 Michal Brylinski1,2*
Michal Brylinski1,2*- 1Department of Biological Sciences, Louisiana State University, Baton Rouge, LA, USA
- 2Center for Computation and Technology, Louisiana State University, Baton Rouge, LA, USA
Protein threading is widely used in the prediction of protein structure and the subsequent functional annotation. Most threading approaches employ similar criteria for the template identification for use in both protein structure and function modeling. Using structure similarity alone might result in a high false positive rate in protein function inference, which suggests that selecting functional templates should be subject to a different set of constraints. In this study, we extend the functionality of eThread, a recently developed approach to meta-threading, focusing on the optimal selection of functional templates. We optimized the selection of template proteins to cover a broad spectrum of protein molecular function: ligand, metal, inorganic cluster, protein, and nucleic acid binding. In large-scale benchmarks, we demonstrate that the recognition rates in identifying templates that bind molecular partners in similar locations are very high, typically 70–80%, at the expense of a relatively low false positive rate. eThread also provides useful insights into the chemical properties of binding molecules and the structural features of binding. For instance, the sensitivity in recognizing similar protein-binding interfaces is 58% at only 18% false positive rate. Furthermore, in comparative analysis, we demonstrate that meta-threading supported by machine learning outperforms single-threading approaches in functional template selection. We show that meta-threading effectively detects many facets of protein molecular function, even in a low-sequence identity regime. The enhanced version of eThread is freely available as a webserver and stand-alone software at http://www.brylinski.org/ethread.
Introduction
Currently, the most accurate and consequently the most widely used methods for protein structure and function prediction build on homology, i.e., they use information educed from related proteins. As demonstrated in the recent Critical Assessment of Protein Structure Prediction (CASP) experiment, the top performing groups in tertiary structure and function prediction categories use various template-based methods (Moult et al., 2009). Typically, the first step in comparative protein structure modeling or function inference is the identification of suitable templates in available databases, such as the Protein Data Bank (PDB; Berman et al., 2000). Here, the simplest approaches detect template proteins using sequence comparisons; however, these methods are generally limited to the high levels of sequence identity (Rost, 2002).
To address this issue, a number of methods have been developed to search for low-sequence identity templates that can be used to construct the structural model of a target protein or to infer its molecular function. Nevertheless, because of the complex and equivocal relations between protein sequence, structure, and function, template-based modeling in the “twilight zone” of sequence similarity (Rost, 1999) may result in a high false positive rate. This problem can be addressed by introducing various scoring functions and filters. For example, a sequence profile score, pairwise interaction potential, environmental fitness, secondary structure compatibility and their linear combinations are popular scoring functions widely used in threading and fold recognition. To provide templates for protein structure prediction, many methods, such as SP3 (Zhou and Zhou, 2005) and SPARKS2 (Zhou and Zhou, 2005), first apply a combined scoring function to assign the score to each structure in the template library and then use a Z-score filter to identify these templates that are likely structurally similar to the target. Similar criteria are commonly used for the template identification in protein function prediction. For example, to collect functional templates, FINDSITE (Brylinski and Skolnick, 2008) and FINDSITE-metal (Brylinski and Skolnick, 2011) use a threading Z-score of ≥4, a threshold that is also used in the detection of structural templates in TASSER (Zhang and Skolnick, 2004b). A similar method, @TOME-2, employs meta-threading to detect template proteins, which are subsequently used in both structure modeling and function inference (Pons and Labesse, 2009). Other approaches such as 3DLigandSite (Wass et al., 2010) or I-TASSER (Roy et al., 2010) use structural alignments to detect ligand-bound templates for function prediction.
It has been demonstrated that using structure similarity alone may result in a high false positive rate in protein function inference (Brylinski and Skolnick, 2010). Historically, template identification algorithms, such as threading or fold recognition, were designed to detect structurally similar templates for protein structure prediction. Therefore, template selection by structure-driven threading or structure alignment may provide false information on protein function. Recently, we developed eThread, an accurate meta-threading procedure for the identification of structurally related templates for the template-based modeling of proteins (Brylinski and Feinstein, 2012; Brylinski and Lingam, 2012). Here, we extend its functionality to also include the optimal selection of functional templates, which cover a broad range of protein function: ligand, metal, inorganic cluster, protein, and nucleic acid binding. We show that state-of-the-art threading algorithms effectively detect many aspects of protein molecular function using low-homology templates, thus should provide practical assistance to evolution/structure-based function inference of proteins.
Methods
Datasets
Five datasets were compiled in this study, which comprise ligand-, metal-, and iron/sulfur-binding proteins as well as protein–protein and protein–DNA macromolecular complexes. Metal- and Fe/S-binding proteins were directly identified in the PDB (Berman et al., 2000). Following metals were included in the dataset: Ca, Co, Cu, Fe, Mg, Mn, and Zn. Iron–sulfur clusters are made up of at least two iron and two sulfur atoms. The set of protein–ligand complexes was obtained from the Protein-Small Molecule Database (Wallach and Lilien, 2009), which provides a convenient resource for studies focusing on protein–small molecule interactions. Small organic compounds non-covalently bound to proteins and composed of 7–60 heavy atoms were included in the dataset. Protein–protein and protein–DNA complexes were downloaded from the PISA database of macromolecular assemblies (Krissinel and Henrick, 2007). For protein–protein complexes, the minimum number of interfacial residues was set to 20, for protein–DNA assemblies, only DNA strands with at least 10 nucleotides are considered. In each dataset, the redundancy was removed at 40% pairwise sequence identity using PISCES (Wang and Dunbrack, 2003). Furthermore, we included only proteins 50–600 residues in length. This procedure resulted in 6,895, 6,610, 209, 8,155, and 440 ligand-, metal-, Fe/S-, protein-, and DNA-binding proteins, respectively. The lists of benchmarking proteins are provided as Supplementary Materials.
Functional Templates
For each protein target in a given dataset, functional templates are defined as these proteins that structurally align onto the target with a statistically significant TM (template modeling)-score of ≥0.4 (Zhang and Skolnick, 2004a) and bind their partners in a similar location. Here, we use distance thresholds of 4, 2, 3, 6, and 6 Å for ligands, metal, iron–sulfur clusters, protein, and DNA interfaces, respectively. The distances are measured upon the global template-to-target superposition generated by fr-TM-align, a structure alignment program (Pandit and Skolnick, 2008). The ratio of the number of positives and negatives across ligand-, metal-, Fe/S-, protein-, and DNA-binding proteins is 0.24, 0.10, 0.87, 0.10, and 0.31, respectively. In addition, we also assess the chemical and geometrical conservation of bound molecules, i.e., the template- and target-bound metals are of the same type, the pairwise Tanimoto coefficient (Tanimoto, 1958), TC, calculated for Daylight 1024-bit SMILES strings between the template- and target-bound ligands is ≥0.5, or the nucleotide composition of the template- and target-bound DNA is similar (AT- or GC-rich). For protein–protein complexes, we assess the local structural similarity of binding interfaces using iAlign with a significant interfacial score, IS (interface similarity)-score, of ≥0.191 (Gao and Skolnick, 2010). According to these criteria, the positives/negatives ratio for ligand-, metal-, Fe/S-, protein-, and DNA-binding proteins is 0.21, 0.70, 1.17, 0.21, and 1.16, respectively.
Meta-Threading by eThread
To identify functional templates, we use eThread (Brylinski and Lingam, 2012), which integrates ten state-of-the-art protein threading/fold recognition algorithms: CSI-BLAST (Biegert and Soding, 2009), COMPASS (Sadreyev and Grishin, 2003), HHpred (Soding, 2005), HMMER (Eddy, 1998), pfTools (Bucher et al., 1996), pGenThreader (Lobley et al., 2009), SAM-T2K (Hughey and Krogh, 1996), SP3 (Zhou and Zhou, 2005), SPARKS2 (Zhou and Zhou, 2005), and Threader (Jones et al., 1992). eThread was originally design to detect structural templates using machine learning and a set of feature vectors composed of individual threading scores. Here, we extend this functionality to include ligand-, metal-, Fe/S-, protein-, and DNA-binding probability estimates. Specifically, for each aspect of molecular function, we constructed two machine learning models with different levels of optimization to assess whether a particular template (1) binds its partners in a similar location, and (2) binds chemically similar molecules and/or the binding mode is similar. We refer to these models as Location and Features, respectively. We use a Naïve Bayes classifier (NBC) to combine individual threading scoring functions into a single probabilistic score. In this classifier, the real-value attributes are modeled by a Gaussian distribution, i.e., the classifier first estimates a normal distribution for each threading component by computing the mean and standard deviation of the training data in that class, which is then used to estimate the posterior probabilities during classification. Both classifiers, Location and Features, are independently trained on threading scores. The accuracy of template selection is assessed using twofold cross validation; Pearson’s chi-squared test applied to each individual scoring function confirmed that both subsets are characterized by the same central tendency and dispersion measures as the whole dataset. Thus, considering a maximum sequence identity of 40% between any two dataset proteins, this protocol provides a sufficient cross validation. Furthermore, the imposed pairwise sequence identity threshold automatically excludes close homologs from benchmarks.
Example Proteins
We selected the following representative examples: recombinant A. aegerita lectin complexed with lactose (PDB-ID: 2zgm), amino acid acyl-carrier protein ligase 1 from B. japonicum bound to zinc (PDB-ID: 3mey), NADH-quinone oxidoreductase from T. thermophiles complexed with Fe4S4 (PDB-ID: 2fug), transcription factor PU.1 from mouse bound to DNA (PDB-ID: 1pue), and a homodimer of hypothetical transposase from S. tokodaii (PDB-ID: 2ec2). In parentheses are the PDB IDs of weakly homologous (<40% sequence identity) templates identified by eThread for 2zgm (1c1l, 1d2s, 1g86, 1gzw, 1hdk, 1is3, 1kel, 1kjl, 1kjr, 1lhu, 1lhw, 1ngx, 1qfm, 1qkq, 1slt, 1t2q, 1w6o, 1w6p, 2bkl, 2d03, 2d6m, 2eak, 2eal, 2nmo, 2ny1, 2r0h, 2vno, 2wkk, 2wt0, 2wt1, 2xg3, 2z3z, 2zaa, 2zab, 2zac, 2zhk, 2zhl, 2zhm, 3a71, 3a72, 3ap6, 3ap7, 3gal, 3h3l, 3htl, 3nv3, 3nv4, 3o4h, 4gal); 3mey (1b8a, 1e1t, 1e22, 1e24, 1eqr, 1evk, 1evl, 1fyf, 1kog, 1nyq, 1qf6, 1x54, 1x55, 2cim, 2cja, 2i4o, 2j3m, 2q7e, 2q7g, 2rhq, 2rhs, 2xgt, 2xti, 2zcd, 2zce, 2zin, 3a31, 3a74, 3bju, 3e9h, 3e9i, 3nem, 3qtc); 2fug (1cc1, 1e3d, 1fp4, 1frf, 1frv, 1g20, 1g21, 1h1l, 1h2a, 1h2r, 1l5h, 1l9g, 1m1n, 1m1y, 1m34, 1mio, 1n2c, 1qgu, 1qh1, 1qh8, 1ui0, 1ui1, 1vk2, 1yqw, 1yrq, 2a5h, 2afh, 2afi, 2afk, 2d3y, 2ddg, 2dp6, 2frv, 2min, 2wpn, 2xdq, 3aek, 3aet, 3k1a, 3min, 3myr, 3pdi); 1pue (1if1, 1j59, 1k79, 1lb2, 1o3s, 1run, 1t2k, 1xsd, 1yo5, 2cgp, 3e54, 3jtg); and 2ec2 (1a9n, 1hr6, 1y13, 2a6m, 2a6o, 2ar9, 2vic, 2vih, 2vju, 3a4i, 3a74, 3bju, 3lmb).
Results and Discussion
We evaluate the performance of eThread in template selection using five datasets that comprise ligand-, metal-, and iron/sulfur-binding proteins as well as protein–protein and protein–DNA complexes. For each target protein, we identify in the PDB library structurally similar proteins that produce statistically significant structure alignment with a TM-score of ≥0.4. These templates that bind molecular partners in similar locations are considered positives with the remaining categorized as negatives. In the subsequent analysis, we examine another requirement for being a positive, which is either a significant chemical similarity of bound molecules for small ligands and metal ions, a similar nucleotide composition for protein–DNA complexes, or a similar interfacial geometry for protein–protein assemblies. The extended version of eThread employs highly tuned machine learning models to provide a set of probability estimates for various aspects of protein molecular function. These are primarily used to assess whether a particular template binds its partners in a similar location; moreover, another set of probabilities estimate whether the template-bound compounds are chemically similar and/or the binding mode between macromolecules is the same. It is important to note that we explore remote evolutionary relationships between protein, excluding those templates that share>40% sequence identity with the target.
Figure 1 shows the accuracy of template identification by eThread for all considered aspects of molecular function. First, we assess the recognition of these templates that bind their partners in similar locations [green ROC (receiver operating characteristic) curves]. The recognition rates are very high, typically 70–80%, at the expense of a relatively low false positive rate with tight 95% confidence bounds (Kestler, 2001). For instance, the true and false positive rate for the selection of weakly homologous Fe/S-binding templates is 0.72 and 0.30, respectively. Many existing approaches to evolution/structure-based binding site prediction cluster the centers of mass of molecules bound to the identified templates upon the global template-to-target superposition (Pons and Labesse, 2009; Roy et al., 2010; Wass et al., 2010). The posterior probabilities estimated by eThread can be used as weight factors in clustering and the subsequent site ranking. This would allow for more precise binding site identification as well as for the improved ranking of predicted binding sites.
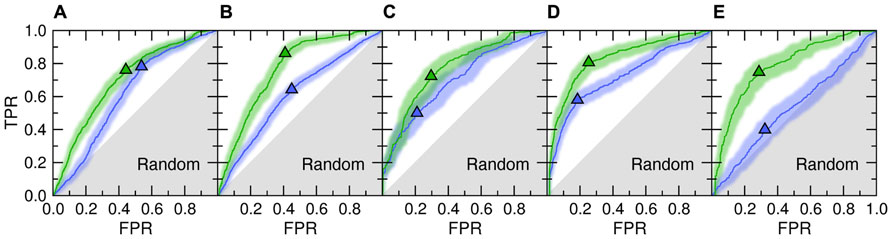
FIGURE 1. ROC plots for template identification by meta-threading andmachine learning. The following aspects of molecular function are considered: (A) ligand-, (B) metal-, (C) Fe/S-, (D) protein-, and (E) DNA-binding. Two different NBC models are trained to detect functionally related proteins that bind their partners in similar locations (Location, green) and bind chemically similar molecules or the binding mode is similar (Feature, blue). Triangles depict optimal cut-off points; solid lines and transparent bands are the ROC curve and its non-parametric two-sided confidence bounds, respectively. Gray area corresponds to prediction accuracy no better than random. TPR, true positive rate; FPR, false positive rate.
Despite the fact that only remote evolutionary relationships between proteins are being explored in the presented benchmarks, meta-threading was found to provide useful insights into the chemical properties of bound molecules and the interfacial geometry; this is shown in Figure 1 as blue ROC curves. This feature can be used to improve the prediction of the chemical properties of binding ligands and iron–sulfur clusters or the type of binding metal. Here, the most accurate is the recognition of similar protein-binding interfaces with a sensitivity of 58% at only 18% false positive rate. This can be beneficially exploited further to extend the target coverage and to improve the accuracy of approaches to protein docking by interfacial similarity (Sinha et al., 2010). The only aspect of molecular function that cannot be predicted by meta-threading is the nucleotide composition of bound DNA (note that the locations of DNA-binding sites are predicted with a high accuracy). This is due to the fact that many DNA-binding proteins associate with non-specific DNA sequences and slide along to search for the specific DNA target sequence (Halford and Marko, 2004).
The area under ROC is a commonly used method to assess the overall performance of ranking approaches. In Table 1, we report the area under the ROC curve for individual threading methods compared to that of eThread. In the majority of cases, the performance of eThread is notably better than that of single-threading algorithms, particularly in the prediction of the chemical properties of binding molecules. The exceptions are HHpred and pfTools in predicting metal-binding sites and binding metals [area under the curve (AUC) of 0.783 and 0.619, respectively], SP3 and SPARKS in detecting DNA-binding aspects (AUC of 0.795 and 0.538, respectively), and COMPASS in recognizing protein–protein interfacial geometry (AUC of 0.732). Table 2 extends this analysis further by accounting for the so-called “early recognition problem.” By analogy to virtual screening, where one requires active compounds to be ranked as high as possible, template identification approaches also should rank good templates early in an ordered list. BEDROC (Boltzmann-enhanced discrimination of ROC) is a generalization of ROC developed specifically to analyze methods that need to segregate positives toward the front of a rank-ordered list (Truchon and Bayly, 2007). As shown in Table 2, except for HHpred in predicting metal-binding sites (BEDROC of 0.723) and DNA feature prediction, which is close to random for all methods, eThread most efficiently tackles the problem of the early recognition of functionally related templates across all binding datasets. These results are finally supported by Z-statistics calculated by the Wilcoxon rank-sum (aka Mann–Whitney U) test. Table 3 shows that only SP3 in the identification of DNA-binding sites (Z-score of 23.18) gives better Z-statistics than eThread.
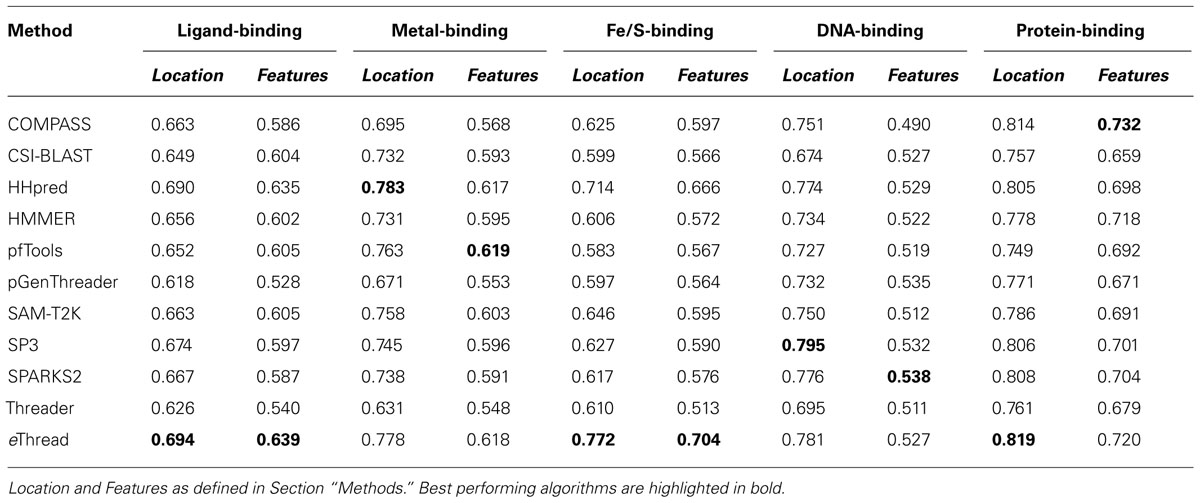
TABLE 1. Area under the ROC curve for individual threading methods compared to that of eThread.
TABLE 2. BEDROC scores calculated for individual threading methods compared to that of eThread.
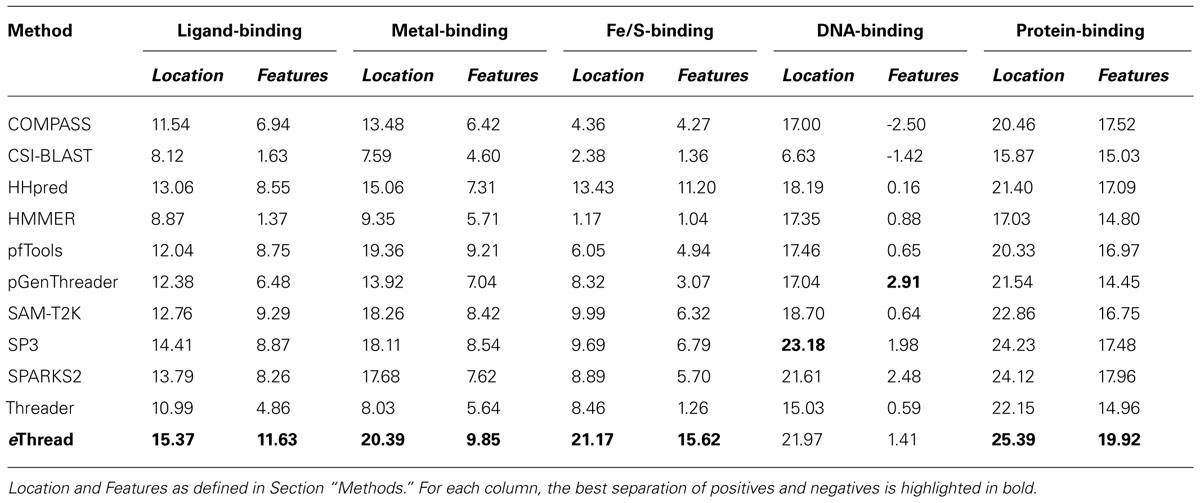
TABLE 3. Z-statistics calculated by the Wilcoxon rank-sum test for individual threading methods compared to that of eThread.
Tables 1,2, and 3 also clearly indicate performance differences between individual methods. For instance, COMPASS, HHpred, and SP3 are the most effective single-threading algorithms in the prediction of protein-, metal-, and DNA-binding sites, respectively. In view of the fact that meta-threading puts much higher demands for high-performance computing resources (Brylinski and Feinstein, 2012), these results provide useful guidelines for selecting a single-threading method with respect to a particular functional aspect when computational speed is critical. Nevertheless, this comparative analysis perspicuously demonstrates that eThread, which uses meta-threading and machine learning models constructed separately for different facets of protein molecular function, systematically outperforms single-threading methods providing higher overall accuracy.
To illustrate how weakly homologous templates identified by eThread pick out binding sites, we selected five representative examples, one for each aspect of molecular function: recombinant lectin (rAAL) complexed with lactose, amino acid acyl-carrier protein ligase 1 (aa:CP) bound to zinc, NADH-quinone oxidoreductase (NDH-1) complexed with Fe4S4, transcription factor PU.1 bound to DNA, and a homodimer of hypothetical transposase (tnpA). In Figure 2, upon the global template-to-target superposition, these templates that are predicted to have a binding site in a similar location are shown in green. Red spots indicate template binding sites, which are below the optimal probability threshold, i.e., are predicted to bind molecules in different locations. In all cases, there are very few false positives and false negatives; the consensus amongst templates predicted to bind their molecular partners in a similar location precisely identifies the correct binding site. Note that for aa:CP and NDH-1, a simple majority voting would result in the incorrect prediction of the binding site location.
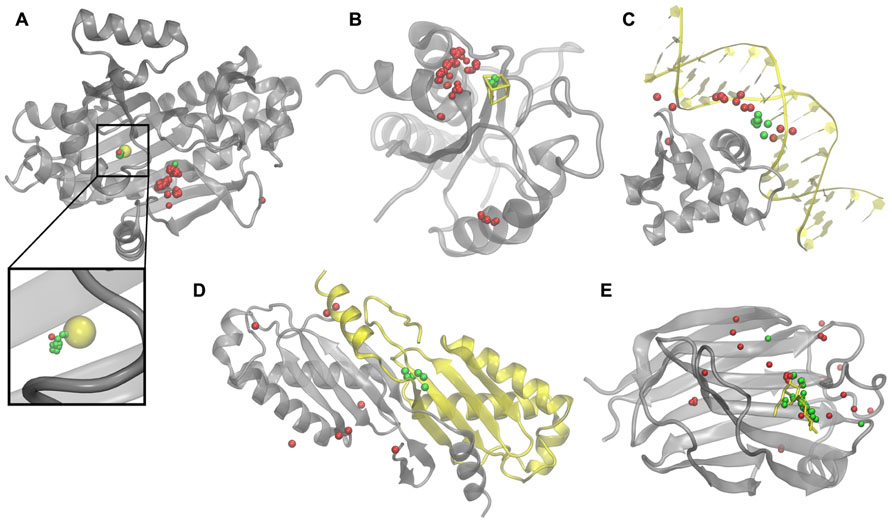
FIGURE 2. Examples of binding sites in weakly homologous templates. Functional templates are identified by eThread for (A) aa:CP, (B) NDH-1, (C)PU.1, (D) tnpA, and (E) rAAL. Target structures and bound molecular partners are shown in gray and yellow, respectively. Template structures (not shown) are superimposed onto the target; binding sites predicted to be in similar and different locations are colored in green and red, respectively.
Conclusion
We extended the functionality of eThread, a recently developed meta-threading approach (Brylinski and Lingam, 2012), to address the problem of identifying weakly homologous templates for function annotation in the “twilight zone” of sequence similarity. This method successfully covers many facets of molecular function including the interactions between proteins and small molecules, metal ions, inorganic clusters, nucleic acids as well as other proteins. It provides not only the location on a protein surface where the interactions take place, but also useful insights into the chemical properties of binding molecules and the geometrical aspects of binding. Such information can be straightforwardly incorporated into any existing evolution/structure-based algorithm for binding site prediction and functional annotation. Moreover, binding pocket prediction can be extended further to include additional downstream analyses, e.g., binding site comparison and mining. For example, Pocket-Surfer (Chikhi et al., 2010) and Patch-Surfer (Sael and Kihara, 2010, 2012) identify ligand molecules that bind to predicted pockets by comparing their geometrical and physicochemical characteristics with a database of known binding pockets. These algorithms are likely to benefit from the improved accuracy of upstream pocket detection.
eThread is freely available to the community at http://www.brylinski.org/ethread as a webserver and a stand-alone software distribution. The latter can be installed in a local environment for high-throughput computations and the integration with existing structure-based protein function inference pipelines. The documentation includes detailed installation instructions and illustrative examples.
Confilct of interest statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the Louisiana Board of Regents through the Board of Regents Support Fund [contract LEQSF(2012-15)-RD-A-05] and Oak Ridge Associated Universities (ORAU) through the 2012 Ralph E. Powe Junior Faculty Enhancement Award. Portions of this research were conducted with high-performance computational resources provided by Louisiana State University (HPC@LSU, http://www.hpc.lsu.edu) and the Louisiana Optical Network Institute (LONI, http://www.loni.org/).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Bioinformatics_and_Computational_Biology/10.3389/fgene.2013.00118/abstract
References
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The Protein Data Bank. Nucleic Acids Res. 28, 235–242. doi: 10.1093/nar/28.1.235
Biegert, A., and Soding, J. (2009). Sequence context-specific profiles for homology searching. Proc. Natl. Acad. Sci. U.S.A. 106, 3770–3775. doi: 10.1073/pnas.0810767106
Brylinski, M., and Feinstein, W. P. (2012). Setting up a meta-threading pipeline for high-throughput structural bioinformatics: eThread software distribution, walkthrough and resource profiling. J. Comput. Sci. Syst. Biol. 6, 001–010. doi: 10.4172/jcsb.1000094
Brylinski, M., and Lingam, D. (2012). eThread: a highly optimized machine learning-based approach to meta-threading and the modeling of protein tertiary structures. PLoS ONE 7:e50200. doi: 10.1371/journal.pone.0050200
Brylinski, M., and Skolnick, J. (2008). A threading-based method (FINDSITE) for ligand-binding site prediction and functional annotation. Proc. Natl. Acad. Sci. U.S.A. 105, 129–134. doi: 10.1073/pnas.0707684105
Brylinski, M., and Skolnick, J. (2010). Comparison of structure-based and threading-based approaches to protein functional annotation. Proteins 78, 118–134. doi: 10.1002/prot.22566
Brylinski, M., and Skolnick, J. (2011). FINDSITE-metal: integrating evolutionary information and machine learning for structure-based metal-binding site prediction at the proteome level. Proteins 79, 735–751. doi: 10.1002/prot.22913
Bucher, P., Karplus, K., Moeri, N., and Hofmann, K. (1996). A flexible motif search technique based on generalized profiles. Comput. Chem. 20, 3–23. doi: 10.1016/S0097-8485(96)80003-9
Chikhi, R., Sael, L., and Kihara, D. (2010). Real-time ligand binding pocket database search using local surface descriptors. Proteins 78, 2007–2028. doi: 10.1002/prot.22715
Eddy, S. R. (1998). Profile hidden Markov models. Bioinformatics 14, 755–763. doi: 10.1093/bioinformatics/14.9.755
Gao, M., and Skolnick, J. (2010). iAlign: a method for the structural comparison of protein–protein interfaces. Bioinformatics 26, 2259–2265. doi: 10.1093/bioinformatics/btq404
Halford, S. E., and Marko, J. F. (2004). How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 32, 3040–3052. doi: 10.1093/nar/gkh624
Hughey, R., and Krogh, A. (1996). Hidden Markov models for sequence analysis: extension and analysis of the basic method. Comput. Appl. Biosci. 12, 95–107. doi: 10.1093/bioinformatics/12.2.95
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). A new approach to protein fold recognition. Nature 358, 86–89. doi: 10.1038/358086a0
Kestler, H. A. (2001). ROC with confidence – a Perl program for receiver operator characteristic curves. Comput. Methods Programs Biomed. 64, 133–136. doi: 10.1016/S0169-2607 (00)00098-5
Krissinel, E., and Henrick, K. (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797. doi: 10.1016/j.jmb.2007.05.022
Lobley, A., Sadowski, M. I., and Jones, D. T. (2009). pGenTHREADER and pDomTHREADER: new methods for improved protein fold recognition and superfamily discrimination. Bioinformatics 25, 1761–1767. doi: 10.1093/bioinformatics/btp302
Moult, J., Fidelis, K., Kryshtafovych, A., Rost, B., and Tramontano, A. (2009). Critical assessment of methods of protein structure prediction – Round VIII. Proteins 77(Suppl. 9), 1–4. doi: 10.1002/prot.22589
Pandit, S. B., and Skolnick, J. (2008). Fr-TM-align: a new protein structural alignment method based on fragment alignments and the TM-score. BMC Bioinformatics 9:531. doi: 10.1186/1471-2105-9-531
Pons, J. L., and Labesse, G. (2009). @TOME-2: a new pipeline for comparative modeling of protein–ligand complexes. Nucleic Acids Res. 37, W485–W491. doi: 10.1093/nar/gkp368
Rost, B. (1999). Twilight zone of protein sequence alignments. Protein Eng. 12, 85–94. doi: 10.1093/protein/12.2.85
Rost, B. (2002). Enzyme function less conserved than anticipated. J. Mol. Biol. 318, 595–608. doi: 10.1016/S0022-2836(02)00016-5
Roy, A., Kucukural, A., and Zhang, Y. (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc.5, 725–738. doi: 10.1038/nprot.2010.5
Sadreyev, R., and Grishin, N. (2003). COMPASS: a tool for comparison of multiple protein alignments with assessment of statistical significance. J. Mol. Biol. 326, 317–336. doi: 10.1016/S0022-2836(02)01371-2
Sael, L., and Kihara, D. (2010). Binding ligand prediction for proteins using partial matching of local surface patches. Int. J. Mol. Sci. 11, 5009–5026. doi: 10.3390/ijms11125009
Sael, L., and Kihara, D. (2012). Detecting local ligand-binding site similarity in non homologous proteins by surface patch comparison. Proteins 80, 1177–1195. doi: 10.1002/prot.24018
Sinha, R., Kundrotas, P. J., and Vakser, I. A. (2010). Docking by structural similarity at protein–protein interfaces. Proteins 78, 3235–3241. doi: 10.1002/prot.22812
Soding, J. (2005). Protein homology detection by HMM–HMM comparison. Bioinformatics 21, 951–960. doi: 10.1093/bioinformatics/bti125
Tanimoto, T. T. (1958). An Elementary Mathematical Theory of Classification and Prediction. IBM Internal Report. New York, IBM.
Truchon, J. F., and Bayly, C. I. (2007). Evaluating virtual screening methods: good and bad metrics for the “early recognition” problem. J. Chem. Inf. Model. 47, 488–508. doi: 10.1021/ci600426e
Wallach, I., and Lilien, R. (2009). The protein-small-molecule database, a non-redundant structural resource for the analysis of protein–ligand binding. Bioinformatics 25, 615–620. doi: 10.1093/bioinformatics/btp035
Wang, G., and Dunbrack, R. L. Jr. (2003). PISCES: a protein sequence culling server. Bioinformatics 19, 1589–1591. doi: 10.1093/bioinformatics/btg224
Wass, M. N., Kelley, L. A., and Sternberg, M. J. (2010). 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res. 38, W469–W473. doi: 10.1093/nar/gkq406
Zhang, Y., and Skolnick, J. (2004a). Scoring function for automated assessment of protein structure template quality. Proteins 57, 702–710. doi: 10.1002/prot.20264
Zhang, Y., and Skolnick, J. (2004b). Tertiary structure predictions on a comprehensive benchmark of medium to large size proteins. Biophys. J. 87, 2647–2655. doi: 10.1529/biophysj.104.045385
Keywords: protein function inference, template-based modeling, protein meta-threading, ligand-binding, metal-binding, iron/sulfur-binding, protein-protein interactions, protein-DNA interactions
Citation: Brylinski M (2013) Unleashing the power of meta-threading for evolution/structure-based function inference of proteins. Front. Genet. 4:118. doi: 10.3389/fgene.2013.00118
Received: 20 April 2013; Accepted: 04 June 2013;
Published online: 19 June 2013.
Edited by:
Fengfeng Zhou, Shenzhen Institutes of Advanced Technology, ChinaReviewed by:
Franca Fraternali, King’s College London, UKYanjie Wei, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, China
Wencai Ma, The University of Texas MD Anderson Cancer Center, USA
Copyright: © 2013 Brylinski. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Michal Brylinski, Department of Biological Sciences, Louisiana State University, 202 Life Sciences Building, Baton Rouge, LA 70803, USA e-mail: michal@brylinski.org