
- 1Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart, Germany
- 2University of Tübingen, Tübingen, Germany
Breast cancer is a complex disease which is provoked by a multitude of exogenous and endogenous factors including genetic variations. Recent genome-wide association studies identified a set of more than 18 novel low penetrant susceptibility loci, however, a limitation of this powerful approach is the hampered analysis of polymorphisms in DNA sequences with a high degree of similarity to other genes or pseudo genes. Since this common feature affects the majority of the highly polymorphic genes encoding phase I and II enzymes the retrieval of specific genotype data requires adapted amplification methods. With regard to breast cancer these genes are of certain interest due to their involvement in the metabolism of carcinogens like exogenous genotoxic compounds or steroid hormones. The present review summarizes the observed effects of functional genetic variants of phase I and II enzymes in well designed case control studies to shed light on their contribution to breast cancer risk.
Introduction
The implementation of cost effective high-throughput genotyping methods enables the determination of genotypes at large scale and fast pace. These improvements are prerequisite of the in depth investigation of the polygenetic basis of complex diseases. Prominent examples are genome-wide association studies which led to the identification of novel breast cancer risk factors such as polymorphisms in FGFR2, CCND1, TOX3, MAP3K1, LSP1, CDKN2A, and 2B (Easton et al., 2007; Lambrechts et al., 2012). However, a shortcoming of this comprehensive approach is the exclusion of the majority of genes encoding phase I and II enzymes, because their special genomic architecture hampers the assessment of accurate genotype data. It is necessary to overcome this limitation due to the fact that functional genetic variations in these genes are known to alter expression, activity, and stability of the encoded enzymes causing defective inactivation and excretion of hormones as well as environmental toxicants (Thompson and Ambrosone, 2000; Reszka et al., 2006). Thus, it is of high relevance to understand the potential impact of these polymorphisms in pathogenic processes such as carcinogenesis. In addition, these phase I and II genes play a pivotal role in activation and metabolism of drugs with the potential to trigger therapy response as well as occurrence of adverse side effects (Meyer et al., 2012). With respect to breast cancer pharmacogenetic investigations revealed the impact of a genetical determined poor metabolizer phenotype of the phase I enzyme cytochrome P450 (CYP) 2D6 and tamoxifen treatment outcome (Schroth et al., 2009). This finding has been a matter of debate due to reports on conflicting results that seem to be based on inaccurate genotype data (Brauch et al., 2012). Amongst others this finding underlines the need of specific genotyping methodologies for genes encoding metabolic enzymes. This review will focus on studies investigating the role of genetic variants of phase I and II enzymes in breast cancer risk that used validated genotyping methods.
Breast Cancer Risk
Breast cancer is a multifactorial disease and it is known that the carcinogenic process is affected by several endogenous as well as exogenous factors (Rebbeck et al., 1997). In this respect, steroid hormones play a pivotal role (Key et al., 2002b). Epidemiological studies indicated an increased breast cancer risk in women with prolonged exposure to sex hormones, e.g., early menarche and late menopause (Henderson and Feigelson, 2000; Clemons and Goss, 2001). Moreover, observational studies revealed the risk effect of exogenous hormones such as postmenopausal hormone replacement therapy (HRT; Rossouw et al., 2002; Beral and Million Women Study Collaborators, 2003; Pesch et al., 2005; Flesch-Janys et al., 2008) and oral contraceptives (Collaborative Group on Hormonal Factors in Breast Cancer, 1996; Kahlenborn et al., 2006). The strong correlation between circulation steroid hormones and breast cancer risk is supported by an observation of a two-fold increased risk for women with elevated sex hormone levels (Key et al., 2002a; Eliassen et al., 2006). A functional explanation of these findings comes from in vitro and in vivo studies that indicated initiation, promotion, and progression of breast tumorigenesis by estrogens and their metabolites (Nandi et al., 1995; Yue et al., 2003; Turan et al., 2004). This effect has been attributed to estrogen-induced gene expression of factors involved in cell growth and division (Liu and Lin, 2004) as well as genotoxic action of metabolic compounds such as 4-hydroxy catechol estrogens and estrogen-3,4-quinones (Yager and Davidson, 2006). Moreover, progesterone adds to hormone-induced carcinogenesis by promotion of estrogen synthesis, estrogen receptor expression, and cell proliferation (Poutanen et al., 1995; Shyamala et al., 2002; Moore et al., 2006; Pawlak and Wiebe, 2007). Beyond hormonal factors environmental carcinogens, e.g., tobacco smoke, or genetic factors, e.g., mutations and polymorphisms contribute to breast cancer susceptibility. A genetic basis of breast cancer has been suggested by family studies indicating a two-fold increased risk in the first-degree relatives of women with the disease (Collaborative Group on Hormonal Factors in Breast Cancer, 2001). In the 1990s, the two major breast cancer susceptibility genes BRCA1 and BRCA2 were identified (Miki et al., 1994; Wooster et al., 1995) revealing that harmful mutations in these genes confer to a cumulative disease risk by age 70 years of 65 and 45%, respectively (Antoniou et al., 2003). In the following years further genetic factors with different penetrance and frequency have been described. As of today less than 5% of familial breast cancer were attributed to high penetrance breast cancer genes BRCA1, BRCA2, PTEN, MSH2, STK11, CDH1, and TP53 (Wooster and Weber, 2003; Malone et al., 2006; Walsh et al., 2006) and rare genetic variants at ATM, CHEK2, BRIP, NBN, RAD50, or PALB2 that jointly confer an approximately two-fold increased risk (Meijers-Heijboer et al., 2002; The CHEK2 Breast Cancer Case-Control Consortium, 2004; Rahman et al., 2007). Recent genome-wide association studies revealed strong evidence for more than 18 common breast cancer susceptibility alleles including FGFR2, CCND1, TNRC9, MAP3K1, and LSP1 (Cox et al., 2007; Easton et al., 2007; Lambrechts et al., 2012). Most of these genes are related to DNA repair, cell cycle control, apoptosis, cell growth, and division, representing the most important pathways for the protection of cells against carcinogenic processes. However, the lack of observed risk associations with phase I and II enzymes is potentially based on their exclusion from genome-wide association studies due to hampered assay design or poor quality data which is reflected by the low coverage of these genes in current genotyping arrays (Gamazon et al., 2012).
The Role of Phase I and II Enzymes in Carcinogenesis
Phase I and II enzymes are of particular interest with respect to breast cancer due to their involvement in the metabolism of steroid hormones, chemical carcinogens, and other environmental toxicants (Thompson and Ambrosone, 2000; Reszka et al., 2006). In phase I reaction substrates usually undergo reduction, oxidation, or hydroxylation yielding more polar metabolites; the predominant mediators of this phase are cytochrome P450 (CYP) enzymes (Guengerich, 1999). In most cases phase I metabolism is followed by phase II conjugation reactions. During phase II exogenous or endogenous compounds or their phase I metabolites are conjugated to a more polar molecule, a process that usually produces inactive and water soluble compounds which can be easily excreted by urine or bile (Smith et al., 1994; Turesky, 2004). Conjugating enzymes include glutathione-S-transferases (GSTs), sulfotransferases (SULTs), uridine diphosphate-glucuronosyltransferases (UGTs), N-acetyltransferases (NATs), and Methyltransferases. The combined phase I and II metabolism is mainly a detoxification and elimination process, however, both phases bear the risk of formation of toxic and highly reactive compounds which can induce or promote serious health problems such as cancer (Smith et al., 1994; Windmill et al., 1997). Thus, altered activity of metabolic enzyme holds the potential to increase the exposure to carcinogenic compounds and consequently the risk of tumor formation (Brockstedt et al., 2002).
Challenges of Genotyping
The majority of phase I and II enzymes are encoded by related genes which constitute gene families and subfamilies depending on their degree of sequence similarities. This particular genomic architecture hampers specific genotyping due to the potential co-amplification of homolog gene sequences. Therefore, the establishment of accurate analysis methods requires primer selection by eye inspection, adapted amplification protocols, and verification of genotype calls by an independent method (Justenhoven et al., 2010). An example for the particular need of an appropriate genotyping procedure is the analysis of the SULT1A1 638 G > A (rs9282861) polymorphism. The human SULT1A subfamily comprises three genes SULT1A1, SULT1A2, and SULT1A3 which are located in close proximity on the short arm of chromosome 16 and share sequence similarities of more than 90% (Hempel et al., 2005). Due to these remarkable homologies the selection of applicable primers which enable specific amplification of the SULT1A1 638 G > A region is difficult (Figure 1). Usually automatic assay design tools generate inappropriate primers for such sequences which lead to simultaneous amplification of all members of a gene subfamily resulting in incorrect genotype calls due to abundance of the referent allele (Figure 2A). Valid assays include the identification of primer binding sites in unique DNA regions of the respective gene and adapted annealing temperatures, only such highly selective amplification conditions assure correct genotype calls (Figure 2B). Other gene families and subfamilies with a similar degree of sequence homologies are known for CYP3A, CYP2C, GSTs, as well as NATs and UGTs (Salinas and Wong, 1999; Gellner et al., 2001; Tukey and Strassburg, 2001; Nelson et al., 2004; Sim et al., 2008). So far individual assays for some of these polymorphisms have been established by researchers, e.g., for CYP3A (Justenhoven et al., 2010; The MARIE-GENICA Consortium on Genetic Susceptibility for Menopausal Hormone Therapy Related Breast Cancer Risk, 2010), CYP2D6 (Schaeffeler et al., 2003; Morike et al., 2008), CYP2C19 (Justenhoven et al., 2012), GST, UGT, and SULT1A (The MARIE-GENICA Consortium on Genetic Susceptibility for Menopausal Hormone Therapy Related Breast Cancer Risk, 2010) as well as companies (e.g., Applied Biosystems and Third Wave Technologies)1 , 2. Moreover, particular panels and arrays for the genetic analysis of metabolic enzymes and transporters have been developed within recent years: the AmpliChip® CYP P450 Test3, the DMET Plus Panel DNA Chip4, VeraCode ADME Core Panel5, and the iPLEX ADME PGx Panel6. These tools were initially launched to support pharmacogenomic testing in clinical research and diagnostics, however, their coverage of relevant genes is still incomplete but they provide a convenient basis for a variety of investigations dealing with diverse health issues.

Figure 1. Sequence homologies among the three members of the SULT1A gene subfamily located at chromosome 16 (NT_010393.16). A DNA fragment of 484 base pairs shows: the genetic variants SULT1A1 638 G > A (rs9282861, bold) and 667 A > G (rs1801030, bold/italic) as well as 100 base pairs upstream and downstream from these loci. Comparison of the DNA sequences shows that these genes differ only in a small of number of nucleotides (marked in gray) indicating sequence similarities of more than 90% between SULT1A1, SULT1A2, and SULT1A3.
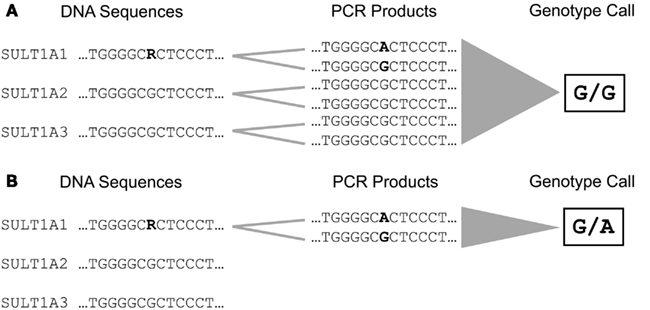
Figure 2. Amplification and genotyping of the DNA sequence comprising the SULT1A1 638 G > A (rs9282861) polymorphism. (A) The selection of unspecific primer binding sites lead to simultaneous amplification of SULT1A1, SULT1A2, and SULT1A3 due to their high degree of sequence homology. This results in accumulation of amplification products carrying the referent G allele leading to an incorrect genotype call for rs9282861 (homozygous GG). (B) Selection of primer binding sites specific for SULT1A1 enables amplification of the rs9282861 sequence region only resulting in correct determination of the genotype (heterozygous GA).
Phase I and II Enzymes in Association with Breast Cancer Risk
Candidate gene approaches provide evidence for a particular role of metabolic enzymes in breast carcinogenesis. As of yet only a few studies analyzed the impact of polymorphisms in genes with high sequence homologies, whereas genes like CYP1A1 and CYP1B1 have been studied intensely (Economopoulos and Sergentanis, 2010; Sergentanis and Economopoulos, 2010). Therefore, this review focuses on those genes which are usually underrepresented in association studies due to technical issues. Literature search was done by PubMed7 using the key words “breast cancer polymorphism phase I,” “breast cancer polymorphism phase II,” “breast cancer polymorphism CYP” “breast cancer polymorphism UGT,” “breast cancer polymorphism SULT,” “breast cancer polymorphism GST,” and “breast cancer polymorphism NAT” in August 2012. In a next step studies analyzing associations between the respective polymorphisms and breast cancer risk factors or breast tumor characteristics were selected on the basis of study size, i.e., inclusion of more than 500 cases and 500 controls, DNA extracted from blood, validation of genotyping results by an independent method or meta analyses on summary data of at least five independent studies.
Significant associations, with p < 0.05 or 95% confidence interval not including 1.0, between polymorphic loci in genes encoding phase I and II enzymes and breast cancer risk are summarized in Table 1. It has been shown that functional genetic variants of the CYP2C19 are associated with overall breast cancer risk and HRT-related breast cancer risk (Gan et al., 2011; Justenhoven et al., 2012). It is of note that these findings in two independent studies show similar effects. The variant CYP2C19*3 (rs57081121) which lead to a decreased activity of the CYP2C19 has been associated with increased risk in Asians (Gan et al., 2011) and the variant CYP2C19*17 (rs12248560) causing an ultra rapid metabolizer phenotype leads to a decreased HRT-related breast cancer risk in Europeans (Justenhoven et al., 2012). It is known that CYP2C19 catabolizes estrogens and progesterone (Yamazaki and Shimada, 1997; Cheng et al., 2001; Cribb et al., 2006) and the reported results suggest that increased metabolic activity of the CYP2C19 lowers endogenous hormone levels leading to a decreased risk.
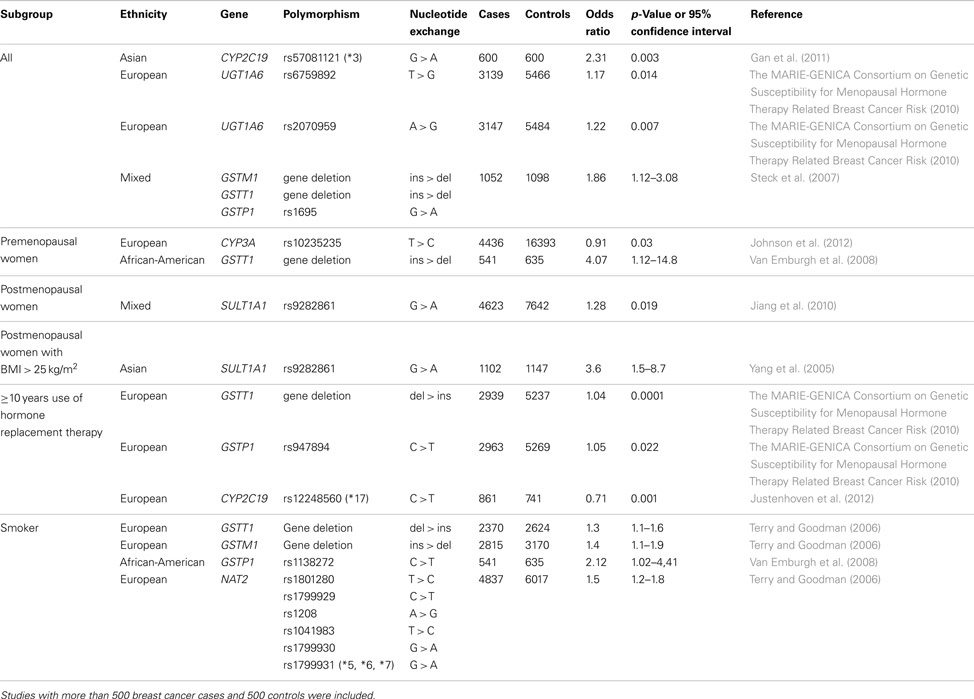
Table 1. Polymorphisms in phase I and II enzymes associated with breast cancer risk.
The polymorphism rs10235235 located the non-coding region of the CYP3A locus has been associated with breast cancer risk in premenopausal women (Johnson et al., 2012). It would be of particular interest to follow-up this finding in independent case control collection and functional studies to understand the observed effect of this variant, because other genetic polymorphisms with known functional consequence located in CYP3A4, CYP3A5, CYP3A7, and CYP3A43 showed no association with breast cancer risk (The MARIE-GENICA Consortium on Genetic Susceptibility for Menopausal Hormone Therapy Related Breast Cancer Risk, 2010).
Two functional genetic variants rs6759892 and rs2070959 which are located in the UGT1A6 have been suggested to affect overall breast cancer risk. These variants did not show any association with hormonal factors (The MARIE-GENICA Consortium on Genetic Susceptibility for Menopausal Hormone Therapy Related Breast Cancer Risk, 2010), therefore, the risk effect is may be based on the role of UGT1A6 in the metabolism of exogenous compounds such as potential carcinogenic drug and food ingredients (Harding et al., 1988; Bock and Kohle, 2005).
It has been reported that the deletion of the GSTM1 and GSTT1 gene as well as the variant allele of the GSTP1 rs1695 polymorphism impact overall breast cancer risk (Steck et al., 2007). Subgroup analyses showed an association of the GSTT1 gene deletion and the GSTP1 rs947894 variant with HRT-related breast cancer susceptibility (The MARIE-GENICA Consortium on Genetic Susceptibility for Menopausal Hormone Therapy Related Breast Cancer Risk, 2010). Moreover, the GSTT1 deletion seems to affect breast cancer risk in premenopausal women (Van Emburgh et al., 2008). These observed effects of GST variants on hormone-related tumorigenesis is may be based on decreased conjugation of genotoxic estrogen quinones leading to elevated levels of DNA damage (Strange et al., 2001; Hachey et al., 2003). In addition, the GSTM1 and GSTT1 deletion as well as the GSTP1 rs1138272 variant, were suggested to affect tobacco smoke-related breast cancer risk (Terry and Goodman, 2006) pointing to the potentially critical role of GSTs in the elimination of exogenous carcinogenic compounds such as polycyclic aromatic hydrocarbons (Hayes and Pulford, 1995).
The SULT1A1 rs9282861 polymorphism has been associated with breast cancer risk in postmenopausal women, in particular with BMI > 25 kg/m2, suggesting a modifying effect of the variant allele on endogenous sex hormone exposure (Yang et al., 2005; Jiang et al., 2010).
It has been reported that the variant NAT2 alleles rs1801280, rs1799929, rs1208, rs1041983, rs1799930, and rs1799931 lead to an increased smoking-related breast cancer which supports the hypothesis that slow acetylators may suffer greater exposure to tobacco carcinogens (Terry and Goodman, 2006).
Phase I and II Enzymes and Breast Tumor Characteristics
Only a few well designed studies investigated the association between phase I and II enzymes and histo-pathological characteristics of breast tumors (Table 2). One study reported an association between the rs61469810 polymorphism of CYP3A43 (CYP3A43*2A) and poorly differentiated breast tumors which may be explained by a potential contribution of the variant allele to increased sex hormone levels (Justenhoven et al., 2010). Another investigation suggested that the rs1058930 polymorphism of CYP2C8 (CYP2C8*4) affects lymph node status of breast cancer patients (Jernstrom et al., 2009). The variant allele is known to lower metabolic activity of the encoded enzyme, however, the authors stated that an impact of the CYP2C9*2 allele which is in linkage disequilibrium with CYP2C8*4 cannot be excluded (Jernstrom et al., 2009).
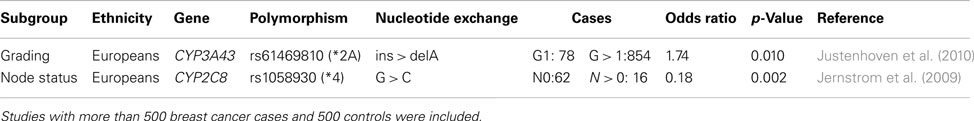
Table 2. Polymorphisms in phase I and II enzymes associated with histo-pathological characteristics of breast tumor.
Conclusion
Genetic variations of phase I and II enzymes alter their activity or protein biosynthesis leading to defective detoxification and elimination of carcinogenic compounds. Due to a high degree of DNA sequence similarity among genes of subfamilies accurate genotyping requires elaborated methods and exhaustive quality control. Until now a few well designed studies give insights into the effect of polymorphisms in metabolic enzymes on breast cancer risk and point to their crucial action in steroid hormone catabolism. These finding underline the pivotal role of sex hormones in the regulation of proliferation, differentiation, and apoptosis as critical pathways for onset and progression of breast cancer (Schindler et al., 1998; Gruber et al., 2002; Seeger et al., 2003; Gadducci et al., 2005). However, a usual short coming is the publication bias related to findings without significant effect. Taken together, the prediction of breast cancer risk on polymorphisms of phase I and II enzymes is in its initial stage and prospective studies including different ethnic groups are needed in order to achieve genotyping based reliable risk determination. Recent developments of gene panels and arrays provide the technical basis for further assessment of the impact of variations in metabolic genes as well as gene–gene and gene-exposure interactions. Overall, comprehensive investigations of multiple genetic, endogenous, and exogenous factors will promote the understanding of the molecular mechanisms of breast carcinogenesis and support the improvement of prevention strategies.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Robert Bosch Foundation Stuttgart, Germany.
Footnotes
References
Antoniou, A., Pharoah, P. D., Narod, S., Risch, H. A., Eyfjord, J. E., Hopper, J. L., et al. (2003). Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am. J. Hum. Genet. 72, 1117–1130.
Beral, V.Million Women Study Collaborators. (2003). Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 362, 419–427.
Bock, K. W., and Kohle, C. (2005). UDP-glucuronosyltransferase 1A6: structural, functional, and regulatory aspects. Methods Enzymol. 400, 57–75.
Brauch, H., Schroth, W., Goetz, M. P., Muerdter, T. E., Winter, S., Ingle, J. N., et al. (2012). Tamoxifen use in postmenopausal breast cancer: CYP2D6 matters. J. Clin. Oncol. [Epub ahead of print].
Brockstedt, U., Krajinovic, M., Richer, C., Mathonnet, G., Sinnett, D., Pfau, W., et al. (2002). Analyses of bulky DNA adduct levels in human breast tissue and genetic polymorphisms of cytochromes P450 (CYPs), myeloperoxidase (MPO), quinone oxidoreductase (NQO1), and glutathione S-transferases (GSTs). Mutat. Res. 516, 41–47.
Cheng, Z. N., Shu, Y., Liu, Z. Q., Wang, L. S., Ou-Yang, D. S., and Zhou, H. H. (2001). Role of cytochrome P450 in estradiol metabolism in vitro. Acta Pharmacol. Sin. 22, 148–154.
Clemons, M., and Goss, P. (2001). Estrogen and the risk of breast cancer. N. Engl. J. Med. 344, 276–285.
Collaborative Group on Hormonal Factors in Breast Cancer. (1996). Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet 347, 1713–1727.
Collaborative Group on Hormonal Factors in Breast Cancer. (2001). Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet 358, 1389–1399.
Cox, A., Dunning, A. M., Garcia-Closas, M., Balasubramanian, S., Reed, M. W., Pooley, K. A., et al. (2007). A common coding variant in CASP8 is associated with breast cancer risk. Nat. Genet. 39, 352–358.
Cribb, A. E., Knight, M. J., Dryer, D., Guernsey, J., Hender, K., Tesch, M., et al. (2006). Role of polymorphic human cytochrome P450 enzymes in estrone oxidation. Cancer Epidemiol. Biomarkers Prev. 15, 551–558.
Easton, D. F., Pooley, K. A., Dunning, A. M., Pharoah, P. D., Thompson, D., Ballinger, D. G., et al. (2007). Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447, 1087–1093.
Economopoulos, K. P., and Sergentanis, T. N. (2010). Three polymorphisms in cytochrome P450 1B1 (CYP1B1) gene and breast cancer risk: a meta-analysis. Breast Cancer Res. Treat. 122, 545–551.
Eliassen, A. H., Missmer, S. A., Tworoger, S. S., and Hankinson, S. E. (2006). Endogenous steroid hormone concentrations and risk of breast cancer: does the association vary by a woman’s predicted breast cancer risk? J. Clin. Oncol. 24, 1823–1830.
Flesch-Janys, D., Slanger, T., Mutschelknauss, E., Kropp, S., Obi, N., Vettorazzi, E., et al. (2008). Risk of different histological types of postmenopausal breast cancer by type and regimen of menopausal hormone therapy. Int. J. Cancer 123, 933–941.
Gadducci, A., Biglia, N., Sismondi, P., and Genazzani, A. R. (2005). Breast cancer and sex steroids: critical review of epidemiological, experimental, and clinical investigations on etiopathogenesis, chemoprevention and endocrine treatment of breast cancer. Gynecol. Endocrinol. 20, 343–360.
Gamazon, E. R., Skol, A. D., and Perera, M. A. (2012). The limits of genome-wide methods for pharmacogenomic testing. Pharmacogenet. Genomics 22, 261–272.
Gan, C. Q., Wang, X. Y., Cao, Y. D., Ye, W. X., Liu, H., and Sun, Y. Y. (2011). Association of CYP2C19*3 gene polymorphism with breast cancer in Chinese women. Genet. Mol. Res. 10, 3514–3519.
Gellner, K., Eiselt, R., Hustert, E., Arnold, H., Koch, I., Haberl, M., et al. (2001). Genomic organization of the human CYP3A locus: identification of a new, inducible CYP3A gene. Pharmacogenetics 11, 111–121.
Gruber, C. J., Tschugguel, W., Schneeberger, C., and Huber, J. C. (2002). Production and actions of estrogens. N. Engl. J. Med. 346, 340–352.
Guengerich, F. P. (1999). Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu. Rev. Pharmacol. Toxicol. 39, 1–17.
Hachey, D. L., Dawling, S., Roodi, N., and Parl, F. F. (2003). Sequential action of phase I and II enzymes cytochrome p450 1B1 and glutathione S-transferase P1 in mammary estrogen metabolism. Cancer Res. 63, 8492–8499.
Harding, D., Fournel-Gigleux, S., Jackson, M. R., and Burchell, B. (1988). Cloning and substrate specificity of a human phenol UDP-glucuronosyltransferase expressed in COS-7 cells. Proc. Natl. Acad. Sci. U.S.A. 85, 8381–8385.
Hayes, J. D., and Pulford, D. J. (1995). The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. Biol. 30, 445–600.
Hempel, N., Negishi, M., and McManus, M. E. (2005). Human SULT1A genes: cloning and activity assays of the SULT1A promoters. Meth. Enzymol. 400, 147–165.
Jernstrom, H., Bageman, E., Rose, C., Jonsson, P. E., and Ingvar, C. (2009). CYP2C8 and CYP2C9 polymorphisms in relation to tumour characteristics and early breast cancer related events among 652 breast cancer patients. Br. J. Cancer 101, 1817–1823.
Jiang, Y., Zhou, L., Yan, T., Shen, Z., Shao, Z., and Lu, J. (2010). Association of sulfotransferase SULT1A1 with breast cancer risk: a meta-analysis of case-control studies with subgroups of ethnic and menopausal statue. J. Exp. Clin. Cancer Res. 29, 101.
Johnson, N., Walker, K., Gibson, L. J., Orr, N., Folkerd, E., Haynes, B., et al. (2012). CYP3A variation, premenopausal estrone levels, and breast cancer risk. J. Natl. Cancer Inst. 104, 657–669.
Justenhoven, C., Obazee, O., Winter, S., Couch, F. J., Olson, J. E., Hall, P., et al. (2012). The postmenopausal hormone replacement therapy-related breast cancer risk is decreased in women carrying the CYP2C19*17 variant. Breast Cancer Res. Treat. 131, 347–350.
Justenhoven, C., Winter, S., Hamann, U., Haas, S., Fischer, H. P., Pesch, B., et al. (2010). The frameshift polymorphism CYP3A43_74_delA is associated with poor differentiation of breast tumors. Cancer 116, 5358–5364.
Kahlenborn, C., Modugno, F., Potter, D. M., and Severs, W. B. (2006). Oral contraceptive use as a risk factor for premenopausal breast cancer: a meta-analysis. Mayo Clin. Proc. 81, 1290–1302.
Key, T., Appleby, P., Barnes, I., Reeves, G., Endogenous Hormones and Breast Cancer Collaborative Group. (2002a). Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J. Natl. Cancer Inst. 94, 606–616.
Key, T. J., Allen, N. E., Spencer, E. A., and Travis, R. C. (2002b). The effect of diet on risk of cancer. Lancet 360, 861–868.
Lambrechts, D., Truong, T., Justenhoven, C., Humphreys, M. K., Wang, J., Hopper, J. L., et al. (2012). 11q13 is a susceptibility locus for hormone receptor positive breast cancer. Hum. Mutat. 33, 1123–1132.
Liu, S., and Lin, Y. C. (2004). Transformation of MCF-10A human breast epithelial cells by zeranol and estradiol-17 beta. Breast J. 10, 514–521.
Malone, K. E., Daling, J. R., Doody, D. R., Hsu, L., Bernstein, L., Coates, R. J., et al. (2006). Prevalence and predictors of BRCA1 and BRCA2 mutations in a Population-based study of breast cancer in white and black American women ages 35 to 64 years. Cancer Res. 66, 8297–8308.
Meijers-Heijboer, H., van den Ouweland, A., Klijn, J., Wasielewski, M., de Snoo, A., Oldenburg, R., et al. (2002). Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in non carriers of BRCA1 or BRCA2 mutations. Nat. Genet. 31, 55–59.
Meyer, U. A., Zanger, U. M., and Schwab, M. (2012). Omics and drug response. Annu. Rev. Pharmacol. Toxicol. [Epub ahead of print].
Miki, Y., Swensen, J., Shattuck-Eidens, D., Futreal, P. A., Harshman, K., Tavtigian, S., et al. (1994). A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266, 66–71.
Moore, M. R., Spence, J. B., Kiningham, K. K., and Dillon, J. L. (2006). Progestin inhibition of cell death in human breast cancer cell lines. J. Steroid Biochem. Mol. Biol. 98, 218–227.
Morike, K., Kivisto, K. T., Schaeffeler, E., Jagle, C., Igel, S., Drescher, S., et al. (2008). Propafenone for the prevention of atrial tachyarrhythmias after cardiac surgery: a randomized, double-blind placebo-controlled trial. Clin. Pharmacol. Ther. 84, 104–110.
Nandi, S., Guzman, R. C., and Yang, J. (1995). Hormones and mammary carcinogenesis in mice, rats, and humans: a unifying hypothesis. Proc. Natl. Acad. Sci. U.S.A. 92, 3650–3657.
Nelson, D. R., Zeldin, D. C., Hoffman, S. M., Maltais, L. J., Wain, H. M., and Nebert, D. W. (2004). Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 14, 1–18.
Pawlak, K. J., and Wiebe, J. P. (2007). Regulation of estrogen receptor (ER) levels in MCF-7 cells by progesterone metabolites. J. Steroid Biochem. Mol. Biol. 107, 172–179.
Pesch, B., Ko, Y., Brauch, H., Hamann, U., Harth, V., Rabstein, S., et al. (2005). Factors modifying the association between hormone-replacement therapy and breast cancer risk. Eur. J. Epidemiol. 20, 699–711.
Poutanen, M., Isomaa, V., Peltoketo, H., and Vihko, R. (1995). Role of 17 beta-hydroxysteroid dehydrogenase type 1 in endocrine and intracrine estradiol biosynthesis. J. Steroid Biochem. Mol. Biol. 55, 525–532.
Rahman, N., Seal, S., Thompson, D., Kelly, P., Renwick, A., Elliott, A., et al. (2007). PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 39, 165–167.
Rebbeck, T. R., Walker, A. H., Phelan, C. M., Godwin, A. K., Buetow, K. H., Garber, J. E., et al. (1997). Defining etiologic heterogeneity in breast cancer using genetic biomarkers. Prog. Clin. Biol. Res. 396, 53–61.
Reszka, E., Wasowicz, W., and Gromadzinska, J. (2006). Genetic polymorphism of xenobiotic metabolising enzymes, diet, and cancer susceptibility. Br. J. Nutr. 96, 609–619.
Rossouw, J. E., Anderson, G. L., Prentice, R. L., LaCroix, A. Z., Kooperberg, C., Stefanick, M. L., et al. (2002). Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 288, 321–333.
Salinas, A. E., and Wong, M. G. (1999). Glutathione S-transferases – a review. Curr. Med. Chem. 6, 279–309.
Schaeffeler, E., Schwab, M., Eichelbaum, M., and Zanger, U. M. (2003). CYP2D6 genotyping strategy based on gene copy number determination by TaqMan real-time PCR. Hum. Mutat. 22, 476–485.
Schindler, A. E., Campagnoli, C., Druckmann, R., Huber, J., Pasqualini, J. R., Schweppe, K. W., et al. (1998). Aspects of progestin activity on the breast. European progestin club. Maturitas 29, 61–65.
Schroth, W., Goetz, M. P., Hamann, U., Fasching, P. A., Schmidt, M., Winter, S., et al. (2009). Association between CYP2D6 polymorphisms and outcomes among women with early stage breast cancer treated with tamoxifen. JAMA 302, 1429–1436.
Seeger, H., Wallwiener, D., and Mueck, A. O. (2003). The effect of progesterone and synthetic progestins on serum- and estradiol-stimulated proliferation of human breast cancer cells. Horm. Metab. Res. 35, 76–80.
Sergentanis, T. N., and Economopoulos, K. P. (2010). Four polymorphisms in cytochrome P450 1A1 (CYP1A1) gene and breast cancer risk: a meta-analysis. Breast Cancer Res. Treat. 122, 459–469.
Shyamala, G., Chou, Y. C., Louie, S. G., Guzman, R. C., Smith, G. H., and Nandi, S. (2002). Cellular expression of estrogen and progesterone receptors in mammary glands: regulation by hormones, development and aging. J. Steroid Biochem. Mol. Biol. 80, 137–148.
Sim, E., Lack, N., Wang, C. J., Long, H., Westwood, I., Fullam, E., et al. (2008). Arylamine N-acetyltransferases: structural and functional implications of polymorphisms. Toxicology 254, 170–183.
Smith, C. A., Smith, G., and Wolf, C. R. (1994). Genetic polymorphisms in xenobiotic metabolism. Eur. J. Cancer 30A, 1921–1935.
Steck, S. E., Gaudet, M. M., Britton, J. A., Teitelbaum, S. L., Terry, M. B., Neugut, A. I., et al. (2007). Interactions among GSTM1, GSTT1 and GSTP1 polymorphisms, cruciferous vegetable intake, and breast cancer risk. Carcinogenesis 28, 1954–1959.
Strange, R. C., Spiteri, M. A., Ramachandran, S., and Fryer, A. A. (2001). Glutathione-S-transferase family of enzymes. Mutat. Res. 482, 21–26.
Terry, P. D., and Goodman, M. (2006). Is the association between cigarette smoking and breast cancer modified by genotype? A review of epidemiologic studies and meta-analysis. Cancer Epidemiol. Biomarkers Prev. 15, 602–611.
The CHEK2 Breast Cancer Case-Control Consortium. (2004). CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am. J. Hum. Genet. 74, 1175–1182.
The MARIE-GENICA Consortium on Genetic Susceptibility for Menopausal Hormone Therapy Related Breast Cancer Risk. (2010). Genetic polymorphisms in phase I and phase II enzymes and breast cancer risk associated with menopausal hormone therapy in postmenopausal women. Breast Cancer Res. Treat. 119, 463–474.
Thompson, P. A., and Ambrosone, C. (2000). Molecular epidemiology of genetic polymorphisms in estrogen metabolizing enzymes in human breast cancer. J. Natl. Cancer Inst. Monographs 27, 125–134.
Tukey, R. H., and Strassburg, C. P. (2001). Genetic multiplicity of the human UDP-glucuronosyltransferases and regulation in the gastrointestinal tract. Mol. Pharmacol. 59, 405–414.
Turan, V. K., Sanchez, R. I., Li, J. J., Li, S. A., Reuhl, K. R., Thomas, P. E., et al. (2004). The effects of steroidal estrogens in ACI rat mammary carcinogenesis: 17beta-estradiol, 2-hydroxyestradiol, 4-hydroxyestradiol, 16alpha-hydroxyestradiol, and 4-hydroxyestrone. J. Endocrinol. 183, 91–99.
Turesky, R. J. (2004). The role of genetic polymorphisms in metabolism of carcinogenic heterocyclic aromatic amines. Curr. Drug Metab. 5, 169–180.
Van Emburgh, B. O., Hu, J. J., Levine, E. A., Mosley, L. J., Perrier, N. D., Freimanis, R. I., et al. (2008). Polymorphisms in CYP1B1, GSTM1, GSTT1, and GSTP1, and susceptibility to breast cancer. Oncol. Rep. 19, 1311–1321.
Walsh, T., Casadei, S., Coats, K. H., Swisher, E., Stray, S. M., Higgins, J., et al. (2006). Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 295, 1379–1388.
Windmill, K. F., McKinnon, R. A., Zhu, X., Gaedigk, A., Grant, D. M., and McManus, M. E. (1997). The role of xenobiotic metabolizing enzymes in arylamine toxicity and carcinogenesis: functional and localization studies. Mutat. Res. 376, 153–160.
Wooster, R., Bignell, G., Lancaster, J., Swift, S., Seal, S., Mangion, J., et al. (1995). Identification of the breast cancer susceptibility gene BRCA2. Nature 378, 789–792.
Yager, J. D., and Davidson, N. E. (2006). Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 354, 270–282.
Yamazaki, H., and Shimada, T. (1997). Progesterone and testosterone hydroxylation by cytochromes P450 2C19, 2C9, and 3A4 in human liver microsomes. Arch. Biochem. Biophys. 346, 161–169.
Yang, G., Gao, Y. T., Cai, Q. Y., Shu, X. O., Cheng, J. R., and Zheng, W. (2005). Modifying effects of sulfotransferase 1A1 gene polymorphism on the association of breast cancer risk with body mass index or endogenous steroid hormones. Breast Cancer Res. Treat. 94, 63–70.
Keywords: breast cancer risk, tumor histo-pathology, phase I and II metabolism, polymorphisms, sequence homology
Citation: Justenhoven C (2012) Polymorphisms of phase I and phase II enzymes and breast cancer risk. Front. Gene. 3:258. doi: 10.3389/fgene.2012.00258
Received: 12 October 2012; Accepted: 05 November 2012;
Published online: 28 November 2012.
Edited by:
Kathrin Klein, Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, GermanyReviewed by:
Inke Regina König, Universität zu Lübeck, GermanyDaniel Frank Carr, University of Liverpool, UK
Copyright: © 2012 Justenhoven. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Christina Justenhoven, Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Auerbachstr. 112, Stuttgart 70376, Germany. e-mail: christina.justenhoven@ikp-stuttgart.de