
- 1 Department of Genetics, The Alexander Silberman Institute of Life Sciences, Faculty of Science, The Hebrew University of Jerusalem, Jerusalem, Israel
- 2 School of Computer Science and Engineering, The Hebrew University of Jerusalem, Jerusalem, Israel
The intron–exon architecture of many eukaryotic genes raises the intriguing question of whether this unique organization serves any function, or is it simply a result of the spread of functionless introns in eukaryotic genomes. In this review, we show that introns in contemporary species fulfill a broad spectrum of functions, and are involved in virtually every step of mRNA processing. We propose that this great diversity of intronic functions supports the notion that introns were indeed selfish elements in early eukaryotes, but then independently gained numerous functions in different eukaryotic lineages. We suggest a novel criterion of evolutionary conservation, dubbed intron positional conservation, which can identify functional introns.
Introduction
Spliceosomal introns are one of the eukaryotic defining characters. With the exception of the highly reduced nucleomorph genome of Hemiselmis andersenii (Lane et al., 2007), introns are found in all fully sequenced eukaryotic genomes, including other nucleomorphs (Gilson et al., 2006). Intron density ranges from a handful in the entire genome of some protists (Mair et al., 2000; Morrison et al., 2007), to about eight per gene in human (Sakharkar et al., 2004).
The presence of introns in a genome is believed to impose substantial burden on the host. First, unlike self-splicing introns, the excision of spliceosomal introns requires a spliceosome, which is among the largest molecular complexes in the cell, comprising 5 snRNAs and more than 150 proteins (Wahl et al., 2009). Intron-bearing genomes must, of course, code for all these proteins and snRNAs. Many eukaryotes even harbor a second class of spliceosomal introns, called U12 introns, that are removed by another spliceosome (the minor spliceosome) whose protein content only partially overlaps with that of the major spliceosome (Will and Luhrmann, 2005). Second, intron transcription is costly in terms of time and energy. The energetic burden is probably tolerable (Lane and Martin, 2010), but an average RNA polymerase II (RNAP II) elongation rate of 60 bases per second (Singh and Padgett, 2009) means that the transcription of some long introns lasts many hours. Third, recognition of splicing junctions by the spliceosome is directed by a host of cis regulatory elements. This makes an organism vulnerable to synonymous (or even non-coding) mutations that otherwise would not have a noticeable effect. Indeed, it is estimated that more than 50% of human genetic disorders are caused by disruption of the normal splicing pattern (Lopez-Bigas et al., 2005; Wang and Cooper, 2007). Finally, malfunction of any of the snRNAs and proteins that are necessary for proper splicing will have a general detrimental effect on the cell.
The recognition of the potentially hazardous nature of introns had initiated a quest for function that would counter these deleterious effects. This had triggered Walter Gilbert to suggest, shortly after the discovery of the introns, what is now known as the intron-early theory (Gilbert, 1987). According to this theory, introns were pivotal in the formation of modern, complex, genes, by allowing for constant shuffling of small, primordial, mini-exons. Hence, introns must have existed in prokaryotes, only to be later eliminated completely from their genomes due to genome streamlining. The accumulation of fully sequenced eukaryotic genomes allowed for high resolution reconstruction of the evolutionary history of introns (Csuros, 2005; Nguyen et al., 2005; Carmel et al., 2007; Csuros et al., 2011). Consequently, the intron-early theory gave way to the view that spliceosomal introns first appeared during the early stages of eukaryogenesis, possibly from self-splicing intron forebears, and that their debut was shortly followed by massive invasion into the eukaryotic nuclear genome (Koonin, 2006, 2009; Martin and Koonin, 2006). It is currently estimated that the last eukaryotic common ancestor was intron-rich, populated with introns whose density was perhaps as high as 50–75% of the intron density in contemporary intron-rich mammals (Carmel et al., 2007; Csuros et al., 2011). According to this view, the first introns were, indeed, deleterious elements, and their spreading in eukaryotic genomes was possible due to severe population bottlenecks (Lynch, 2002; Martin and Koonin, 2006). At later times, episodes of massive intron gains seem to have been rare, generally limited to lineages that experienced significant evolutionary innovations, such as the emergence of opisthokonts (common ancestor of metazoan and fungi), metazoans, and plants (Carmel et al., 2007; Csuros et al., 2011). Many other lineages seem to have gone through phases of massive intron losses, leading to all those present-day intron-poor species.
This evolutionary scenario is compatible with the view that early introns lacked function. However, the mere existence of transcribed gene parts, that are free from selective constraints triggered an increase in genetic diversity that eventually led to the gain of many intron-related functions, up to the point that today they are absolutely essential in intron-rich species, as well as in many intron-poor ones (Lynch, 2007).
One of the best examples to a crucial intronic function in contemporary eukaryotes is the increase in protein abundance of intron-bearing genes. This effect was initially observed in simian vacuolating virus 40 constructs whose protein product was rendered undetectable upon the elimination of their introns (Gruss et al., 1979; Hamer et al., 1979). Using similar viral constructs, it was shown that intron removal already affects the mRNA level. In some cases intron-bearing constructs were expressed up to 400 times more than their intronless counterparts (Buchman and Berg, 1988). Subsequent works reported the same phenomenon to be associated with numerous other introns in many eukaryotic species, suggesting that this intronic function is wide-ranging (Le Hir et al., 2003). In plants, for example, this intronic effect had been widely described, and was even privileged in getting a unique name – intron-mediated enhancement (Mascarenhas et al., 1990; Luehrsen and Walbot, 1991; Akua et al., 2010). In fact, some introns are so efficient in boosting expression levels, that they are regularly included in constructs in order to guarantee high expression (Clark et al., 1993). Some introns were even engineered to this purpose. It was shown, for example, that a hybrid intron made of an adenovirus 5′ splice site and an immunoglobulin G 3′ splice site, is boosting the expression level of various genes in transgenic mice up to 300-fold (Choi et al., 1991).
Large-scale analyses further corroborated these observations. Intron-bearing genes in yeast were shown to produce more mRNA and more protein than intronless genes (Juneau et al., 2006). Similarly, intron-bearing genes in mammals were shown to have higher and broader expression than intronless genes (Shabalina et al., 2010). Reconstruction of the intron–exon evolutionary history in 19 eukaryotes revealed that highly expressed genes tend to have higher intron gain rates (Carmel et al., 2007a).
As we shall see, there is no single mechanism by which introns enhance expression. In many cases, the mechanism is not yet known, but in those cases in which it had been revealed, introns seem to affect virtually any step of mRNA maturation, including transcription initiation, transcription elongation, transcription termination, polyadenylation, nuclear export, and mRNA stability. We view this functional diversity as a reflection of the fact that introns gained this function on many independent occasions in a rather “opportunistic” manner.
In this review, we will show examples to the great variety of functions carried out by introns. We found it illuminating to divide the life span of an intron to five phases, and to separately refer to the functions that are associated with each phase (Figure 1). The first phase is the genomic intron, which is the DNA sequence of the intron. The second phase is the transcribed intron, which is the phase in which the intron is under active transcription. The third phase is the spliced intron, in which the spliceosome is assembled on the intron and is actively excising it. The fourth phase is the excised intron, which is the intronic RNA sequence released upon the completion of the splicing reaction. The final phase is the exon-junction complex (EJC)-harboring transcript, which is the mature mRNA in which the location of the exon–exon junctions is marked by the EJC.
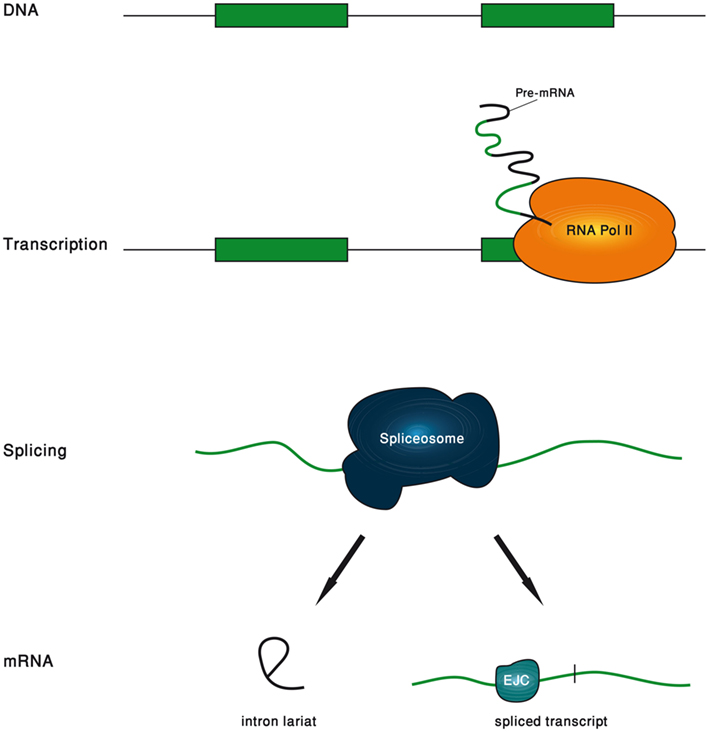
Figure 1. Schematic description of the five phases of an intron life span.
Another distinction that we found useful is between the various intronic properties that mediate the function (Table 1). Sequence-dependent functions are mediated by sequence elements within the intron; length-dependent functions are mediated by the length of the intron, regardless of its nucleotide content; position-dependent functions are mediated by the position of the intron with respect to the exons; and splicing-dependent functions are mediated by the mere fact that splicing had occurred during the maturation of the mRNA.
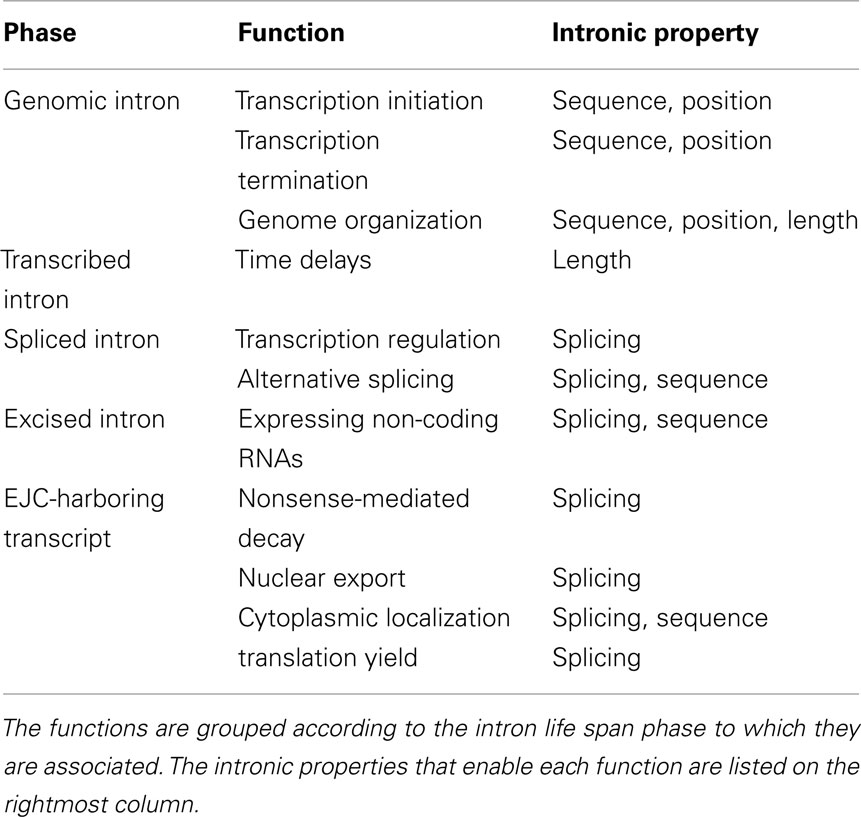
Table 1. Summary of the intronic functions covered in this review.
Functions Associated with the Genomic Intron
At the DNA level introns may be viewed as selection-free sequences within genes. From an evolutionary perspective, such setup is an ideal “evolutionary playground,” whereby almost any mutational tinkering of the intronic sequence is tolerable. In particular, introns have a potential to serve as repositories of cis elements, participating in the regulation of transcription, and genome organization.
Transcription Initiation
Introns modify the expression level of their host gene in many different ways, and underpinning the mechanism is of major challenge in every specific case. In particular, it is often important to determine whether the function is associated with an intronic sequence element, or rather with the spliceosome or any of its numerous satellite proteins. In many cases, the effect on the expression is especially strong for a specific intron, implying that it is its sequence, rather than splicing per se, that underlies the function. For example, Vasil et al. (1989) showed that the first intron of the shrunken-1 (Sh1) locus in maize increased expression at least 10 times more efficiently than other maize introns that they checked. In another experiment, some introns were shown to boost expression level in transgenic mice, when inserted in between a promoter and the intronless rate growth hormone gene, beautifully demonstrating function without being recognized as introns by the spliceosome (Palmiter et al., 1991). Many other studies identified specific intron-hosted DNA elements that regulate transcription initiation. These elements include enhancers (Tourmente et al., 1993; Scohy et al., 2000; Bianchi et al., 2009; Beaulieu et al., 2011), silencers (Tourmente et al., 1993; Gaunitz et al., 2004, 2005), or other elements that modulate the function of the main upstream promoter (Bornstein et al., 1988; Zhang et al., 2011).
In the vast majority of cases, these regulatory elements are found within the 5′-most introns (first introns; Bornstein et al., 1988; Vasil et al., 1989; Tourmente et al., 1993; Scohy et al., 2000; Gaunitz et al., 2004, 2005; Bianchi et al., 2009; Beaulieu et al., 2011; Zhang et al., 2011). Large-scale studies provide further credence to the special regulatory role of first introns, showing that 5′-proximal introns, and especially those in the 5′ UTR, are significantly longer than more distal introns (Bradnam and Korf, 2008). The accepted interpretation of this finding is that these introns are longer because they harbor more cis regulatory sequences, likely related to transcription initiation. This is not the only case that multitude of regulatory elements is suggested as an explanation to long introns. We shall see another example later on, when similar arguments were recruited to explain why alternative exons tend to be flanked by long introns. And yet, the validity of this surmise is questionable. For example, contrary to the expectations, a clear association between intron length and expression breadth in human was not found (Cenik et al., 2010).
Genome-wide analysis in A. thaliana found that promoter-proximal introns that cause expression enhancement are characterized by unique sequence profile, enriched with certain motifs (Rose et al., 2008). Later, such motifs were claimed to have been identified in other plant species (Parra et al., 2011), although a comprehensive survey in rice could not find a correlation between the presence of these motifs and expression boost (Morello et al., 2011).
Some introns do not harbor elements that modify the efficiency of the main promoter, but rather host an alternative promoter that gives rise, when activated, to an isoform with a different transcription start site. For example, Scohy et al. (2000) found an alternative promoter within the first intron of the α-fetoprotein (AFT) gene, bringing about an isoform whose transcription start site is 295 bases downstream of the original transcription start site, and is expressed in the yolk sac and fetal liver. Similarly, Petit et al. (2008) found an SRF-dependent alternative promoter in the second intron of the lipoma preferred partner (LPP) gene, yielding an isoform specific to certain tissues.
Transcription Termination
As will be shown later, splicing is strongly coupled with 3′-end formation. But intronic sequence elements that regulate 3′-end processing in a splicing-independent manner also exist. A well-known example is the second intron of the human β-globin gene. A removal of this intron or its replacement by other introns substantially reduces the efficiency of the 3′-end formation. Moreover, mutants that have defective splicing do have intact 3′-end formation, indicating that there is no coupling between 3′-end processing and the splicing itself. Further experiments with hybrid introns showed that it is a 60-bp-long segment toward the 3′-end of the second intron that enhances the 3′-end processing (Antoniou et al., 1998).
Genome Organization
In an attempt to explain the negative correlation between intron length and expression breadth in multicellular eukaryotes, Vinogradov suggested the “genomic design” hypothesis, stating that introns are longer in tissue-specific genes because they host regulatory elements, and, importantly, because they serve as scaffold elements to assure correct assembly of nucleosomes (Vinogradov, 2004, 2006). Recently, when genome-wide mapping of nucleosome positions became available, several large-scale studies have found that nucleosomes preferentially occupy exons, and are depleted in introns (Schwartz et al., 2009; Spies et al., 2009; Tilgner et al., 2009). This preferential nucleosome coverage of exons was shown to be independent of whether the exon is constitutive or alternative, of its expression level, and of its GC content (Andersson et al., 2009; Nahkuri et al., 2009; Chen et al., 2010). It is currently unknown what drives this nucleosome marking of exons, but it had been suggested that sequence elements near the intron ends function as nucleosome disfavoring elements, pushing the nucleosomes away toward the exons (Schwartz et al., 2009). This exon marking by nucleosomes seems to be interconnected to their marking by specific histone modifications, like H3K36me3 (Andersson et al., 2009; Schwartz et al., 2009), but the full extent of the association between gene architecture, chromatin structure, nucleosome positioning, and histone modifications still has to be clarified (Schwartz and Ast, 2010). These conclusions from large-scale analyses are supported by a few experiments showing that the ability of nucleosomes to form in some genes is severely perturbed when their introns are deleted (Lauderdale and Stein, 1992; Liu et al., 1995).
Nested Genes
Some genes, called nested genes, appear within introns of other genes. The number of nested genes ranges from 158 in human (Yu et al., 2005) to almost 800 in Drosophila (Kumar, 2009). However, in the vast majority of cases nested genes have their own promoters, and their pattern of expression is different from that of their host (Kumar, 2009). Therefore, the presence of nested genes within introns seems a result of stochastic process, only weakly related to the fact that they reside within introns.
Functions Associated with Transcribed Introns
Introns go through transcription just like exons, to form the pre-mRNA. Large-scale transcription studies found that sense transcription is typically accompanied by substantial antisense transcription (Gingeras, 2007). Many seemingly functional antisense elements come from intronic regions (Reis et al., 2005), and may therefore be regarded as intron-hosted RNA genes (see Functions Associated with Excised Introns) that are activated during transcription rather than following intron excision. In this section we would like to focus on a different, very unique function of introns, associated only with the fact that they are transcribed, regardless of their sequence content, or their position, or of the fact that they are later excised from the pre-mRNA.
RNA polymerase II elongation rate had been estimated using various techniques (Ardehali and Lis, 2009). Recent measurement on different regions of nine long human genes found a rather homogeneous rate of 3.8 kb min−1 (Singh and Padgett, 2009), although rates higher than 50 kb min−1 had also been reported (Maiuri et al., 2011). Many introns, therefore, require minutes, hours, and even days to transcribe. This raises the intriguing possibility that introns may serve as tools to orchestrate time delays between activation of a gene, and the appearance of its protein product (Gubb, 1986; Swinburne and Silver, 2008).
Indeed, such a role was nicely demonstrated in the E74 gene that switches on at the beginning of the metamorphosis of D. melanogaster. This complex gene consists of three transcripts, of which the primary one is the 60-kb long E74A gene that matures, after splicing, to a 6-kb mRNA. The gene is induced by the steroid hormone Ecdysone, and appears in the cytoplasm after about an hour from the time of induction. Thummel et al. (1990) measured an elongation rate of RNAP II along this gene of about 1.1 kb min−1, suggesting that it is the introns transcription time alone that underlies this delay.
It is a known theoretical result that negative feedback loops with a time delay may end up in oscillatory behavior. This was demonstrated in an artificial setup by engineering gene networks with time delays, and obtaining expression pulses whose cycle depended on the intron length (Swinburne et al., 2008). But it was also shown in physiological transcripts. The gene Hes7 is cyclically expressed in the presomitic mesoderm and regulates the somite segmentation. It had been recently shown that introns within the mouse Hes7 cause a 19-min delay in transcription, and that without this delay (i.e., if the introns are removed) the oscillations disappear and Hes7 is expressed steadily, leading to severe segmentation defects (Takashima et al., 2011). As expected from a length-dependent intronic function, the total length of all introns in Hes7 was found to be highly conserved across the eukaryotic domain (Seoighe and Korir, 2011). Large-scale analysis of additional 1875 genes identified at least 10 more genes whose total intron length is conserved much more than expected, suggesting a similar role in time delays (Seoighe and Korir, 2011). Interestingly, many of these genes are related to developmental processes, in which negative feedback time delay loops are expected to play an important role (Swinburne and Silver, 2008).
Functions Associated with Spliced Introns
Pre-mRNA splicing is carried out by the spliceosome, that is built from five core snRNAs (U1, U2, U4, U5, and U6), many core proteins, and numerous other satellite proteins (Wahl et al., 2009). The spliceosome is increasingly recognized as a huge cellular machine that carries with it proteins that participate in a host of RNA maturation processes, other than splicing. Here, we will survey functions that come about by the fact that the spliceosome was recruited to the pre-mRNA.
Transcription Regulation
Many studies show that splicing of most of the introns occurs concomitantly with transcription, and that these two cellular processes are strongly coupled (Beyer and Osheim, 1988; LeMaire and Thummel, 1990; Wuarin and Schibler, 1994; Furger et al., 2002; Khodor et al., 2011), mainly through the carboxyl-terminal domain (CTD) of RNAP II (McCracken et al., 1997; Akhtar et al., 2009; Moore and Proudfoot, 2009). In general, RNAP II was shown to be preferentially associated with all of the U1 snRNP core proteins, as well as with some SR-proteins splicing factors (Das et al., 2007). The original interpretation of this finding was that RNAP II brings along factors that facilitate fast spliceosome assembly on the nascent pre-mRNA. Nowadays, however, despite some works that suggest otherwise (Brody et al., 2011), this coupling is generally believed to be bidirectional, in the sense that transcription modulates splicing (see next section), and splicing modulates transcription. It is this latter effect of splicing on transcription that would be the focus of this section. We will show how splicing modulates all phases of transcription, including initiation, elongation, and termination.
Transcription initiation, or re-initiation, is thought to be affected by U1 snRNA. U1 snRNA was shown to associate with TFIIH, a general transcription initiation factor, and to stimulate the rate of formation of the first phosphodiester bond by RNAP II (Kwek et al., 2002). Further research showed that besides TFIIH, two other transcription initiation factors, TFIID and TFIIB, are preferentially associated with donor splice junctions, leading to the hypothesis that 5′-most introns stimulate transcription initiation at the upstream promoter through U1 snRNA-mediated pre-initiation complex assembly at the donor splice site (Damgaard et al., 2008). However, it is not known whether this role of U1 snRNA is related to the role it plays at the spliceosome, or is it a splicing-independent function of U1 snRNA (Jobert et al., 2009).
Splicing was also found to directly promote transcription elongation, through interactions between splicing factors or spliceosomal components and transcription elongation factors. Some experiments suggest that U2 snRNP, apart from its role in the spliceosome, also promotes transcription elongation by interacting with the transcription elongation factors TAT-SF1 and P-TEFb (Fong and Zhou, 2001). The generality of this mechanism is questionable, though, as it could not be reproduced in yeast (McKay and Johnson, 2011). The splicing factor SC35 was also shown to enhance RNAP II elongation of some mammalian genes via interaction with P-TEFb. Actually, it had been shown that SC35 depletion attenuates transcription, and that this defective phenotype can be rescued by adding recombinant SC35 (Lin et al., 2008). The splicing-associated c-Ski-interacting protein (SKIP) was similarly shown to promote RNAP II elongation by associating, yet again, with P-TEFb. In this case, however, SKIP seems to have a function that is independent of its role in splicing (Bres et al., 2005).
At the final stage of transcription, mRNAs undergo 3′-end processing, involving endonucleolytic cleavage and the addition of a poly(A) tail. Splicing was found to modify the efficiency of this mRNA processing stage as well (Millevoi and Vagner, 2010; Proudfoot, 2011). In general, functional coupling between splicing, and in particular of the 3′-most intron, and 3′-end formation had been demonstrated (Rigo and Martinson, 2008). In search for mechanism, at least two snRNPs (U1 and U2) were found to modulate 3′-end processing, in addition to several splicing factors.
U2 snRNP was shown to physically interact with the cleavage/polyadenylation specificity factor (CPSF), and that its presence is required for efficient cleavage. In fact, mutations to the U2 snRNP binding site of the pre-mRNA resulted not only in aberrant splicing, but also in reduced cleavage efficiency (Kyburz et al., 2006). However, it is unknown whether this role of U2 snRNP is linked to its splicing role, because it was later shown that U2 snRNP contributes to 3′-end formation of the intronless non-polyadenylated histone genes (Friend et al., 2007).
While U2 snRNP seems to enhance 3′-end processing, it was found that binding of U1 snRNA upstream of a polyadenylation signal represses 3′-end formation. For example, bovine papillomavirus type 1 genes are expressed only in late stages of the infection. In early stages, expression is repressed by 3′-end formation inhibition caused by U1 snRNA-bound 5′ splice site-like elements upstream of the polyadenylation signal (Furth et al., 1994). It was shown that bases at the 5′-end of the U1 snRNA are critical for this inhibition (Furth et al., 1994), and that mutations in this part of the U1 snRNA repress expression of many endogenous mammalian genes by binding to their terminal exon (Fortes et al., 2003). Recently, using morpholinos to knockdown U1 snRNA in human HeLa cells, it was demonstrated that except for the expected accumulation of unspliced pre-mRNA, premature cleavage, and polyadenylation was observed in numerous pre-mRNAs at cryptic polyadenylation sites, mostly within introns. Interestingly, knockdown of U2 snRNA did not show this effect, suggesting that it may be a splicing-independent function of U1 snRNA (Kaida et al., 2010), which explains the overabundance of U1 snRNA with respect to the other snRNAs. The role of U1 snRNA in repressing 3′-end processing is probably because it brings with it the U1 snRNP proteins that actually mediate the suppression. For example, the inhibition of the 3′-end formation in the bovine papillomavirus type 1 mentioned above was found to be caused by a direct interaction between the U1 snRNP protein U1 70K and the poly(A) polymerase (PAP; Gunderson et al., 1998). The U1 snRNP protein U1A was also found to have similar inhibitory roles by interacting with PAP (Gunderson et al., 1997), although, interestingly, it was also suggested to have stimulating effect on 3′-end processing via interaction with the 160-kDa subunit of CPSF (Lutz et al., 1996).
Further splicing factors have been shown to have an impact on the cleavage/polyadenylation process (Millevoi and Vagner, 2010), such as hnRNP F (Veraldi et al., 2001) and SRP75 (Ko and Gunderson, 2002) that have inhibitory roles, Srm160 with a stimulating role (McCracken et al., 2002), and U2AF65 that probably has a stimulating effect (Millevoi et al., 2002, 2006), although it had also been claimed to have an inhibitory role (Ko and Gunderson, 2002).
Alternative Splicing
Some splice sites are recognized as such by the spliceosome in every tissue, time, and condition. Other splice sites have, at least in certain tissues, times, or conditions, some probability to be missed by the spliceosome, giving rise to alternative splicing. Alternative splicing allows for proteome diversity that much exceeds the number of genes in the genome (Nilsen and Graveley, 2010). One remarkable example is the Dscam gene of D. melanogaster, which potentially generates more than 38,000 isoforms (Schmucker et al., 2000). This means that Dscam’s protein repertoire is larger than the number of genes in the fruit fly! Recent genome-wide analyses based on RNA-seq data found that in human, nearly 95% of the multiexon genes undergo alternative splicing, mostly in a very tissue-specific way (Pan et al., 2008). While alternative splicing is probably widespread in human and in many other eukaryotes, it is still undetermined what fraction of it is functional, and what fraction is simply splicing noise (Graveley, 2001; Lareau et al., 2004; Sorek et al., 2004; Lu et al., 2009).
Proving function of alternative splicing at the systems level is challenging, but specific examples are accumulating (Smith et al., 1989; Stamm et al., 2005). Here, we shall mention just a few. The fibronectin (FN) gene in human is an extracellular matrix protein. It has several different isoforms, some of which have different patterns of localization and slightly different functions in human cells (Demir-Weusten, 2002); The Slo avian gene coding for a K+ channel protein has 576 possible isoforms, of which several are expressed in a specific gradient along the sensory receptor cells of the inner ear, contributing to the highly accurate perception of different sound frequencies in birds (Black, 1998); A beautiful autoregulation based on alternative splicing is demonstrated by the ADAR2 gene, which is a key factor in A-to-I RNA editing. Strikingly, one of the acceptor splice sites in this gene, which has the typical AG dinucleotide at the end of the intron, is preceded by an AA dinucleotide 47 bases upstream. Normally, AA is not recognized as an acceptor splice site, but high levels of ADAR2 edit it to AI, which is recognized as AG, and thus as an acceptor splice site, by the spliceosome. Preference of this new splice site over the original one leads to the production of non-active isoforms of ADAR2, following a decrease in its levels (Rueter et al., 1999).
Conserved alternative exons are orthologous exons that are alternative in several organisms. Likewise, conserved constitutive exons are orthologous exons that are constitutive in several organisms. Human–mouse comparative study showed that 77% of the introns flanking conserved alternative exons are made of long conserved sequences, while the same held for only 17% of the introns flanking conserved constitutive exons (Sorek and Ast, 2003). This observation puts forward the notion that introns host cis regulatory elements that facilitate alternative splicing. Indeed, introns not only passively allow for alternative splicing because of their mere existence, but also actively regulate splicing by hosting splicing regulatory elements (SREs; Schwartz et al., 2008; Wang and Burge, 2008; Hartmann and Valcarcel, 2009). These are short cis motifs that generally bind to splicing factors that enhance or repress the spliceosome assembly on a nearby potential splice site. Some SREs are found within exons, and some are harbored within introns and are divided into intronic splicing silencers (ISSs) and intronic splicing enhancers (ISEs; Havlioglu et al., 2007; Venables, 2007; Culler et al., 2010). For example, Nova-1 is a neuron-specific RNA binding protein that functions mainly in the brain (Ule et al., 2005), and regulates alternative splicing by binding to intronic motifs – such as YCAY – and enhancing splicing of the downstream splice site (Dredge and Darnell, 2003). Fox-1 is another splicing factor that induces exon skipping in heart and skeletal muscles by binding to the intronic motif GCAUG (Jin et al., 2003). In general, ISSs and ISEs are short, degenerate, and of variable distance for the splice site, and are therefore hard to detect and identify, and many putative elements await experimental validation.
Functions Associated with Excised Introns
Once an intron had been excised, it typically becomes part of post-splicing complexes that lead to efficient debranching and degradation (Yoshimoto et al., 2009). But when an RNA gene is embedded within the intron, it is expressed upon intron removal, and outlives its intronic host. Many families of non-coding RNAs (ncRNAs) have been characterized, such as microRNAs (miRNAs), small nucleolar RNAs (snoRNAs), piwi-interacting RNAs (piRNAs), small-interfering RNAs (siRNAs), and various long non-coding RNAs (lncRNAs). Except for piRNAs, Rearick et al. (2011) found that members of these families are preferentially associated with introns in human, leading to the hypothesis that genes may autoregulate their expression by hosting relevant ncRNAs within their introns.
MicroRNA are small ncRNAs of about 22–23 nucleotides that bind to target sites along mRNAs, usually within their 3′ UTRs, and direct them for degradation or translation repression (Bartel, 2009). It is thought that – at least in vertebrates – miRNAs affect thousands of genes, and that in general they form an important layer of regulation (Shalgi et al., 2009; Berezikov, 2011). Roughly half of the human miRNAs lie in intergenic regions and are associated with their own transcriptional promoter. The other half reside within introns, usually lack independent promoter, and are co-expressed with their host gene (Baskerville and Bartel, 2005), potentially regulating its expression by feedback loops (Hinske et al., 2010). It is generally believed that miRNAs are processed from the excised intron, although some evidence points to the possibility that they are processed already on the pre-mRNA (Kim and Kim, 2007).
Usually, miRNAs lie within a long transcriptional unit, called pri-miRNA, that is cleaved by Drosha to a shorter hairpin structure known as pre-miRNA (Lee et al., 2003). The pre-miRNA is then exported to the cytoplasm, where it is cleaved again, this time by Dicer, to form a double-stranded RNA. One of the strands is then associated with the RISC complex to form functional miRNA (Obernosterer et al., 2006). Ruby et al. (2007) reported an alternative miRNA biogenesis pathway. They found that certain debranched introns have the structural features of pre-miRNAs, and that they are generated following splicing without the need to cleave a precursor transcriptional unit by Drosha. These miRNAs that require splicing but not Drosha for their maturation are termed mirtrons. They were first identified in D. melanogaster and C. elegans, but later discovered in mammals, birds, and even plants (Westholm and Lai, 2011).
Small nucleolar RNAs comprise a rather large family of small RNAs, mainly known for their role in posttranscriptional methylation and pseudouridylation of various RNA genes like rRNAs, tRNAs, and snRNAs. Similarly to miRNAs, members of this family can reside in intergenic regions and have their own transcriptional promoter, or dwell in introns and rely on splicing for their maturation (Dieci et al., 2009). In fact, snoRNAs are rather abundant in introns of both vertebrates and insects, where they are processed by the exonucleolytic digestion of debranched introns after their excision from the pre-mRNA (Filipowicz and Pogacic, 2002; Huang et al., 2005). The introns of some ribosome-associated genes were found to host snoRNAs that guide rRNA modifications (Maxwell and Fournier, 1995), but it is generally not the rule that snoRNAs are related to the regulation of their host genes. Strikingly, the sole function of some genes seems to be harboring snoRNAs in their introns, and their mRNA does not look as if it has a protein-coding potential (Tycowski et al., 1996; Bachellerie et al., 2002; Makarova and Kramerov, 2009).
Endogenous siRNAs form yet another family of small RNAs that is involved in the RNA interference pathway and in many other cellular processes such as posttranscriptional gene silencing (Okamura and Lai, 2008). These are double stranded, 20–25 nucleotides long RNA molecules, whose identification is hindered by the abundance of hairpin structures in eukaryotic genomes (Watanabe et al., 2008). The number of verified intronic siRNAs is small, but recent large-scale studies found a large number of potential hairpin endogenous siRNAs within introns in human (Rearick et al., 2011) and rice (O. sativa; Chen et al., 2011).
Introns were also found to host lncRNAs (Rearick et al., 2011). These are RNA genes longer than 200 bases, that have diverse regulatory functions, presumably affecting the expression of protein-coding genes in cis or in trans (Mattick and Gagen, 2001; Wang and Chang, 2011).
Functions Associated with EJC-Harboring Transcripts
In metazoans, the splicing reaction leaves traces in the form of a protein complex deposited 20–24 nucleotides upstream of the exon–exon junction, known as the EJC (Le Hir et al., 2000). It contains four core proteins, MAGO, Y14, eIF4AIII, and MLN51, and many others that are transiently associated with it (Bono and Gehring, 2011). Subject to changes in its composition, the EJC survives from the splicing in the nucleus to the pioneer round of translation in the cytoplasm (Dreyfuss et al., 2002; Tange et al., 2004; Moore, 2005). During all this time, it serves as a memory device, marking the position of excised introns. It had been gradually appreciated that by interacting with many other factors, EJC participates in a range of mRNA-related cellular processes (Wiegand et al., 2003; Figure 2).
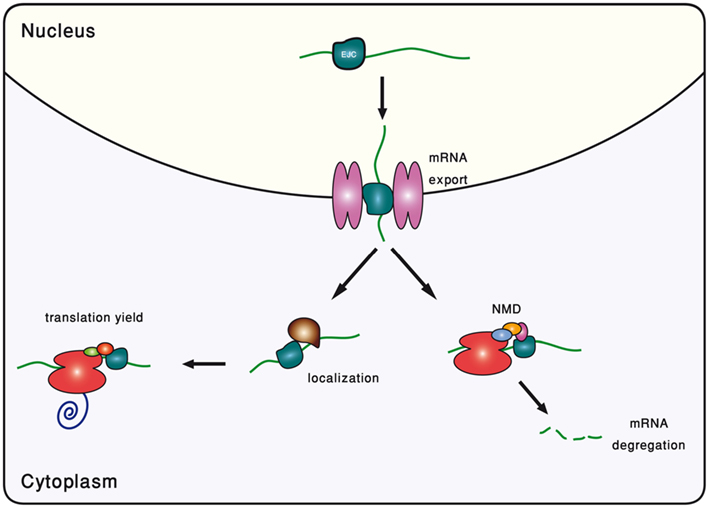
Figure 2. Schematic description of the different roles played by the exon-junction complex (EJC).
Nonsense-Mediated Decay
Nonsense-mediated decay (NMD) is a eukaryotic surveillance mechanism that selectively degrades mRNAs harboring premature termination codons (PTCs). PTCs arise frequently, mostly as a result of mutations in the DNA level, alternative splicing in the RNA level, and errors in transcription. NMD prevents such transcripts from being translated, as otherwise they can give rise to truncated proteins with dominant-negative or deleterious gain-of-function activities (Maquat, 2004; Chang et al., 2007; Silva and Romao, 2009). A major puzzle in the field is what makes a termination codon recognized as premature by NMD. Several properties of the 3′ UTR had been suggested as possible NMD triggers, including sequence motifs, protein context, and the 3′ UTR length (Zhang et al., 1995; Gonzalez et al., 2000; Lykke-Andersen et al., 2000; Amrani et al., 2004; Brogna and Wen, 2009).
In mammals, and possibly in other vertebrates as well (Wittkopp et al., 2009), the dominant form of NMD is splicing-dependent, in which EJCs that are more than 50–55 nucleotides downstream of a termination codon mark it as premature (Cheng et al., 1994; Nagy and Maquat, 1998). Mechanistically, it is believed that NMD is triggered by a phosphorylation–unphosphorylation cycle of the UPF1 protein. The UPF3 protein (which has two paralogs in vertebrates and one copy in invertebrates) is associated with the EJC, to which it recruits the UPF2 protein. Upon transcription termination, the ribosome deposits a complex named SURF on the mRNA, containing the release factors eRF1 and eRF3. These factors recruit unphosphorylated UPF1. In the presence of nearby EJC, and in particular of UPF2 and UPF3, the UPF1 is phosphorylated by SMG-1 (Chang et al., 2007).
Interestingly, NMD may sometimes be linked to alternative splicing. A nice demonstration of such coupling is the autoregulation of the PTB protein. This protein has many functions related to mRNA processing, and is also an hnRNP splicing repressor. It was found that is has two isoforms – one is functional and contains all the exons, and the other lacks exon 11 and as a result has a PTC and is degraded by NMD. Wollerton et al. (2004) found that PTB promotes exon 11 skipping, thereby controlling its own expression level in a negative feedback loop. A few other similar examples have been described (Amor et al., 2010; Durand et al., 2011), in particular in genes that are regulators of alternative splicing (Mitrovich and Anderson, 2000; Sureau et al., 2001; McGlincy and Smith, 2008). In a more general context, however, the extent to which the coupling between NMD and alternative splicing is widespread is debated. In an attempt to explain the high percentage of human alternative transcripts that are NMD targets (Green et al., 2003; Lewis et al., 2003), Hillman et al. (2004) carefully analyzed existing data of mRNA and protein expression in human, and concluded that NMD participates in the regulation of many genes. Using a splicing-sensitive custom microarray Hansen et al. (2009) identified at least 45 genes in Drosophila with an isoform that is NMD-sensitive, leading an NMD-dependent regulation of their expression level. On the other hand, Pan et al. used both mouse and human exon arrays to show that PTC-containing isoforms are expressed at low levels, and thus have no measurable effect on the total abundance of the gene. They supported this finding by knocking down Upf1 in human, and showing that only a minority (6%) of the genes is affected, and that 80% of the PTC-generating alternative splicing events (one third of all alternative splicing events) result in transcripts with low abundance, independent of whether NMD is active or not (Pan et al., 2006).
Nuclear Export
In eukaryotes, mature mRNAs must be exported from the nucleus to the cytoplasm before they can start being translated. Mature nuclear mRNAs bind to mRNA-specific transport factors, and are shuttled through pores in the nucleus membrane, formed by the nuclear pore complexes (Hood and Silver, 1999; Kohler and Hurt, 2007; Le Hir and Seraphin, 2008). A link between splicing and export was sought by comparing export rates of spliced transcripts to that of their unspliced counterparts. The first studies pointed at significantly more efficient export of spliced mRNA in mammals (Ryu and Mertz, 1989) and amphibians (Luo and Reed, 1999), but this was called into question by subsequent works (Rodrigues et al., 2001; Ohno et al., 2002; Lu and Cullen, 2003; Nott et al., 2003). Recently, however, Valencia et al. (2008) introduced intron-bearing and the corresponding intronless constructs into human and mouse cell nuclei, and then used FISH to study the distribution of transcripts across the nuclear and cytoplasmic compartments. They found that spliced transcripts were mostly cytoplasmic, whereas unspliced transcripts were mostly nuclear. Overall, they reported that the kinetics and efficiency of mRNA export of mammalian cells were enhanced 6- to 10-fold by splicing.
The link between splicing and export is presumably caused by the fact that the spliceosome assembly on the pre-mRNA facilitates the recruitment of export factors. This can be done directly by the spliceosome, or by the EJC that is deposited near the exon–exon junction. For example, it was found that the ALY/REF export factor binds mRNAs that have gone through splicing, but is absent from identical mRNAs that were generated from intronless pre-mRNAs (Zhou et al., 2000). In fact, the EJC seems to provide strong binding sites for this export factor (Le Hir et al., 2001). Further work revealed that ALY/REF binds to intronless transcripts too, via a different, splicing-independent, mechanism (Taniguchi and Ohno, 2008). Other examples include the export-associated THO complex which associates with spliced mRNAs but not with unspliced ones (Masuda et al., 2005), and the UAP56 splicing factor which also has a key role in export (Shen, 2009).
Cytoplasmic Localization
Some eukaryotic cellular processes require certain mRNAs to be translated only within a demarcated region of the cell. mRNA localization is achieved with the help of a diverse family of shuttling proteins. Some bind the mRNA cotranscriptionally in the nucleus, while others are recruited in the cytoplasm, right after the nuclear export (Martin and Ephrussi, 2009; Trcek and Singer, 2010; Forget and Chartrand, 2011).
It is believed that the EJC plays an important role in recruiting shuttle proteins to the mRNA. One well-known example is the localization of the oskar mRNA in the cytoplasm of D. melanogaster’s oocytes, which affects germline and abdomen development. Although it has not yet been formally proven that splicing deposits EJCs on Drosophila mRNAs, fly homologs of the EJC proteins Y14 and MAGO were found to be essential for proper localization of oskar during oogenesis (Hachet and Ephrussi, 2001, 2004; Mohr et al., 2001). Moreover, Hachet and Ephrussi (2004) demonstrated that this localization depends on the splicing of the 5′-most intron. They generated constructs of oskar with all possible combinations of its introns, and showed that transcripts that included the first intron were correctly localized, whereas it was not the case in other versions of the gene. It is worth noting that the intron removal did not affect export, as the same amounts of mRNA were obtained as in the wild type. Interestingly, using intronless constructs in eggs led to over two thirds of the embryos to fail to hatch. As expected from an EJC-dependent function, substituting the third intron in place of the first one did not disrupt the proper localization.
Although not an EJC-dependent function, we shall mention here that splicing can affect mRNA localization by inclusion/exclusion of sequence localization signals via alternative splicing and/or alternative polyadenylation. Such sequence signals are thought to drive localization by serving as targets for shuttle proteins. Such sequence elements appear everywhere, but they are particularly abundant within 3′ UTRs (Bullock and Ish-Horowicz, 2001; Gilligan et al., 2011). For example, Horne-Badovinac and Bilder (2008) have shown in Drosophila, that the mRNA of the Stardust protein (sdt), which forms a vital complex for epithelial polarity, is apically localized in the membrane. This localization is a result of an inclusion of the alternative third exon that contains a localization motif. In the absence of this exon, sdt mRNA is uniformly distributed. Regulation of this exon inclusion and exclusion generates a switch, producing the Stardust complex when it is needed during the early stages of epithelial development (Horne-Badovinac and Bilder, 2008). Another illuminating example was found in the mouse’s brain. Brain cells generate two isoforms of the brain-derived neurotrophic factor (BDNF), one with short 3′ UTR and another with long 3′ UTR. An et al. compared BDNF mRNA quantities in different brain regions and found great differences in the relative abundance of the long and the short versions. The long isoform was found to be mainly positioned in the dendrites, while the short isoform was shown to be in the soma (An et al., 2008). More generally, alternative polyadenylation is considered as an important regulator of mRNA localization (Tian et al., 2007; Wu et al., 2011).
Translation Yield
Greater amounts of protein are produced per molecule of spliced mRNA than from otherwise identical mRNA molecules not produced by splicing. In some cases, it was possible to show that this is due to direct effect of splicing on the translation yield (Lee et al., 2009). For example, having an EJC appears to promote mRNA polysome association, which can also be obtained by tethering the EJC proteins Y14, MAGO, and RNPS1 on intronless transcripts (Nott et al., 2004). The mechanism by which EJC promotes translational yield is still unclear. It had been suggested that the EJC proteins Y14 and MAGO, when associated with the cytoplasmic transcript, bind to the PYM protein, which, in turn, binds to the ribosome and therefore serves as a bridge between the EJC and the translation mechanism. Indeed, it had been shown that PYM knockdown reduces translation efficiency of intron-bearing transcripts, but does not affect intronless transcripts (Diem et al., 2007). Another work showed that the EJC recruits the SKAR protein, which, in turn, recruits S6K1 and that together, SKAR and S6K1 increase the translational efficiency of spliced mRNA (Ma et al., 2008).
Greater protein levels can also be a result of splicing conferring enhanced stability to the protein product, or to its mRNA forebear. For example, the Dihydrofolate reductase protein expressed from stably transfected minigenes was found to have a 2.7-fold longer half-life when expressed from an intron-containing construct than from an identical cDNA construct (Tange et al., 2004). Another example is the mouse’s chemokine gene CXCL1. It has been demonstrated that mRNA derived from a transcript that contains introns is significantly more stable than that derived from an intron-free transcript. Only a single intron is required to produce this effect, and the intron position and sequence do not appear to be important. Although the presence of at least one intron modulates the rate of mRNA decay, it does not modulate the nuclear/cytoplasmic distribution, the rate of translation, or the ability of extracellular stimulus to stabilize the mRNA (Zhao and Hamilton, 2007).
Intron Positional Conservation
A fundamental supposition in comparative genomics is that evolutionary conservation is indicative of biological function. This makes the identification of highly conserved genomic regions a chief strategy in looking for function. Evolutionary conservation is mainly identified with sequence conservation, but also with conservation of secondary and tertiary structure of DNA, RNA, and proteins, and with conservation of genome-wide organization (Graur and Li, 2000). The success of this strategy notwithstanding, it is increasingly recognized that many functional elements – mostly non-coding – still evade detection (Fisher et al., 2006; Birney et al., 2007). In this review we have developed the idea that introns invaded in great numbers to early eukaryotic genomes as slightly deleterious selfish elements, but later gained many functions up to the point that today higher eukaryotes cannot survive without them (Lynch, 2007). This fact implies that the level of conservation of intron position may be correlated with the functional importance of this intron.
Analyzing the intron–exon structure – the gene architecture – of orthologous genes makes the comparison of their respective intron positions straightforward (Figure 3). Using such alignments of orthologous genes, it had been noticed that intron positions are sometimes conserved throughout long evolutionary times, in a frequency that is significantly above random expectation (Rogozin et al., 2003; Carmel et al., 2007b). Current intron populations are regarded as a result of intron gain and loss processes. If an intron becomes associated with a function, of whatever type, its chances to be lost will decrease. Therefore, conservation of intron position should be indicative of function of any type, even if the function is not directly related to the intron position.
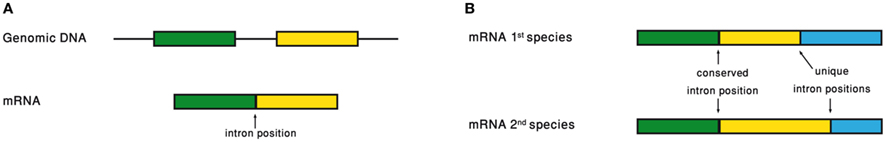
Figure 3. (A) Intron position is defined as the point of intron insertion along the mRNA. (B) Comparison of intron positions between orthologous genes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akhtar, M. S., Heidemann, M., Tietjen, J. R., Zhang, D. W., Chapman, R. D., Eick, D., and Ansari, A. Z. (2009). TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol. Cell 34, 387–393.
Akua, T., Berezin, I., and Shaul, O. (2010). The leader intron of AtMHX can elicit, in the absence of splicing, low-level intron-mediated enhancement that depends on the internal intron sequence. BMC Plant Biol. 10, 93. doi: 10.1186/1471-2229-10-93
Amor, S., Remy, S., Dambrine, G., Le Vern, Y., Rasschaert, D., and Laurent, S. (2010). Alternative splicing and nonsense-mediated decay regulate telomerase reverse transcriptase (TERT) expression during virus-induced lymphomagenesis in vivo. BMC Cancer 10, 571. doi: 10.1186/1471-2407-10-571
Amrani, N., Ganesan, R., Kervestin, S., Mangus, D. A., Ghosh, S., and Jacobson, A. (2004). A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 432, 112–118.
An, J. J., Gharami, K., Liao, G. Y., Woo, N. H., Lau, A. G., Vanevski, F., Torre, E. R., Jones, K. R., Feng, Y., Lu, B., and Xu, B. (2008). Distinct role of long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell 134, 175–187.
Andersson, R., Enroth, S., Rada-Iglesias, A., Wadelius, C., and Komorowski, J. (2009). Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome Res. 19, 1732–1741.
Antoniou, M., Geraghty, F., Hurst, J., and Grosveld, F. (1998). Efficient 3′-end formation of human beta-globin mRNA in vivo requires sequences within the last intron but occurs independently of the splicing reaction. Nucleic Acids Res. 26, 721–729.
Ardehali, M. B., and Lis, J. T. (2009). Tracking rates of transcription and splicing in vivo. Nat. Struct. Mol. Biol. 16, 1123–1124.
Bachellerie, J. P., Cavaille, J., and Huttenhofer, A. (2002). The expanding snoRNA world. Biochimie 84, 775–790.
Baskerville, S., and Bartel, D. P. (2005). Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11, 241–247.
Beaulieu, E., Green, L., Elsby, L., Alourfi, Z., Morand, E. F., Ray, D. W., and Donn, R. (2011). Identification of a novel cell type-specific intronic enhancer of macrophage migration inhibitory factor (MIF) and its regulation by mithramycin. Clin. Exp. Immunol. 163, 178–188.
Berezikov, E. (2011). Evolution of microRNA diversity and regulation in animals. Nat. Rev. Genet. 12, 846–860.
Beyer, A. L., and Osheim, Y. N. (1988). Splice site selection, rate of splicing, and alternative splicing on nascent transcripts. Genes Dev. 2, 754–765.
Bianchi, M., Crinelli, R., Giacomini, E., Carloni, E., and Magnani, M. (2009). A potent enhancer element in the 5′-UTR intron is crucial for transcriptional regulation of the human ubiquitin C gene. Gene 448, 88–101.
Birney, E., Stamatoyannopoulos, J. A., Dutta, A., Guigo, R., Gingeras, T. R., Margulies, E. H., Weng, Z., Snyder, M., Dermitzakis, E. T., Thurman, R. E., Kuehn, M. S., Taylor, C. M., Neph, S., Koch, C. M., Asthana, S., Malhotra, A., Adzhubei, I., Greenbaum, J. A., Andrews, R. M., Flicek, P., Boyle, P. J., Cao, H., Carter, N. P., Clelland, G. K., Davis, S., Day, N., Dhami, P., Dillon, S. C., Dorschner, M. O., Fiegler, H., Giresi, P. G., Goldy, J., Hawrylycz, M., Haydock, A., Humbert, R., James, K. D., Johnson, B. E., Johnson, E. M., Frum, T. T., Rosenzweig, E. R., Karnani, N., Lee, K., Lefebvre, G. C., Navas, P. A., Neri, F., Scj Parker, Sabo, P. J., Sandstrom, R., Shafer, A., Vetrie, D., Weaver, M., Wilcox, S., Yu, M., Collins, F. S., Dekker, J., Lieb, J. D., Tullius, T. D., Crawford, G. E., Sunyaev, S., Noble, W. S., Dunham, I., Dutta, A., Guigo, R., Denoeud, F., Reymond, A., Kapranov, P., Rozowsky, J., Zheng, D. Y., Castelo, R., Frankish, A., Harrow, J., Ghosh, S., Sandelin, A., Hofacker, I. L., Baertsch, R., Keefe, D., Flicek, P., Dike, S., Cheng, J., Hirsch, H. A., Sekinger, E. A., Lagarde, J., Abril, J. F., Shahab, A., Flamm, C., Fried, C., Hackermuller, J., Hertel, J., Lindemeyer, M., Missal, K., Tanzer, A., Washietl, S., Korbel, J., Emanuelsson, O., Pedersen, J. S., Holroyd, N., Taylor, R., Swarbreck, D., Matthews, N., Dickson, M. C., Thomas, D. J., Weirauch, M. T., Gilbert, J., Drenkow, J., Bell, I., Zhao, X., Srinivasan, K. G., Sung, W. K., Ooi, H. S., Chiu, K. P., Foissac, S., Alioto, T., Brent, M., Pachter, L., Tress, M. L., Valencia, A., Choo, S. W., Choo, C. Y., Ucla, C., Manzano, C., Wyss, C., Cheung, E., Clark, T. G., Brown, J. B., Ganesh, M., Patel, S., Tammana, H., Chrast, J., Henrichsen, C. N., Kai, C., Kawai, J., Nagalakshmi, U., Wu, J. Q., Lian, Z., Lian, J., Newburger, P., Zhang, X. Q., Bickel, P., Mattick, J. S., Carninci, P., Hayashizaki, Y., Weissman, S., Dermitzakis, E. T., Margulies, E. H., Hubbard, T., Myers, R. M., Rogers, J., Stadler, P. F., Lowe, T. M., Wei, C. L., Ruan, Y. J., Snyder, M., Birney, E., Struhl, K., Gerstein, M., Antonarakis, S. E., Gingeras, T. R., Brown, J. B., Flicek, P., Fu, Y. T., Keefe, D., Birney, E., Denoeud, F., Gerstein, M., Green, E. D., Kapranov, P., Karaoz, U., Myers, R. M., Noble, W. S., Reymond, A., Rozowsky, J., Struhl, K., Siepel, A., Stamatoyannopoulos, J. A., Taylor, C. M., Taylor, J., Thurman, R. E., Tullius, T. D., Washietl, S., Zheng, D. Y., Liefer, L. A., Wetterstrand, K. A., Good, P. J., Feingold, E. A., Guyer, , Collins, F. S., Margulies, E. H., Cooper, G. M., Asimenos, G., Thomas, D. J., Dewey, C. N., Siepel, A., Birney, E., Keefe, D., Hou, M. M., Taylor, J., Nikolaev, S., Montoya, , Burgos, J.-I., Loytynoja, A., Whelan, S., Pardi, F., Massingham, T., Brown, J. B., Huang, H. Y., Zhang, N. R., Bickel, P., Holmes, I., Mullikin, J. C., Ureta-Vidal, A., Paten, B., Seringhaus, M., Church, D., Rosenbloom, K., Kent, W. J., Stone, E. A., Gerstein, M., Antonarakis, S. E., Batzoglou, S., Goldman, N., Hardison, R. C., Haussler, D., Miller, W., Pachter, L., Green, E. D., Sidow, A., Weng, Z. P., Trinklein, N. D., Fu, Y. T., Zdd Zhang, , Karaoz, U., Barrera, L., Stuart, R., Zheng, D. Y., Ghosh, S., Flicek, P., King, D. C., Taylor, J., Ameur, A., Enroth, S., Bieda, M. C., Koch, C. M., Hirsch, H. A., Wei, C. L., Cheng, J., Kim, J., Bhinge, A. A., Giresi, P. G., Jiang, N., Liu, J., Yao, F., Sung, W. K., Chiu, K. P., Vega, V. B., Cwh Lee, , Ng, P., Shahab, A., Sekinger, E. A., Yang, A., Moqtaderi, Z., Zhu, Z., Xu, X. Q., Squazzo, S., Oberley, M. J., Inman, D., Singer, M. A., Richmond, T. A., Munn, K. J., Rada-Iglesias, A., Wallerman, O., Komorowski, J., Clelland, G. K., Wilcox, S., Dillon, S. C., Andrews, R. M., Fowler, J. C., Couttet, P., James, K. D., Lefebvre, G. C., Bruce, A. W., Dovey, O. M., Ellis, P. D., Dhami, P., Langford, C. F., Carter, N. P., Vetrie, D., Kapranov, P., Nix, D. A., Bell, I., Patel, S., Rozowsky, J., Euskirchen, G., Hartman, S., Lian, J., Wu, J. Q., Urban, A. E., Kraus, P., Van Calcar, S., Heintzman, N., Kim, T. H., Wang, K., Qu, C. X., Hon, G., Luna, R., Glass, C. K., Rosenfeld, M. G., Aldred, S. F., Cooper, S. J., Halees, A., Lin, J. M., Shulha, H. P., Zhang, X. L., Xu, , Jns Haidar, , Yu, Y., Birney, E., Weissman, S., Ruan, Y. J., Lieb, J. D., Iyer, V. R., Green, R. D., Gingeras, T. R., Wadelius, C., Dunham, I., Struhl, K., Hardison, R. C., Gerstein, M., Farnham, P. J., Myers, R. M., Ren, B., Snyder, M., Thomas, D. J., Rosenbloom, K., Harte, R. A., Hinrichs, A. S., Trumbower, H., Clawson, H., Hillman-Jackson, J., Zweig, A. S., Smith, K., Thakkapallayil, A., Barber, G., Kuhn, R. M., Karolchik, D., Haussler, D., Kent, W. J., Dermitzakis, E. T., Armengol, L., Bird, C. P., Clark, T. G., Cooper, G. M., De Bakker, P. I., Kern, A. D., Lopez-Bigas, N., Martin, J. D., Stranger, B. E., Thomas, D. J., Woodroffe, A., Batzoglou, S., Davydov, E., Dimas, A., Eyras, E., Hallgrimsdottir, I. B., Hardison, R. C., Huppert, J., Sidow, A., Taylor, J., Trumbower, H., Zody, M. C., Guigo, R., Mullikin, J. C., Abecasis, G. R., Estivill, X., Birney, E., Bouffard, G. G., Guan, X. B., Hansen, N. F., Idol, J. R., Maduro, V. V. B., Maskeri, B., McDowell, J. C., Park, M., Thomas, P. J., Young, A. C., Blakesley, R. W., Muzny, D. M., Sodergren, E., Wheeler, D. A., Worley, K. C., Jiang, H. Y., Weinstock, G. M., Gibbs, R. A., Graves, T., Fulton, R., Mardis, E. R., Wilson, R. K., Clamp, M., Cuff, J., Gnerre, S., Jaffe, D. B., Chang, J. L., Lindblad-Toh, K., Lander, E. S., Koriabine, M., Nefedov, M., Osoegawa, K., Yoshinaga, Y., Zhu, B. L., De Jong, P. J., and Encode Project Consortium. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816.
Black, D. L. (1998). Splicing in the inner ear: a familiar tune, but what are the instruments? Neuron 20, 165–168.
Bono, F., and Gehring, N. H. (2011). Assembly, disassembly and recycling: the dynamics of exon junction complexes. RNA Biol. 8, 24–29.
Bornstein, P., McKay, J., Liska, D. J., Apone, S., and Devarayalu, S. (1988). Interactions between the promoter and first intron are involved in transcriptional control of alpha 1(I) collagen gene expression. Mol. Cell. Biol. 8, 4851–4857.
Bradnam, K. R., and Korf, I. (2008). Longer first introns are a general property of eukaryotic gene structure. PLoS ONE 3, e3093. doi: 10.1371/journal.pone.0003093
Bres, V., Gomes, N., Pickle, L., and Jones, K. A. (2005). A human splicing factor, SKIP, associates with P-TEFb and enhances transcription elongation by HIV-1 Tat. Genes Dev. 19, 1211–1226.
Brody, Y., Neufeld, N., Bieberstein, N., Causse, S. Z., Bohnlein, E. M., Neugebauer, K. M., Darzacq, X., and Shav-Tal, Y. (2011). The in vivo kinetics of RNA polymerase II elongation during co-transcriptional splicing. PLoS Biol. 9, e1000573. doi: 10.1371/journal.pbio.1000573
Brogna, S., and Wen, J. (2009). Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Mol. Biol. 16, 107–113.
Buchman, A. R., and Berg, P. (1988). Comparison of intron-dependent and intron-independent gene expression. Mol. Cell. Biol. 8, 4395–4405.
Bullock, S. L., and Ish-Horowicz, D. (2001). Conserved signals and machinery for RNA transport in Drosophila oogenesis and embryogenesis. Nature 414, 611–616.
Carmel, L., Rogozin, I. B., Wolf, Y. I., and Koonin, E. V. (2007a). Evolutionarily conserved genes preferentially accumulate introns. Genome Res. 17, 1045–1050.
Carmel, L., Rogozin, I. B., Wolf, Y. I., and Koonin, E. V. (2007b). Patterns of intron gain and conservation in eukaryotic genes. BMC Evol. Biol. 7, 192. doi: 10.1186/1471-2148-7-192
Carmel, L., Wolf, Y. I., Rogozin, I. B., and Koonin, E. V. (2007). Three distinct modes of intron dynamics in the evolution of eukaryotes. Genome Res. 17, 1034–1044.
Cenik, C., Derti, A., Mellor, J. C., Berriz, G. F., and Roth, F. P. (2010). Genome-wide functional analysis of human 5’ untranslated region introns. Genome Biol. 11, R29.
Chang, Y. F., Imam, J. S., and Wilkinson, M. F. (2007). The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 76, 51–74.
Chen, D., Meng, Y., Yuan, C., Bai, L., Huang, D., Lv, S., Wu, P., Chen, L. L., and Chen, M. (2011). Plant siRNAs from introns mediate DNA methylation of host genes. RNA 17, 1012–1024.
Chen, W., Luo, L., and Zhang, L. (2010). The organization of nucleosomes around splice sites. Nucleic Acids Res. 38, 2788–2798.
Cheng, J., Belgrader, P., Zhou, X., and Maquat, L. E. (1994). Introns are cis effectors of the nonsense-codon-mediated reduction in nuclear mRNA abundance. Mol. Cell. Biol. 14, 6317–6325.
Choi, T., Huang, M., Gorman, C., and Jaenisch, R. (1991). A generic intron increases gene expression in transgenic mice. Mol. Cell. Biol. 11, 3070–3074.
Clark, A. J., Archibald, A. L., McClenaghan, M., Simons, J. P., Wallace, R., and Whitelaw, C. B. (1993). Enhancing the efficiency of transgene expression. Philos. Trans. R. Soc. Lond. B Biol. Sci. 339, 225–232.
Csuros, M. (2005). “Likely scenarios of intron evolution,” in RECOMB WS on Comparative Genomics 2005, eds A. Mclysaght, and D. H. Huson (Dublin: Springer), 47–60.
Csuros, M., Rogozin, I. B., and Koonin, E. V. (2011). A detailed history of intron-rich eukaryotic ancestors inferred from a global survey of 100 complete genomes. PLoS Comput. Biol. 7, e1002150. doi: 10.1371/journal.pcbi.1002150
Culler, S. J., Hoff, K. G., Voelker, R. B., Berglund, J. A., and Smolke, C. D. (2010). Functional selection and systematic analysis of intronic splicing elements identify active sequence motifs and associated splicing factors. Nucleic Acids Res. 38, 5152–5165.
Damgaard, C. K., Kahns, S., Lykke-Andersen, S., Nielsen, A. L., Jensen, T. H., and Kjems, J. (2008). A 5′ splice site enhances the recruitment of basal transcription initiation factors in vivo. Mol. Cell 29, 271–278.
Das, R., Yu, J., Zhang, Z., Gygi, M. P., Krainer, A. R., Gygi, S. P., and Reed, R. (2007). SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol. Cell 26, 867–881.
Demir-Weusten, A. Y. (2002). Distribution of different Fibronectin isoforms in the extracellular matrices of human term Placenta. Turk J. Med. Sci. 32, 445–456.
Dieci, G., Preti, M., and Montanini, B. (2009). Eukaryotic snoRNAs: a paradigm for gene expression flexibility. Genomics 94, 83–88.
Diem, M. D., Chan, C. C., Younis, I., and Dreyfuss, G. (2007). PYM binds the cytoplasmic exon-junction complex and ribosomes to enhance translation of spliced mRNAs. Nat. Struct. Mol. Biol. 14, 1173–1179.
Dredge, B. K., and Darnell, R. B. (2003). Nova regulates GABA(A) receptor gamma2 alternative splicing via a distal downstream UCAU-rich intronic splicing enhancer. Mol. Cell. Biol. 23, 4687–4700.
Dreyfuss, G., Kim, V. N., and Kataoka, N. (2002). Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell. Biol. 3, 195–205.
Durand, C., Roeth, R., Dweep, H., Vlatkovic, I., Decker, E., Schneider, K. U., and Rappold, G. (2011). Alternative splicing and nonsense-mediated RNA decay contribute to the regulation of SHOX expression. PLoS ONE 6, e18115. doi: 10.1371/journal.pone.0018115
Filipowicz, W., and Pogacic, V. (2002). Biogenesis of small nucleolar ribonucleoproteins. Curr. Opin. Cell Biol. 14, 319–327.
Fisher, S., Grice, E. A., Vinton, R. M., Bessling, S. L., and McCallion, A. S. (2006). Conservation of RET regulatory function from human to zebrafish without sequence similarity. Science 312, 276–279.
Fong, Y. W., and Zhou, Q. (2001). Stimulatory effect of splicing factors on transcriptional elongation. Nature 414, 929–933.
Forget, A., and Chartrand, P. (2011). Cotranscriptional assembly of mRNP complexes that determine the cytoplasmic fate of mRNA. Transcription 2, 86–90.
Fortes, P., Cuevas, Y., Guan, F., Liu, P., Pentlicky, S., Jung, S. P., Martinez-Chantar, M. L., Prieto, J., Rowe, D., and Gunderson, S. I. (2003). Inhibiting expression of specific genes in mammalian cells with 5′ end-mutated U1 small nuclear RNAs targeted to terminal exons of pre-mRNA. Proc. Natl. Acad. Sci. U.S.A. 100, 8264–8269.
Friend, K., Lovejoy, A. F., and Steitz, J. A. (2007). U2 snRNP binds intronless histone pre-mRNAs to facilitate U7-snRNP-dependent 3′ end formation. Mol. Cell 28, 240–252.
Furger, A., O’Sullivan, J. M., Binnie, A., Lee, B. A., and Proudfoot, N. J. (2002). Promoter proximal splice sites enhance transcription. Genes Dev. 16, 2792–2799.
Furth, P. A., Choe, W. T., Rex, J. H., Byrne, J. C., and Baker, C. C. (1994). Sequences homologous to 5′ splice sites are required for the inhibitory activity of papillomavirus late 3′ untranslated regions. Mol. Cell. Biol. 14, 5278–5289.
Gaunitz, F., Deichsel, D., Heise, K., Werth, M., Anderegg, U., and Gebhardt, R. (2005). An intronic silencer element is responsible for specific zonal expression of glutamine synthetase in the rat liver. Hepatology 41, 1225–1232.
Gaunitz, F., Heise, K., and Gebhardt, R. (2004). A silencer element in the first intron of the glutamine synthetase gene represses induction by glucocorticoids. Mol. Endocrinol. 18, 63–69.
Gilligan, P. C., Kumari, P., Lim, S., Cheong, A., Chang, A., and Sampath, K. (2011). Conservation defines functional motifs in the squint/nodal-related 1 RNA dorsal localization element. Nucleic Acids Res. 39, 3340–3349.
Gilson, P. R., Su, V., Slamovits, C. H., Reith, M. E., Keeling, P. J., and McFadden, G. I. (2006). Complete nucleotide sequence of the chlorarachniophyte nucleomorph: nature’s smallest nucleus. Proc. Natl. Acad. Sci. U.S.A. 103, 9566–9571.
Gonzalez, C. I., Ruiz-Echevarria, M. J., Vasudevan, S., Henry, M. F., and Peltz, S. W. (2000). The yeast hnRNP-like protein Hrp1/Nab4 marks a transcript for nonsense-mediated mRNA decay. Mol. Cell 5, 489–499.
Graur, D., and Li, W.-H. (2000). Fundamentals of Molecular Evolution, 2nd Edn. Sunderland, MA: Sinauer Associates.
Graveley, B. R. (2001). Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 17, 100–107.
Green, R. E., Lewis, B. P., Hillman, R. T., Blanchette, M., Lareau, L. F., Garnett, A. T., Rio, D. C., and Brenner, S. E. (2003). Widespread predicted nonsense-mediated mRNA decay of alternatively-spliced transcripts of human normal and disease genes. Bioinformatics 19(Suppl. 1), i118–i121.
Gruss, P., Lai, C. J., Dhar, R., and Khoury, G. (1979). Splicing as a requirement for biogenesis of functional 16S mRNA of simian virus 40. Proc. Natl. Acad. Sci. U.S.A. 76, 4317–4321.
Gubb, D. (1986). Intron-delay and the precision of expression of homoeotic gene products in Drosophila. Dev. Genet. 7, 119–131.
Gunderson, S. I., Polycarpou-Schwarz, M., and Mattaj, I. W. (1998). U1 snRNP inhibits pre-mRNA polyadenylation through a direct interaction between U1 70K and poly(A) polymerase. Mol. Cell 1, 255–264.
Gunderson, S. I., Vagner, S., Polycarpou-Schwarz, M., and Mattaj, I. W. (1997). Involvement of the carboxyl terminus of vertebrate poly(A) polymerase in U1A autoregulation and in the coupling of splicing and polyadenylation. Genes Dev. 11, 761–773.
Hachet, O., and Ephrussi, A. (2001). Drosophila Y14 shuttles to the posterior of the oocyte and is required for oskar mRNA transport. Curr. Biol. 11, 1666–1674.
Hachet, O., and Ephrussi, A. (2004). Splicing of oskar RNA in the nucleus is coupled to its cytoplasmic localization. Nature 428, 959–963.
Hamer, D. H., Smith, K. D., Boyer, S. H., and Leder, P. (1979). SV40 recombinants carrying rabbit beta-globin gene coding sequences. Cell 17, 725–735.
Hansen, K. D., Lareau, L. F., Blanchette, M., Green, R. E., Meng, Q., Rehwinkel, J., Gallusser, F. L., Izaurralde, E., Rio, D. C., Dudoit, S., and Brenner, S. E. (2009). Genome-wide identification of alternative splice forms down-regulated by nonsense-mediated mRNA decay in Drosophila. PLoS Genet. 5, e1000525. doi: 10.1371/journal.pgen.1000525
Hartmann, B., and Valcarcel, J. (2009). Decrypting the genome’s alternative messages. Curr. Opin. Cell Biol. 21, 377–386.
Havlioglu, N., Wang, J., Fushimi, K., Vibranovski, M. D., Kan, Z., Gish, W., Fedorov, A., Long, M., and Wu, J. Y. (2007). An intronic signal for alternative splicing in the human genome. PLoS ONE 2, e1246. doi: 10.1371/journal.pone.0001246
Hillman, R. T., Green, R. E., and Brenner, S. E. (2004). An unappreciated role for RNA surveillance. Genome Biol. 5, R8.
Hinske, L. C., Galante, P. A., Kuo, W. P., and Ohno-Machado, L. (2010). A potential role for intragenic miRNAs on their hosts’ interactome. BMC Genomics 11, 533. doi: 10.1186/1471-2164-11-533
Hood, J. K., and Silver, P. A. (1999). In or out? Regulating nuclear transport. Curr. Opin. Cell Biol. 11, 241–247.
Horne-Badovinac, S., and Bilder, D. (2008). Dynein regulates epithelial polarity and the apical localization of stardust A mRNA. PLoS Genet. 4, e8. doi: 10.1371/journal.pgen.0040008
Huang, Z. P., Zhou, H., He, H. L., Chen, C. L., Liang, D., and Qu, L. H. (2005). Genome-wide analyses of two families of snoRNA genes from Drosophila melanogaster, demonstrating the extensive utilization of introns for coding of snoRNAs. RNA 11, 1303–1316.
Jin, Y., Suzuki, H., Maegawa, S., Endo, H., Sugano, S., Hashimoto, K., Yasuda, K., and Inoue, K. (2003). A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J. 22, 905–912.
Jobert, L., Pinzon, N., Van Herreweghe, E., Jady, B. E., Guialis, A., Kiss, T., and Tora, L. (2009). Human U1 snRNA forms a new chromatin-associated snRNP with TAF15. EMBO Rep. 10, 494–500.
Juneau, K., Miranda, M., Hillenmeyer, M. E., Nislow, C., and Davis, R. W. (2006). Introns regulate RNA and protein abundance in yeast. Genetics 174, 511–518.
Kaida, D., Berg, M. G., Younis, I., Kasim, M., Singh, L. N., Wan, L., and Dreyfuss, G. (2010). U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 468, 664–668.
Khodor, Y. L., Rodriguez, J., Abruzzi, K. C., Tang, C. H., Marr, M. T. II, and Rosbash, M. (2011). Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev. 25, 2502–2512.
Ko, B., and Gunderson, S. I. (2002). Identification of new poly(A) polymerase-inhibitory proteins capable of regulating pre-mRNA polyadenylation. J. Mol. Biol. 318, 1189–1206.
Kohler, A., and Hurt, E. (2007). Exporting RNA from the nucleus to the cytoplasm. Nat. Rev. Mol. Cell. Biol. 8, 761–773.
Koonin, E. V. (2006). The origin of introns and their role in eukaryogenesis: a compromise solution to the introns-early versus introns-late debate? Biol. Direct 1, 22.
Koonin, E. V. (2009). Intron-dominated genomes of early ancestors of eukaryotes. J. Hered. 100, 618–623.
Kwek, K. Y., Murphy, S., Furger, A., Thomas, B., O’Gorman, W., Kimura, H., Proudfoot, N. J., and Akoulitchev, A. (2002). U1 snRNA associates with TFIIH and regulates transcriptional initiation. Nat. Struct. Biol. 9, 800–805.
Kyburz, A., Friedlein, A., Langen, H., and Keller, W. (2006). Direct interactions between subunits of CPSF and the U2 snRNP contribute to the coupling of pre-mRNA 3′ end processing and splicing. Mol. Cell 23, 195–205.
Lane, C. E., van den Heuvel, K., Kozera, C., Curtis, B. A., Parsons, B. J., Bowman, S., and Archibald, J. M. (2007). Nucleomorph genome of Hemiselmis andersenii reveals complete intron loss and compaction as a driver of protein structure and function. Proc. Natl. Acad. Sci. U.S.A. 104, 19908–19913.
Lareau, L. F., Green, R. E., Bhatnagar, R. S., and Brenner, S. E. (2004). The evolving roles of alternative splicing. Curr. Opin. Struct. Biol. 14, 273–282.
Lauderdale, J. D., and Stein, A. (1992). Introns of the chicken ovalbumin gene promote nucleosome alignment in vitro. Nucleic Acids Res. 20, 6589–6596.
Le Hir, H., Gatfield, D., Izaurralde, E., and Moore, M. J. (2001). The exon-exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J. 20, 4987–4997.
Le Hir, H., Izaurralde, E., Maquat, L. E., and Moore, M. J. (2000). The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon-exon junctions. EMBO J. 19, 6860–6869.
Le Hir, H., Nott, A., and Moore, M. J. (2003). How introns influence and enhance eukaryotic gene expression. Trends Biochem. Sci. 28, 215–220.
Lee, H. C., Choe, J., Chi, S. G., and Kim, Y. K. (2009). Exon junction complex enhances translation of spliced mRNAs at multiple steps. Biochem. Biophys. Res. Commun. 384, 334–340.
Lee, Y., Ahn, C., Han, J., Choi, H., Kim, J., Yim, J., Lee, J., Provost, P., Radmark, O., Kim, S., and Kim, V. N. (2003). The nuclear RNase III Drosha initiates microRNA processing. Nature 425, 415–419.
LeMaire, M. F., and Thummel, C. S. (1990). Splicing precedes polyadenylation during Drosophila E74A transcription. Mol. Cell. Biol. 10, 6059–6063.
Lewis, B. P., Green, R. E., and Brenner, S. E. (2003). Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc. Natl. Acad. Sci. U.S.A. 100, 189–192.
Lin, S., Coutinho-Mansfield, G., Wang, D., Pandit, S., and Fu, X. D. (2008). The splicing factor SC35 has an active role in transcriptional elongation. Nat. Struct. Mol. Biol. 15, 819–826.
Liu, K., Sandgren, E. P., Palmiter, R. D., and Stein, A. (1995). Rat growth hormone gene introns stimulate nucleosome alignment in vitro and in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 92, 7724–7728.
Lopez-Bigas, N., Audit, B., Ouzounis, C., Parra, G., and Guigo, R. (2005). Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 579, 1900–1903.
Lu, H., Lin, L., Sato, S., Xing, Y., and Lee, C. J. (2009). Predicting functional alternative splicing by measuring RNA selection pressure from multigenome alignments. PLoS Comput. Biol. 5, e1000608. doi: 10.1371/journal.pcbi.1000608
Lu, S., and Cullen, B. R. (2003). Analysis of the stimulatory effect of splicing on mRNA production and utilization in mammalian cells. RNA 9, 618–630.
Luehrsen, K. R., and Walbot, V. (1991). Intron enhancement of gene expression and the splicing efficiency of introns in maize cells. Mol. Gen. Genet. 225, 81–93.
Luo, M. J., and Reed, R. (1999). Splicing is required for rapid and efficient mRNA export in metazoans. Proc. Natl. Acad. Sci. U.S.A. 96, 14937–14942.
Lutz, C. S., Murthy, K. G., Schek, N., O’Connor, J. P., Manley, J. L., and Alwine, J. C. (1996). Interaction between the U1 snRNP-A protein and the 160-kD subunit of cleavage-polyadenylation specificity factor increases polyadenylation efficiency in vitro. Genes Dev. 10, 325–337.
Lykke-Andersen, J., Shu, M. D., and Steitz, J. A. (2000). Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 103, 1121–1131.
Lynch, M. (2002). Intron evolution as a population-genetic process. Proc. Natl. Acad. Sci. U.S.A. 99, 6118–6123.
Ma, X. M., Yoon, S. O., Richardson, C. J., Julich, K., and Blenis, J. (2008). SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell 133, 303–313.
Mair, G., Shi, H., Li, H., Djikeng, A., Aviles, H. O., Bishop, J. R., Falcone, F. H., Gavrilescu, C., Montgomery, J. L., Santori, M. I., Stern, L. S., Wang, Z., Ullu, E., and Tschudi, C. (2000). A new twist in trypanosome RNA metabolism: cis-splicing of pre-mRNA. RNA 6, 163–169.
Maiuri, P., Knezevich, A., De Marco, A., Mazza, D., Kula, A., McNally, J. G., and Marcello, A. (2011). Fast transcription rates of RNA polymerase II in human cells. EMBO Rep. 12, 1280–1285.
Makarova, J. A., and Kramerov, D. A. (2009). Analysis of C/D box snoRNA genes in vertebrates: The number of copies decreases in placental mammals. Genomics 94, 11–19.
Maquat, L. E. (2004). Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell. Biol. 5, 89–99.
Martin, K. C., and Ephrussi, A. (2009). mRNA localization: gene expression in the spatial dimension. Cell 136, 719–730.
Martin, W., and Koonin, E. V. (2006). Introns and the origin of nucleus-cytosol compartmentalization. Nature 440, 41–45.
Mascarenhas, D., Mettler, I. J., Pierce, D. A., and Lowe, H. W. (1990). Intron-mediated enhancement of heterologous gene expression in maize. Plant Mol. Biol. 15, 913–920.
Masuda, S., Das, R., Cheng, H., Hurt, E., Dorman, N., and Reed, R. (2005). Recruitment of the human TREX complex to mRNA during splicing. Genes Dev. 19, 1512–1517.
Mattick, J. S., and Gagen, M. J. (2001). The evolution of controlled multitasked gene networks: the role of introns and other noncoding RNAs in the development of complex organisms. Mol. Biol. Evol. 18, 1611–1630.
Maxwell, E. S., and Fournier, M. J. (1995). The small nucleolar RNAs. Annu. Rev. Biochem. 64, 897–934.
McCracken, S., Fong, N., Yankulov, K., Ballantyne, S., Pan, G., Greenblatt, J., Patterson, S. D., Wickens, M., and Bentley, D. L. (1997). The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature 385, 357–361.
McCracken, S., Lambermon, M., and Blencowe, B. J. (2002). SRm160 splicing coactivator promotes transcript 3′-end cleavage. Mol. Cell. Biol. 22, 148–160.
McGlincy, N. J., and Smith, C. W. (2008). Alternative splicing resulting in nonsense-mediated mRNA decay: what is the meaning of nonsense? Trends Biochem. Sci. 33, 385–393.
McKay, S. L., and Johnson, T. L. (2011). An investigation of a role for U2 snRNP spliceosomal components in regulating transcription. PLoS ONE 6, e16077. doi: 10.1371/journal.pone.0016077
Millevoi, S., Geraghty, F., Idowu, B., Tam, J. L., Antoniou, M., and Vagner, S. (2002). A novel function for the U2AF 65 splicing factor in promoting pre-mRNA 3′-end processing. EMBO Rep. 3, 869–874.
Millevoi, S., Loulergue, C., Dettwiler, S., Karaa, S. Z., Keller, W., Antoniou, M., and Vagner, S. (2006). An interaction between U2AF 65 and CF I(m) links the splicing and 3′ end processing machineries. EMBO J. 25, 4854–4864.
Millevoi, S., and Vagner, S. (2010). Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res. 38, 2757–2774.
Mitrovich, Q. M., and Anderson, P. (2000). Unproductively spliced ribosomal protein mRNAs are natural targets of mRNA surveillance in C. elegans. Genes Dev. 14, 2173–2184.
Mohr, S. E., Dillon, S. T., and Boswell, R. E. (2001). The RNA-binding protein Tsunagi interacts with Mago Nashi to establish polarity and localize oskar mRNA during Drosophila oogenesis. Genes Dev. 15, 2886–2899.
Moore, M. J. (2005). From birth to death: the complex lives of eukaryotic mRNAs. Science 309, 1514–1518.
Moore, M. J., and Proudfoot, N. J. (2009). Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136, 688–700.
Morello, L., Giani, S., Troina, F., and Breviario, D. (2011). Testing the IMEter on rice introns and other aspects of intron-mediated enhancement of gene expression. J. Exp. Bot. 62, 533–544.
Morrison, H. G., McArthur, A. G., Gillin, F. D., Aley, S. B., Adam, R. D., Olsen, G. J., Best, A. A., Cande, W. Z., Chen, F., Cipriano, M. J., Davids, B. J., Dawson, S. C., Elmendorf, H.G., Hehl, A. B., Holder, M. E., Huse, S. M., Kim, U. U., Lasek-Nesselquist, E., Manning, G., Nigam, A., Nixon, J. E. J., Palm, D., Passamaneck, N. E., Prabhu, A., Reich, C. I., Reiner, D. S., Samuelson, J., Svard, S. G., and Sogin, M. L. (2007). Genomic minimalism in the early diverging intestinal parasite Giardia lamblia. Science 317, 1921–1926.
Nagy, E., and Maquat, L. E. (1998). A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci. 23, 198–199.
Nahkuri, S., Taft, R. J., and Mattick, J. S. (2009). Nucleosomes are preferentially positioned at exons in somatic and sperm cells. Cell Cycle 8, 3420–3424.
Nguyen, H. D., Yoshihama, M., and Kenmochi, N. (2005). New maximum likelihood estimators for eukaryotic intron evolution. PLoS Comput. Biol. 1, e79. doi: 10.1371/journal.pcbi.0010079
Nilsen, T. W., and Graveley, B. R. (2010). Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463.
Nott, A., Le Hir, H., and Moore, M. J. (2004). Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Genes Dev. 18, 210–222.
Nott, A., Meislin, S. H., and Moore, M. J. (2003). A quantitative analysis of intron effects on mammalian gene expression. RNA 9, 607–617.
Obernosterer, G., Leuschner, P. J., Alenius, M., and Martinez, J. (2006). Post-transcriptional regulation of microRNA expression. RNA 12, 1161–1167.
Ohno, M., Segref, A., Kuersten, S., and Mattaj, I. W. (2002). Identity elements used in export of mRNAs. Mol. Cell 9, 659–671.
Okamura, K., and Lai, E. C. (2008). Endogenous small interfering RNAs in animals. Nat. Rev. Mol. Cell. Biol. 9, 673–678.
Palmiter, R. D., Sandgren, E. P., Avarbock, M. R., Allen, D. D., and Brinster, R. L. (1991). Heterologous introns can enhance expression of transgenes in mice. Proc. Natl. Acad. Sci. U.S.A. 88, 478–482.
Pan, Q., Saltzman, A. L., Kim, Y. K., Misquitta, C., Shai, O., Maquat, L. E., Frey, B. J., and Blencowe, B. J. (2006). Quantitative microarray profiling provides evidence against widespread coupling of alternative splicing with nonsense-mediated mRNA decay to control gene expression. Genes Dev. 20, 153–158.
Pan, Q., Shai, O., Lee, L. J., Frey, B. J., and Blencowe, B. J. (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415.
Parra, G., Bradnam, K., Rose, A. B., and Korf, I. (2011). Comparative and functional analysis of intron-mediated enhancement signals reveals conserved features among plants. Nucleic Acids Res. 39, 5328–5337.
Petit, M. M., Lindskog, H., Larsson, E., Wasteson, P., Athley, E., Breuer, S., Angstenberger, M., Hertfelder, D., Mattsson, E., Nordheim, A., Nelander, S., and Lindahl, P. (2008). Smooth muscle expression of lipoma preferred partner is mediated by an alternative intronic promoter that is regulated by serum response factor/myocardin. Circ. Res. 103, 61–69.
Proudfoot, N. J. (2011). Ending the message: poly(A) signals then and now. Genes Dev. 25, 1770–1782.
Rearick, D., Prakash, A., McSweeny, A., Shepard, S. S., Fedorova, L., and Fedorov, A. (2011). Critical association of ncRNA with introns. Nucleic Acids Res. 39, 2357–2366.
Reis, E. M., Louro, R., Nakaya, H. I., and Verjovski-Almeida, S. (2005). As antisense RNA gets intronic. OMICS 9, 2–12.
Rigo, F., and Martinson, H. G. (2008). Functional coupling of last-intron splicing and 3′-end processing to transcription in vitro: the poly(A) signal couples to splicing before committing to cleavage. Mol. Cell. Biol. 28, 849–862.
Rodrigues, J. P., Rode, M., Gatfield, D., Blencowe, B. J., Carmo-Fonseca, M., and Izaurralde, E. (2001). REF proteins mediate the export of spliced and unspliced mRNAs from the nucleus. Proc. Natl. Acad. Sci. U.S.A. 98, 1030–1035.
Rogozin, I. B., Wolf, Y. I., Sorokin, A. V., Mirkin, B. G., and Koonin, E. V. (2003). Remarkable interkingdom conservation of intron positions and massive, lineage-specific intron loss and gain in eukaryotic evolution. Curr. Biol. 13, 1512–1517.
Rose, A. B., Elfersi, T., Parra, G., and Korf, I. (2008). Promoter-proximal introns in Arabidopsis thaliana are enriched in dispersed signals that elevate gene expression. Plant Cell 20, 543–551.
Ruby, J. G., Jan, C. H., and Bartel, D. P. (2007). Intronic microRNA precursors that bypass Drosha processing. Nature 448, 83–86.
Rueter, S. M., Dawson, T. R., and Emeson, R. B. (1999). Regulation of alternative splicing by RNA editing. Nature 399, 75–80.
Ryu, W. S., and Mertz, J. E. (1989). Simian virus 40 late transcripts lacking excisable intervening sequences are defective in both stability in the nucleus and transport to the cytoplasm. J. Virol. 63, 4386–4394.
Sakharkar, M. K., Chow, V. T., and Kangueane, P. (2004). Distributions of exons and introns in the human genome. In silico Biol. (Gedrukt) 4, 387–393.
Schmucker, D., Clemens, J. C., Shu, H., Worby, C. A., Xiao, J., Muda, M., Dixon, J. E., and Zipursky, S. L. (2000). Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell 101, 671–684.
Schwartz, S., and Ast, G. (2010). Chromatin density and splicing destiny: on the cross-talk between chromatin structure and splicing. EMBO J. 29, 1629–1636.
Schwartz, S., Meshorer, E., and Ast, G. (2009). Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 16, 990–995.
Schwartz, S. H., Silva, J., Burstein, D., Pupko, T., Eyras, E., and Ast, G. (2008). Large-scale comparative analysis of splicing signals and their corresponding splicing factors in eukaryotes. Genome Res. 18, 88–103.
Scohy, S., Gabant, P., Szpirer, C., and Szpirer, J. (2000). Identification of an enhancer and an alternative promoter in the first intron of the alpha-fetoprotein gene. Nucleic Acids Res. 28, 3743–3751.
Seoighe, C., and Korir, P. K. (2011). Evidence for intron length conservation in a set of mammalian genes associated with embryonic development. BMC Bioinformatics 12(Suppl. 9), S16. doi: 10.1186/1471-2105-12-S9-S16
Shabalina, S. A., Ogurtsov, A. Y., Spiridonov, A. N., Novichkov, P. S., Spiridonov, N. A., and Koonin, E. V. (2010). Distinct patterns of expression and evolution of intronless and intron-containing mammalian genes. Mol. Biol. Evol. 27, 1745–1749.
Shalgi, R., Brosh, R., Oren, M., Pilpel, Y., and Rotter, V. (2009). Coupling transcriptional and post-transcriptional miRNA regulation in the control of cell fate. Aging (Albany NY) 1, 762–770.
Shen, H. (2009). UAP56 – a key player with surprisingly diverse roles in pre-mRNA splicing and nuclear export. BMB Rep. 42, 185–188.
Silva, A. L., and Romao, L. (2009). The mammalian nonsense-mediated mRNA decay pathway: to decay or not to decay! Which players make the decision? FEBS Lett. 583, 499–505.
Singh, J., and Padgett, R. A. (2009). Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol. 16, 1128–1133.
Smith, C. W., Patton, J. G., and Nadal-Ginard, B. (1989). Alternative splicing in the control of gene expression. Annu. Rev. Genet. 23, 527–577.
Sorek, R., and Ast, G. (2003). Intronic sequences flanking alternatively spliced exons are conserved between human and mouse. Genome Res. 13, 1631–1637.
Sorek, R., Shamir, R., and Ast, G. (2004). How prevalent is functional alternative splicing in the human genome? Trends Genet. 20, 68–71.
Spies, N., Nielsen, C. B., Padgett, R. A., and Burge, C. B. (2009). Biased chromatin signatures around polyadenylation sites and exons. Mol. Cell 36, 245–254.
Stamm, S., Ben-Ari, S., Rafalska, I., Tang, Y., Zhang, Z., Toiber, D., Thanaraj, T. A., and Soreq, H. (2005). Function of alternative splicing. Gene 344, 1–20.
Sureau, A., Gattoni, R., Dooghe, Y., Stevenin, J., and Soret, J. (2001). SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J. 20, 1785–1796.
Swinburne, I. A., Miguez, D. G., Landgraf, D., and Silver, P. A. (2008). Intron length increases oscillatory periods of gene expression in animal cells. Genes Dev. 22, 2342–2346.
Swinburne, I. A., and Silver, P. A. (2008). Intron delays and transcriptional timing during development. Dev. Cell 14, 324–330.
Takashima, Y., Ohtsuka, T., Gonzalez, A., Miyachi, H., and Kageyama, R. (2011). Intronic delay is essential for oscillatory expression in the segmentation clock. Proc. Natl. Acad. Sci. U.S.A. 108, 3300–3305.
Tange, T. O., Nott, A., and Moore, M. J. (2004). The ever-increasing complexities of the exon junction complex. Curr. Opin. Cell Biol. 16, 279–284.
Taniguchi, I., and Ohno, M. (2008). ATP-dependent recruitment of export factor Aly/REF onto intronless mRNAs by RNA helicase UAP56. Mol. Cell. Biol. 28, 601–608.
Thummel, C. S., Burtis, K. C., and Hogness, D. S. (1990). Spatial and temporal patterns of E74 transcription during Drosophila development. Cell 61, 101–111.
Tian, B., Pan, Z., and Lee, J. Y. (2007). Widespread mRNA polyadenylation events in introns indicate dynamic interplay between polyadenylation and splicing. Genome Res. 17, 156–165.
Tilgner, H., Nikolaou, C., Althammer, S., Sammeth, M., Beato, M., Valcarcel, J., and Guigo, R. (2009). Nucleosome positioning as a determinant of exon recognition. Nat. Struct. Mol. Biol. 16, 996–1001.
Tourmente, S., Chapel, S., Dreau, D., Drake, M. E., Bruhat, A., Couderc, J. L., and Dastugue, B. (1993). Enhancer and silencer elements within the first intron mediate the transcriptional regulation of the beta 3 tubulin gene by 20-hydroxyecdysone in Drosophila Kc cells. Insect Biochem. Mol. Biol. 23, 137–143.
Trcek, T., and Singer, R. H. (2010). The cytoplasmic fate of an mRNP is determined cotranscriptionally: exception or rule? Genes Dev. 24, 1827–1831.
Tycowski, K. T., Shu, M. D., and Steitz, J. A. (1996). A mammalian gene with introns instead of exons generating stable RNA products. Nature 379, 464–466.
Ule, J., Ule, A., Spencer, J., Williams, A., Hu, J. S., Cline, M., Wang, H., Clark, T., Fraser, C., Ruggiu, M., Zeeberg, B. R., Kane, D., Weinstein, J. N., Blume, J., and Darnell, R. B. (2005). Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 37, 844–852.
Valencia, P., Dias, A. P., and Reed, R. (2008). Splicing promotes rapid and efficient mRNA export in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 105, 3386–3391.
Vasil, V., Clancy, M., Ferl, R. J., Vasil, I. K., and Hannah, L. C. (1989). Increased gene expression by the first intron of maize shrunken-1 locus in grass species. Plant Physiol. 91, 1575–1579.
Veraldi, K. L., Arhin, G. K., Martincic, K., Chung-Ganster, L. H., Wilusz, J., and Milcarek, C. (2001). hnRNP F influences binding of a 64-kilodalton subunit of cleavage stimulation factor to mRNA precursors in mouse B cells. Mol. Cell. Biol. 21, 1228–1238.
Vinogradov, A. E. (2004). Compactness of human housekeeping genes: selection for economy or genomic design? Trends Genet. 20, 248–253.
Vinogradov, A. E. (2006). “Genome design” model: evidence from conserved intronic sequence in human-mouse comparison. Genome Res. 16, 347–354.
Wahl, M. C., Will, C. L., and Luhrmann, R. (2009). The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718.
Wang, G. S., and Cooper, T. A. (2007). Splicing in disease: disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 8, 749–761.
Wang, K. C., and Chang, H. Y. (2011). Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914.
Wang, Z., and Burge, C. B. (2008). Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14, 802–813.
Watanabe, T., Totoki, Y., Toyoda, A., Kaneda, M., Kuramochi-Miyagawa, S., Obata, Y., Chiba, H., Kohara, Y., Kono, T., Nakano, T., Azim Surani, M., Sakaki, Y., and Sasaki, H. (2008). Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 453, 539–543.
Westholm, J. O., and Lai, E. C. (2011). Mirtrons: microRNA biogenesis via splicing. Biochimie 93, 1897–1904.
Wiegand, H. L., Lu, S., and Cullen, B. R. (2003). Exon junction complexes mediate the enhancing effect of splicing on mRNA expression. Proc. Natl. Acad. Sci. U.S.A. 100, 11327–11332.
Will, C. L., and Luhrmann, R. (2005). Splicing of a rare class of introns by the U12-dependent spliceosome. Biol. Chem. 386, 713–724.
Wittkopp, N., Huntzinger, E., Weiler, C., Sauliere, J., Schmidt, S., Sonawane, M., and Izaurralde, E. (2009). Nonsense-mediated mRNA decay effectors are essential for zebrafish embryonic development and survival. Mol. Cell. Biol. 29, 3517–3528.
Wollerton, M. C., Gooding, C., Wagner, E. J., Garcia-Blanco, M. A., and Smith, C. W. (2004). Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol. Cell 13, 91–100.
Wu, X., Liu, M., Downie, B., Liang, C., Ji, G., Li, Q. Q., and Hunt, A. G. (2011). Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc. Natl. Acad. Sci. U.S.A. 108, 12533–12538.
Wuarin, J., and Schibler, U. (1994). Physical isolation of nascent RNA chains transcribed by RNA polymerase II: evidence for cotranscriptional splicing. Mol. Cell. Biol. 14, 7219–7225.
Yoshimoto, R., Kataoka, N., Okawa, K., and Ohno, M. (2009). Isolation and characterization of post-splicing lariat-intron complexes. Nucleic Acids Res. 37, 891–902.
Zhang, G. R., Li, X., Cao, H., Zhao, H., and Geller, A. I. (2011). The vesicular glutamate transporter-1 upstream promoter and first intron each support glutamatergic-specific expression in rat postrhinal cortex. Brain Res. 1377, 1–12.
Zhang, S., Ruiz-Echevarria, M. J., Quan, Y., and Peltz, S. W. (1995). Identification and characterization of a sequence motif involved in nonsense-mediated mRNA decay. Mol. Cell. Biol. 15, 2231–2244.
Zhao, C., and Hamilton, T. (2007). Introns regulate the rate of unstable mRNA decay. J. Biol. Chem. 282, 20230–20237.
Keywords: intron function, gene architecture, intron–exon structure, intron positional conservation, expression regulation, non-coding RNAs, exon-junction complex, splicing
Citation: Chorev M and Carmel L (2012) The function of introns. Front. Gene. 3:55. doi: 10.3389/fgene.2012.00055
Received: 19 February 2012; Paper pending published: 05 March 2012;
Accepted: 26 March 2012; Published online: 13 April 2012.
Edited by:
Galina Glazko, University of Arkansas for Medical Sciences, USAReviewed by:
Boris L. Zybailov, University of Arkansas Medical Sciences, USAAncha Baranova, George Mason University, USA
Copyright: © 2012 Chorev and Carmel. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Liran Carmel, Department of Genetics, The Alexander Silberman Institute of Life Sciences, Faculty of Science, The Hebrew University of Jerusalem, Edmond J. Safra Campus, Givat Ram, Jerusalem 91904, Israel. e-mail: carmell@cc.huji.ac.il