
- Department of General Pediatrics, Münster University Children’s Hospital, Münster, Germany
Generalized arterial calcification of infancy (GACI) is associated with biallelic mutations in ENPP1 in the majority of cases, whereas mutations in ABCC6 (ATP-binding cassette subfamily C number 6) are known to cause pseudoxanthoma elasticum (PXE). However, ABCC6 mutations account for a significant subset of GACI cases, and ENPP1 mutations can also be associated with PXE lesions. Based on the considerable overlap of GACI and PXE, both entities appear to reflect two ends of a clinical spectrum of ectopic calcification rather than two distinct disorders. ABCC6 and ENPP1 mutations might lead to alterations of the same physiological pathways.
GACI and ENPP1 Mutations
Generalized arterial calcification of infancy (GACI; OMIM208000) is a rare autosomal recessive disease, which is characterized by severe calcification of the internal elastic lamina in large- and medium-sized arteries associated with intimal proliferation leading to arterial stenoses and heart failure within the first months of life. Although survival to adulthood has been reported, GACI is often lethal in the first 6 months of life. In the past, few patients survived the neonatal period (Moran, 1975; Morton, 1978), whereas more recently, patients treated with bisphosphonates have experienced a more favorable outcome (Rutsch et al., 2008; Ramjan et al., 2009). Some patients may also develop hypophosphatemic rickets with hyperphosphaturia, a finding associated with improved survival beyond infancy in patients with GACI (Rutsch et al., 2008; Levy-Litan et al., 2010; Lorenz-Depiereux et al., 2010). The disease has been found to be caused by inactivating mutations in ENPP1 (MIM 173335; Rutsch et al., 2003). Mutations in ENPP1 have been identified as the underlying defect in about 75% of the cases of GACI (Rutsch et al., 2008, 2011). ENPP1 encodes the ecto-nucleotide pyrophosphatase/phosphodiesterase 1 (NPP1), a cell surface protein that catalyzes the hydrolysis of ATP to AMP and extracellular inorganic pyrophosphate (PPi; Rutsch et al., 2001; Goding et al., 2003). PPi potently inhibits hydroxyapatite crystal deposition and growth and regulates chondrogenesis, collagen I expression and synthesis, and other cell differentiation processes (Bollen et al., 2000; Goding et al., 2003).
PXE and ABCC6 Mutations
Pseudoxanthoma elasticum (PXE; OMIM 264800) is a hereditary, autosomal recessive, multisystemic disease characterized by ectopic mineralization and fragmentation of elastic fibers of soft connective tissues such as the skin, the retina, and the arterial blood vessels. The clinical manifestations of classic PXE center on the skin, the eyes, and the cardiovascular system. The primary cutaneous lesions are small, yellowish papules on the neck and in large flexural areas, and these lesions progressively coalesce to form larger plaques, and skin folding occasionally develops. The eyes are frequently involved by calcification of Bruch’s membrane leading to angioid streaks, and bleeding from the choroidal vessels can result in loss of visual acuity and, occasionally, in central blindness. The cardiovascular manifestations derive from mineralization of arterial blood vessels, and include gastrointestinal bleeding, intermittent claudication, hypertension, and sometimes early myocardial infarcts. Additionally, PXE can manifest with gastrointestinal hemorrhage and abnormal tissue mineralization in different organs, including the liver, kidneys, spleen, breast, and testes (Li et al., 2009; Plomp et al., 2010). Although dermatological signs are common, the main burden of PXE results from the complications in the visual and cardiovascular systems (Hu et al., 2003). Cutaneous and eye involvement usually occurs in adolescence, but may appear earlier in childhood. Cardiovascular complications usually develop later, in mid-adulthood (Naouri et al., 2009). The prevalence of PXE is estimated to 1/25,000 to 1/75,000 in the general population (Chassaing et al., 2005; Li et al., 2009). Mutations in the ABCC6 (ATP-binding cassette subfamily C number 6) gene are demonstrated in about 66–97% of patients who are genotyped (Bergen et al., 2000; Le Saux et al., 2000, 2001; Miksch et al., 2005; Chassaing et al., 2007; Vanakker et al., 2008). The ABCC6-transported substrate or substrates, which modulate arterial calcification and other phenotypic changes of PXE, are not known, and hepatic abnormalities that have effects on calcification-regulating plasma proteins such as fetuin have been suggested to at least partially mediate the pathogenesis of PXE (Hendig et al., 2006).
Generalized arterial calcification of infancy and PXE have been considered to be two distinct entities in the past and have been primarily linked to mutations in ENPP1 and ABCC6, respectively. But recent findings indicate that GACI and PXE might be more closely related than previously thought.
The First Case of GACI and PXE in One Family
Recently, we reported on a family with two brothers born to unrelated parents. The elder developed uncomplicated PXE in adolescence. Interestingly, the younger brother died after his second myocardial infarction at 15 months of age. Autopsy demonstrated calcifications of the endocardium, with extensive calcifications of the coronary arteries and of medium-sized arteries and the aorta, leading to the diagnosis of GACI. We performed molecular genetic analyses in the family. Unfortunately, no DNA of the deceased younger brother with GACI was available. The elder brother had two heterozygous missense mutations of ABCC6. Each mutation was inherited from one of his heterozygous asymptomatic parents. However, no ENPP1 mutations were found in the three living family members (Le Boulanger et al., 2010).
This case was the first one suggesting a correlation between PXE and GACI. We hypothesized that GACI could be independent of ENPP1, but related to ABCC6 mutations and that on the other hand PXE could be related to ENPP1 mutations.
Patients with GACI Carry Mutations in ABCC6
Based on this case of GACI and PXE in one family with ABCC6 mutations, we sequenced the ABCC6 gene in 30 patients with a typical GACI phenotype but without disease-causing ENPP1 mutations. In 14 of these patients, we detected pathogenic mutations in ABCC6 (biallelic mutations in eight patients, monoallelic mutations in six patients). This study showed that biallelic mutations in the ABCC6 gene account for a substantial number of typical GACI cases (Nitschke et al., 2012). The fact that even monoallelic mutations in ABCC6 were associated with the severe phenotype of GACI cannot fully be explained on the basis of autosomal recessive inheritance. However, mutations of other disease-associated genes have not been ruled out so far.
Patients with GACI and PXE Carry Mutations in ENPP1
Three of our GACI patients, who showed extensive calcifications of the large- and medium-sized arteries, arterial stenoses, and periarticular calcifications in infancy, carried biallelic ENPP1 mutations. These patients developed clinical features of PXE in childhood between 5 and 8 years of age. The patients showed angioid streaks and typical pseudoxanthomatous skin lesions (Nitschke et al., 2012). Most recently, one additional 2-year-old patient with a relatively mild form of GACI developed PXE with pseudoxanthomatous lesions of the neck, inguinal folds, and lower abdomen. The patient was also found to harbor a homozygous missense mutation in ENPP1 (Li et al., 2012).
Genocopy and Phenocopy in GACI and PXE
GACI and PXE have been considered to be two distinct entities in the past and have been primarily linked to ENPP1 and ABCC6, respectively. But based on the overlap of genotype and phenotype of GACI and PXE, both entities appear to reflect two ends of a clinical spectrum of ectopic calcification and other organ pathologies, rather than two distinct disorders (Figure 1). It was shown, that biallelic mutations in ABCC6 account for a significant number of typical GACI cases, which involve widespread arterial calcifications, arterial stenoses, periarticular calcifications, and hypophosphatemic rickets. ABCC6 mutations can be associated with a much more severe phenotype, including death in infancy from myocardial infarction, than was previously known. We conclude that the phenotypic spectrum of diseases associated with ABCC6 mutations is much broader than was previously assumed. In fact, the infantile phenotype of patients carrying ABCC6 mutations can be indistinguishable from the phenotype associated with ENPP1 mutations. The fact that the same ABCC6 mutations can cause the severe GACI phenotype associated with death in early infancy and the relatively mild phenotype of PXE warrants further explanation. Because of the difficulty with charting a clear pattern of inheritance to phenotype, it is likely that mutations in other disease-associated genes may play a role here.
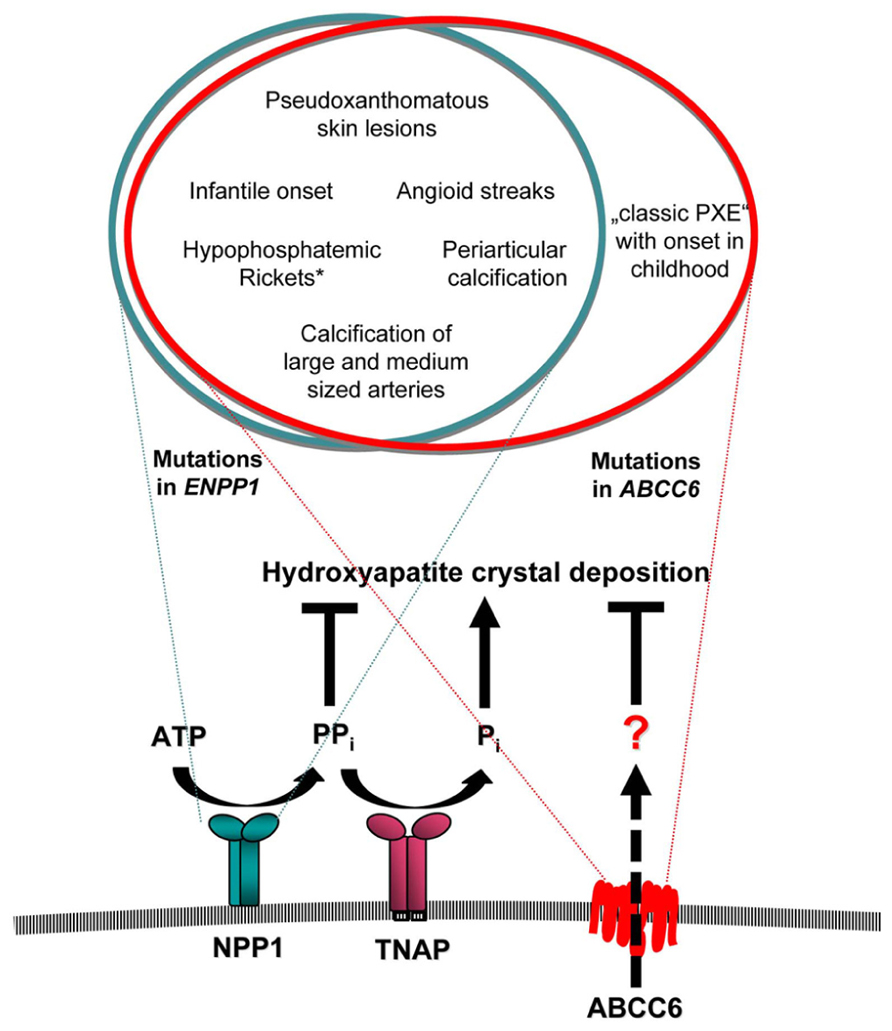
FIGURE 1. Overlap of clinical manifestations associated with mutations in ENPP1 and ABCC6. NPP1 and ABCC6 serve as a minor component in the function of a network of factors that exert balanced effects to promote and suppress arterial calcification. The transmembrane ectoenzyme NPP1 generates AMP and PPi from ATP. PPi is hydrolyzed by the tissue non-specific alkaline phosphatase (TNAP) to generate Pi, which is a component of hydroxyapatite crystal deposition and plays a role in the regulation of osteoblast differentiation. PPi suppresses hydroxyapatite deposition and inhibits ectopic chondrogenesis (and modulates artery calcification by other effects). The role of ABCC6 has to be defined. Mutations of either ABCC6 or ENPP1 can cause the severe phenotype of GACI, which frequently leads to death within the first year of life. While mutations in ENPP1 can also cause typical pseudoxanthomatous skin lesions and angioid streaks of the retina in children with GACI, who survived the critical period of infancy, the later onset of “classic PXE” phenotype without GACI was only observed in patients with mutations in ABCC6. *Hypophosphatemic rickets has been observed frequently in patients with ENPP1 mutations, but was observed only in one proband carrying a mutation in ABCC6 on one allele.
Up to date, four patients who presented with GACI and carried biallelic ENPP1 mutations developed the clinical manifestation of PXE in childhood. Symptoms included angioid streaks and histologically proven calcifications of elastic skin fibers. Thus, given the poor prognosis of severe GACI, affected patients might die of the cardiovascular complications of the disease before they develop typical signs of PXE. This might be the reason that no previous case of GACI has been described in the PXE literature. Also, many PXE characteristics, including angioid streaks of the retina and peau d’orange skin lesions might frequently be overlooked in the clinical examinations of GACI patients. Hence, the true number of patients carrying ENPP1 mutations and showing PXE lesions might be higher. In summary, these findings show that mutations in the different genes ENPP1 and ABCC6 can lead to similar pathophysiological consequences and that GACI and PXE do not simply represent two distinct disorders. They rather represent a spectrum of different peculiarities of ectopic calcification. It can therefore be hypothesized that the pathophysiology of ENPP1 and ABCC6 related disorders is based on common downstream mechanisms.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Frank Rutsch and Yvonne Nitschke were supported by a grant from the Interdisciplinary Center for Clinical Research (IZKF) Münster and by the Deutsche Forschungsgemeinschaft.
References
Bergen, A. A., Plomp, A. S., Schuurman, E. J., Terry, S., Breuning, M., Dauwerse, H., et al. (2000). Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet. 25, 228–231.
Bollen, M., Gijsbers, R., Ceulemans, H., Stalmans, W., and Stefan, C. (2000). Nucleotide pyrophosphatases/phosphodiesterases on the move. Crit. Rev. Biochem. Mol. Biol. 35, 393–432.
Chassaing, N., Martin, L., Bourthoumieu, S., Calvas, P., and Hovnanian, A. (2007). Contribution of ABCC6 genomic rearrangements to the diagnosis of pseudoxanthoma elasticum in French patients. Hum. Mutat. 28, 1046.
Chassaing, N., Martin, L., Calvas, P., Le, B. M., and Hovnanian, A. (2005). Pseudoxanthoma elasticum: a clinical, pathophysiological and genetic update including 11 novel ABCC6 mutations. J. Med. Genet. 42, 881–892.
Goding, J. W., Grobben, B., and Slegers, H. (2003). Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim. Biophys. Acta 1638, 1–19.
Hendig, D., Schulz, V., Arndt, M., Szliska, C., Kleesiek, K., and Gotting, C. (2006). Role of serum fetuin-A, a major inhibitor of systemic calcification, in pseudoxanthoma elasticum. Clin. Chem. 52, 227–234.
Hu, X., Plomp, A., Wijnholds, J., Ten, B. J., van Soest, S., van den Born, A., et al. (2003). ABCC6/MRP6 mutations: further insight into the molecular pathology of pseudoxanthoma elasticum. Eur. J. Hum. Genet. 11, 215–224.
Le Boulanger, G., Labreze, C., Croue, A., Schurgers, L. J., Chassaing, N., Wittkampf, T., et al. (2010). An unusual severe vascular case of pseudoxanthoma elasticum presenting as generalized arterial calcification of infancy. Am. J. Med. Genet. A152A, 118–123.
Le Saux, O., Beck, K., Sachsinger, C., Silvestri, C., Treiber, C., Goring, H. H., et al. (2001). A spectrum of ABCC6 mutations is responsible for pseudoxanthoma elasticum. Am. J. Hum. Genet. 69, 749–764.
Le Saux, O., Urban, Z., Tschuch, C., Csiszar, K., Bacchelli, B., Quaglino, D., et al. (2000). Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 25, 223–227.
Levy-Litan, V., Hershkovitz, E., Avizov, L., Leventhal, N., Bercovich, D., Chalifa-Caspi, V., et al. (2010). Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am. J. Hum. Genet. 86, 273–278.
Li, Q., Jiang, Q., Pfendner, E., Váradi, A., and Uitto, J. (2009). Pseudoxanthoma elasticum: clinical phenotypes, molecular genetics and putative pathomechanisms. Exp. Dermatol. 18, 1–11.
Li, Q., Schumacher, W., Jablonski, D., Siegel, D., and Uitto, J. (2012). Cutaneous features of pseudoxanthoma elasticum in a patient with generalized arterial calcification of infancy due to a homozygous missense mutation in the ENPP1 gene. Br. J. Dermatol. 166, 1107–1111.
Lorenz-Depiereux, B., Schnabel, D., Tiosano, D., Häusler, G., and Strom, T. M. (2010). Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am. J. Hum. Genet. 86, 267–272.
Miksch, S., Lumsden, A., Guenther, U. P., Foernzler, D., Christen-Zäch, S., Daugherty, C., et al. (2005). Molecular genetics of pseudoxanthoma elasticum: type and frequency of mutations in ABCC6. Hum. Mutat. 26, 235–248.
Moran, J. J. (1975). Idiopathic arterial calcification of infancy: a clinicopathologic study. Pathol. Annu. 10, 393–417.
Naouri, M., Boisseau, C., Bonicel, P., Daudon, P., Bonneau, D., Chassaing, N., et al. (2009). Manifestations of pseudoxanthoma elasticum in childhood. Br. J. Dermatol. 161, 635–639.
Nitschke, Y., Baujat, G., Botschen, U., Wittkampf, T., du Moulin, M., Stella, J., et al. (2012). Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am. J. Hum. Genet. 90, 25–39.
Plomp, A. S., Toonstra, J., Bergen, A. A., van Dijk, M. R., and de Jong, P. T. (2010). Proposal for updating the pseudoxanthoma elasticum classification system and a review of the clinical findings. Am. J. Med. Genet. 152A, 1049–1058.
Ramjan, K. A., Roscioli, T., Rutsch, F., Sillence, D., and Munns, C. F. (2009). Generalized arterial calcification of infancy: treatment with bisphosphonates. Nat. Clin. Pract. Endocrinol. Metab. 5, 167–172.
Rutsch, F., Boyer, P., Nitschke, Y., Ruf, N., Lorenz-Depierieux, B., Witt-kampf, T., et al. (2008). Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ. Cardiovasc. Genet. 1, 133–140.
Rutsch, F., Nitschke, Y., and Terkeltaub, R. (2011). Genetics in arterial calcification: pieces of a puzzle and cogs in a wheel. Circ. Res. 109, 578–592.
Rutsch, F., Ruf, N., Vaingankar, S., Toliat, M. R., Suk, A., Höhne, W., et al. (2003). Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat. Genet. 34, 379–381.
Rutsch, F., Vaingankar, S., Johnson, K., Goldfine, I., Maddux, B., Schauerte, P., et al. (2001). PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am. J. Pathol. 158, 543–554.
Keywords: ENPP1, ABCC6, GACI, PXE, arterial calcification
Citation: Nitschke Y and Rutsch F (2012) Generalized arterial calcification of infancy and pseudoxanthoma elasticum: two sides of the same coin. Front. Gene. 3:302. doi: 10.3389/fgene.2012.00302
Received: 25 September 2012; Paper pending published: 22 October 2012;
Accepted: 05 December 2012; Published online: 24 December 2012.
Edited by:
Olivier M. Vanakker, Ghent University Hospital, BelgiumReviewed by:
Scott H. Harrison, North Carolina Agricultural and Technical State University, USATianxiao Huan, Framingham Heart Study, USA
Copyright: © 2012 Nitschke and Rutsch. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Frank Rutsch, Department of General Pediatrics, Münster University Children’s Hospital, Albert-Schweitzer-Campus 1, Gbde. A1, D-48149 Münster, Germany. e-mail: rutschf@ukmuenster.de