
- 1Friedrich Miescher Institute for Biomedical Research, Basel, Switzerland
- 2Department of Biological Sciences, Wayne State University, Detroit, MI, USA
- 3Institut für Populationsgenetik, Vetmeduni Vienna, Vienna, Austria
- 4Wissenschaftskolleg zu Berlin, Institute for Advanced Study, Berlin, Germany
- 5Institute of Molecular Genetics, Russian Academy of Sciences, Moscow, Russia
An animal’s survival strongly depends on its ability to maintain homeostasis in response to the changing quality of its external and internal environment. This is achieved through intracellular and intercellular communication within and among different tissues. One of the organ systems that plays a major role in this communication and the maintenance of homeostasis is the nervous system. Here we highlight different aspects of the neuronal inputs and outputs of pathways that affect aging and longevity. Accordingly, we discuss how sensory inputs influence homeostasis and lifespan through the modulation of different types of neuronal signals, which reflects the complexity of the environmental cues that affect physiology. We also describe feedback, compensatory, and feed-forward mechanisms in these longevity-modulating pathways that are necessary for homeostasis. Finally, we consider the temporal requirements for these neuronal processes and the potential role of natural genetic variation in shaping the neurobiology of aging.
Introduction
The study of aging is the study of an open system, where tissues and organs within the whole animal regularly exchange information not only with each other but also with their external environment during the course of the animal’s lifespan. These exchanges in information allow the animal to maintain a stable internal environment, known as homeostasis, which is necessary for survival amid the constant flux in the animal’s external environment. An important node within this flow of information is the nervous system, which serves as an interface between the animal’s external and internal environments. Not surprisingly, neuronal signaling activities and their regulation have a major influence on the animal’s survival and aging process. Here we address the role of the nervous system in maintaining homeostasis and its consequent impact on longevity and aging.
Signaling Networks: Intracellular, Intercellular, and Interorgan Communication in Homeostatic Maintenance – The Influence on Lifespan
The nervous system is a network of specialized cells that relay information between different organ systems and the environment. Sensory neurons perceive environmental cues, whose information are transmitted to non-neuronal tissues either directly or indirectly via neural circuits that consist of interneurons and/or other types of neurons, like motor neurons. These intercellular and interorgan communications involve different types of signaling molecules that range from small molecule neurotransmitters to neuropeptides and hormones (Figure 1; reviewed in Alcedo et al., 2010). Indeed, consistent with the findings that the nervous system affects longevity, the processing of environmental information by sensory neurons and the corresponding neural circuitries can modulate hormonal secretions that maintain homeostasis (Figure 1; reviewed in Alcedo et al., 2010).
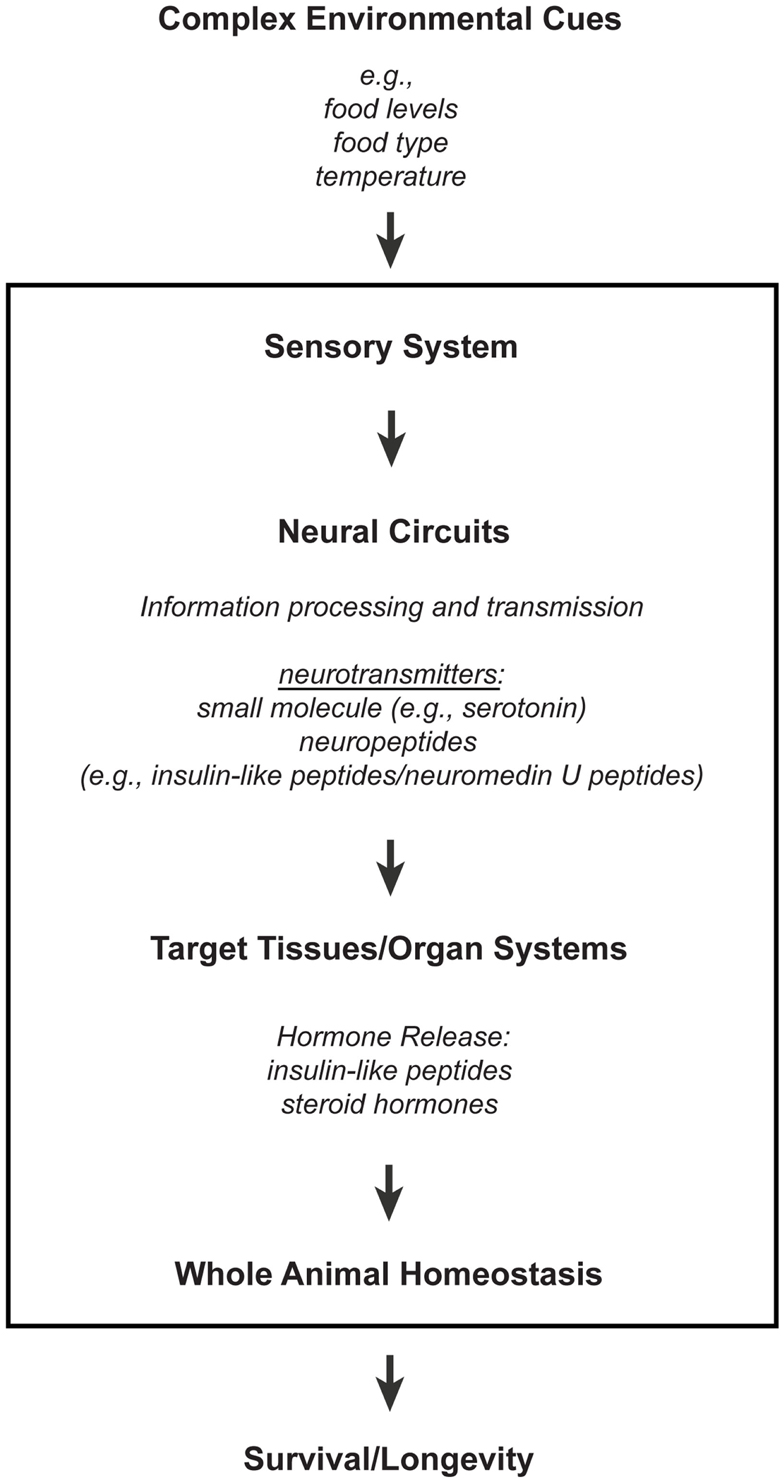
Figure 1. A model for how the nervous system processes environmental information through neuronal and non-neuronal circuits to maintain homeostasis for optimal survival. Information processing of environmental inputs at the neuronal level will involve the function of (1) small molecule neurotransmitters and neuropeptides, such as insulin-like peptides, (2) stress-sensing pathways, and (3) mitochondria-associated signals. These neuronal signaling outputs will in turn target other tissues to regulate the production of secondary signals, like hormones, and thus promote homeostasis and longevity. Insulin-like peptides can function either as short-range peptide neurotransmitters (Chen et al., 2013) or as peptide hormones.
Sensory Influence on Homeostasis and Lifespan
Sensory perception can alter a number of physiological processes, from circadian clocks (Wurtman et al., 1963, 1964; la Fleur et al., 2001; Challet et al., 2003; Ha et al., 2006), developmental plasticity (Bargmann and Horvitz, 1991; Schackwitz et al., 1996) and metabolism (Zafra et al., 2006; Greer et al., 2008) to reproduction (Yoon et al., 2005), and stress responses (Prahlad et al., 2008). Similarly, sensory neurons have been found to affect lifespan in the nematode worm C. elegans (Apfeld and Kenyon, 1999; Alcedo and Kenyon, 2004; Bishop and Guarente, 2007; Lee and Kenyon, 2009) and in the fruit fly Drosophila (Libert et al., 2007; Poon et al., 2010). This influence on lifespan involves positive or negative inputs from gustatory, olfactory, and thermosensory neurons that can modulate the activities of different peptide or steroid hormones (Apfeld and Kenyon, 1999; Alcedo and Kenyon, 2004; Libert et al., 2007; Lee and Kenyon, 2009), which would in turn presumably affect different homeostatic mechanisms (reviewed in Fielenbach and Antebi, 2008; Kenyon, 2010). The above studies demonstrating the sensory influence on C. elegans and Drosophila lifespan have been reviewed in greater detail by Jeong et al. (2012), as part of this Research Topic.
The nature of some of these neurons suggests that some of the cues that affect lifespan are food-derived, which agrees with the observation that some olfactory inputs are involved in the lifespan effects of restricting food intake levels (Libert et al., 2007), a phenomenon that is commonly known as calorie restriction (Klass, 1977; Weindruch and Walford, 1988). However, the longevity-promoting effects of food-level restriction are linked to changes in feeding rates, delayed development, and decreased reproduction (Klass, 1977; Weindruch and Walford, 1988). In contrast, the sensory influence on lifespan does not always correlate with the sensory effects on feeding behaviors, development, and reproduction (Apfeld and Kenyon, 1999; Alcedo and Kenyon, 2004; Poon et al., 2010), which suggests that the sensory system will affect lifespan through more than one mechanism. This would be expected since different types of sensory neurons can perceive a wide variety of environmental cues, ranging from temperature (Lee and Kenyon, 2009; Xiao et al., 2013) or the inherent complexity of food sources (Libert et al., 2007; Maier et al., 2010; Poon et al., 2010) to other types of cues, many of which can potentially alter organismal homeostasis and affect lifespan.
Recently, the sensory system has been shown to promote another form of dietary influence on lifespan – dependence on food-type/composition, which is distinct from the lifespan effects of food-level restriction (Maier et al., 2010). This is consistent with the previous observation that only a subset of gustatory and olfactory neurons affects lifespan in a given environment (Alcedo and Kenyon, 2004), i.e., the presence of a specific set of lifespan-influencing cues in some food sources will only be detected by a specific set of sensory neurons. Indeed, this is supported by the recent identification of a monocarboxylate-like transporter (MCT-1) that mediates the lifespan effects of only certain sensory neurons, suggesting that MCT-1 will transport some, but not all, small metabolites (Gaglia et al., 2012).
The sensory influence on lifespan via food-type recognition has also been shown to involve the activities of specific neuropeptide signaling pathways under certain environmental conditions (Maier et al., 2010). For example, a neuropeptide neuromedin U pathway processes food-type information that alters C. elegans lifespan, independent of food intake levels (Maier et al., 2010). Considering that many species have a large repertoire of neuropeptide ligands and receptors, many of which are expressed in the nervous system (Bargmann, 1998; Strand, 1999), these neuropeptide signaling pathways could presumably process distinct sets of sensory information into physiological responses that would optimize survival.
Modulation of Lifespan and Aging by Neuronal Insulin/IGF Signaling
The sensory influence on lifespan can be mediated by insulin/insulin-like peptides (ILPs) and their corresponding signaling pathway(s), IIS (Apfeld and Kenyon, 1999; Alcedo and Kenyon, 2004), which are also known to play a central role in regulating various aspects of growth, development, metabolism, and reproduction. Indeed, among the molecular pathways known to affect longevity, IIS is probably the best-known, and perhaps the most important, mainly due to its major, evolutionarily conserved effects on lifespan in various model organisms, from invertebrates to mammals (reviewed in Tatar et al., 2003; Taguchi and White, 2008; Partridge et al., 2011). Here we provide a brief overview of recent studies suggesting that, among the many tissues affected by this endocrine pathway, IIS action in the central nervous system (CNS) is of special importance for modulating aging and longevity (reviewed in Broughton and Partridge, 2009).
IIS in the CNS has essentially two roles in aging. On the one hand, it can have local, neuroprotective effects in the CNS itself, for example, by promoting neuronal survival under neurodegenerative conditions (Chrysis et al., 2001; Schubert et al., 2004; Plum et al., 2005; Bateman and McNeill, 2006). On the other hand, in response to environmental cues, some of which could be food-derived, CNS-acting factors could regulate the production and release of ILPs, which in turn systemically act to influence whole-organismal aging. Here we focus on such CNS-mediated, lifespan-promoting effects of reduced IIS in worms, flies, and mice (reviewed in Tatar et al., 2003; Fielenbach and Antebi, 2008; Alcedo et al., 2010).
The worm C. elegans has 40 genes that are predicted to encode ILPs, many of which are expressed in sensory neurons and interneurons and can function as ligands for the insulin receptor ortholog DAF-2 (Pierce et al., 2001; Li et al., 2003; Cornils et al., 2011). Consistent with the notion that sensory neurons produce and release ILPs that regulate lifespan by influencing IIS in remote tissues, mutations that cause defects in ciliated sensory neurons or targeted ablation of gustatory and olfactory neurons extend lifespan in a manner that is fully or partially dependent on DAF-16/FOXO, a forkhead transcription factor downstream of IIS that becomes activated when IIS is reduced (Apfeld and Kenyon, 1999; Alcedo and Kenyon, 2004; Shen et al., 2010). The central role of the CNS in the IIS modulation of longevity is further underscored by the fact that the extended lifespan due to mutations in daf-2 and age-1/PI-3K, a central kinase downstream of DAF-2, can be largely or fully rescued, when wild-type daf-2 or age-1 is expressed in the neurons of the corresponding mutants (Wolkow et al., 2000; Iser et al., 2007). In contrast, neuronal activity of DAF-16/FOXO seems to be less important for lifespan extension in animals with impaired IIS (Libina et al., 2003; Iser et al., 2007; also, see below). However, expression of the microRNA mir-71 in the nervous system mediates the lifespan extension in germline-ablated worms in a fashion that depends upon intestinal DAF-16 activity, revealing a complex signaling interaction between the CNS, the intestine, and the gonad in IIS-mediated lifespan regulation (Boulias and Horvitz, 2012).
Work in the fruit fly Drosophila melanogaster reveals remarkable parallels to these observations in worms. In the adult fly, three out of seven distinct ILPs are produced in specialized median neurosecretory cells (also called insulin-producing cells, IPCs) in the pars intercerebralis of the CNS (Rulifson et al., 2002; Grönke et al., 2010), and ablation of the IPCs significantly extends lifespan (Wessells et al., 2004; Broughton et al., 2005; Haselton et al., 2010), presumably due to reduced levels of ILP2, ILP3, and ILP5 (Broughton et al., 2008; Grönke et al., 2010). Consistent with these observations, several factors that regulate the production and/or release of ILPs affect IIS and lifespan. These factors include the metabotropic GABA receptors or uncoupling proteins (UCPs) expressed in the IPCs (Fridell et al., 2009; Humphrey et al., 2009; Enell et al., 2010) and short neuropeptide F (sNPF) expressed in the CNS (Lee et al., 2008, 2009). In addition, downregulation of p53 in the IPCs extends lifespan by reducing ILP levels and inhibiting PI-3K activity in peripheral tissues (Bauer et al., 2007). Similarly, the stress-responsive Jun kinase (JNK) in the IPCs promotes longevity by downregulating ILP2 through activation of FOXO (Wang et al., 2005). In contrast, and similar to the above-mentioned findings in C. elegans, activation of FOXO in the CNS, either pan-neuronally, in the neurolemma or in glial cells, is not sufficient to extend lifespan, whereas its downregulation in head fat body tissues promotes longevity (Hwangbo et al., 2004).
In mammals, the CNS also seems to play an important role in regulating the production and release of insulin-like hormones, although the bulk of insulin or IGF-1 is produced outside the brain. For example, mice with certain mutations affecting the so-called hypothalamic-pituitary-somatotropic growth hormone (GH-IIS) axis, known to regulate the release of insulin/insulin-like hormones, are long-lived, presumably due to downregulation of IIS (reviewed in Tatar et al., 2003; Holzenberger et al., 2004; Berryman et al., 2008). More direct evidence for a role of IIS in affecting mammalian lifespan via the nervous system comes from studies with transgenic or mutant mice with impaired IIS. Mice with a brain-specific deletion of the insulin receptor substrate-2 (Irs2) locus are 14% longer lived than control mice, despite being hyperinsulinemic, obese, and insulin-resistant (Taguchi et al., 2007). Similarly, partial genetic inactivation of the IGF-1 receptor (IGF-1R) gene in the embryonic mouse brain inhibits GH and IGF-1 signaling after birth, which leads to growth retardation, small adult size, metabolic changes, and prolonged mean lifespan (Kappeler et al., 2008).
While much future work remains to be done for a detailed understanding of the underlying regulatory mechanisms, the available studies in worms, flies, and mice to date clearly show that neuroendocrine processes in the CNS are critically important for modulating the lifespan effects of IIS.
The Effects of Neuronal Stress-Sensing Pathways on Lifespan and Aging
The nervous system not only perceives a variety of environmental stressors but also integrates these information, which are then converted into appropriate physiological and behavioral adaptive responses. Below we discuss two such examples and their possible consequent effects on lifespan.
Exposure to acute stress, like heat, heavy metals, or toxins, can lead to proteotoxicity, as a result of protein misfolding within the animal (reviewed in Åkerfelt et al., 2010). To survive such insults, the animal activates its heat shock response, which is mediated by the heat shock transcription factor 1 (HSF-1; (Hsu et al., 2003; Morley and Morimoto, 2004; Cohen et al., 2006). For example, Kourtis et al. (2012) have shown that HSF-1 is required to protect the animal against cytotoxicity that is induced by thermal or other stresses through activation of the small heat shock protein HSP-16.1. This mechanism, which also protects against neurodegeneration, has been found to be conserved across species (Kourtis et al., 2012). Since thermosensory neurons and their associated neuronal circuitry can regulate the C. elegans heat shock responses non-autonomously (Prahlad et al., 2008; Prahlad and Morimoto, 2011), it is possible that the sensory regulation of the HSF-1/HSP-16.1 response is similarly conserved.
However, HSF-1 activity promotes longevity not only in the presence, but also in the absence, of acute stress (Hsu et al., 2003; Morley and Morimoto, 2004). Intriguingly, protein misfolding, whether it is mediated (Morley et al., 2002; van Ham et al., 2010) or not (David et al., 2010) by polyglutamine repeats, increases with age. This suggests that protein aggregation is inherent with age and is not restricted to a subset of proteins that have been implicated in diseases like neurodegeneration (David et al., 2010). Hence, given the role of HSF-1 in promoting protein disaggregation (Cohen et al., 2006), it is not surprising that HSF-1 activity in multiple tissues affects lifespan even in the absence of acute stress (Hsu et al., 2003; Morley and Morimoto, 2004).
Animals also employ different sensors for different types of gases that are required and/or affect important physiological processes. Some examples are the mechanisms through which animals perceive oxygen levels within their environment. For example, environmental oxygen is sensed by specific soluble guanylyl cyclases (sGCs) in specific sensory neurons in C. elegans and Drosophila (Cheung et al., 2005; Chang et al., 2006; Rogers et al., 2006; Vermehren-Schmaedick et al., 2010). These sGCs regulate the aerotactic behaviors of the animals: C. elegans prefers 7–11% ambient oxygen and is repelled by hypoxic (<5% O2) and hyperoxic (>14% O2) environments (Cheung et al., 2005; Chang et al., 2006; Rogers et al., 2006); whereas Drosophila larvae prefer a more restricted range of O2 concentration (∼21%) (Vermehren-Schmaedick et al., 2010). Besides the sGC-expressing neurons, the avoidance of hyperoxia, i.e., in C. elegans, also depends on the activities of neurons that sense pain and neurons that integrate information about food availability and population density (Chang et al., 2006; Rogers et al., 2006). Thus, these different sensory neurons together allow the animals to generate rapid behavioral responses to ambient O2, so that they can migrate to environments with the optimal O2 levels necessary for their survival. At present, none of the sGCs are known to affect lifespan, unlike the receptor guanylyl cyclases for which a few have been reported to inhibit longevity (Murphy et al., 2003; Alcedo and Kenyon, 2004).
There are also many other cells that respond to O2, albeit more slowly, through the hypoxia-inducible transcription factor HIF-1, which modifies the activities of the above O2-sensing neurons and existing neural circuitries (Chang and Bargmann, 2008; Pocock and Hobert, 2010). Hypoxic activation of HIF-1 shifts the animal’s preferences to lower oxygen concentrations and eliminates the dependence on some neurons, e.g., those that integrate information about food and population density, in promoting O2-dependent responses (Chang and Bargmann, 2008). Interestingly, this HIF-1 effect requires that it acts coordinately in neuronal and gonadal cells (Chang and Bargmann, 2008), whose outputs are known to affect lifespan (Apfeld and Kenyon, 1999; Hsin and Kenyon, 1999; Wolkow et al., 2000; Broughton et al., 2005; Flatt et al., 2008).
Indeed, the HIF-1 pathway has been recently found to influence C. elegans lifespan and that these lifespan effects depend on environmental context (Chen et al., 2009; Mehta et al., 2009; Zhang et al., 2009; Lee et al., 2010; Leiser et al., 2011). Particularly, loss of hif-1 can extend C. elegans lifespan at higher temperatures (25°C; Chen et al., 2009; Leiser et al., 2011) or shorten lifespan at lower temperatures (20°C; Mehta et al., 2009; Lee et al., 2010). Since O2 perception can be modulated by food-derived information (Chang et al., 2006; Rogers et al., 2006; Chang and Bargmann, 2008; Pocock and Hobert, 2010), these temperature-dependent effects of HIF-1 may also reflect differences in the animal’s bacterial food sources grown at 25 versus 20°C. Consistent with this idea, an interaction between HIF-1 and the food-dependent TOR pathway has been observed in affecting lifespan (Chen et al., 2009). Likewise, because O2-sensing is also subject to population density (Chang et al., 2006; Rogers et al., 2006), the hif-1 lifespan effects observed by Zhang et al. (2009) might reflect the higher density of animals used in their assays. Thus, HIF-1 function nicely illustrates how environmental context and its perception can modulate the effects of a signaling pathway on lifespan.
The Role of Mitochondria in Brain Aging and Longevity
Mitochondria are among the most important cellular organelles that contribute to the aging process, mainly through respiratory chain dysfunction, changes in redox status, or by generating reactive oxygen species (ROS; Humphries et al., 2006; Mattson, 2006). It is therefore not surprising that the nervous system exhibits a highly active mitochondrial metabolism, especially because of the high energetic demands associated with processes such as ion homeostasis, neurotransmission, or the firing of action potentials.
Indeed in mammals, structural impairments in mitochondrial DNA and an age-dependent reduction in brain mitochondrial function are correlated with the age-dependent decrease in cognitive function and neuromuscular coordination (reviewed in Bishop et al., 2010; Escames et al., 2010; Chakrabarti et al., 2011; Yin et al., 2012). Similarly, mitochondrial dysfunction has been implicated in neurodegenerative diseases (reviewed in Eckert et al., 2011; Reddy and Reddy, 2011; Swerdlow, 2011; Troulinaki and Bano, 2012; Yin et al., 2012), although it remains unclear whether the functional changes seen in the healthily aging brain are distinct from the pathological processes associated with neurodegenerative diseases. The empirical evidence at hand today thus suggests that neuronal mitochondria play an important role in maintaining organismal homeostasis and in influencing aging.
Several observations support the importance of proper neuronal mitochondrial function for lifespan and healthy aging. As mentioned previously, expression of human mitochondrial UCPs, which can uncouple mitochondrial respiration from ATP synthesis, in the neurons of adult flies extends lifespan (Fridell et al., 2005, 2009; Humphrey et al., 2009). This effect is likely to occur through reduced secretion of ILPs (Fridell et al., 2009; Humphrey et al., 2009), since the human UCP2 is known to regulate insulin secretion (Zhang et al., 2001). Interestingly, while moderate levels of neuronal UCP expression lengthen lifespan (Fridell et al., 2005; Humphrey et al., 2009), high levels have the opposite effect (Humphrey et al., 2009; Figure 2). This is reminiscent of previous studies that show a mild reduction of mitochondrial function can extend lifespan, whereas a strong functional impairment shortens lifespan (Rea et al., 2007). Therefore, hypothetically, mild mitochondrial dysfunction may cause (1) a change in levels of ROS production, e.g., a decrease that ensures preservation of DNA and protein structures or a mild increase that leads to compensatory mechanisms, or (2) a change in the types of ROS produced, which would then stimulate the expression of longevity-promoting genes. Together these data suggest that the increased lifespan associated with mild impairment of neuronal mitochondrial function (Dillin et al., 2002a; Rea and Johnson, 2003; Morrow et al., 2004; Fridell et al., 2005, 2009; Conti et al., 2006; Rea et al., 2007; Copeland et al., 2009; Humphrey et al., 2009; Lee et al., 2010; Figure 2) represents a compensatory mechanism that enables the maintenance of homeostasis.
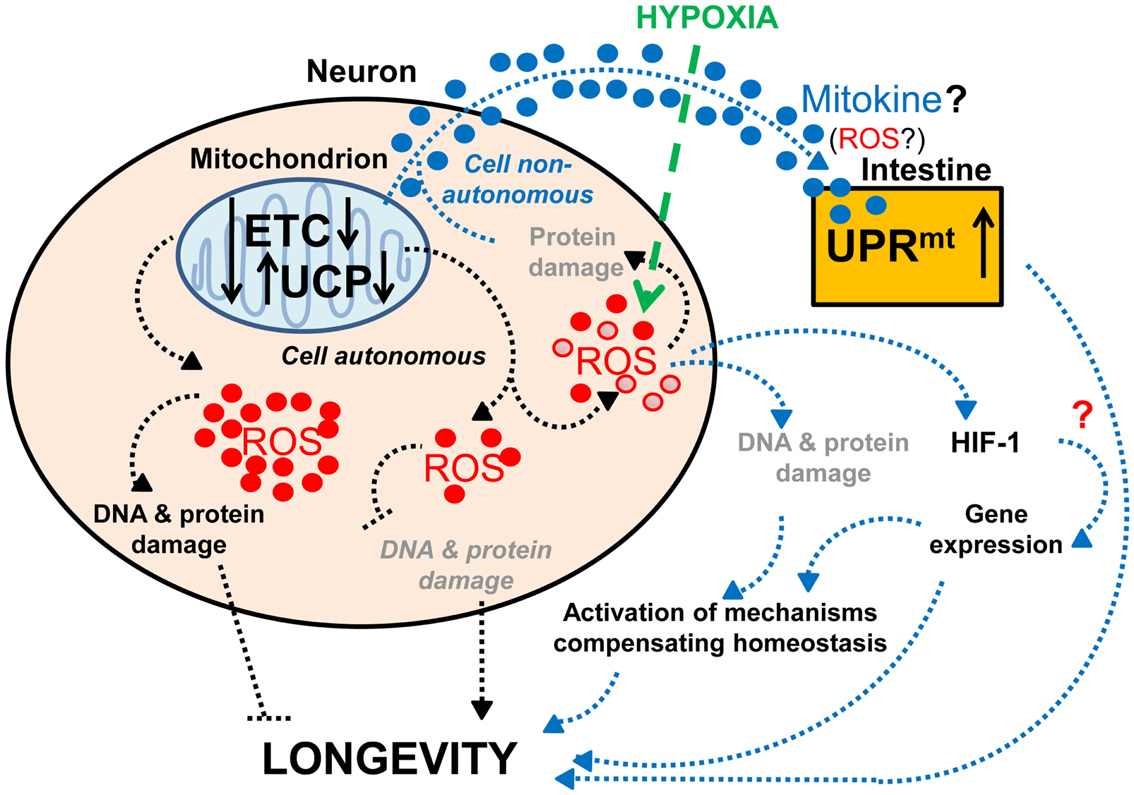
Figure 2. Effects of neuronal mitochondrial UCP and the electron transport chain on longevity. Lifespan is modulated by altered mitochondrial function in neurons: a lower level of UCP and electron transport chain (ETC) expression lengthens lifespan, whereas a higher level of UCP and ETC expression has the opposite effect on lifespan (Fridell et al., 2005; Rea et al., 2007; Copeland et al., 2009; Humphrey et al., 2009; Durieux et al., 2011). The lifespan increase observed with mild mitochondrial dysfunction may hypothetically be due to (1) a decrease in ROS production and DNA and protein damage (denoted in gray and italics) or (2) a mild increase in ROS production and DNA and protein damage (denoted in gray), which can activate compensatory mechanisms. Alternatively, mitochondria-dependent lifespan increases might also be due to other compensatory mechanisms induced by a change in the types of ROS produced (red • versus red ^). Neuronal mitochondrial dysfunction can also induce a cell non-autonomous UPRmt in intestinal cells and lead to lifespan extension, via a proposed mitokine, like ROS (Durieux et al., 2011). However, intestinal UPRmt response is necessary but not sufficient to promote longevity (Durieux et al., 2011). Since HIF-1 activates survival genes in response to hypoxia and a mild inhibition of mitochondrial ETC, which involves an increase in ROS levels (Lee et al., 2010), it is tempting to speculate about the possible role of HIF-1 in this process (denoted by a red “?”).
A reduction of the function of the mitochondrial respiratory chain in the nervous system has also been shown to induce a mitochondria-specific unfolded protein response (UPRmt) in intestinal cells and to extend lifespan (Durieux et al., 2011; Figure 2). Interestingly, a similar impairment of mitochondrial function in muscle cells can also induce UPRmt, but this does not cause lifespan extension (Durieux et al., 2011), which could suggest that UPRmt by itself is not sufficient for promoting longevity. On the other hand, the induction of UPRmt has been found to be necessary for the long-life phenotype due to reduced mitochondrial respiration (Durieux et al., 2011). Thus, these findings suggest that mitochondrial dysfunction in neurons extends lifespan by producing an unknown signal that acts together with the UPRmt-inducing signal. While the nature of this additional signal remains unknown, it is tempting to speculate about the possible role of the HIF-1 pathway in this process. Indeed, HIF-1 not only modifies neuronal activities (Chang and Bargmann, 2008; Pocock and Hobert, 2010), but also promotes longevity in response to mild inhibition of mitochondrial respiration through increased ROS levels (Lee et al., 2010). Although the longevity-promoting effects of increased ROS (Lee et al., 2010) contradict a previous hypothesis that ROS would shorten lifespan through increased oxidative damage (Humphries et al., 2006; Mattson, 2006), this observation is consistent with the more recent hypothesis of mitohormesis, where higher ROS subsequently leads to increased stress resistance (Schulz et al., 2007). Alternatively, it is conceivable that certain types of ROS act as signaling molecules to activate survival pathways (Bishop et al., 2010; Lee et al., 2010; Durieux et al., 2011).
As the major source of ROS, the mitochondria are intimately involved in crosstalk among different pathways. Not surprisingly, mitochondrial activity is also regulated by major pathways that affect longevity, including the IIS, TOR, and JNK signaling pathways (reviewed in Troulinaki and Bano, 2012, as part of this Research Topic). Indeed, the ROS-mediated induction of JNK activity (Wang et al., 2005), which leads to translocation of JNK from the cytoplasm to the mitochondria, has been proposed to be of fundamental importance in the transduction of cytosolic signals to the mitochondria in the aging mammalian brain Schroeter et al., 2003; Eminel et al., 2004; Zhou et al., 2008, 2009).
Reactive oxygen species signaling itself also modulates mitochondrial homeostasis, which involves constant remodeling of this organelle, i.e., through mitochondrial fusion, fission, and autophagy (reviewed in Lemasters, 2005; Lee et al., 2012; Palikaras and Tavernarakis, 2012; Liesa and Shirihai, 2013). Such remodeling, which is tightly regulated, appears to be an adaptive response to the cell’s energy expenditure and demands (reviewed in Liesa and Shirihai, 2013). However, mitochondrial fusion and fission have also been proposed to distribute damaged organelle components across the cell’s mitochondrial network, whereas mitochondrial autophagy, known as mitophagy, removes highly damaged mitochondria (reviewed in Lemasters, 2005; Lee et al., 2012; Palikaras and Tavernarakis, 2012). Thus, an increase in ROS levels can shift the balance between fusion and fission to mitophagy (reviewed in Lemasters, 2005; Lee et al., 2012; Palikaras and Tavernarakis, 2012). Interestingly, mitophagy requires genes that have been implicated in the neurodegenerative Parkinson’s disease, i.e., the serine/threonine kinase PINK1 and the E3 ubiquitin ligase Parkin, where PINK1 senses the damaged mitochondria and recruits Parkin to induce mitophagy (Narendra et al., 2008, 2010). Thus, dysregulation of mitochondrial remodeling, including mitophagy, through excess ROS, likely contributes to the onset and progression of several age-associated neurodegenerative diseases (reviewed in Batlevi and La Spada, 2011; Palikaras and Tavernarakis, 2012).
Feedback, Compensatory, and Feed-Forward Mechanisms in Longevity-Modulating Pathways
The studies discussed above point to the existence of major feedback mechanisms within the nervous system. Feedback loops are critically important in regulating physiology and metabolism, particularly with respect to homeostasis, and are often controlled by hormones (reviewed in Baker and Thummel, 2007; Leopold and Perrimon, 2007; Fielenbach and Antebi, 2008; Rajan and Perrimon, 2011; Hill et al., 2012). Notably, many such endocrine feedback mechanisms are thought to modulate aging and lifespan (Tatar et al., 2003; Murphy et al., 2007; Fielenbach and Antebi, 2008; Broughton and Partridge, 2009; Karpac and Jasper, 2009; Karpac et al., 2009; Tazearslan et al., 2009; Landis and Murphy, 2010), and the nervous system has been implicated in several of them (Hwangbo et al., 2004; Broughton et al., 2008; Flatt et al., 2008; Grönke et al., 2010; Alic et al., 2011; Boulias and Horvitz, 2012). Here, we focus on a few examples of feedback mechanisms that involve IIS and the nervous system.
A first example concerns the communication between adipose tissue and the brain via IIS. Hwangbo et al. (2004) found that in D. melanogaster overexpression of FOXO in the head fat body (equivalent of mammalian liver and adipose) extends lifespan and – remarkably – reduces the levels of ILP2 produced in the IPCs of the CNS, suggesting that lifespan extension is caused by FOXO-mediated negative feedback regulation of neural ILP production. This is consistent with the observation that ablation of IPCs extends lifespan (Wessells et al., 2004; Broughton et al., 2005), probably due to lowered levels of the ILP2, ILP3, and ILP5 ligands (Broughton et al., 2008; Grönke et al., 2010). Moreover, these findings are particularly interesting in view of the fact that a humoral factor produced by the fat body has been found to remotely control insulin secretion from the IPCs (Geminard et al., 2009; Tatar, 2009), yet whether this factor itself modulates lifespan remains unknown.
Another example is the existence of endocrine communication between the gonad and the brain. Similar to previous findings in C. elegans (Hsin and Kenyon, 1999; Arantes-Oliveira et al., 2002), Flatt et al. (2008) found that ablation of germline stem cells (GSCs) extends Drosophila lifespan. However, despite evidence of impaired IIS in peripheral tissues, fly GSC ablation also upregulates the production of ILP2, ILP3, and ILP5 in the brain IPCs (Flatt et al., 2008). Since neurally produced ILPs are known to bind to the insulin-like receptor (InR) on GSCs to regulate GSC proliferation in the gonad (LaFever and Drummond-Barbosa, 2005; Hsu et al., 2008), it is tempting to speculate that GSCs in the gonad exert negative feedback on ILP production in the brain. Although the nature of the signal that relays this communication remains unknown, a promising candidate may be IMP-L2, an insulin-binding protein. IMP-L2, which is expressed in the germline niche, among other tissues (Terry et al., 2006), limits the availability of free ILPs by sequestering them away from the InR, thereby antagonizing systemic IIS (Honegger et al., 2008). Interestingly, this protein is upregulated in germline-less, long-lived flies exhibiting ILP overproduction (Flatt et al., 2008). Moreover, similar to the phenotypes seen in germline-less flies, the Partridge group has shown that direct upregulation of IMP-L2 itself extends lifespan and increases ILP2, ILP3, and ILP5 levels, whereas genetic deletion of the ilp2, ilp3, and ilp5 loci decreases IMP-L2 (Grönke et al., 2010; Alic et al., 2011). Together these observations support the hypothesis that IMP-L2 is part of a gonad-brain signaling circuit that regulates neural ILP levels (Figure 3).
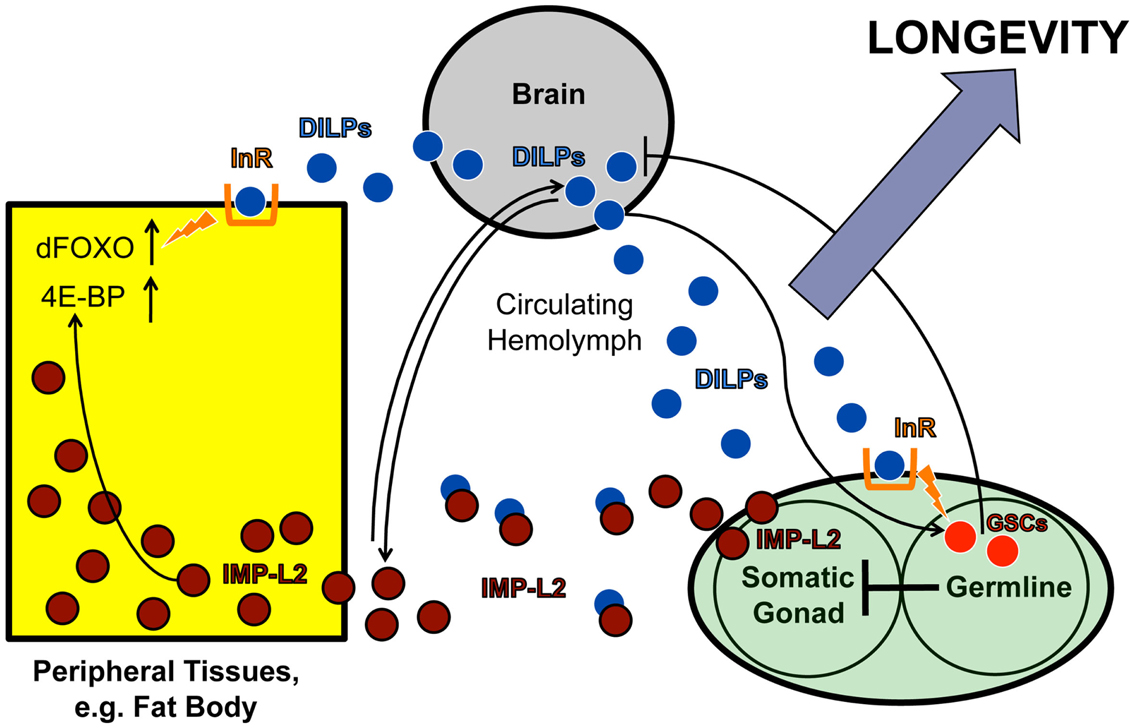
Figure 3. IMP-L2-mediated endocrine feedback loop between brain and ovary. Model of the endocrine feedback loop between brain and ovary mediated by the ILP-binding protein IMP-L2, based on findings in LaFever and Drummond-Barbosa (2005), Flatt et al. (2008), and Alic et al. (2011). ILPs produced in the brain bind to the ovarian InR and stimulate GSC proliferation. GSC proliferation likely downregulates ILP production in the IPCs since GSC ablation causes ILP transcription to increase, suggesting the existence of a negative feedback loop between the brain and ovarian tissues. This putative feedback loop might be mediated, at least in part, by the ILP-binding protein, IMP-L2, which is known to inhibit aspects of insulin signaling. Remarkably, GSC ablation results in a strong upregulation of IMP-L2. Consistent with this observation, GSC ablation and IMP-L2 overexpression cause very similar phenotypes: in both cases, flies exhibit increased lifespan, upregulation of ilp2, ilp3, and ilp5, and increased expression of DAF-16/FOXO targets (such as 4E-BP), although other aspects of DAF-16/FOXO activity (e.g., subcellular localization and phosphorylation status) remain unaltered. Together this suggests that the long-lifespan phenotype of GSC-ablated flies is mediated by IMP-L2, which in turn modulates insulin signaling. See text for further details.
While the detailed consequences for physiology, and in particular for aging and longevity, are in most cases still unknown, feedback mechanisms also occur at the level of transcriptional regulation. For example, some of the seven different Drosophila ILPs demonstrate feedback regulation of each other (Figure 4): IPC-expressed ilp3 is required for the normal expression of ilp2 and ilp5 in the IPCs, whereas knockdown of ilp2 leads to upregulation of ilp3 and ilp5 expression in the IPCs (Broughton et al., 2008; Grönke et al., 2010). Similar feedback loops also exist for other components of IIS: Drosophila FOXO (dFOXO), which is activated when InR signaling is downregulated, activates the transcription of InR (Puig et al., 2003; Puig and Tjian, 2005).
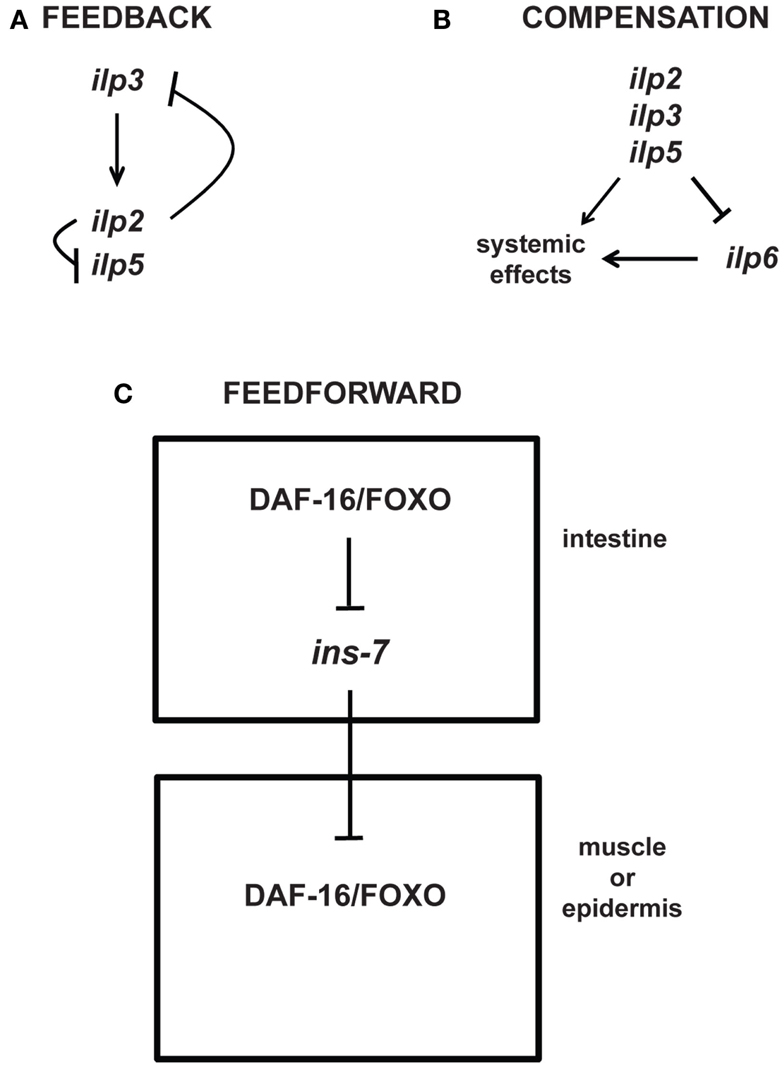
Figure 4. Feedback, compensatory, and feed-forward mechanisms in the longevity-modulating insulin signaling pathway. (A) Neuronally produced Drosophila insulin-like peptides exhibit feedback regulation among each other (Broughton et al., 2008; Grönke et al., 2010). (B) The systemic activities of the Drosophila neuronal ilp2, ilp3, and ilp5 can be compensated by the systemic activity of the ilp6 produced from the head fat body (Grönke et al., 2010). (C) C. elegans ILP signaling between tissues (i.e., intestine to muscle or epidermis) involves feed-forward regulation via transcriptional inhibition of the ILP ins-7 (Murphy et al., 2007).
Intriguingly, besides feedback loops, the genetic study of aging is also beginning to uncover other types of regulatory motifs, e.g., compensatory and feed-forward regulatory mechanisms. For instance, upregulation of the fat body-specific ilp6 seems to compensate for the loss of the brain-specific ilp2, ilp3, and ilp5 (Figure 4; Grönke et al., 2010). Moreover, like in Drosophila, C. elegans exhibits feed-forward regulation between ILPs (Murphy et al., 2007). Increased DAF-16/FOXO activity in a specific tissue is shown to increase the activity of DAF-16/FOXO in other tissues through feed-forward regulation that requires the inhibition of the ILP ins-7 expression in the C. elegans intestine (Figure 4; Murphy et al., 2003, 2007). Thus, these studies beautifully exemplify the complexity of existing feedback, compensatory, and feed-forward mechanisms that may be relevant for modulating aging and longevity.
Temporal Requirements of Longevity-Influencing Genes
The experimental data available today suggest that adult-expressed neuronal genes may have important effects on aging and longevity (e.g., reviewed in Broughton and Partridge, 2009). However, to what extent genes that regulate the development of the nervous system and its circuitry also influence homeostasis and longevity remains presently unclear. Interestingly, several data support the notion that early-life environmental influences might have “carry-over” effects into adulthood and might thus impact lifespan (Gavrilov and Gavrilova, 2011; Saino et al., 2012). For example, putative biomarkers of aging that affect gene activity and chromosome structure at an early age have been shown to predict life expectancy (Baeriswyl et al., 2009; Pincus and Slack, 2010; Heidinger et al., 2012). Similarly, newly emerging data from C. elegans show that age-related behaviors are associated with distinct transcriptomes and that the statistical analysis of these aggregate gene expression profiles can predict age and health states (Golden et al., 2008). Thus, such data tempt one to speculate that genes involved in developmental canalization (or “robustness”) might also have long-term effects on physiological homeostasis and somatic maintenance later in life. This canalization has been predicted to be a generic feature of developmental gene networks (Siegal and Bergman, 2002; Flatt, 2005).
A particularly plausible mechanism underlying these “carry-over” effects on adult lifespan is pleiotropic gene action, whereby one gene’s effect during development differs from its effect in adulthood, i.e., the same gene variant might have pleiotropic roles in affecting development versus lifespan (e.g., see Dillin et al., 2002b). On the other hand, a gene could also have different lifespan effects that depend on its temporal activity, as has been observed with the overexpression of different p53 constructs in Drosophila: this can lead to different lifespan effects in females and males depending on whether expression was driven during development versus adulthood (Waskar et al., 2009). Indeed, several key lifespan modulators, like the mitochondrial electron transport chain, microRNAs, HSF-1, and FOXO, can have “carry-over” effects on adult lifespan when manipulated (e.g., overexpressed or silenced) during early larval development and/or early adulthood (Dillin et al., 2002a,b; Giannakou et al., 2007; Rea et al., 2007; Durieux et al., 2011; Pincus et al., 2011; Volovik et al., 2012). Other examples are the age-dependent expression changes in neocortical genes, which not only play a role during development but also in altered neocortical function that is observed during age-related cognitive decline and brain dysfunction (reviewed in Huffman, 2012, as part of this Research Topic).
The distinct functional roles of pleiotropic genes during development versus aging are also demonstrated by the uncoupling of their gene functions between these two processes (Chen et al., 2007; Shen et al., 2009; Thyagarajan et al., 2010). In some cases, strong loss-of-function (or null) mutations have been found to affect embryonic development in C. elegans, whereas weaker mutant alleles of the same gene have been shown to affect adult lifespan (Kenyon et al., 1993; Kimura et al., 1997; Gems et al., 1998; Boehm and Slack, 2005), suggesting that essential developmental genes can have deleterious effects late in life. To neutralize these late-acting deleterious effects, Liu et al. (2012) have shown that miRNA signaling is involved in specifically silencing a set of these developmental genes in adulthood, thereby restricting the pleiotropic “carry-over” effects of such genes. This is exemplified by the miRNA miR-34-mediated silencing of the steroid pathway gene E74A in Drosophila adults to maintain brain integrity and viability (Liu et al., 2012).
Another obvious mechanism that might play a role in “carry-over” effects on lifespan and aging are epigenetic modifications. Experiments in rodents, for instance, have shown that experiences during sensitive periods of brain development influence DNA methylation patterns, which in turn could alter gene transcription throughout life and promote specific phenotypic outcomes (Roth and Sweatt, 2011). In a similar vein, the “heterochromatin loss model of aging” posits that heterochromatin domains that are set up early in embryogenesis are gradually lost with age, which results in aberrant and age-associated gene expression patterns (Villeponteau, 1997). In support of this hypothesis, genetic manipulation of HP1 levels and JAK/STAT signaling suggests that heterochromatin formation contributes to the prevention of premature aging (Larson et al., 2012). These are intriguing preliminary observations and it will be interesting to learn more about the role of epigenetic changes in aging and lifespan in future work.
Evolutionary Implications of Longevity-Modulating Neuronal Mechanisms
Although the classical evolutionary theory of aging posits that aging should be affected by different mechanisms in different species (Williams, 1957; Reznick, 2005), recent studies suggest that several pathways have conserved effects on longevity (reviewed in Partridge and Gems, 2002; Tatar et al., 2003; Kenyon, 2005; Partridge et al., 2005; Smith et al., 2008; Flatt and Schmidt, 2009; Fontana et al., 2010; Nakagawa et al., 2012; Wuttke et al., 2012). Whereas lifespan can vary by several orders of magnitude across different species (Finch, 1990; Stearns, 1992; Nabholz et al., 2008; Li and de Magalhães, 2011), the molecular underpinnings of longevity have so far been mainly studied in a few short-lived and genetically tractable model systems, suggesting that our current understanding of the mechanisms of aging might be biased (Deweerdt, 2012). Moreover, while many of the conserved, pleiotropic signaling pathways implicated in aging have neuronal roles, not all of these functions might directly impinge on aging. Therefore, the extent to which the neuronal mechanisms of longevity are evolutionarily conserved remains largely unclear.
A recent study directly comparing gene expression profiles during aging in mouse, rhesus macaque and human brains indicates that only a small subset of the age-dependent expression changes might be conserved (Loerch et al., 2008). These few genes include the neuroprotective gene apolipoprotein D (APOD), which is robustly upregulated with age in all three species and whose two Drosophila homologs are known to affect lifespan (Ruiz et al., 2011). Another example is the calcium/calmodulin-dependent protein kinase IV (CAMK4), which has been shown to regulate synaptic plasticity (Ho et al., 2000) and is downregulated with age in all three species (Loerch et al., 2008). In contrast, most genes did not show a consistent age-dependent pattern across species, leading the authors to conclude that humans and rhesus macaques have greatly diverged from mice as demonstrated by a dramatic increase in age-dependent repression of human and macaque neuronal genes (Loerch et al., 2008). While these results indicate that the neuronal mechanisms of aging and longevity might not be highly conserved among different taxa, a study by Fonseca et al. (2005) provides a remarkable counter-example. Across a range of terrestrial, freshwater, marine, tropical, and temperate arthropods, whose lifespans vary by three orders of magnitude, the neuronal deposition of lipofuscin, a lipid-protein aggregate, is highly correlated with lifespan. This suggests that age-dependent damage accumulation in the brain might be the primary driver of senescence (Fonseca et al., 2005).
Similarly, at the microevolutionary or intraspecies level, it is still unclear whether natural variation in lifespan is based on allelic variation within the same genes and pathways that have already been previously found to affect longevity in laboratory studies of mutant or transgenic model organisms (Flatt, 2004; Paaby and Schmidt, 2008; Flatt and Schmidt, 2009). On the one hand, some studies have failed to confirm the lifespan effects of natural variants of candidate longevity genes (Geiger-Thornsberry and Mackay, 2004). On the other hand, there is increasing evidence that genetic variation in candidate longevity genes might indeed contribute to variation in lifespan, as well as life history traits, in natural populations (Schmidt et al., 2000; Paaby and Schmidt, 2008; Suh et al., 2008; Paaby et al., 2010; Rose et al., 2010, 2011; Pijpe et al., 2011; Luisi et al., 2012).
A particularly striking example of such a variant is the gene FOXO3A, a human ortholog of Drosophila FOXO and C. elegans DAF-16. Several independent studies of natural polymorphisms in FOXO3A in Japanese, German, French, Italian, and Han Chinese populations have found that specific variants in this gene are associated with exceptional longevity among human centenarians (Willcox et al., 2008; Anselmi et al., 2009; Flachsbart et al., 2009; Li et al., 2009; Pawlikowska et al., 2009; Soerensen et al., 2010; Zeng et al., 2010). Although one cannot rule out a certain level of ascertainment bias, these results suggest that FOXO not only plays a functional role in regulating lifespan in laboratory model organisms, but that naturally occurring alleles can also have measurable effects on lifespan. Similar associations between natural polymorphisms and human longevity have been identified for IGF-1R (Suh et al., 2008). Likewise, evidence from Drosophila indicates that natural alleles in the InR locus do affect life history traits that are closely linked to longevity (Paaby et al., 2010).
Finally, a similar pattern appears to be emerging with regard to natural variants of genes involved in the neuronal regulation of lifespan: correlations have been found between longevity and genes that function in (1) neuronal development (Rybina and Pasyukova, 2010; Walter et al., 2011), (2) in neural circuitry (De Benedictis et al., 1998; De Luca et al., 2001, 2003; Carbone et al., 2006), or (3) in the uncoupling process in neuronal tissues (Rose et al., 2011).
Conclusions and Perspectives
Here we have provided a review of the recent knowledge about the neuronal inputs and outputs that affect aging and longevity, mainly by focusing on the latest work in genetically tractable model organisms, such as flies, worms, and mice. Even though many details remain to be discovered, it is amply clear today that aging and longevity are profoundly influenced by neuronal activities. Indeed, given that the nervous system (especially, the neuroendocrine system) is intimately involved in regulating an animal’s physiology, e.g., its homeostasis and survival, in response to environmental changes, such a role for this organ system in the aging process is not surprising, both from a physiological and evolutionary perspective. Yet numerous difficult puzzles remain to be solved in future work. For example, with regard to IIS, we know that downregulation of this pathway can have positive effects on lifespan; however, at the same time such downregulation can severely impair neuronal survival and CNS function in old age (also, see discussion in Broughton and Partridge, 2009). Perhaps these distinct effects of IIS on animal physiology could depend on the tissue- or temporal-specific activities of the pathway. Hence, these pleiotropic effects of IIS highlight our need for a much better understanding of how, why, and when “brain aging” and “organismal aging” are exactly coupled or decoupled. More generally, understanding the developmental “carry-over” effects on adult lifespan will require us to gain further insight into the tissue-, age-, and stage-specificity of the neuronal effects on aging and longevity. Similarly, our current knowledge of the intricate interactions involved in the neuronal regulation of aging and longevity is still extremely rudimentary. For instance, not much is known about the interactions between different “longevity” pathways in the brain, or how different tissues (such as the gonad or adipose tissue) cross-talk with the CNS in the modulation of whole-organism lifespan. Thus, despite the fact that recent years have witnessed a lot of progress in this area, there are clearly very exciting times and novel discoveries ahead in the elucidation of the neuronal aspects of aging and longevity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Joy Alcedo has been supported by the Novartis Research Foundation, the Swiss National Science Foundation (SNF, 31003A_134958) and Wayne State University. Thomas Flatt acknowledges support from the Austrian Science Foundation (FWF, P21498-B11), the Swiss National Science Foundation (SNF, PP00P3_133641), and the Wissenschaftskolleg zu Berlin. Elena G. Pasyukova was supported by the Presidium of the Russian Academy of Sciences and the Russian Foundation for Basic Research (#12-04-01182-a).
References
Åkerfelt, M., Morimoto, R. I., and Sistonen, L. (2010). Heat shock factors: integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 11, 545–555.
Alcedo, J., and Kenyon, C. (2004). Regulation of C. elegans longevity by specific gustatory and olfactory neurons. Neuron 41, 45–55.
Alcedo, J., Maier, W., and Ch’ng, Q. (2010). “Sensory influence on homeostasis and lifespan: molecules and circuits,” in Protein Metabolism and Homeostasis in Aging, ed. N. Tavernarakis (Austin, TX: Landes Bioscience), 197–210.
Alic, N., Hoddinott, M. P., Vinti, G., and Partridge, L. (2011). Lifespan extension by increased expression of the Drosophila homologue of the IGFBP7 tumour suppressor. Aging Cell 10, 137–147.
Anselmi, C. V., Malovini, A., Roncarati, R., Novelli, V., Villa, F., Condorelli, G., et al. (2009). Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuv. Res. 12, 95–104.
Apfeld, J., and Kenyon, C. (1999). Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature 402, 804–809.
Arantes-Oliveira, N., Apfeld, J., Dillin, A., and Kenyon, C. (2002). Regulation of life-span by germ-line stem cells in Caenorhabditis elegans. Science 295, 502–505.
Baeriswyl, S., Diard, M., Mosser, T., Leroy, M., Manière, X., Taddei, F., et al. (2009). Modulation of aging profiles in isogenic populations of Caenorhabditis elegans by bacteria causing different extrinsic mortality rates. Biogerontology 11, 53–65.
Baker, K. D., and Thummel, C. S. (2007). Diabetic larvae and obese flies – emerging studies of metabolism in Drosophila. Cell Metab. 6, 257–266.
Bargmann, C. I., and Horvitz, H. R. (1991). Control of larval development by chemosensory neurons in Caenorhabditis elegans. Science 251, 1243–1246.
Bateman, J. M., and McNeill, H. (2006). Insulin/IGF signalling in neurogenesis. Cell. Mol. Life Sci. 63, 1701–1705.
Batlevi, Y., and La Spada, A. R. (2011). Mitochondrial autophagy in neural function, neurodegenerative disease, neuron cell death, and aging. Neurobiol. Dis. 43, 46–51.
Bauer, J. H., Chang, C., Morris, S. N., Hozier, S., Andersen, S., Waitzman, J. S., et al. (2007). Expression of dominant-negative Dmp53 in the adult fly brain inhibits insulin signaling. Proc. Natl. Acad. Sci. U.S.A. 104, 13355–13360.
Berryman, D. E., Christiansen, J. S., Johannsson, G., Thorner, M. O., and Kopchick, J. J. (2008). Role of the GH/IGF-1 axis in lifespan and healthspan: lessons from animal models. Growth Horm. IGF Res. 18, 455–471.
Bishop, N. A., and Guarente, L. (2007). Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature 447, 545–549.
Bishop, N. A., Lu, T., and Yankner, B. A. (2010). Neural mechanisms of ageing and cognitive decline. Nature 464, 529–535.
Boehm, M., and Slack, F. (2005). A developmental timing microRNA and its target regulate life span in C. elegans. Science 310, 1954–1957.
Boulias, K., and Horvitz, H. R. (2012). The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO. Cell Metab. 15, 439–450.
Broughton, S., Alic, N., Slack, C., Bass, T., Ikeya, T., Vinti, G., et al. (2008). Reduction of DILP2 in Drosophila triages a metabolic phenotype from lifespan revealing redundancy and compensation among DILPs. PLoS ONE 3:e3721. doi:10.1371/journal.pone.0003721
Broughton, S., and Partridge, L. (2009). Insulin/IGF-like signalling, the central nervous system and aging. Biochem. J. 418, 1–12.
Broughton, S. J., Piper, M. D. W., Ikeya, T., Bass, T. M., Jacobsen, J., Driege, Y., et al. (2005). Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc. Natl. Acad. Sci. U.S.A. 102, 3105–3110.
Carbone, M. A., Jordan, K. W., Lyman, R. F., Harbison, S. T., Leips, J., Morgan, T. J., et al. (2006). Phenotypic variation and natural selection at catsup, a pleiotropic quantitative trait gene in Drosophila. Curr. Biol. 16, 912–919.
Chakrabarti, S., Munshi, S., Kalpita Banerjee, R., Ishita Guha Thakurta, I. G., Sinha, M., and Bagh, M. B. (2011). Mitochondrial dysfunction during brain aging: role of oxidative stress and modulation by antioxidant supplementation. Aging Dis. 2, 242–256.
Challet, E., Caldelas, I., Graff, C., and Pévet, P. (2003). Synchronization of the molecular clockwork by light- and food-related cues in mammals. Biol. Chem. 384, 711–719.
Chang, A. J., and Bargmann, C. I. (2008). Hypoxia and the HIF-1 transcriptional pathway reorganize a neuronal circuit for oxygen-dependent behavior in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 105, 7321–7326.
Chang, A. J., Chronis, N., Karow, D. S., Marletta, M. A., and Bargmann, C. I. (2006). A distributed chemosensory circuit for oxygen preference in C. elegans. PLoS Biol. 4:e274. doi:10.1371/journal.pbio.0040274
Chen, D., Pan, K. Z., Palter, J. E., and Kapahi, P. (2007). Longevity determined by developmental arrest genes in Caenorhabditis elegans. Aging Cell 6, 525–533.
Chen, D., Thomas, E. L., and Kapahi, P. (2009). HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis elegans. PLoS Genet. 5:e1000486. doi:10.1371/journal.pgen.1000486
Chen, Z., Hendricks, M., Cornils, A., Maier, W., Alcedo, J., and Zhang, Y. (2013). Two insulin-like peptides antagonistically regulate aversive olfactory learning in C. elegans. Neuron 77, 572–585.
Cheung, B. H. H., Cohen, M., Rogers, C., Albayram, O., and de Bono, M. (2005). Experience-dependent modulation of C. elegans behavior by ambient oxygen. Curr. Biol. 15, 905–917.
Chrysis, D., Calikoglu, A. S., Ye, P., and D’Ercole, A. J. (2001). Insulin-like growth factor-I overexpression attenuates cerebellar apoptosis by altering the expression of Bcl family proteins in a developmentally specific manner. J. Neurosci. 21, 1481–1489.
Cohen, E., Bieschke, J., Perciavalle, R. M., Kelly, J. W., and Dillin, A. (2006). Opposing activities protect against age-onset proteotoxicity. Science 313, 1604–1610.
Conti, B., Sanchez-Alavez, M., Winsky-Sommerer, R., Morale, M. C., Lucero, J., Brownell, S., et al. (2006). Transgenic mice with a reduced core body temperature have an increased life span. Science 314, 825–828.
Copeland, J. M., Cho, J., Lo, T., Hur, J. H., Bahadorani, S., Arabyan, T., et al. (2009). Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 19, 1591–1598.
Cornils, A., Gloeck, M., Chen, Z., Zhang, Y., and Alcedo, J. (2011). Specific insulin-like peptides encode sensory information to regulate distinct developmental processes. Development 138, 1183–1193.
David, D. C., Ollikainen, N., Trinidad, J. C., Cary, M. P., Burlingame, A. L., and Kenyon, C. (2010). Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 8:e1000450. doi:10.1371/journal.pbio.1000450
De Benedictis, G., Carotenuto, L., Carrieri, G., De Luca, M., Falcone, E., Rose, G., et al. (1998). Gene/longevity association studies at four autosomal loci (REN, THO, PARP, SOD2). Eur. J. Hum. Genet. 6, 534–541.
De Luca, M., Rose, G., Bonafè, M., Garasto, S., Greco, V., Weir, B. S., et al. (2001). Sex-specific longevity associations defined by tyrosine hydroxylase-insulin-insulin growth factor 2 haplotypes on the 11p15.5 chromosomal region. Exp. Gerontol. 36, 1663–1671.
De Luca, M., Roshina, N. V., Geiger-Thornsberry, G. L., Lyman, R. F., Pasyukova, E. G., and Mackay, T. F. C. (2003). Dopa decarboxylase (Ddc) affects variation in Drosophila longevity. Nat. Genet. 34, 429–433.
Dillin, A., Hsu, A. L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A. G., et al. (2002a). Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401.
Dillin, A., Crawford, D. K., and Kenyon, C. (2002b). Timing requirements for insulin/IGF-1 signaling in C. elegans. Science 298, 830–834.
Durieux, J., Wolff, S., and Dillin, A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91.
Eckert, A., Schmitt, K., and Götz, J. (2011). Mitochondrial dysfunction – the beginning of the end in Alzheimer’s disease? Separate and synergistic modes of tau and amyloid-β toxicity. Alzheimers Res. Ther. 3, 15. doi:10.1186/alzrt74
Eminel, S., Klettner, A., Roemer, L., Herdegen, T., and Waetzig, V. (2004). JNK2 translocates to the mitochondria and mediates cytochrome c release in PC12 cells in response to 6-hydroxydopamine. J. Biol. Chem. 279, 55385–55392.
Enell, L. E., Kapan, N., Söderberg, J. A. E., Kahsai, L., and Nässel, D. R. (2010). Insulin signaling, lifespan and stress resistance are modulated by metabotropic GABA receptors on insulin producing cells in the brain of Drosophila. PLoS ONE 5:e15780. doi:10.1371/journal.pone.0015780
Escames, G., López, A., García, J. A., García, L., Acuña-Castroviejo, D., García, J. J., et al. (2010). The role of mitochondria in brain aging and the effects of melatonin. Curr. Neuropharmacol. 8, 182–193.
Fielenbach, N., and Antebi, A. (2008). C. elegans dauer formation and the molecular basis of plasticity. Genes Dev. 22, 2149–2165.
Finch, C. E. (1990). Longevity, Senescence, and the Genome. Chicago: The University of Chicago Press.
Flachsbart, F., Caliebe, A., Kleindorp, R., Blanché, H., von Eller-Eberstein, H., Nikolaus, S., et al. (2009). Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc. Natl. Acad. Sci. U.S.A. 106, 2700–2705.
Flatt, T. (2004). Assessing natural variation in genes affecting Drosophila lifespan. Mech. Ageing Dev. 125, 155–159.
Flatt, T., Min, K.-J., D’Alterio, C., Villa-Cuesta, E., Cumbers, J., Lehmann, R., et al. (2008). Drosophila germ-line modulation of insulin signaling and lifespan. Proc. Natl. Acad. Sci. U.S.A. 105, 6368–6373.
Flatt, T., and Schmidt, P. S. (2009). Integrating evolutionary and molecular genetics of aging. Biochim. Biophys. Acta 1790, 951–962.
Fonseca, D. B., Brancato, C. L., Prior, A. E., Shelton, P. M., and Sheehy, M. R. (2005). Death rates reflect accumulating brain damage in arthropods. Proc. Biol. Sci. 272, 1941–1947.
Fontana, L., Partridge, L., and Longo, V. D. (2010). Extending healthy life span – from yeast to humans. Science 328, 321–328.
Fridell, Y. W., Hoh, M., Kréneisz, O., Hosier, S., Chang, C., Scantling, D., et al. (2009). Increased uncoupling protein (UCP) activity in Drosophila insulin-producing neurons attenuates insulin signaling and extends lifespan. Aging (Albany, NY) 1, 699–713.
Fridell, Y. W., Sanchez_Blanco, A., Silvia, B. A., and Helfand, S. L. (2005). Targeted expression of the human uncoupling protein 2 (hUCP2) to adult neurons extends life span in the fly. Cell Metab. 1, 145–152.
Gaglia, M. M., Jeong, D.-E., Ryu, E.-A., Lee, D., Kenyon, C., and Lee, S.-J. (2012). Genes that act downstream of sensory neurons to influence longevity, dauer formation, and pathogen responses in Caenorhabditis elegans. PLoS Genet. 8:e1003133. doi:10.1371/journal.pgen.1003133
Gavrilov, L. A., and Gavrilova, N. S. (2011). Season of birth and exceptional longevity: comparative study of American centenarians, their siblings, and spouses. J. Aging Res. 2011:104616. doi:10.4061/2011/104616
Geiger-Thornsberry, G. L., and Mackay, T. F. C. (2004). Quantitative trait loci affecting natural variation in Drosophila longevity. Mech. Ageing Dev. 125, 179–189.
Geminard, C., Rulifson, E. J., and Leopold, P. (2009). Remote control of insulin secretion by fat cells in Drosophila. Cell Metab. 10, 199–207.
Gems, D., Sutton, A. J., Sundermeyer, M. L., Albert, P. S., King, K. V., Edgley, M. L., et al. (1998). Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics 150, 129–155.
Giannakou, M. E., Goss, M., Jacobson, J., Vinti, G., Leevers, S. J., and Partridge, L. (2007). Dynamics of the action of dFOXO on adult mortality in Drosophila. Aging Cell 6, 429–438.
Golden, T. R., Hubbard, A., Dando, C., Herren, M., and Melov, S. (2008). Age-related behaviors have distinct transcriptional profiles in C. elegans. Aging Cell 7, 850–865.
Greer, E. R., Pérez, C. L., Van Gilst, M. R., Lee, B. H., and Ashrafi, K. (2008). Neural and molecular dissection of a C. elegans sensory circuit that regulates fat and feeding. Cell Metab. 8, 118–131.
Grönke, S., Clarke, D. F., Broughton, S., Andrews, T. D., and Partridge, L. (2010). Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 6:e1000857. doi:10.1371/journal.pgen.1000857
Ha, E., Yim, S.-V., Chung, J.-H., Yoon, K.-S., Kang, I., Cho, Y. H., et al. (2006). Melatonin stimulates glucose transport via insulin receptor substrate-1/phosphatidylinositol 3-kinase pathway in C2C12 murine skeletal muscle cells. J. Pineal Res. 41, 67–72.
Haselton, A., Sharmin, E., Schrader, J., Sah, M., Poon, P., and Fridell, Y. W. (2010). Partial ablation of adult Drosophila insulin-producing neurons modulates glucose homeostasis and extends life span without insulin resistance. Cell Cycle 9, 3063–3071.
Heidinger, B. J., Blount, J. D., Boner, W., Griffiths, K., Metcalfe, N. B., and Monaghan, P. (2012). Telomere length in early life predicts lifespan. Proc. Natl. Acad. Sci. U.S.A. 109, 1743–1748.
Hill, R. W., Wyse, G. A., and Anderson, M. (2012). Animal Physiology, 3rd Edn. Sunderland, MA: Sinauer Associates, Inc.
Ho, N., Liauw, J. A., Blaeser, F., Wei, F., Hanissian, S., Muglia, L. M., et al. (2000). Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J. Neurosci. 20, 6459–6472.
Holzenberger, M., Kappeler, L., and De Magalhaes Filho, C. (2004). IGF-1 signaling and aging. Exp. Gerontol. 39, 1761–1764.
Honegger, B., Galic, M., Kohler, K., Wittwer, F., Brogiolo, W., Hafen, E., et al. (2008). Imp-L2, a putative homolog of vertebrate IGF-binding protein 7, counteracts insulin signaling in Drosophila and is essential for starvation resistance. J. Biol. 7:10. doi:10.1186/jbiol72
Hsin, H., and Kenyon, C. (1999). Signals from the reproductive system regulate the lifespan of C. elegans. Nature 399, 362–366.
Hsu, A. L., Murphy, C. T., and Kenyon, C. (2003). Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300, 1142–1145.
Hsu, H. J., LaFever, L., and Drummond-Barbosa, D. (2008). Diet controls normal and tumorous germline stem cells via insulin-dependent and -independent mechanisms in Drosophila. Dev. Biol. 313, 700–712.
Huffman, K. (2012). The developing, aging neocortex: how genetics and epigenetics influence early developmental patterning and age-related change. Front. Genet. 3:212. doi:10.3389/fgene.2012.00212
Humphrey, D. M., Toivonen, J. M., Giannakou, M., Partridge, L., and Brand, M. D. (2009). Expression of human uncoupling protein-3 in Drosophila insulin-producing cells increases insulin-like peptide (DILP) levels and shortens lifespan. Exp. Gerontol. 44, 316–327.
Humphries, K. M., Szweda, P. A., and Szweda, L. I. (2006). Aging: a shift from redox regulation to oxidative damage. Free Radic. Res. 40, 1239–1243.
Hwangbo, D. S., Gershman, B., Tu, M. P., Palmer, M., and Tatar, M. (2004). Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 429, 562–566.
Iser, W. B., Gami, M. S., and Wolkow, C. A. (2007). Insulin signaling in Caenorhabditis elegans regulates both endocrine-like and cell-autonomous outputs. Dev. Biol. 303, 434–447.
Jeong, D.-E., Artan, M., Seo, K., and Lee, S.-J. (2012). Regulation of lifespan by chemosensory and thermosensory systems: findings in invertebrates and their implications in mammalian aging. Front. Genet. 3:218. doi:10.3389/fgene.2012.00218
Kappeler, L., De Magalhaes Filho, C., Dupont, J., Leneuve, P., Cervera, P., Périn, L., et al. (2008). Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol. 6:e254. doi:10.1371/journal.pbio.0060254
Karpac, J., Hull-Thompson, J., Falleur, M., and Jasper, H. (2009). JNK signaling in insulin-producing cells is required for adaptive responses to stress in Drosophila. Aging Cell 8, 288–295.
Karpac, J., and Jasper, H. (2009). Insulin and JNK: optimizing metabolic homeostasis and lifespan. Trends Endocrinol. Metab. 20, 100–106.
Kenyon, C., Chang, J., Gensch, E., Rudner, A., and Tabtiang, R. (1993). A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464.
Kimura, K. D., Tissenbaum, H. A., Liu, Y., and Ruvkun, G. (1997). Daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942–694.
Klass, M. R. (1977). Aging in the nematode Caenorhabditis elegans: major biological and environmental factors influencing life span. Mech. Ageing Dev. 6, 413–429.
Kourtis, N., Nikoletopoulou, V., and Tavernarakis, N. (2012). Small heat-shock proteins protect from heat-stroke-associated neurodegeneration. Nature 490, 213–218.
la Fleur, S. E., Kalsbeek, A., Wortel, J., van der Vliet, J., and Buijs, R. M. (2001). Role for the pineal and melatonin in glucose homeostasis: pinealectomy increases night-time glucose concentrations. J. Neuroendocrinol. 13, 1025–1032.
LaFever, L., and Drummond-Barbosa, D. (2005). Direct control of germline stem cell division and cyst growth by neural insulin in Drosophila. Science 309, 1071–1073.
Landis, J. N., and Murphy, C. T. (2010). Integration of diverse inputs in the regulation of Caenorhabditis elegans DAF-16/FOXO. Dev. Dyn. 239, 1405–1412.
Larson, K., Yan, S. J., Tsurumi, A., Liu, J., Zhou, J., Gaur, K., et al. (2012). Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet. 8:e1002473. doi:10.1371/journal.pgen.1002473
Lee, J., Giordano, S., and Zhang, J. (2012). Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem. J. 441, 523–540.
Lee, K. S., Hong, S. H., Kim, A. K., Ju, S. K., Kwon, O. Y., and Yu, K. (2009). Processed short neuropeptide F peptides regulate growth through the ERK-insulin pathway in Drosophila melanogaster. FEBS Lett. 583, 2573–2577.
Lee, K. S., Kwon, O. Y., Lee, J. H., Kwon, K., Min, K. J., Jung, S. A., et al. (2008). Drosophila short neuropeptide F signalling regulates growth by ERK-mediated insulin signalling. Nat. Cell Biol. 10, 468–475.
Lee, S. J., Hwang, A. B., and Kenyon, C. (2010). Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 20, 2131–2136.
Lee, S. J., and Kenyon, C. (2009). Regulation of the longevity response to temperature by thermosensory neurons in Caenorhabditis elegans. Curr. Biol. 19, 715–722.
Leiser, S. F., Begun, A., and Kaeberlein, M. (2011). HIF-1 modulates longevity and healthspan in a temperature-dependent manner. Aging Cell 10, 318–326.
Lemasters, J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 8, 3–5.
Leopold, P., and Perrimon, N. (2007). Drosophila and the genetics of the internal milieu. Nature 450, 186–188.
Li, W., Kennedy, S. G., and Ruvkun, G. (2003). daf-28 Encodes a C. elegans insulin superfamily member that is regulated by environmental cues and acts in the DAF-2 signaling pathway. Genes Dev. 17, 844–858.
Li, Y., and de Magalhães, J. P. (2011). Accelerated protein evolution analysis reveals genes and pathways associated with the evolution of mammalian longevity. Age (Dordr.) 35, 301–314.
Li, Y., Wang, W. J., Cao, H., Lu, J., Wu, C., Hu, F. Y., et al. (2009). Genetic association of FOXO1A and FOXO3A with longevity trait in Han Chinese populations. Hum. Mol. Genet. 18, 4897–4904.
Libert, S., Zwiener, J., Chu, X., VanVoorhies, W., Roman, G., and Pletcher, S. D. (2007). Regulation of Drosophila life span by olfaction and food-derived odors. Science 315, 1133–1137.
Libina, N., Berman, J. R., and Kenyon, C. (2003). Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115, 489–502.
Liesa, M., and Shirihai, O. S. (2013). Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 17, 491–506.
Liu, N., Landreh, M., Cao, K., Abe, M., Hendriks, G. J., Kennerdell, J. R., et al. (2012). The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 482, 519–523.
Loerch, P. M., Lu, T., Dakin, K. A., Vann, J. M., Isaacs, A., Geula, C., et al. (2008). Evolution of the aging brain transcriptome and synaptic regulation. PLoS ONE 3:e3329. doi:10.1371/journal.pone.0003329
Luisi, P., Alvarez-Ponce, D., Dall’Olio, G. M., Sikora, M., Bertranpetit, J., and Laayouni, H. (2012). Network-level and population genetics analysis of the insulin/TOR signal transduction pathway across human populations. Mol. Biol. Evol. 29, 1379–1392.
Maier, W., Adilov, B., Regenass, M., and Alcedo, J. (2010). A neuromedin U receptor acts with the sensory system to modulate food type-dependent effects on C. elegans lifespan. PLoS Biol. 8:e1000376. doi:10.1371/journal.pbio.1000376
Mattson, M. P. (2006). Neuronal life-and-death signaling, apoptosis, and neurodegenerative disorders. Antioxid. Redox Signal. 8, 1997–2006.
Mehta, R., Steinkraus, K. A., Sutphin, G. L., Ramos, F. J., Shamieh, L. S., Huh, A., et al. (2009). Proteasomal regulation of the hypoxic response modulates aging in C. elegans. Science 324, 1196–1198.
Morley, J. F., Brignull, H. R., Weyers, J. J., and Morimoto, R. I. (2002). The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 99, 10417–10422.
Morley, J. F., and Morimoto, R. I. (2004). Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol. Biol. Cell 15, 657–664.
Morrow, G., Samson, M., Michaud, S., and Tanguay, R. M. (2004). Overexpression of the small mitochondrial Hsp22 extends Drosophila lifespan and increases resistance to oxidative stress. FASEB J. 18, 598–609.
Murphy, C. T., Lee, S. J., and Kenyon, C. (2007). Tissue entrainment by feedback regulation of insulin gene expression in the endoderm of Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 104, 19046–19050.
Murphy, C. T., McCarroll, S., Bargmann, C., Fraser, A., Kamath, R. S., Ahringer, J., et al. (2003). Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277–284.
Nabholz, B., Glémin, S., and Galtier, N. (2008). Strong variations of mitochondrial mutation rate across mammals – the longevity hypothesis. Mol. Biol. Evol. 25, 120–130.
Nakagawa, S., Lagisz, M., Hector, K. L., and Spencer, H. G. (2012). Comparative and meta-analytic insights into life-extension via dietary restriction. Aging Cell 11, 401–409.
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803.
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8:e1000298. doi:10.1371/journal.pbio.1000298
Paaby, A. B., Blacket, M. J., Hoffmann, A. A., and Schmidt, P. S. (2010). Identification of a candidate adaptive polymorphism for Drosophila life history by parallel independent clines on two continents. Mol. Ecol. 19, 760–774.
Paaby, A. B., and Schmidt, P. S. (2008). Functional significance of allelic variation at methuselah, an aging gene in Drosophila. PLoS ONE 3:e1987. doi:10.1371/journal.pone.0001987
Palikaras, K., and Tavernarakis, N. (2012). Mitophagy in neurodegeneration and aging. Front. Genet. 3:297. doi:10.3389/fgene.2012.00297
Partridge, L., and Gems, D. (2002). Mechanisms of ageing: public or private? Nat. Rev. Genet. 3, 165–175.
Partridge, L., Alic, N., Bjedov, I., and Piper, M. D. W. (2011). Ageing in Drosophila: the role of the insulin/Igf and TOR signalling network. Exp. Gerontol. 46, 376–381.
Partridge, L., Gems, D., and Withers, D. J. (2005). Sex and death: what is the connection? Cell 120, 461–472.
Pawlikowska, L., Hu, D., Huntsman, S., Sung, A., Chu, C., Chen, J., et al. (2009). Association of common genetic variation in the insulin/IGF1 signaling pathway with human longevity. Aging Cell 8, 460–472.
Pierce, S. B., Costa, M., Wisotzkey, R., Devadhar, S., Homburger, S. A., Buchman, A. R., et al. (2001). Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes Dev. 15, 672–686.
Pijpe, J., Pul, N., van Duijn, S., Brakefield, P. M., and Zwaan, B. J. (2011). Changed gene expression for candidate ageing genes in long-lived Bicyclus anynana butterflies. Exp. Gerontol. 46, 426–434.
Pincus, Z., and Slack, F. J. (2010). Developmental biomarkers of aging in Caenorhabditis elegans. Dev. Dyn. 239, 1306–1314.
Pincus, Z., Smith-Vikos, T., and Slack, F. J. (2011). MicroRNA predictors of longevity in Caenorhabditis elegans. PLoS Genet. 7:e1002306. doi:10.1371/journal.pgen.1002306
Plum, L., Schubert, M., and Bruning, J. C. (2005). The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 16, 59–65.
Pocock, R., and Hobert, O. (2010). Hypoxia activates a latent circuit for processing gustatory information in C. elegans. Nat. Neurosci. 13, 610–614.
Poon, P. C., Kuo, T.-H., Linford, N. J., Roman, G., and Pletcher, S. D. (2010). Carbon dioxide sensing modulates lifespan and physiology in Drosophila. PLoS Biol. 8:e1000356. doi:10.1371/journal.pbio.1000356
Prahlad, V., Cornelius, T., and Morimoto, R. I. (2008). Regulation of the cellular heat shock response in Caenorhabditis elegans by thermosensory neurons. Science 320, 811–814.
Prahlad, V., and Morimoto, R. I. (2011). Neuronal circuitry regulates the response of Caenorhabditis elegans to misfolded proteins. Proc. Natl. Acad. Sci. U.S.A. 108, 14204–14209.
Puig, O., Marr, M. T. II, Ruhf, M. L., and Tjian, R. (2003). Control of cell number by Drosophila FOXO: downstream and feedback regulation of the insulin receptor pathway. Genes Dev. 17, 2006–2020.
Puig, O., and Tjian, R. (2005). Transcriptional feedback control of insulin receptor by dFOXO/FOXO1. Genes Dev. 19, 2435–2446.
Rajan, A., and Perrimon, N. (2011). Drosophila as a model for interorgan communication: lessons from studies on energy homeostasis. Dev. Cell 21, 29–31.
Rea, S., and Johnson, T. E. (2003). A metabolic model for life span determination in Caenorhabditis elegans. Dev. Cell 5, 197–203.
Rea, S. L., Ventura, N., and Johnson, T. E. (2007). Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 5:e259. doi:10.1371/journal.pbio.0050259
Reddy, P. H., and Reddy, T. P. (2011). Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr. Alzheimer Res. 8, 393–409.
Reznick, D. N. (2005). The genetic basis of aging: an evolutionary biologist’s perspective. Sci. Aging Knowledge Environ. 2005, e7.
Rogers, C., Persson, A., Cheung, B., and de Bono, M. (2006). Behavioral motifs and neural pathways coordinating O2 responses and aggregation in C. elegans. Curr. Biol. 16, 649–659.
Rose, G., Crocco, P., De Rango, F., Montesanto, A., and Passarino, G. (2011). Further support to the uncoupling-to-survive theory: the genetic variation of human UCP genes is associated with longevity. PLoS ONE 6:e29650. doi:10.1371/journal.pone.0029650
Rose, G., Romeo, G., Dato, S., Crocco, P., Bruni, A. C., Hervonen, A., et al. (2010). Somatic point mutations in mtDNA control region are influenced by genetic background and associated with healthy aging: a GEHA study. PLoS ONE 5:e13395. doi:10.1371/journal.pone.0013395
Roth, T. L., and Sweatt, J. D. (2011). Annual research review: epigenetic mechanisms and environmental shaping of the brain during sensitive periods of development. J. Child Psychol. Psychiatry 52, 398–408.
Ruiz, M., Sanchez, D., Canal, I., Acebes, A., and Ganfornina, M. D. (2011). Sex-dependent modulation of longevity by two Drosophila homologues of human apolipoprotein D, GLaz and NLaz. Exp. Gerontol. 46, 579–589.
Rulifson, E. J., Kim, S. K., and Nusse, R. (2002). Ablation of insulin-producing neurons in flies: growth and diabetic phenotypes. Science 296, 1118–1120.
Rybina, O. Y., and Pasyukova, E. G. (2010). A naturally occurring polymorphism at Drosophila melanogaster Lim3 locus, a homolog of human LHX3/4, affects Lim3 transcription and fly lifespan. PLoS ONE 5:e12621. doi:10.1371/journal.pone.0012621
Saino, N., Romano, M., Ambrosini, R., Rubolini, D., Boncoraglio, G., Caprioli, M., et al. (2012). Longevity and lifetime reproductive success of barn swallow offspring are predicted by their hatching date and phenotypic quality. J. Anim. Ecol. 81, 1004–1012.
Schackwitz, W. S., Inoue, T., and Thomas, J. H. (1996). Chemosensory neurons function in parallel to mediate a pheromone response in C. elegans. Neuron 17, 719–728.
Schmidt, P. S., Duvernell, D. D., and Eanes, W. F. (2000). Adaptive evolution of a candidate gene for aging in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 97, 10861–10865.
Schroeter, H., Boyd, C. S., Ahmed, R., Spencer, J. P., Duncan, R. F., Rice-Evans, C., et al. (2003). c-Jun N-terminal kinase (JNK)-mediated modulation of brain mitochondria function: new target proteins for JNK signalling in mitochondrion-dependent apoptosis. Biochem. J. 372, 359–369.
Schubert, M., Gautam, D., Surjo, D., Ueki, K., Baudler, S., Schubert, D., et al. (2004). Role for neuronal insulin resistance in neurodegenerative diseases. Proc. Natl. Acad. Sci. U.S.A. 101, 3100–3105.
Schulz, T. J., Zarse, K., Voigt, A., Urban, N., Birringer, M., and Ristow, M. (2007). Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 6, 280–293.
Shen, J., Ford, D., Landis, G. N., and Tower, J. (2009). Identifying sexual differentiation genes that affect Drosophila life span. BMC Geriatr. 9:56 doi:10.1186/1471-2318-9-56
Shen, L. L., Du, M., Lin, X. F., Cai, T., and Wang, D. Y. (2010). Genes required for the functions of olfactory AWA neuron regulate the longevity of Caenorhabditis elegans in an insulin/IGF signaling-dependent fashion. Neurosci. Bull. 26, 91–103.
Siegal, M. L., and Bergman, A. (2002). Waddington’s canalization revisited: developmental stability and evolution. Proc. Natl. Acad. Sci. U.S.A. 99, 10528–10532.
Smith, E. D., Tsuchiya, M., Fox, L. A., Dang, N., Hu, D., Kerr, E. O., et al. (2008). Quantitative evidence for conserved longevity pathways between divergent eukaryotic species. Genome Res. 18, 564–570.
Soerensen, M., Dato, S., Christensen, K., McGue, M., Stevnsner, T., Bohr, V. A., et al. (2010). Replication of an association of variation in the FOXO3A gene with human longevity using both case-control and longitudinal data. Aging Cell 9, 1010–1017.
Strand, F. L. (1999). Neuropeptides – Regulators of Physiological Processes. Cambridge, MA: MIT Press.
Suh, Y., Atzmon, G., Cho, M. O., Hwang, D., Liu, B., Leahy, D. J., et al. (2008). Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc. Natl. Acad. Sci. U.S.A. 105, 3438–3442.
Swerdlow, R. H. (2011). Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta 1812, 1630–1639.
Taguchi, A., Wartschow, L. M., and White, M. F. (2007). Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science 317, 369–372.
Taguchi, A., and White, M. F. (2008). Insulin-like signaling, nutrient homeostasis, and life span. Annu. Rev. Physiol. 70, 191–212.
Tatar, M., Bartke, A., and Antebi, A. (2003). The endocrine regulation of aging by insulin-like signals. Science 299, 1346–1351.
Tazearslan, C., Ayyadevara, S., Bharill, P., and Shmookler Reis, R. J. (2009). Positive feedback between transcriptional and kinase suppression in nematodes with extraordinary longevity and stress resistance. PLoS Genet. 5:e1000452. doi:10.1371/journal.pgen.1000452
Terry, N. A., Tulina, N., Matunis, E., and DiNardo, S. (2006). Novel regulators revealed by profiling Drosophila testis stem cells within their niche. Dev. Biol. 294, 246–257.
Thyagarajan, B., Blaszczak, A. G., Chandler, K. J., Watts, J. L., Johnson, W. E., and Graves, B. J. (2010). ETS-4 is a transcriptional regulator of life span in Caenorhabditis elegans. PLoS Genet. 6:e1001125. doi:10.1371/journal.pgen.1001125
Troulinaki, K., and Bano, D. (2012). Mitochondrial deficiency: a double-edged sword for aging and neurodegeneration. Front. Genet. 3:244. doi:10.3389/fgene.2012.00244
van Ham, T. J., Holmberg, M. A., van der Goot, A. T., Teuling, E., Garcia-Arencibia, M., Kim, H. E., et al. (2010). Identification of MOAG-4/SERF as a regulator of age-related proteotoxicity. Cell 142, 601–612.
Vermehren-Schmaedick, A., Ainsley, J. A., Johnson, W. A., Davies, S.-A., and Morton, D. B. (2010). Behavioral responses to hypoxia in Drosophila larvae are mediated by atypical soluble guanylyl cyclases. Genetics 186, 183–196.
Volovik, Y., Maman, M., Dubnikov, T., Bejerano-Sagie, M., Joyce, D., Kapernick, E. A., et al. (2012). Temporal requirements of heat shock factor-1 for longevity assurance. Aging Cell 11, 491–499.
Walter, S., Atzmon, G., Demerath, E. W., Garcia, M. E., Kaplan, R. C., Kumari, M., et al. (2011). A genome-wide association study of aging. Neurobiol. Aging 32, 2109.e15–28.
Wang, M. C., Bohmann, D., and Jasper, H. (2005). JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell 121, 115–125.
Waskar, M., Landis, G. N., Shen, J., Curtis, C., Tozer, K., Abdueva, D., et al. (2009). Drosophila melanogaster p53 has developmental stage-specific and sex-specific effects on adult life span indicative of sexual antagonistic pleiotropy. Aging (Albany, NY) 1, 903–936.
Weindruch, R., and Walford, R. L. (1988). The Retardation of Aging and Disease by Dietary Restriction. Springfield, IL: C. C. Thomas.
Wessells, R. J., Fitzgerald, E., Cypser, J. R., Tatar, M., and Bodmer, R. (2004). Insulin regulation of heart function in aging fruit flies. Nat. Genet. 36, 1275–1281.
Willcox, B. J., Donlon, T. A., He, Q., Chen, R., Grove, J. S., Yano, K., et al. (2008). FOXO3A genotype is strongly associated with human longevity. Proc. Natl. Acad. Sci. U.S.A. 105, 13987–13992.
Williams, G. C. (1957). Pleiotropy, natural selection, and the evolution of senescence. Evolution 11, 398–411.
Wolkow, C. A., Kimura, K. D., Lee, M. S., and Ruvkun, G. (2000). Regulation of C. elegans life-span by insulinlike signaling in the nervous system. Science 290, 147–150.
Wurtman, R. J., Axelrod, J., and Fischer, J. E. (1964). Melatonin synthesis in the pineal gland: effect of light mediated by the sympathetic nervous system. Science 143, 1328–1329.
Wurtman, R. J., Axelrod, J., and Phillips, L. S. (1963). Melatonin synthesis in the pineal gland: control by light. Science 142, 1071–1073.
Wuttke, D., Connor, R., Vora, C., Craig, T., Li, Y., Wood, S., et al. (2012). Dissecting the gene network of dietary restriction to identify evolutionarily conserved pathways and new functional genes. PLoS Genet. 8:e1002834. doi:10.1371/journal.pgen.1002834
Xiao, R., Zhang, B., Dong, Y., Gong, J., Xu, T., Jianfeng, L., et al. (2013). A genetic program promotes C. elegans longevity at cold temperatures via a thermosensitive TRP channel. Cell 152, 806–817.
Yin, F., Boveris, A., and Cadenas, E. (2012). Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid. Redox Signal. doi:10.1089/ars.2012.4774
Yoon, H., Enquist, L. W., and Dulac, C. (2005). Olfactory inputs to hypothalamic neurons controlling reproduction and fertility. Cell 123, 669–682.
Zafra, M. A., Molina, F., and Puerto, A. (2006). The neural/cephalic phase reflexes in the physiology of nutrition. Neurosci. Biobehav. Rev. 30, 1032–1044.
Zeng, Y., Cheng, L., Chen, H., Cao, H., Hauser, E. R., Liu, Y., et al. (2010). Effects of FOXO genotypes on longevity: a biodemographic analysis. J. Gerontol. A Biol. Sci. Med. Sci. 65, 1285–1299.
Zhang, C. Y., Baffy, G., Perret, P., Krauss, S., Peroni, O., Grujic, D., et al. (2001). Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell 105, 745–755.
Zhang, Y., Shao, Z., Zhai, Z., Shen, C., and Powell-Coffman, J. A. (2009). The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS ONE 4:e6348. doi:10.1371/journal.pone.0006348
Zhou, Q., Lam, P. Y., Han, D., and Cadenas, E. (2008). c-Jun N-terminal kinase regulates mitochondrial bioenergetics by modulating pyruvate dehydrogenase activity in primary cortical neurons. J. Neurochem. 104, 325–335.
Keywords: aging, longevity, homeostasis, brain, nervous system, neuroendocrine system
Citation: Alcedo J, Flatt T and Pasyukova EG (2013) Neuronal inputs and outputs of aging and longevity. Front. Genet. 4:71. doi: 10.3389/fgene.2013.00071
Received: 01 March 2013; Accepted: 13 April 2013;
Published online: 06 May 2013.
Edited by:
Nektarios Tavernarakis, University of Crete, Foundation for Research and Technology – Hellas, GreeceReviewed by:
Vassiliki Nikoletopoulou, Institute of Molecular Biology and Biotechnology, GreeceMaria Markaki, Foundation for Research and Technology – Hellas, Greece
Marta Artal Sanz, University Pablo de Olavide, Spain
Copyright: © 2013 Alcedo, Flatt and Pasyukova. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Joy Alcedo, Department of Biological Sciences, Wayne State University, 5047 Gullen Mall, Detroit, MI 48202, USA. e-mail: joy.alcedo@wayne.edu;
Thomas Flatt, Department of Ecology and Evolution, University of Lausanne, UNIL Sorge, Biophore, CH-1015 Lausanne, Switzerland. e-mail: thomas.flatt@unil.ch;
Elena G. Pasyukova, Institute of Molecular Genetics, Russian Academy of Sciences, 2 Kurchatov Square, Moscow 123182, Russia. e-mail: egpas@rambler.ru
†Present address: Thomas Flatt, Department of Ecology and Evolution, University of Lausanne, Lausanne, Switzerland.