
- 1 Human Nutrition Research Centre, Institute for Ageing and Health, Newcastle University, Newcastle upon Tyne, UK
- 2 Human Nutrition Unit, Department of Oncology, University of Sheffield, Sheffield, UK
Increasing evidence supports the developmental origins of adult health and disease hypothesis which argues for a causal relationship between adverse early life nutrition and increased disease risk in adulthood. Modulation of epigenetic marks, e.g., DNA methylation and consequential altered gene expression, has been proposed as a mechanism mediating these effects. Via its role as a methyl donor, dietary folate supply may influence DNA methylation. As aberrant methylation is an early event in colorectal cancer (CRC) pathogenesis, we hypothesized low maternal and/or post-weaning folate intake may influence methylation of genes involved in CRC development. We investigated the effects of maternal folate depletion during pregnancy and lactation on selected gene methylation in the small intestine of wild type (WT) and Apc+/Min mice at weaning and as adults. We also investigated the effects of folate depletion post-weaning on gene methylation in adult mice. Female C57Bl6/J mice were fed low or normal folate diets from mating with Apc+/Min males to the end of lactation. A sub-set of offspring were killed at weaning. Remaining offspring were weaned on to low or normal folate diets, resulting in four treatment groups of Apc+/Min and WT mice. p53 was more methylated in weaning and adult WT compared with Apc+/Min mice (p > 0.001). Igf2 and Apc were hypermethylated in adult Apc+/Min compared with WT mice (p = 0.004 and 0.012 respectively). Low maternal folate reduced p53 methylation in adults (p = 0.04). Low post-weaning folate increased Apc methylation in Apc+/Min mice only (p = 0.008 for interaction). These observations demonstrate that folate depletion in early life can alter epigenetic marks in a gene-specific manner. Also, the differential effects of altered folate supply on DNA methylation in WT and Apc+/Min mice suggest that genotype may modulate epigenetic responses to environmental cues and may have implications for the development of personalized nutrition.
Introduction
The developmental origins of health and disease (DOHaD) hypothesis proposes that exposures during early life modulate disease risk in adulthood. Indeed, there is substantial evidence for an association between lower birth weight and increased risk of type 2 diabetes, coronary heart disease, and hypertension, which has been attributed to poor nutrition in utero (Barker, 2004). These observations indicate the potential for a degree of plasticity during development which allows the fetal phenotype to be altered in response to environment cues (Bateson et al., 2004) in ways that may prepare it for the anticipated post-natal environment (Gluckman et al., 2005). To persist into adulthood and to affect disease risk, these environmentally orchestrated programming events must “mark” the animal at a molecular, cellular, and/or tissue level in ways that are sustained for much of the life-course and which impact on the processes which lead to the development of disease.
Epigenetic marks, including DNA methylation and covalent histone modifications, are established during embryonic and fetal development and constitute a rich information source layered on top of the DNA sequence. These marks contribute to the regulation of transcription, allowing cell specific gene expression which is essential for cell differentiation (Bird, 2002). DNA methylation, the most commonly investigated epigenetic mark, can be altered in offspring in response to maternal nutrition and these changes are associated with changes in gene expression and in the phenotype of the progeny (Waterland and Jirtle, 2003; Lillycrop et al., 2005; Dolinoy et al., 2006).
Since folate is a key dietary source of methyl groups for synthesis of S-adenosyl methionine (SAM) – the universal methyl donor – it is an attractive candidate nutrient for the modulation of DNA methylation. Furthermore, many studies highlight the effects of maternal folate status on DNA methylation in offspring (Lillycrop et al., 2005; Sinclair et al., 2007; Kim et al., 2009; Steegers-Theunissen et al., 2009; McKay et al., 2011).
As aberrant DNA methylation is known to be an early event in the pathogenesis of colorectal cancer (CRC; Feinberg et al., 2006), we hypothesized that maternal folate supply may influence CRC development in adult offspring through manipulation of the epigenome in utero. Indeed, we have reported global DNA hypomethylation in the small intestine (SI) of adult mice born to folate-depleted mothers compared with those born to folate replete mothers (McKay et al., 2011). Furthermore, we established that maternal folate depletion altered DNA methylation in a gene-specific manner in the fetal gut (McKay et al., unpublished data), suggesting that some epigenetic marks in developing offspring are modifiable in response to altered maternal folate intake. Understanding these early epigenetic events may help to define appropriate standards for maternal nutrition during pregnancy to optimize adult gut health in the next generation.
Apc+/Min mice, which carry a STOP mutation at codon 850 in the Apc gene and develop multiple intestinal neoplasms spontaneously, have been used widely as a model of CRC (Su et al., 1992). Following depleted or normal maternal folate supply from mating until the end of lactation, we investigated the methylation status of candidate genes in Apc+/Min and in wild type (WT) littermates to explore the potential effects of a nutritional insult (reduced folate supply) during early life on DNA methylation in the context of risk of CRC. For this purpose, we selected candidate genes implicated in the development of CRC and which have exhibited methylation changes in CRC (Igf2, p53, p16, and Apc; Burri et al., 2001; Feinberg et al., 2006; Segditsas et al., 2008; Baba et al., 2010). Furthermore, to investigate the potentially cumulative effects of both maternal and post-weaning folate depletion (a double nutritional insult), we also examined DNA methylation in adult Apc+/Min and WT mice fed the low folate diet from weaning.
Materials and Methods
Animal Housing and Husbandry
All animal procedures were approved by the Newcastle University Ethics Review Committee and the Home Office. Animals were housed in the Comparative Biology Centre (Newcastle University) at 20–22°C with 12 h light/dark cycles. Fresh water was available ad libitum. Experimental diets were based on the AIN-93G rodent diets (Reeves et al., 1993) and have been described previously (McKay et al., 2008). Mating trios [two C57BL/6J females (8 weeks old), one Apc+/Min male) were offered 6 g/day/mouse of a normal folate (2 mg folic acid/kg diet) or low folate (0.4 mg folic acid/kg diet) diet. Pregnant females were re-caged and offered 10 g/day/mouse of their allocated diet throughout pregnancy. At 2 weeks post-partum, diet quantity was increased (20 g/day/mouse).
At weaning (mean 32 days post-partum), offspring were genotyped (as described in McKay et al., 2008) and a randomly chosen sub-set of offspring, and all dams were killed for sample collection. The remaining offspring were re-caged (one to four per cage) and randomly assigned to a post-weaning diet of either normal folate (2 mg folic acid/kg diet) or low folate (0.4 mg folic acid/kg diet) diet (6 g/day/mouse). At an average of 96 days old, mice were killed for sample collection.
The effects of a low folate diet during pregnancy and/or post-weaning on the outcome of pregnancy, offspring growth, folate status, and tumor number and size (in Apc+/Min mice), have been reported previously (McKay et al., 2008).
Sample Collection
Animals were anesthetized using gaseous isoflurane followed by cervical dislocation. The entire SI from the stomach to the cecum was removed and cut into two sections of equal length, the proximal and the terminal SI. These sections were then opened longitudinally, washed with PBS, and examined for tumors before being wrapped in foil and snap frozen in liquid nitrogen. Samples were stored at −80°C until DNA was extracted.
DNA Extraction and Gene-Specific DNA Methylation Assessment
DNA was extracted (including RNase treatment) using a Qiagen DNA mini kit (Qiagen-51306) following the manufacturer’s protocol from normal appearing (i.e., an area of the gut where no macroscopic tumors were present) proximal SI tissue of juvenile and adult Apc+/Min and WT mice.
Two micrograms of genomic DNA were bisulfite treated, purified, desulfonated, and ethanol precipitated as detailed in Belshaw et al. (2004). A one-twentieth volume was used as a template in PCR reactions to amplify CpG rich regions of p53, Igf2, Apc, and p16 genes using primers detailed in Table 1. Nested PCR reactions were required to amplify p53 and Igf2, therefore from the initial PCR, using F1 and R1 primers, 2 μl was transferred to a subsequent PCR reaction using F2 and R2 primers (see Table 1). All PCR reactions were carried out in 20 μl volume and contained 1 Unit HotStarTaq (Qiagen) and 0.75 pmol of each forward and reverse primer. Other reagents used for specific PCR reaction are detailed in Table 1. Cycling conditions were 95°C for 15 min × 1, 35 cycles of 95°C for 30 s, annealing temperature (see Table 1) for 30 s, 72°C for 30 s or 1 min (Table 1) followed by a final 10 min extension at 72°C, and then held at 4°C. CpG methylation status of the four genes was then determined by a combined bisulfite restriction analysis (COBRA) assay (Xiong and Laird, 1997). PCR products were digested overnight with the appropriate restriction enzyme (Table 1). Digested DNA was separated by gel electrophoresis (3% agarose) and gels stained with 0.01% SYBR green I in 1× TBE for 4 h. DNA SYBR green fluorescence was captured using a UV camera (UVItech Limited) and band intensities were quantified using UVIband software (UVItech Limited). The percentage DNA methylation was then calculated from the relative intensities of the digested and undigested PCR products.
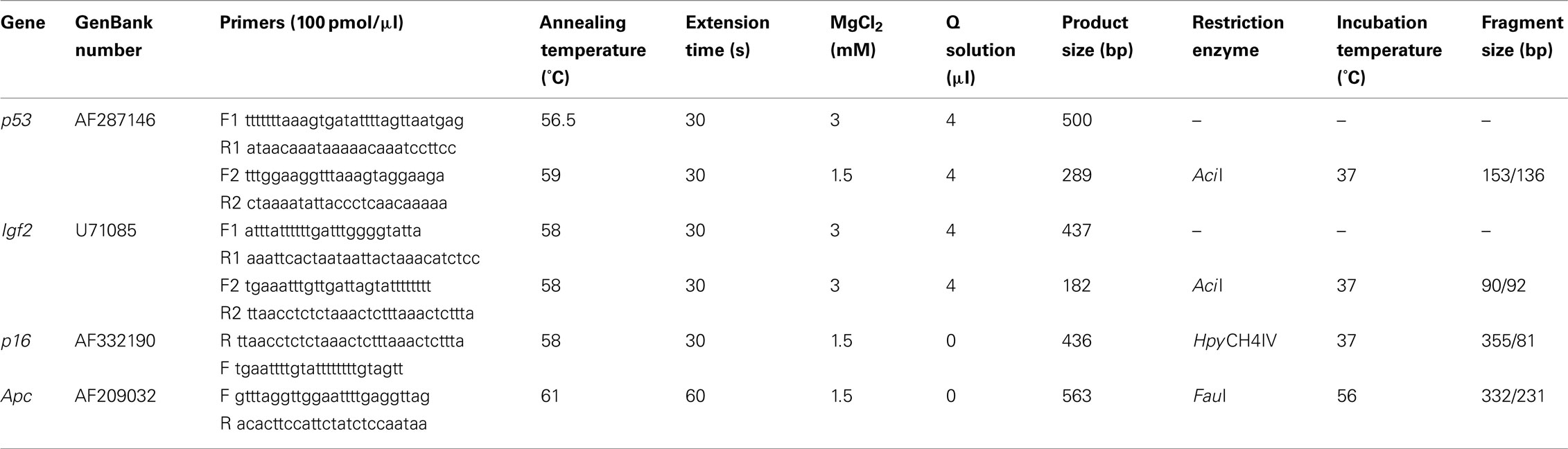
Table 1. PCR and enzyme digestion details for COBRA assays to detect gene-specific DNA methylation.
Statistical Analysis
Data distributions from this 2 × 2 factorially designed study (two levels of maternal folate supply and two levels of folate supply to the offspring post-weaning) were examined by the Kolmogorov–Smirnov test. All data sets were normally distributed. Analysis of variance was used to examine the effects of sex, genotype, and maternal and post-weaning diets, and interactions between maternal and post-weaning folate supply, on methylation of selected genes in the SI of weaning aged and adult mice.
Results
Effects of Sex and Genotype on Gene-Specific DNA Methylation in SI
There were no differences between male and female mice in methylation at any of the loci investigated in offspring killed at either weaning or in adulthood (aged 96 days).
At weaning there was no difference in percentage methylation at the Igf2, p16, or Apc loci in Apc+/Min compared with WT mice but the p53 locus was significantly (p < 0.001) more methylated in WT mice (Figure 1).
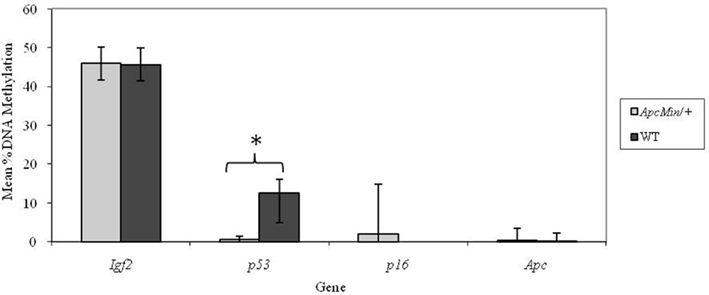
Figure 1. Effect of genotype on gene-specific DNA methylation (%) in the proximal SI of weaning mice (mean age 32 days). n = 10 for all groups. Error bars represent 95% confidence intervals. *p < 0.05.
In adult mice, genotype did not affect DNA methylation at the p16 locus. As we observed at weaning, methylation at the p53 locus was significantly (p < 0.001) higher in WT mice (Figure 2) in adulthood. Furthermore, both the Igf2 and Apc loci were more highly methylated in Apc+/Min mice compared with WT mice (p = 0.004 and 0.012 respectively).
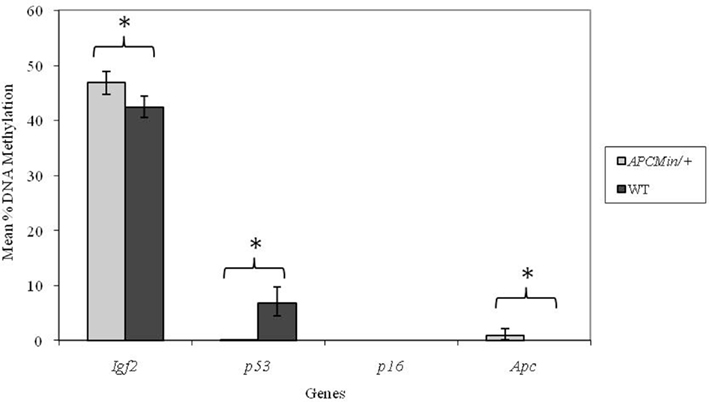
Figure 2. Effect of genotype on gene-specific DNA methylation (%) in the proximal SI of adult mice (mean age 96 days). Igf2, n = 20 and 21; p53, n = 20 and 20; p16, n = 19 and 19; and Apc, n = 19 and 18 for Apc+/Min and WT mice respectively. Error bars represent 95% confidence intervals. *p < 0.05.
Effects of Low Maternal Dietary Folate on Gene-Specific DNA Methylation
No significant effects of low maternal dietary folate during pregnancy and lactation were observed on DNA methylation at the loci investigated in weaning mice (data not shown).
However, adult mice born to low folate fed mothers had a lower percentage methylation at the p53 locus compared with mice born to normal folate fed mothers (p = 0.04; Figure 3). There were no detectable effects (p > 0.05) of low maternal folate intake at other loci in adult mice (Figure 3).
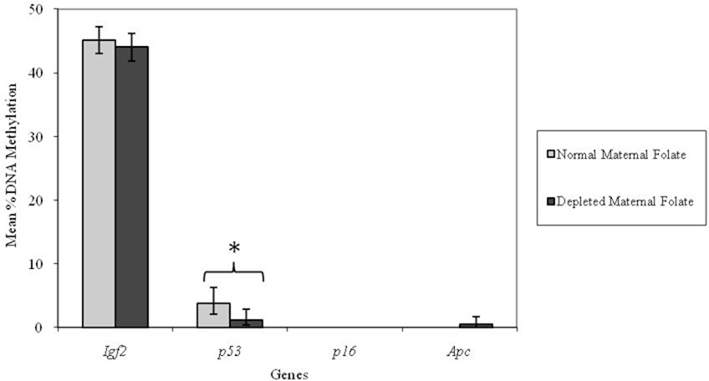
Figure 3. Effect of depleted versus normal maternal dietary folate supply on gene-specific DNA methylation (%) in proximal SI of adult offspring (both Apc+/Min and WT mice combined; mean age 96 days). Igf2, n = 21 and 20; p53, n = 20 and 20; p16, n = 20 and 18; and Apc, n = 19 and 18 for normal and folate-depleted maternal diets respectively. Error bars represent 95% confidence intervals. *p < 0.05.
Effects of Low Post-Weaning Dietary Folate on Gene-Specific DNA Methylation
Altering dietary folate supply from weaning had no significant effect on DNA methylation at the Igf2, p53, or p16 loci in adult mice (Figure 4). However, the Apc locus was found to be unmethylated in adult mice fed normal folate post-weaning diets, but methylated in mice fed folate-depleted diets. This difference in mean Apc methylation was statistically significant (p = 0.009; Figure 4). Furthermore, there was a significant interaction between genotype and post-weaning diet for methylation at the Apc locus (p = 0.008). Mean methylation of the Apc gene in Apc+/Min mice fed the folate-depleted diet post-weaning was 4.2% whereas methylation was undetectable in Apc+/Min and WT mice fed the normal folate diet. There were no significant interactions between maternal and post-weaning folate supply for methylation at any of the loci investigated.
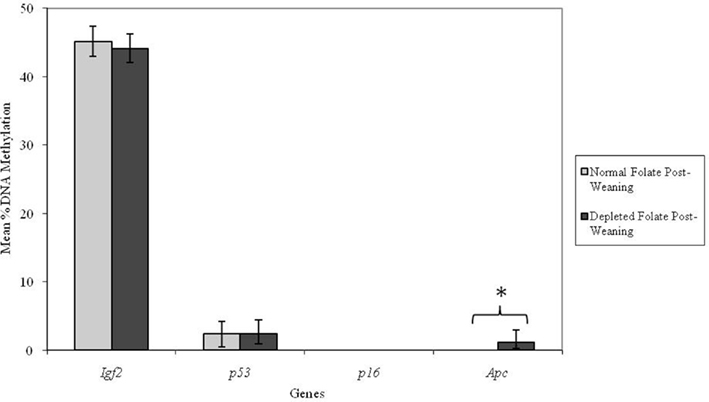
Figure 4. Effect of dietary folate supply from weaning on gene-specific DNA methylation (%) in proximal SI of adult offspring (both Apc+/Min and WT mice combined; mean age 96 days). Igf2, n = 21 and 20; p53, n = 20 and 20; p16, n = 20 and 18; and Apc, n = 19 and 18 for normal and folate-depleted diets respectively. Error bars represent 95% confidence intervals. *p < 0.05.
Discussion
Aberrant DNA methylation is a cardinal feature of many cancers (Watanabe and Maekawa, 2010). Although abnormal DNA methylation patterns are an early event in CRC development, it is currently unclear whether the observed changes in DNA methylation are causal for cancer development or a consequence of it. In the study described here, we report hypomethylation at the p53 locus in the SI of Apc+/Min mice compared with WT mice at weaning. At this age (mean 32 days), no macroscopic lesions were apparent in the SI so the observed difference in p53 methylation appears to precede tumor development in this model. Furthermore, the difference in p53 methylation between Apc+/Min and WT mice was also observed in adults (mean 96 days of age), indicating that these epigenetic changes are sustained during the period of development of macroscopic tumors (see McKay et al., 2008 for details of tumor size and multiplicity). Previously, hypomethylation within exons 5–8 of the p53 gene has been associated with colon tumorigenesis in the DMH rat model (Kim et al., 1996) and p53 hypomethylation has been associated with increased mutation rates within this gene (Tornaletti and Pfeifer, 1995). In addition, loss of function of the p53 tumor suppressor gene is common in the progression of late adenoma to carcinoma in humans (Fearon, 2011). In our study, mice with genetically inherited loss of function of one Apc allele had lower methylation at the p53 locus than their WT littermates. To our knowledge, this is the first report of a collateral epigenetic effect on a tumor suppressor gene (p53) resulting from a germ-line mutation in another tumor suppressor gene (Apc). Such effects might be anticipated in tumors per se but the present discovery is all the more remarkable because it was observed in macroscopically normal tissue. It will be important to seek confirmation of this finding in independent studies and to investigate the mechanism responsible. Since reduced p53 methylation is expected to be accompanied by increased expression of the corresponding tumor suppressor gene and hence greater resistance to tumorigenesis, the sustained lower p53 methylation in the tumor-prone Apc+/Min mice is surprising. It is possible that the altered p53 methylation is unconnected mechanistically with tumor development. This would be expected if the methylation change at the specific CpG site probed by the present COBRA assay does not signal a change in p53 transcription. Further investigation of the functional consequences of this apparently novel genotype:epigenetic interaction is warranted.
In contrast to our findings of genotype-related p53 hypomethylation, we observed that the Igf2 and Apc genes were more highly methylated in adult Apc+/Min mice compared with WT mice. Hypermethylation at the Apc locus is widely documented in human CRC (Venkatachalam et al., 2010) and may contribute to the loss of function through silencing of the second (non-mutated) allele of this tumor suppressor gene described as the gate-keeper of the colorectum (Kwong and Dove, 2009). Both hypo- and hypermethylation at the Igf2 locus have been reported in human cases of CRC (Issa et al., 1996; Nakagawa et al., 2001; Cui et al., 2002; Baba et al., 2010) with loss of imprinting at this locus believed to contribute to tumor growth (Cui et al., 2002).
Although dietary and other exposures during fetal and early post-natal life might be expected to influence risk of many complex diseases in adulthood, to date there is limited and conflicting evidence of associations between early life exposures and adult cancer incidence (Johnson et al., 2009). It is plausible that environmental factors that alter DNA methylation patterns during development may be early events influencing cancer development in later life, particularly as aberrant DNA methylation patterns are associated with most cancers (Watanabe and Maekawa, 2010) and is an early event in CRC (Feinberg et al., 2006). We therefore hypothesized that reducing maternal folate supply during pregnancy and lactation would stress the developing mouse’s capacity to methylate cellular macromolecules. This may therefore alter patterns of DNA methylation during fetal and early post-natal development. If these putative epigenetic derangements occurred in genes critical for cancer development, e.g., tumor suppressor genes then the maternal dietary insult might influence CRC risk in adult offspring. Indeed it was reported recently that early life exposure to famine was associated with the decreased risk of developing a CpG island methylator phenotype (CIMP) CRC (Hughes et al., 2009). Since the presence of a CIMP tumor is indicative of epigenetic instability coupled with transcriptional silencing of gene expression and microsatellite instability (Ferguson et al., 2004; Slattery et al., 2009), these findings suggest that early life exposures may indeed result in persistent epigenetic alterations that may influence risk of CRC development in later life. In the present study, we did not observe any differences in response to maternal dietary folate supply in methylation of our panel of cancer-related genes when examined in mice at weaning (mean 32 days of age). However, in adult offspring (mean age 96 days) we observed reduced methylation at the p53 locus (but no changes in the other three genes investigated) in offspring born to mothers fed the low folate diet. Previously we reported reduced genomic DNA methylation in the SI of adult offspring in response to low maternal folate intake (McKay et al., 2011) and we have reported elsewhere that the response to maternal folate supply was gene, and CpG site, specific in the fetal gut (McKay et al., unpublished data). Our observations in the present study support these earlier reports since we found that methylation changes in the offspring in response to reduced maternal folate supply are gene-specific. Taken together, these data strengthen the hypothesis that maternal folate intake modulates epigenomic patterns in the offspring. Furthermore, the changes reported here in respect of p53 methylation may indicate that such exposures have the potential to influence gut health in adulthood via epigenetic programming of some cancer related genes.
We report here that low post-weaning dietary folate intake caused hypermethylation at the Apc locus in Apc+/Min mice only. Although increased methylation in response to decreased folate intake may seem paradoxical, hypermethylation at the Apc locus has been reported in association with consumers of low folate/high alcohol in human cases of sporadic CRC (van Engeland et al., 2003). That this effect on Apc methylation occurred in Apc+/Min mice only (not in WT mice), suggests that there may be aberrations in the methylation machinery within SI of these tumor-predisposed mice. Since folate depletion is predicted to reduce cellular SAM supply, this will have implications for resource (methyl group) allocation to competing pathways within the cell and it may be that such competition, rather than derangements in the DNA methylation machinery per se, is responsible for the altered Apc methylation that we have observed. Indeed, in human CRC, hypermethylation of the APC promoter by folate depletion has been hypothesized to reduce expression of the gene (Esteller et al., 2000), which may be sufficient to initiate tumor development.
Finally, our observation of gene-specific responses to altered folate supply indicate that some genomic loci appear to be more labile than others in response to environmental cues such as altered nutrition. For example, it is well documented that metastable epialleles are epigenetically labile (“epilabile”) in response to folate and other dietary factors (Waterland and Jirtle, 2003; Dolinoy et al., 2006; Waterland et al., 2006). Currently, relatively little is known about the characteristics of DNA domains which make them more or less epilabile. In this study we measured DNA methylation of a panel of genes known to be involved in the development of CRC and sensitive to methylation changes in CRC. Of the four genes we studied, methylation of p53 was altered by maternal folate intake in both Apc+/Min and WT mice whereas Apc methylation was altered in Apc+/Min mice only, in response to low folate intake after weaning. The COBRA assay probes a single CpG site and it remains to be discovered whether the CpG site investigated here is characteristic of all the cytosine residues in the corresponding CpG island. It would be useful to extend this analysis using additional methodologies such as bisulfite sequencing or pyrosequencing which can measure several CpG sites within a given DNA domain. The only gene for which we did not observe any variation in DNA methylation was p16. Although aberrant DNA methylation of this gene has been observed in several human cancers (Auerkari, 2006), p16 appears to be largely unmethylated in non-neoplastic tissue (Belshaw et al., 2008). In agreement with these findings, in the present study we analyzed DNA from macroscopically normal mucosa and found that p16 methylation was undetectable in most samples – only very low levels of p16 methylation were detected in Apc+/Min mice at weaning. The present study adopted a candidate gene approach and it is also possible that other genes may also have altered DNA methylation in response to maternal and/or post-weaning dietary folate depletion. Now that we have established proof of principle that reduced folate supply in early life can modulate gene methylation, further studies using genome-wide approaches such as methylation arrays or next generation sequencing are warranted to identify “epilabile” loci and to investigate both the molecular and overall health consequences of such epigenetic events.
In summary, the results presented here provide proof of principle that reduced maternal folate supply during pregnancy and lactation and/or reduced folate supply from weaning can alter the methylation of genes involved in CRC development. In particular, we have shown that reduced maternal folate intake during pregnancy and lactation alters p53 methylation in the adult offspring. In addition, we have reported the apparently novel observation that genetically inherited loss of function of one Apc allele resulted in lower methylation at the p53 locus. To our knowledge this is the first reported observation of a collateral epigenetic effect on a tumor suppressor gene (p53) resulting from a germ-line mutation in another tumor suppressor gene (Apc). It will be important to replicate these novel findings in independent studies.
Contribution
The study was designed by John C. Mathers and Elizabeth A. Williams and laboratory work was undertaken by Jill A. McKay. All authors contributed to data analysis. Jill A. McKay and John C. Mathers wrote the manuscript with contributions from Elizabeth A. Williams. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Adele Kitching, CBC, Newcastle University for technical assistance. This project was funded by a BBSRC studentship held by Jill A. McKay (01/A1/D/17951) and by a World Cancer Research Fund (2001/37) grant (held by John C. Mathers and Elizabeth A. Williams). In addition, John C. Mathers and Jill A. McKay acknowledge support for the current analysis from the BBSRC through grant BB/ G007993/1.
References
Auerkari, E. I. (2006). Methylation of tumor suppressor genes p16(INK4a), p27(Kip1) and E-cadherin in carcinogenesis. Oral Oncol. 42, 5–13.
Baba, Y., Nosho, K., Shima, K., Huttenhower, C., Tanaka, N., Hazra, A., Giovannucci, E. L., Fuchs, C. S., and Ogino, S. (2010). Hypomethylation of the IGF2 DMR in colorectal tumors, detected by bisulfite pyrosequencing, is associated with poor prognosis. Gastroenterology 139, 1855–1864.
Barker, D. J. (2004). The developmental origins of well-being. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 1359–1366.
Bateson, P., Barker, D., Clutton-Brock, T., Deb, D., D’Udine, B., Foley, R. A., Gluckman, P., Godfrey, K., Kirkwood, T., Lahr, M. M., Mcnamara, J., Metcalfe, N. B., Monaghan, P., Spencer, H. G., and Sultan, S. E. (2004). Developmental plasticity and human health. Nature 430, 419–421.
Belshaw, N. J., Elliott, G. O., Foxall, R. J., Dainty, J. R., Pal, N., Coupe, A., Garg, D., Bradburn, D. M., Mathers, J. C., and Johnson, I. T. (2008). Profiling CpG island field methylation in both morphologically normal and neoplastic human colonic mucosa. Br. J. Cancer 99, 136–142.
Belshaw, N. J., Elliott, G. O., Williams, E. A., Bradburn, D. M., Mills, S. J., Mathers, J. C., and Johnson, I. T. (2004). Use of DNA from human stools to detect aberrant CpG island methylation of genes implicated in colorectal cancer. Cancer Epidemiol. Biomarkers Prev. 13, 1495–1501.
Burri, N., Shaw, P., Bouzourene, H., Sordat, I., Sordat, B., Gillet, M., Schorderet, D., Bosman, F. T., and Chaubert, P. (2001). Methylation silencing and mutations of the p14ARF and p16INK4a genes in colon cancer. Lab. Invest. 81, 217–229.
Cui, H., Onyango, P., Brandenburg, S., Wu, Y., Hsieh, C. L., and Feinberg, A. P. (2002). Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 62, 6442–6446.
Dolinoy, D. C., Weidman, J. R., Waterland, R. A., and Jirtle, R. L. (2006). Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ. Health Perspect. 114, 567–572.
Esteller, M., Sparks, A., Toyota, M., Sanchez-Cespedes, M., Capella, G., Peinado, M. A., Gonzalez, S., Tarafa, G., Sidransky, D., Meltzer, S. J., Baylin, S. B., and Herman, J. G. (2000). Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res. 60, 4366–4371.
Feinberg, A. P., Ohlsson, R., and Henikoff, S. (2006). The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 7, 21–33.
Ferguson, L. R., Karunasinghe, N., and Philpott, M. (2004). Epigenetic events and protection from colon cancer in New Zealand. Environ. Mol. Mutagen. 44, 36–43.
Gluckman, P. D., Hanson, M. A., Morton, S. M., and Pinal, C. S. (2005). Life-long echoes – a critical analysis of the developmental origins of adult disease model. Biol. Neonate 87, 127–139.
Hughes, L. A., Van Den Brandt, P. A., De Bruine, A. P., Wouters, K. A., Hulsmans, S., Spiertz, A., Goldbohm, R. A., De Goeij, A. F., Herman, J. G., Weijenberg, M. P., and van Engeland, M. (2009). Early life exposure to famine and colorectal cancer risk: a role for epigenetic mechanisms. PLoS ONE 4, e7951. doi: 10.1371/journal.pone.0007951.
Issa, J. P., Vertino, P. M., Boehm, C. D., Newsham, I. F., and Baylin, S. B. (1996). Switch from monoallelic to biallelic human IGF2 promoter methylation during aging and carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 93, 11757–11762.
Johnson, K. J., Springer, N. M., Bielinsky, A. K., Largaespada, D. A., and Ross, J. A. (2009). Developmental origins of cancer. Cancer Res. 69, 6375–6377.
Kim, J. M., Hong, K., Lee, J. H., Lee, S., and Chang, N. (2009). Effect of folate deficiency on placental DNA methylation in hyperhomocysteinemic rats. J. Nutr. Biochem. 20, 172–176.
Kim, Y. I., Pogribny, I. P., Salomon, R. N., Choi, S. W., Smith, D. E., James, S. J., and Mason, J. B. (1996). Exon-specific DNA hypomethylation of the p53 gene of rat colon induced by dimethylhydrazine. Modulation by dietary folate. Am. J. Pathol. 149, 1129–1137.
Kwong, L. N., and Dove, W. F. (2009). APC and its modifiers in colon cancer. Adv. Exp. Med. Biol. 656, 85–106.
Lillycrop, K. A., Phillips, E. S., Jackson, A. A., Hanson, M. A., and Burdge, G. C. (2005). Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J. Nutr. 135, 1382–1386.
McKay, J. A., Waltham, K. J., Williams, E. A., and Mathers, J. C. (2011). Folate depletion during pregnancy and lactation reduces genomic DNA methylation in murine adult offspring. Genes Nutr. 6, 189–196.
McKay, J. A., Williams, E. A., and Mathers, J. C. (2008). Gender-specific modulation of tumorigenesis by folic acid supply in the Apc mouse during early neonatal life. Br. J. Nutr. 99, 550–558.
Nakagawa, H., Chadwick, R. B., Peltomaki, P., Plass, C., Nakamura, Y., and De La Chapelle, A. (2001). Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc. Natl. Acad. Sci. U.S.A. 98, 591–596.
Reeves, P. G., Nielsen, F. H., and Fahey, G. C. Jr. (1993). AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J. Nutr. 123, 1939–1951.
Segditsas, S., Sieber, O. M., Rowan, A., Setien, F., Neale, K., Phillips, R. K., Ward, R., Esteller, M., and Tomlinson, I. P. (2008). Promoter hypermethylation leads to decreased APC mRNA expression in familial polyposis and sporadic colorectal tumours, but does not substitute for truncating mutations. Exp. Mol. Pathol. 85, 201–206.
Sinclair, K. D., Allegrucci, C., Singh, R., Gardner, D. S., Sebastian, S., Bispham, J., Thurston, A., Huntley, J. F., Rees, W. D., Maloney, C. A., Lea, R. G., Craigon, J., Mcevoy, T. G., and Young, L. E. (2007). DNA methylation, insulin resistance, and blood pressure in offspring determined by maternal periconceptional B vitamin and methionine status. Proc. Natl. Acad. Sci. U.S.A. 104, 19351–19356.
Slattery, M. L., Wolff, R. K., Curtin, K., Fitzpatrick, F., Herrick, J., Potter, J. D., Caan, B. J., and Samowitz, W. S. (2009). Colon tumor mutations and epigenetic changes associated with genetic polymorphism: insight into disease pathways. Mutat. Res. 660, 12–21.
Steegers-Theunissen, R. P., Obermann-Borst, S. A., Kremer, D., Lindemans, J., Siebel, C., Steegers, E. A., Slagboom, P. E., and Heijmans, B. T. (2009). Periconceptional maternal folic acid use of 400 μg per day is related to increased methylation of the IGF2 gene in the very young child. PLoS ONE 4, e7845. doi: 10.1371/journal.pone.0007845.
Su, L. K., Kinzler, K. W., Vogelstein, B., Preisinger, A. C., Moser, A. R., Luongo, C., Gould, K. A., and Dove, W. F. (1992). Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256, 668–670.
Tornaletti, S., and Pfeifer, G. P. (1995). Complete and tissue-independent methylation of CpG sites in the p53 gene: implications for mutations in human cancers. Oncogene 10, 1493–1499.
van Engeland, M., Weijenberg, M. P., Roemen, G. M., Brink, M., De Bruine, A. P., Goldbohm, R. A., Van Den Brandt, P. A., Baylin, S. B., De Goeij, A. F., and Herman, J. G. (2003). Effects of dietary folate and alcohol intake on promoter methylation in sporadic colorectal cancer: the Netherlands cohort study on diet and cancer. Cancer Res. 63, 3133–3137.
Venkatachalam, R., Ligtenberg, M. J., Hoogerbrugge, N., De Bruijn, D. R., Kuiper, R. P., and Geurts Van Kessel, A. (2010). The epigenetics of (hereditary) colorectal cancer. Cancer Genet. Cytogenet. 203, 1–6.
Waterland, R. A., Dolinoy, D. C., Lin, J. R., Smith, C. A., Shi, X., and Tahiliani, K. G. (2006). Maternal methyl supplements increase offspring DNA methylation at Axin Fused. Genesis 44, 401–406.
Waterland, R. A., and Jirtle, R. L. (2003). Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 23, 5293–5300.
Keywords: folate, gene-specific DNA methylation, in utero, Apc+/Min mouse, CRC
Citation: McKay JA, Williams EA and Mathers JC (2011) Effect of maternal and post-weaning folate supply on gene-specific DNA methylation in the small intestine of weaning and adult Apc+/Min and wild type mice. Front. Gene. 2:23. doi: 10.3389/fgene.2011.00023
Received: 05 March 2011;
Accepted: 11 May 2011;
Published online: 23 May 2011.
Edited by:
Beverly Sara Muhlhausler, The University of Adelaide, AustraliaReviewed by:
Lawrence P. Reynolds, North Dakota State University, USAMichael Carey Satterfield, Texas A&M University, USA
Copyright: © 2011 McKay, Williams and Mathers. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jill A. McKay, Human Nutrition Research Centre, Institute for Ageing and Health, Newcastle University, M2020, 2nd Floor, Cookson Building, Framlington Place, Newcastle upon Tyne NE2 4HH, UK. e-mail: jill.mckay@ncl.ac.uk