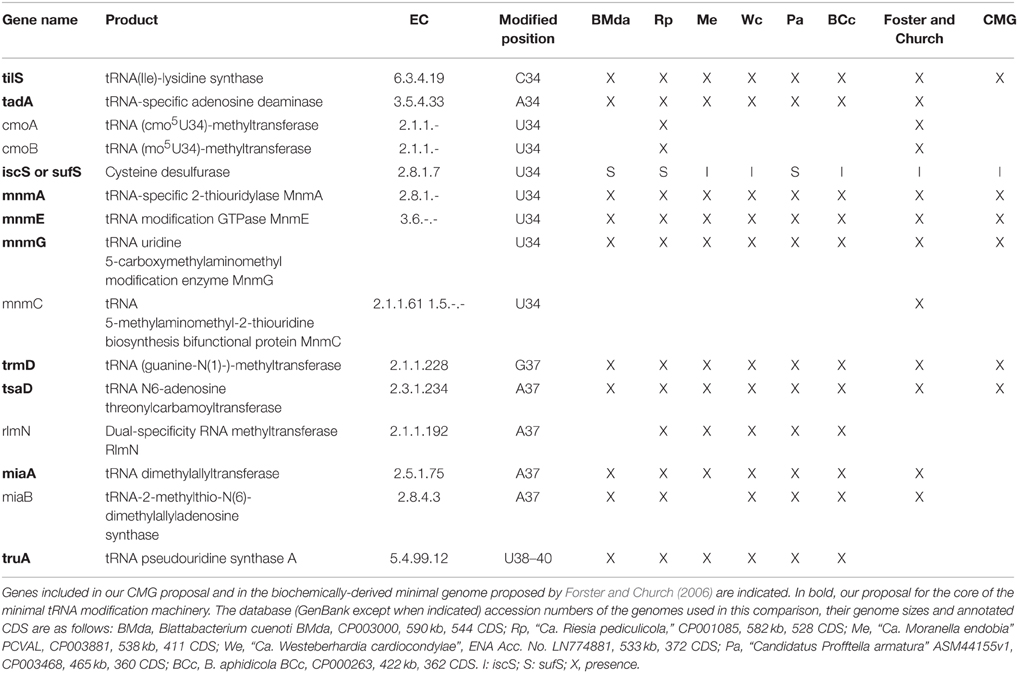
Table 2. Genes involved in tRNA modifications in the extended anticodon region (nucleotides 34–39) present in insect endosymbionts with highly reduced genomes but that still retain a full set of tRNAs.
A Diversity of Metabolic Modes in Cells with Reduced Genomes
The heterotrophic mode (i.e., a metabolism that uses organic compounds as a source of carbon, energy, and electrons) is widespread among insect endosymbionts. Endosymbiont metabolisms supply essential nutrients to their insect hosts living on nutrient-deficient diets (Moya et al., 2008). This is the case for bacteria in insects feeding on phloem sap (deficient in some amino acids) or animal blood (poor in some vitamins). There is also the case of endosymbionts expanding the metabolic abilities of some omnivore insects, affecting the capacity to manage available nitrogen reserves during periods of nutrient scarcity. In all above-mentioned examples, the metabolic mode of the endosymbiont is strictly heterotrophic and shows auxotrophies for some intermediary metabolites or nutrients provided by the host. Take the case of cockroaches and their endosymbiont, the flavobacterium Blattabacterium cuenoti. Cockroaches are omnivore animals but the endosymbiont located in the fat body has the ability to provide essential amino acids. In addition, the bacterial urease present in the endosymbiont can hydrolyze the urea provided by the host uricolytic pathway, completing the whole transformation from uric acid accumulated in specialized cells to ammonia. But the astonishing feature is that Blattabacterium lacks the capacity of synthesizing glutamine, one of the main nitrogen suppliers to metabolism. Thus, ammonia from the bacterium must be incorporated to glutamine by the host, which in turn must supply this amino acid to the endosymbiont (Patiño-Navarrete et al., 2014).
In all known cases of heterotrophic endosymbionts, the metabolic machinery is reduced to a minimum, just enough to provide the essential nutrients to the host, but at the cost of lacking some other important processes, including pathways such as those for the de novo synthesis of nitrogen bases or even the bioenergetic supply of ATP in what could be considered a higher stage of heterotrophy (Moran and Bennett, 2014). Hence, genome reduction during the symbiont domestication implies the emergence of auxotrophies or metabolic dependencies that have the potential of being switches for host control of the bacterial population. This phenomenon has been studied in certain detail in the symbioses between Leguminosae plants and nitrogen fixing bacteria, where the endosymbionts are auxotrophs for some amino acids supplied by the host (Prell and Poole, 2006). Host dependency can be extreme in the case of intracellular pathogens, as Mycoplasma, adapted to metabolic parasitism. Metabolic modeling can reveal additional features of the adaptation to the intracellular milieu. Wodke et al. (2013) quantitatively analyzed the intermediary and energetic metabolism of M. pneumoniae using a genome-wide, constraint-based metabolic model (iJW145, in silico model including 145 genes), combined with experimental validations and systematic searches of literature. A remarkable result was the observation that M. pneumoniae invests most of its energy (i.e., ATP) in cell homeostasis rather than in growth, coherently with its adaptive strategy to parasitic intracellular life.
At the other end of the metabolic spectrum there are autotrophic endosymbionts that can fix carbon from CO2 using electrons and energy from inorganic sources, i.e., they are chemolithoautotrohs. There is a wide diversity of symbiotic associations between chemosynthetic bacteria and marine animals that colonize different environments (such as cold seeps, shallow-water coastal sediments, and deep-sea hydrothermal vents), and involving members of at least six animal phyla that harbor endosymbiotic bacteria (Dubilier et al., 2008). Among the best characterized systems there are the bacterial endosymbionts present in marine invertebrates such as the gutless worm from marine sediments Olavius algarvensis (Woyke et al., 2006; Kleiner et al., 2012), and several deep-sea vent inhabitants, including the tubeworm Riftia pachyptila (Markert et al., 2007; Robidart et al., 2008), clams from the genus Calyptogena (Kuwahara et al., 2007; Newton et al., 2007; Roeselers et al., 2010), and scaly-foot gastropods or armored snails (tentatively named “Crysomallon squamiferum”) living near black smoker chimneys (Nakagawa et al., 2014).
In general, although the genomes of the chemosynthetic endosymbionts are reduced in comparison to their closest free-living relatives, the genomic reduction is not as dramatic as the observed in the heterotrophic endosymbionts (Table 1). For instance, the genome from B. aphidicola BCc, the cedar aphid endosymbiont, is 9.2% the size of the genome from the phylogenetically close, free-living bacterium E. coli K12 (4.64 Mb; Pérez-Brocal et al., 2006). In contrast, the 1.16-Mb genome from “Candidatus Ruthia magnifica,” endosymbiont of the clam Calyptogena magnifica, is ca. 48% the size of the genome of a close free-living bacterium, the sulfur-oxidizing gammaproteobacterium Thiomicrospira crunogena (2.43 Mb; Scott et al., 2006).
Different experimental approaches, including metagenomics, metatranscriptomics, metaproteomics, metabolomics, and stable carbon isotope labeling, have been used to characterize the carbon fixation pathways operative in intracellular bacteria (Dubilier et al., 2008). Thus, the bacterial consortium residing in O. algarvensis, composed by sulfide-oxidizing gammaproteobacteria (at least two species) and sulfate-reducing deltaproteobacteria (at least two species), fix carbon through the Calvin-Benson cycle (gammaproteobacteria), and the net synthesis of acetyl-CoA (Wood-Ljungdahl pathway inferred in the deltaproteobacteria members of the consortium; Kleiner et al., 2012). On the other hand, Riftia gammaproteobacterial endosymbiont (“Candidatus Endoriftia persephone”) reduces CO2 with electrons from H2S, using both the Calvin-Bandon cycle and the reductive tricarboxylic acid cycle (or reverse Krebs cycle; Robidart et al., 2008). The Calvin-Benson cycle is the autotrophic pathway identified in the gammaproteobacteria endosymbionts from both Calyptogena clams (Kuwahara et al., 2007; Newton et al., 2007) and the scaly-foot gastropods (Nakagawa et al., 2014). In contrast to the above mentioned auxotrophies observed in all heterotrophic endosymbionts, the inferred metabolic networks from the different chemosynthetic endosymbionts indicate that, in general, all of them appear to be able (by themselves or in conjunction with accompanying endosymbionts) to synthesize the complete repertoire of essential metabolites needed by the host with the uptake of only very simple precursors, such as CO2 as carbon source and H2S as a source of electrons and energy.
As stated before, abundant free-living members of marine bacterioplankton, like the methylotrophic β-proteobacterium member of the OM43 clade (strain HTCC2181), the α-proteobacterium “Candidatus Pelagibacter ubique,” and the cyanobacterium Prochlorococcus marinus also exhibit small genomes (Table 1), and the streamlining selection mechanism through elimination of unnecessary DNA has been proposed (Dufresne et al., 2005; Giovannoni et al., 2005, 2008). In addition to large effective population sizes, bacterioplankton thrives usually in habitats that are poor in macronutrients. The minimization of the genome alleviates the demand of N and P, and allows decreasing cell sizes, thus improving the surface-to-volume ratio and, hence, the efficiency of nutrient uptake. It also has been proposed that the heterotrophic members of bacterioplankton, such as HTCC2181 and “Ca. Pelagibacter ubique,” follow the strategy of reducing the metabolic repertoire whereas specializing on scarce carbon sources, like C1 compounds (Giovannoni et al., 2008). On the other hand, Tripp et al. (2010) described a photoheterotrophic nitrogen-fixing cyanobacterium with a 1.4-Mb genome (Table 1). The incompleteness of its metabolic pathways suggested the existence of some symbiotic relationship with other microorganisms, which were later identified as picoeukaryotes, probably unicellular algae (Thompson et al., 2012; Krupke et al., 2014).
In the ultra-small bacteria (i.e., free-living bacteria with a cell diameter < 0.2 μm), with short genomes (from 0.7 to 1.2 Mb) and incomplete metabolic networks that have been described in acetate-amended groundwater (Kantor et al., 2013; Luef et al., 2015), albeit the annotated genomes contain a substantial amount of proteins with unknown function (around 20%), a diversity of fermentative metabolisms can be predicted. There are also diverse enzymes dealing with the metabolism of complex sugars and electron transport, as well as the utilization of amino acids and nucleotides. However, there is a conspicuous absence of biosynthetic pathways for most amino acids, nucleotides, and lipids. Thus, the authors suggest that these microorganisms are auxotrophs for some essential metabolites, although it cannot be completely ruled out that they harbor some novel (unknown) biosynthetic pathways (Kantor et al., 2013). Nevertheless, the presence of numerous nucleases, proteases, and membrane transporters, points to the possibility of ecological interactions between microorganisms that compensate their incomplete metabolic networks (Kantor et al., 2013; Luef et al., 2015). Corroborating previous findings, recent description of CPR organisms showed that their tricarboxylic acid cycle, nucleotide, and amino acid biosynthesis pathways, electron transport chains and even ATP synthase complex, are incomplete in most cases (Brown et al., 2015). Thus, sharing metabolisms, even in free-living microorganisms, emerges as a strategy in reductive genome evolution.
Taken together, we find natural reduced genomes either in bacteria adapted to intracellular life or in free-living microorganisms adapted to specific habitats like oligotrophic water. In both cases, and looking at the streamlining and the Black Queen hypotheses, it follows the emergence of metabolic sharing as a strong force toward genome reduction, and that metabolic consortia are an extended way of living that has been underestimated up to now. Thus, loss of apparently essential metabolic functions is compensated by metabolic complementation by other endosymbiont and/or the host cell (in the case of intracellular bacteria) or by other partners living in the same place (prokaryotic and/or eukaryotic species). Looking at small bacteria sharing the metabolism, it is reasonable to propose that a quasi-universal genetic machinery (e.g., note the difficulties in defining a truly universal translation machinery) can be supported by many different combinations of metabolic reactions depending on the chemical/biochemical composition of the ecosystem.
A Hierarchy of Minimal Cells Based on Metabolic Complexity
It is sensible to assume that metabolic complexity and environmental chemical complexity are inversely correlated. In a seminal work, Morowitz (1992) discussed an ecologically-dependent simplicity. This author considers that the simplicity observed in many extant microorganisms per se does not imply primitiveness. On the contrary, it is the result of long paths of evolutionary adaptations. This is more obvious for the many independent cases of ancestral bacterial infections in insect lineages that give rise to the extant endosymbionts. Morowitz (1992) proposed two different notions of cell simplicity, depending on the chemical complexity of the environment. Thus, it is apparent that Mycoplasmatales have reduced to a minimum the necessary metabolic network for survival in a chemically complex environment—either the intracellular milieu or the minimal medium for this particular organism prepared by the experimentalist. On the other hand, to look for truly autonomous organisms among present-day biota, a different type of simplicity is needed, a kind of ecological minimum: a microorganism displaying the least demands on the environment, i.e., cells endowed with a chemolithoautotrophic metabolism. Such searches lead Morowitz (1992) to cyanobacteria that exhibit complex metabolic networks with the capacity to build biomass from simple molecules such as CO2 and N2. The reduced metabolic networks observed in small cells of bacterioplankton and the ultra-small bacteria found in biostimulated aquifers, are additional examples of simplicity likely achieved by a strategy of metabolic complementation or trophic dependences with other metabolic networks. In summary, considering the diversity of environmental dependence in natural populations of microorganisms and consortia, from full heterotropy to autotrophy, it is reasonable to suggest that there is a trade-off between metabolic network complexity and environmental chemical complexity (i.e., minimal growth medium or other accessible metabolic networks): the simpler the medium, the more complex the metabolism. In this sense, a hierarchy of minimal cells may be defined, ranking from a strong chemical dependence on the environment to freedom to thrive in nutrient-poor environments (see footnote 2 in Luisi et al., 2006).
One way to approach the measure of metabolic complexity uses stoichiometric analysis and graph-theory based methods to estimate the level of redundancy in metabolic networks. Using stoichiometric analysis of elementary flux modes (EFM), Gabaldón et al. (2007) showed that 49 out of 50 enzymatic steps of a minimalist metabolism were essential for maintaining the stoichiometric coherence. Thus, this metabolic network had only one redundant chemical transformation revealing its true minimalist character. A survey of the literature gives us some examples of functional stoichiometric analysis of natural small metabolic networks (based on flux balance analysis, FBA). The results of diverse in silico knock-out experiments are presented in Figure 1. As expected, a negative correlation of the proportion of essential genes vs. the number of protein-coding genes is observed. If we take gene essentiality as a proxy of network fragility, small metabolic networks are more fragile against gene loss, probably due to a decrease in functional redundancy. In other words, the number of alternative pathways able to rescue for a specific deleted enzymatic step is drastically reduced and the consequent fragility of the network could limit its usefulness as a support (or chassis) in synthetic biology projects. A ladder of small networks can be observed in Figure 1, from that of an autonomous free-living bacterium (e.g., E. coli) up to the theoretical minimalist heterotrophic network (Gabaldón et al., 2007), throughout the stages of obligate intracellular symbionts, such as M. pneumoniae (Wodke et al., 2013), B. cuenoti (González-Domenech et al., 2012), and B. aphidicola (Thomas et al., 2009; Belda et al., 2012), and the secondary endosymbiont Sodalis glossinidius and its putative evolutionary precursor (Belda et al., 2012). It would be of interest to model natural free-living cells with incomplete metabolisms to examine their relative position in this hierarchy.
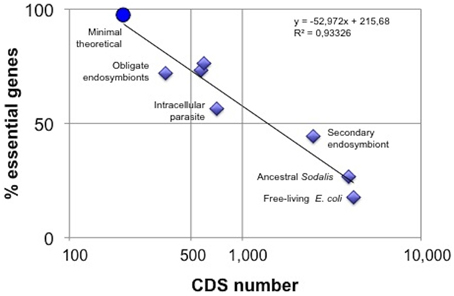
Figure 1. Network fragility increases with metabolic minimization. Gene essentiality was determined in in silico knock-out experiments using Flux Balance Analysis (FBA) on metabolic models inferred from complete genomes, except for the minimal theoretical network, based on CMG (Gil et al., 2004), where Elementary Flux Mode analysis was used. From right to left, the data points correspond to E. coli (Belda et al., 2012), ancestral and extant S. glossinidius network (Belda et al., 2012), M. pneumoniae (Wodke et al., 2013), Blattabacterium (González-Domenech et al., 2012), B. aphidicola BAp (Thomas et al., 2009), B. aphidicola BCc (Belda et al., 2012), and the minimal theroretical metabolism (Gabaldón et al., 2007). CDS, protein coding sequences.
Xavier et al. (2014) pointed out that genome size hardly correlates with some indirect measures of complexity, like the interactome size or the doubling time. A better approach to define complexity could be the measure of the metabolic network size. Gabaldón et al. (2007, 2008) described the correlation between the total number of CDS in the genome and several topological properties and graph-theory parameters of the corresponding inferred metabolic networks such as its size (number of nodes/metabolites, diameter) and node clustering. Xavier et al. (2014) noted also the correlation between the number of CDS, and the number of reactions in networks. But what seems more interesting is their use of the number of chemical components in the minimal medium of a species as a proxy of the biosynthetic abilities and the metabolic complexity of the corresponding microorganism. The negative correlation observed with genome size, at least for genomes < 3 Mb in size, is compatible with the above mentioned notion of a hierarchy of metabolic complexities among cells with reduced genomes. We propose that stoichiometric analysis of the hypothetical minimalist metabolisms, obtained by assembling extant reactions and pathways, could quantitatively approach this concept.
Conclusions and Perspectives
From an academic point of view, reductive genomic evolution is a perfect illustration of how DNA evolution is constrained by metabolic performances. The classical “central dogma” of molecular biology establishes the preeminence of genes over the rest of elements in the chain of information transmission (Crick, 1958; Morange, 2008). However, an insightful discussion by de Lorenzo (2014) shows that current biochemical knowledge demands an expansion of the canonical view of the “central dogma” where metabolism takes a leading role, as the ultimate source of materials for the construction of the rest of the involved elements (DNA, RNA, proteins). In other words, the metabolome is a product of the proteome and the genome/transcriptome/proteome is a product of the metabolome (Cornish-Bowden et al., 2007). But de Lorenzo (2014) thinking goes further to the observation that one of the driving forces in bacterial evolution is the exploration/exploitation of new chemical landscapes. Small cells with small genomes show that this goal can be achieved by reductive evolution throughout the sharing of metabolic networks.
Looking ahead to future applications of minimal cells in synthetic biology for industrial purposes, the discussion on what is an ideal chassis—a natural, robust cell or a minimized version— is still open (Juhas, 2015 and references therein). More research is needed regarding gene essentiality and also on the dependence of minimal cell performance from environmental conditions. In this work we have emphasized that there is a hierarchy of minimal cells depending on the chemical composition of the medium. Around a central core of essential genes, almost universally conserved, and devoted to genetic information management, there is a shell of chemical reactions constituting a network whose complexity is inversely correlated with environmental chemical complexity. Thus, in the future, the requirements of growing particular minimal cells—or even consortia of minimized cells—could be formulated from the deduced correspondence between the minimal metabolic network and the medium composition throughout appropriated modeling and simulation methods.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial support from the European Commission (ST-FLOW, grant agreement 289326), Spanish Government (grant reference: BFU2012-39816-C02-01, co-financed by FEDER funds and Ministerio de Economía y Competitividad) and Generalitat Valenciana (grant reference: PROMETEOII/2014/065) is gratefully acknowledged. We also want to acknowledge the comments of V. de Crézy-Lagard and J. Glass to the 2014 version of the CMG (Gil, 2014), which inspired part of this work, and the useful comments and criticisms of the reviewers.
References
Albertsen, M., Hugenholtz, P., Skarshewski, A., Nielsen, K. L., Tyson, G. W., and Nielsen, P. H. (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotech. 31, 533–538. doi: 10.1038/nbt.2579
Batut, B., Knibbe, C., Marais, G., and Daubin, V. (2014). Reductive genome evolution at both ends of the bacterial population size spectrum. Nat. Rev. Microbiol. 12, 541–550. doi: 10.1038/nrmicro3331
Belda, E., Silva, F. J., Peretó, J., and Moya, A. (2012). Metabolic networks of Sodalis glossinidius: a systems biology approach to reductive evolution. PLoS ONE 7:e30652. doi: 10.1371/journal.pone.0030652
Bennett, G. M., and Moran, N. A. (2013). Small, smaller, smallest: the origins and evolution of ancient dual symbioses in a phloem-feeding insect. Genome Biol. Evol. 5, 1675–1688. doi: 10.1093/gbe/evt118
Brown, C. T., Hug, L. A., Thomas, B. C., Sharon, I., Castelle, C. J., Singh, A., et al. (2015). Unusual biology across a group comprising more than 15% of domain bacteria. Nature 523, 208–211. doi: 10.1038/nature14486
Christen, B., Abeliuk, E., Collier, J. M., Kalogeraki, V. S., Passarelli, B., Coller, J. A., et al. (2011). The essential genome of a bacterium. Mol. Syst. Biol. 7, 528. doi: 10.1038/msb.2011.58
Cornish-Bowden, A., Cárdenas, M. L., Letelier, J. C., and Soto-Andrade, J. (2007). Beyond reductionism: metabolic circularity as a guiding vision for a real biology of systems. Proteomics 7, 839–845. doi: 10.1002/pmic.200600431
Dandekar, T., Huynen, M., Regula, J. T., Ueberle, B., Zimmermann, C. U., Andrade, M. A., et al. (2000). Re-annotating the Mycoplasma pneumoniae genome sequence: adding value, function and reading frames. Nucleic Acids Res. 28, 3278–3288. doi: 10.1093/nar/28.17.3278
de Crécy-Lagard, V., Marck, C., Brochier-Armanet, C., and Grosjean, H. (2007). Comparative RNomics and modomics in mollicutes: prediction of gene function and evolutionary implications. IUBMB Life 59, 634–658. doi: 10.1080/15216540701604632
de Crécy-Lagard, V., Marck, C., and Grosjean, H. (2012). Decoding in Candidatus Riesia pediculicola, close to a minimal tRNA modification set? Trends Cell Mol. Biol. 7, 11–34.
de la Escosura, A., Briones, C., and Ruiz-Mirazo, K. (2015). The systems perspective at the crossroads between chemistry and biology. J. Theor. Biol. 381, 11–22. doi: 10.1016/j.jtbi.2015.04.036
de Lorenzo, V. (2014). From the selfish gene to selfish metabolism: revisiting the central dogma. Bioessays 36, 226–235. doi: 10.1002/bies.201300153
Deutsch, C., El Yacoubi, B., de Crécy-Lagard, V., and Iwata-Reuyl, D. (2012). Biosynthesis of threonylcarbamoyl adenosine (t6A), a universal tRNA nucleoside. J. Biol. Chem. 287, 13666–13673. doi: 10.1074/jbc.M112.344028
Dubilier, N., Bergin, C., and Lott, C. (2008). Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat. Rev. Microbiol. 6, 725–740. doi: 10.1038/nrmicro1992
Dufresne, A., Garczarek, L., and Partensky, F. (2005). Accelerated evolution associated with genome reduction in a free-living prokaryote. Genome Biol. 6:R14. doi: 10.1186/gb-2005-6-2-r14
Fleischmann, R. D., Adams, M. D., White, O., Clayton, R. A., Kirkness, E. F., Kerlavage, A. R., et al. (1995). Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269, 496–512. doi: 10.1126/science.7542800
Forster, A. C., and Church, G. M. (2006). Towards synthesis of a minimal cell. Mol. Syst. Biol. 2:45. doi: 10.1038/msb4100090
Fraser, C. M., Gocayne, J. D., White, O., Adams, M. D., Clayton, R. A., Fleischmann, R. D., et al. (1995). The minimal gene complement of Mycoplasma genitalium. Science 270, 397–404. doi: 10.1126/science.270.5235.397
Gabaldón, T., Gil, R., Peretó, J., Latorre, A., and Moya, A. (2008). “The core of a minimal gene set: insight form natural reduced genomes,” in Protocells: Bridging Nonliving and Living Matter, eds S. Rasmussen, M. A. Bedau, L. Chen, D. Deamer, D. C. Krakauer, N. H. Packard, and P. F. Stadler (Boston: MIT Press), 347–366. doi: 10.7551/mitpress/9780262182683.003.0016
Gabaldón, T., Peretó, J., Montero, F., Gil, R., Latorre, A., and Moya, A. (2007). Structural analyses of a hypothetical minimal metabolism. Philos. Trans. R. Soc. Lond. B Biol. Sci. 362, 1751–1762. doi: 10.1098/rstb.2007.2067
Gánti, T. (2003). The Principles of Life. With commentaries by J. Griesemer and E. Szathmáry. Oxford: Oxford University Press.
Gil, R. (2011). “Minimal cell,” in Encyclopedia of Astrobiology, eds M. Gargaud, R. Amils, J. C. Quintanilla, H. J. Cleaves II, W. M. Irvine, D. L. Pinti, and M. Viso (Berlin: Springer Science & Business Media), 1065–1066. doi: 10.1007/978-3-642-11274-4_1000
Gil, R. (2014). “The minimal gene-set machinery,” in Encyclopedia of Molecular Cell Biology and Molecular Medicine: Synthetic Biology, 2nd Edn., ed R. A. Meyers (Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA), 1–36. doi: 10.1002/3527600906.mcb.20130079
Gil, R., Latorre, A., and Moya, A. (2010). “Evolution of prokaryote-animal symbiosis from a genomics perspective,” in (Endo)Symbiotic Methanogenic Archaea. Microbiology Monographs, Vol. 19, ed J. H. P. Hackstein (Berlin: Springer-Verlag), 207–233. doi: 10.1007/978-3-642-13615-3_11
Gil, R., Silva, F. J., Peretó, J., and Moya, A. (2004). Determination of the core of a minimal bacterial gene set. Microbiol. Mol. Biol. Rev. 68, 518–537. doi: 10.1128/MMBR.68.3.518-537.2004
Gil, R., Silva, F. J., Zientz, E., Delmotte, F., González-Candelas, F., Latorre, A., et al. (2003). The genome sequence of Blochmannia floridanus: comparative analysis of reduced genomes. Proc. Natl. Acad. Sci. U.S.A. 100, 9388–9393. doi: 10.1073/pnas.1533499100
Giovannoni, S. J., Cameron Thrash, J., and Temperton, B. (2014). Implications of streamlining theory for microbial ecology. ISME J. 8, 1553–1565. doi: 10.1038/ismej.2014.60
Giovannoni, S. J., Hayakawa, D. H., Tripp, H. J., Stingl, U., Givan, S. A., Cho, J. C., et al. (2008). The small genome of an abundant coastal ocean methylotroph. Environ. Microbiol. 10, 1771–1782. doi: 10.1111/j.1462-2920.2008.01598.x
Giovannoni, S. J., Tripp, H. J., Givan, S., Podar, M., Vergin, K. L., Baptista, D., et al. (2005). Genome streamlining in a cosmopolitan oceanic bacterium. Science 309, 1242–1245. doi: 10.1126/science.1114057
Glass, J. I., Assad-Garcia, N., Alperovich, N., Yooseph, S., Lewis, M. R., Maruf, M., et al. (2006). Essential genes of a minimal bacterium. Proc. Nat. Acad. Sci. U.S.A. 103, 425–430. doi: 10.1073/pnas.0510013103
González-Domenech, C. M., Belda, E., Patiño-Navarrete, R., Moya, A., Peretó, J., and Latorre, A. (2012). Metabolic stasis in an ancient symbiosis: genome-scale metabolic networks from two Blattabacterium cuenoti strains, primary endosymbionts of cockroaches. BMC Microbiol. 12(Suppl. 1):S5. doi: 10.1186/1471-2180-12-S1-S5
Grosjean, H., Breton, M., Sirand-Pugnet, P., Tardy, F., Thiaucourt, F., Citti, C., et al. (2014). Predicting the minimal translation apparatus: lessons from the reductive evolution of Mollicutes. PLoS Genet. 10:e1004363. doi: 10.1371/journal.pgen.1004363
He, X., McLean, J. S., Edlund, A., Yooseph, S., Hall, A. P., Liu, S. Y., et al. (2015). Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc. Natl. Acad. Sci. U.S.A. 112, 244–249. doi: 10.1073/pnas.1419038112
Jacob, A. I., Köhrer, C., Davies, B. W., RajBhandary, U. L., and Walker, G. C. (2013). Conserved bacterial RNase YbeY plays key roles in 70S ribosome quality control and 16S rRNA maturation. Mol. Cell 49, 427–438. doi: 10.1016/j.molcel.2012.11.025
Jensen, R. A. (1976). Enzyme recruitment in evolution of new function. Annu. Rev. Microbiol. 30, 409–425. doi: 10.1146/annurev.mi.30.100176.002205
Juhas, M. (2015). On the road to synthetic life: the minimal cell and genome-scale engineering. Crit. Rev. Biotechnol. doi: 10.3109/07388551.2014.989423. [Epub ahead of print].
Kantor, R. S., Wrighton, K. C., Handley, K. M., Sharon, I., Hug, L. A., Castelle, C. J., et al. (2013). Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. MBio 4, e00708–e00713. doi: 10.1128/mbio.00708-13
Katz, C., Cohen-Or, I., Gophna, U., and Ron, E. Z. (2010). The ubiquitous conserved glycopeptidase Gcp prevents accumulation of toxic glycated proteins. MBio 1, e00195–e00110. doi: 10.1128/mBio.00195-10
Khersonsky, O., and Tawfik, D. S. (2010). Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu. Rev. Biochem. 79, 471–505. doi: 10.1146/annurev-biochem-030409-143718
Kim, J., Kershner, J. P., Novikov, Y., Shoemaker, R. K., and Copley, S. D. (2010). Three serendipitous pathways in E. coli can bypass a block in pyridoxal-5′-phosphate synthesis. Mol. Syst. Biol. 6, 436. doi: 10.1038/msb.2010.88
Kimura, S., and Suzuki, T. (2010). Fine-tuning of the ribosomal decoding center by conserved methyl-modifications in the Escherichia coli 16S rRNA. Nucleic Acids Res. 38, 1341–1352. doi: 10.1093/nar/gkp1073
Kirkness, E. F., Haas, B. J., Sun, W. L., Braig, H. R., Perotti, M. A., Clark, J. M., et al. (2010). Genome sequences of the human body louse and its primary endosymbiont provide insights into the permanent parasitic lifestyle. Proc. Natl. Acad. Sci. U.S.A. 107, 12168–12173. doi: 10.1073/pnas.1003379107
Klasson, L., and Andersson, S. G. (2004). Evolution of minimal-gene-sets in host-dependent bacteria. Trends Microbiol. 12, 37–43. doi: 10.1016/j.tim.2003.11.006
Klein, A., Schrader, L., Gil, R., Manzano-Marín, A., Flórez, L., Wheeler, D., et al. (2015). A novel intracellular mutualistic bacterium in the invasive ant Cardiocondyla obscurior. ISME J. doi: 10.1038/ismej.2015.119. [Epub ahead of print].
Kleiner, M., Wentrup, C., Lott, C., Teeling, H., Wetzel, S., Young, J., et al. (2012). Metaproteomics of a gutless marine worm and its symbiotic microbial community reveal unusual pathways for carbon and energy use. Proc. Natl. Acad. Sci. U.S.A. 109, E1173–E1182. doi: 10.1073/pnas.1121198109
Krupke, A., Lavik, G., Halm, H., Fuchs, B. M., Amann, R. I., and Kuypers, M. M. M. (2014). Distribution of a consortium between unicellular algae and the N2 fixing cyanobacterium UCYN-A in the North Atlantic Ocean. Environ. Microbiol. 16, 3153–3167. doi: 10.1111/1462-2920.12431
Kühner, S., van Noort, V., Betts, M. J., Leo-Macias, A., Batisse, C., Rode, M., et al. (2009). Proteome organization in a genome-reduced bacterium. Science 326, 1235–1240. doi: 10.1126/science.1176343
Kurata, T., Nakanishi, S., Hashimoto, M., Taoka, M., Yamazaki, Y., Isobe, T., et al. (2015). Novel essential gene involved in 16S rRNA processing in Escherichia coli. J. Mol. Biol. 427, 955–965. doi: 10.1016/j.jmb.2014.12.013
Kuwahara, H., Yoshida, T., Takaki, Y., Shimamura, S., Nishi, S., Harada, M., et al. (2007). Reduced genome of the thioautotrophic intracellular symbiont in a deep-sea clam, Calyptogena okutanii. Curr. Biol. 17, 881–886. doi: 10.1016/j.cub.2007.04.039
Lazcano, A., Díaz-Villagómez, E., Mills, T., and Oró, J. (1995). On the levels of enzymatic substrate specificity: implications for the early evolution of metabolic pathways. Adv. Space Res. 15, 345–356. doi: 10.1016/S0273-1177(99)80106-9
Lluch-Senar, M., Delgado, J., Chen, W. H., Lloréns-Rico, V., O'Reilly, F. J., Wodke, J. A., et al. (2015). Defining a minimal cell: essentiality of small ORFs and ncRNAs in a genome-reduced bacterium. Mol. Syst. Biol. 11, 780. doi: 10.15252/msb.20145558
López-Madrigal, S., Latorre, A., Porcar, M., Moya, A., and Gil, R. (2011). Complete genome sequence of “Candidatus Tremblaya princeps” strain PCVAL, an intriguing translational machine below the living-cell status. J. Bacteriol. 193, 5587–5588. doi: 10.1128/JB.05749-11
Luef, B., Frischkorn, K. R., Wrighton, K. C., Holman, H. Y. N., Birarda, G., Thomas, B. C., et al. (2015). Diverse uncultivated ultra-small bacterial cells in groundwater. Nat. Comm. 6, 6372. doi: 10.1038/ncomms7372
Luisi, P. L., Ferri, F., and Stano, P. (2006). Approaches to semi-synthetic minimal cells: a review. Naturwissenschaften 93, 1–13. doi: 10.1007/s00114-005-0056-z
Markert, S., Arndt, C., Felbeck, H., Becher, D., Sievert, M., Hügler, M., et al. (2007). Physiological proteomics of the uncultured endosymbiont of >Riftia pachyptila. Science 315, 247–250. doi: 10.1126/science.1132913
Martínez-Cano, D. J., Reyes-Prieto, M., Martínez-Romero, E., Partida-Martínez, L. P., Latorre, A., Moya, A., et al. (2015). Evolution of small prokaryotic genomes. Front. Microbiol. 5:742. doi: 10.3389/fmicb.2014.00742
McCutcheon, J. P., and von Dohlen, C. D. (2011). An interdependent metabolic patchwork in the nested symbiosis of mealybugs. Curr. Biol. 21, 1366–1372. doi: 10.1016/j.cub.2011.06.051
Mears, J. A., Cannone, J. J., Stagg, S. M., Gutell, R. R., Agrawal, R. K., and Harvey, S. C. (2002). Modeling a minimal ribosome based on comparative sequence analysis. J. Mol. Biol. 321, 215–234. doi: 10.1016/S0022-2836(02)00568-5
Mira, A., Ochman, H., and Moran, N. A. (2001). Deletional bias and the evolution of bacterial genomes. Trends Genet. 17, 589–596. doi: 10.1016/S0168-9525(01)02447-7
Moran, N. A. (1996). Accelerated evolution and Muller's rachet in endosymbiotic bacteria. Proc. Natl. Acad. Sci. U.S.A. 93, 2873–2878. doi: 10.1073/pnas.93.7.2873
Moran, N. A., and Bennett, G. M. (2014). The tiniest tiny genomes. Annu. Rev. Microbiol. 68, 195–215. doi: 10.1146/annurev-micro-091213-112901
Morange, M. (2008). What history tell us XIII. Fifty years of the central dogma. J. Biosci. 33, 171–175. doi: 10.1007/s12038-008-0034-7
Morowitz, H. J. (1992). Beginnings of Cellular Life: Metabolism Recapitulates Biogenesis. New Haven: Yale University Press.
Morris, J. J., Lenski, R. E., and Zinser, E. R. (2012). The Black Queen hypothesis: evolution of dependencies through adaptive gene loss. Mbio 3, e00036–e00012. doi: 10.1128/mbio.00036-12
Moya, A., Peretó, J., Gil, R., and Latorre, A. (2008). Learning how to live together: genomic insights into prokaryote-animal symbioses. Nat. Rev. Genet. 9, 218–229. doi: 10.1038/nrg2319
Mushegian, A. (1999). The minimal genome concept. Curr. Opin. Genet. Dev. 9, 709–714. doi: 10.1016/S0959-437X(99)00023-4
Mushegian, A. R., and Koonin, E. V. (1996). A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc. Natl. Acad. Sci. U.S.A. 93, 10268–10273. doi: 10.1073/pnas.93.19.10268
Nakagawa, S., Shimamura, S., Takaki, Y., Suzuki, Y., Murakami, S., Watanabe, T., et al. (2014). Allying with armored snails: the complete genome of gammaproteobacterial endosymbiont. ISME J. 8, 40–51. doi: 10.1038/ismej.2013.131
Newton, I. L. G., Woyke, T., Auchtung, T. A., Dilly, G. F., Dutton, R. J., Fisher, M. C., et al. (2007). The Calyptogena magnifica chemoautotrophic symbiont genome. Science 315, 998–1000. doi: 10.1126/science.1138438
Patiño-Navarrete, R., Piulachs, M. D., Belles, X., Moya, A., Latorre, A., and Peretó, J. (2014). The cockroach Blattella germanica obtains nitrogen from uric acid through a metabolic pathway shared with its bacterial endosymbiont. Biol. Lett. 10:20140407. doi: 10.1098/rsbl.2014.0407
Peretó, J. (2011). “Origin and evolution of metabolisms,” in Origins and Evolution of Life. An Astrobiological Perspective, eds M. Gargaud, P. López-Garcìa, and H. Martin (Cambridge: Cambridge University Press), 270–288. doi: 10.1017/CBO9780511933875.020
Peretó, J. (2012). Out of fuzzy chemistry: from prebiotic chemistry to metabolic networks. Chem. Soc. Rev. 41, 5394–53403. doi: 10.1039/c2cs35054h
Pérez-Brocal, V., Gil, R., Ramos, S., Lamelas, A., Postigo, M., Michelena, J. M., et al. (2006). A small microbial genome: the end of a long symbiotic relationship? Science 314, 312–313. doi: 10.1126/science.1130441
Petrov, A. S., Bernier, C. R., Hsiao, C., Norris, A. M., Kovacs, N. A., Waterbury, C. C., et al. (2014). Evolution of the ribosome at atomic resolution. Proc. Natl. Acad. Sci. U.S.A. 111, 10251–10256. doi: 10.1073/pnas.1407205111
Prell, J., and Poole, P. (2006). Metabolic changes of rhizobia in legume nodules. Trends Microbiol. 14, 161–168. doi: 10.1016/j.tim.2006.02.005
Reyes-Prieto, M., Latorre, A., and Moya, A. (2014). Scanty microbes, the ‘symbionelle’ concept. Environ. Microbiol. 16, 335–338. doi: 10.1111/1462-2920.12220
Robidart, J. C., Bench, S. R., Feldman, R. A., Novoradovsky, A., Podell, S. B., Gaasterland, T., et al. (2008). Metabolic versatility of the Riftia pachyptila endosymbiont revealed through metagenomics. Environ. Microbiol. 10, 727–737. doi: 10.1111/j.1462-2920.2007.01496.x
Roeselers, G., Newton, I. L., Woyke, T., Auchtung, T. A., Dilly, G. F., Dutton, R. J., et al. (2010). Complete genome sequence of Candidatus Ruthia magnifica. Stand. Genomic Sci. 3, 163–173. doi: 10.4056/sigs.1103048
Samayoa, J., Yildiz, F. H., and Karplus, K. (2011). Identification of prokaryotic small proteins using a comparative genomic approach. Bioinformatics 27, 1765–1771. doi: 10.1093/bioinformatics/btr275
Scott, K. M., Sievert, S. M., Abril, F. N., Ball, L. A., Barrett, C. J., Blake, R. A., et al. (2006). The genome of deep-sea vent chemolithoautotroph Thiomicrospira crunogena XCL-2. PLoS Biol. 4:e383. doi: 10.1371/journal.pbio.0040383
Soma, A., Ikeuchi, Y., Kanemasa, S., Kobayashi, K., Ogasawara, N., Ote, T., et al. (2003). An RNA-modifying enzyme that governs both the codon and amino acid specificities of isoleucine tRNA. Mol. Cell 12, 689–698. doi: 10.1016/S1097-2765(03)00346-0
Srinivasan, M., Mehta, P., Yu, Y., Prugar, E., Koonin, E. V., Karzai, A. W., et al. (2011). The highly conserved KEOPS/EKC complex is essential for a universal tRNA modification, t6A. EMBO J. 30, 873–878. doi: 10.1038/emboj.2010.343
Tawfik, D. S. (2010). Messy biology and the origins of evolutionary innovations. Nat. Chem. Biol. 6, 692–696. doi: 10.1038/nchembio.441
Thomas, G. H., Zucker, J., MacDonald, S. J., Sorokin, A., Goryanin, I., and Douglas, A. E. (2009). A fragile metabolic network adapted for cooperation in the symbiotic bacterium Buchnera aphidicola. BMC Syst. Biol. 3:24. doi: 10.1186/1752-0509-3-24
Thompson, A. W., Foster, R. A., Krupke, A., Carter, B. J., Musat, N., Vaulot, D., et al. (2012). Unicellular cyanobacterium symbiotic with a single-celled eukaryotic alga. Science 337, 1546–1550. doi: 10.1126/science.1222700
Tripp, H. J., Bench, S. R., Turk, K. A., Foster, R. A., Desany, B. A., Niazi, F., et al. (2010). Metabolic streamlining in an open-ocean nitrogen-fixing cyanobacterium. Nature 464, 90–94. doi: 10.1038/nature08786
Wodke, J. A., Puchałka, J., Lluch-Senar, M., Marcos, J., Yus, E., Godinho, M., et al. (2013). Dissecting the energy metabolism in Mycoplasma pneumoniae through genome-scale metabolic modeling. Mol. Syst. Biol. 9, 653. doi: 10.1038/msb.2013.6
Woyke, T., Teeling, H., Ivanova, N., Huntemann, M., Richter, M., Gloeckner, F., et al. (2006). Symbiosis insights through metagenomic analysis of a microbial consortium. Nature 443, 950–955. doi: 10.1038/nature05192
Xavier, J. C., Patil, K. R., and Rocha, I. (2014). Systems biology perspectives on minimal and simpler cells. Microbiol. Mol. Biol. Rev. 78, 487–509. doi: 10.1128/MMBR.00050-13
Yčas, M. (1974). On earlier states of the biochemical system. J. Theor. Biol. 44, 145–160. doi: 10.1016/S0022-5193(74)80035-4
Yus, E., Maier, T., Michalodimitrakis, K., van Noort, V., Yamada, T., Chany, W.-H., et al. (2009). Impact of genome reduction on bacterial metabolism and its regulation. Science 326, 1263–1268. doi: 10.1126/science.1177263
Keywords: Black Queen hypothesis, minimal cell, minimal metabolism, Muller's ratchet, streamlining hypothesis, synthetic biology, tRNA post-transcriptional modifications
Citation: Gil R and Peretó J (2015) Small genomes and the difficulty to define minimal translation and metabolic machineries. Front. Ecol. Evol. 3:123. doi: 10.3389/fevo.2015.00123
Received: 17 June 2015; Accepted: 13 October 2015;
Published: 28 October 2015.
Edited by:
Luis Delaye, CINVESTAV, MexicoReviewed by:
Agustino Martinez-Antonio, Centro de Investigacion y de Estudios Avanzados del IPN Department of Mathematics, MexicoYo Suzuki, J. Craig Venter Institute, USA
Copyright © 2015 Gil and Peretó. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosario Gil, rosario.gil@uv.es;
Juli Peretó, pereto@uv.es