 Frank Emmert-Streib
Frank Emmert-Streib Matthias Dehmer
Matthias Dehmer- 1Computational Biology and Machine Learning Laboratory, Faculty of Medicine, Health and Life Sciences, Center for Cancer Research and Cell Biology, School of Medicine, Dentistry and Biomedical Sciences, Queen's University Belfast, Belfast, UK
- 2Institute for Bioinformatics and Translational Research, UMIT, Hall in Tyrol, Austria
In order to establish systems medicine, based on the results and insights from basic biological research applicable for a medical and a clinical patient care, it is essential to measure patient-based data that represent the molecular and cellular state of the patient's pathology. In this paper, we discuss potential limitations of the sole usage of static genotype data, e.g., from next-generation sequencing, for translational research. The hypothesis advocated in this paper is that dynOmics data, i.e., high-throughput data that are capable of capturing dynamic aspects of the activity of samples from patients, are important for enabling personalized medicine by complementing genotype data.
1. Introduction
After the completion of the HUMAN GENOME PROJECT (Lander et al., 2001; Venter et al., 2001; Consortium, International Human Genome Sequencing, 2004) a new era started aiming to bring results from basic biology and biomedical research into the clinic to the patients. This is often called “from bench to bedside” and defines the general idea underlying translational research and its particular realization in the form of personalized medicine. From a practical point of view, in order to accomplish such a translation of basic research results into the daily clinical routine, it is necessary to be able to generate cost-efficient patient data on the molecular and cellular level (Butte, 2008; Lussier et al., 2010). Fortunately, technological progress within the last 15 years has led to a variety of different experimental assays that provide such opportunities, even on the genomic-scale involving large portions of an organism's genes. For example, in biological research different types of “Omics” data (Ghosh and Poisson, 2009; Moreno-Risueno et al., 2010; The ENCODE Project Consortium, 2011), e.g., genomics, transcriptomics, proteomics, metabolomics and epigenomics data (Lee et al., 2002; Förster et al., 2003; Rual et al., 2005; Stelzl et al., 2005; Palsson, 2006; Sechi, 2007; Garbett et al., 2008; Yu et al., 2008) are frequently employed and could, principally, also be used in translational bioinformatics for studying patient data. Instead, currently, one could gain the feeling that genotype data from sequencing technologies, including next-generation DNA sequencing (Mardis, 2008; Shendure and Ji, 2008; Ansorge, 2009; Metzker, 2009), are dominating the discussions and the initial practical endeavours in this context (Alkan et al., 2009; Werner, 2010; Fernald et al., 2011; Zhang et al., 2011; Highnam and Mittelman, 2012; Ziegler et al., 2012). For instance, in Ng et al. (2009) direct-to-consumer (DTC) DNA tests are reviewed that are already offered by companies to identify potential disease risks of patients. Similar examples are presented in Stepanov (2010); Chin et al. (2011) with an emphasize on the utilization of DNA variations. Also, it has been argued that a genetically guided personalized medicine (GPM) has the potential to enable a patient-based treatment by utilizing sequenced DNA information from the individual patients that can be used to influence medical care decisions in the clinical practice (Welch and Kawamoto, 2012).
We would like to emphasize that it is unquestioned that genotype data, as represented for instance by single nucleotide polymorphisms (SNPs) (Collins et al., 1997; Sachidanandam et al., 2001; Wheeler et al., 2007; LaFramboise, 2009), microsatellites or whole genome sequences, provide a valuable source of information for translational bioinformatics and personalized medicine (Fernald et al., 2011). However, in this paper, we discuss potential limitations of approaches that are solely based on genotype data and emphasize the need for considering high-throughput data that are capable of capturing dynamic states and activity levels of physiological conditions of the patients. In order to distinguish such Omics high-throughput data from genotype data, we will term the latter “dynOmics” data.
This paper is organized as follows. In the next section, environmental and epigenetic influences on the genotype are discussed. Further, we characterize the static nature of genotype data. In section 3 we discuss limitation of genotype data as a consequence of the three factors discussed in section 2. In section 4 we define dynOmics data and discuss gene expression and RNA-seq data as a sources of information for such dynamic high-throughput data. Finally, in section 5 we present three application examples that utilize dynOmics data for their analysis. This paper finishes with concluding remarks.
2. Environmental and Epigenetic Factors and the Static Nature of Genotype Data
It is unquestioned that the DNA within biological cells plays an eminent role in the description of the development and evolution of an organism and the transcription regulation of the gene it encodes. Aside from the understanding of such fundamental processes, the usage of genetic information has been proven useful in studying diseases. For instance, DNA copy number variations (CNVs) (Freeman et al., 2006; Pinto et al., 2011) have been used for elucidating their role in complex disorders (McCarroll and Altshuler, 2007). Specifically, in Stephens et al. (2009); Beroukhim et al. (2010) the effect of somatic copy number alternations (SCNA) and rearrangements has been investigated for a variety of different cancer types, including breast cancer, non-small cell lung cancer, and acute lymphoblastic leukaemia. In Beroukhim et al. (2010) 158 significant regions with a focal SCNA have been identified including a large number of sites without known cancer target genes that constitute potential key players in form of tumor suppressor or oncogenes in the more than 20 different cancer types studied. Also, they found that a large majority of SCNAs can be identified in several different cancers revealing a potential similarity of the molecular pathology among these disorders. Further, in Stephens et al. (2009) it has been found that tandem duplications are particularly frequent, which might indicate a specific type of defect in DNA maintenance. A clinical connection between CNVs and patient survival was found in Kresse et al. (2010) by studying malignant fibrous histiocytoma (MFH). This finding is of particular interest because it shows a concrete example for a medical application of CNV for the diagnosis of MFH.
Despite these promising results and applications of genetic information, it is known that the information stored in the DNA alone is not sufficient to understand, and explain, the phenotypic appearance of an organism. The reason for this is that there are genotype-environment interactions that have an important influence on this as well (Falconer and Mackay, 1996; Lynch and Walsh, 1998). This means, usually, it is not possible to map a certain genotype uniquely to a phenotype. This genotype-environment interrelation is well know from genome-wide association studies (GWAS) and leads to a considerable increment in the complexity of the problem (Manolio et al., 2009) if one wants to apply genotype data in the medical and clinical practice, because one needs to control environmental factors. An example of environmental influences are given by mutagens. These physical or chemical agents have the ability to mutate the content of the DNA of an organism and, hence, are capable of changing the transcription of genes and the functioning of biological processes like DNA repair. Particular examples of mutagens are carcinogens, e.g., asbestos, formaldehyde, mustard gas or X-rays, that have been shown to have an influence on the development of cancer and its progression (Soffritti et al., 1989; Murthy and Testa, 1999; Hecht, 2003).
In addition to environmental influences that have an effect on the genetic information, there are epigenetic factors, e.g., DNA methylation (Ehrlich et al., 1982; Law and Jacobsen, 2010), that have also an important influence on the cell function and, hence, possibly on the phenotypic characteristics of an organism. For instance, the gene expression in normal and disease cells is known to be influenced by DNA methylation by controlling the protein-DNA binding (Richardson, 2002; Baylin, 2005; Robertson, 2005). Other examples for epigenetic factors are histone modifications and RNA interference (Egger et al., 2004; Moss and Wallrath, 2007; Ballestar and Esteller, 2008; Djupedal and Ekwall, 2009). So far it is largely unknown to what extend the epigenetic code contributes non-genetic factors to the regulation, control and maintenance of a cellular phenotype (Turner, 2007), although, within recent years important progress has been made (Dawson and Kouzarides, 2012; Sassone-Corsi et al., 2012).
A different problem in using genotype data alone for a medical application is that the DNA represents only static information about a cellular phenotype. This static information is stored in the form of nucleotide sequences representing putative programs, which may be activated under certain signaling, environmental or epigenetic conditions. That means for instance that mutations in coding or non-coding regions may or may not have an influence on the expression of genes or proteins in a particular cell type that effects the phenotype of an organism. Here, we would like to emphasize that the term “static” can also be interpreted as “passive,” because from the content of the DNA alone one cannot conclude on the activity level of its genes.
3. Limitations of Genotype Data
As a consequence of the heterogeneity induced by environmental and epigenetic factors, but also of the static nature of the DNA, there are limitations in the explanatory power of genotype data. Quantitatively, these limitations can be seen from the results of GWAS studies. Typically, GWAS studies lead only to a very small number of putative gene-associations with complex traits that are statistically significant (Sladek et al., 2007; Yeager et al., 2007). In contrast, there is usually a larger number of loci that is right below the significance threshold and, hence, these do not allow for definite conclusions. This implies that such studies suffer from a limited power and an increase is only possible by significantly increasing the number of the participating subjects (McCarthy et al., 2008). Unfortunately, this constitutes enormous practical problems for the organization and initiation of such studies and it cannot be expected that within the next few years larger studies with the required sample sizes are available which could potentially lead to the clarification of the causal involvement of genes in particular complex disorders. On a more fundamental note, we want to briefly remark that even if a locus is significantly associated with a phenotype it is not straight forward to identify the relevant gene(s) in the proximity of that locus that are implicated in the underlying disorder (Pearson and Manolio, 2008; Manolio, 2010). Further, even for significantly associated genes, their causal involvement in the explanation of a clinical phenotype is not guaranteed.
From a practical analysis perspective there is an additional problem provided by the many detectable events (variables) on the genotype-level, for instance in form of SNPs, CNVs or DNA-methylations that do not lead to actual consequences for, e.g., the survival rates of patients or other observable phenotype characteristics. This leads to a non-negligible amount of data that can be seen as genetic noise because it is distorting the analysis. Statistically, this constitutes non-trivial problems for the feature selection and dimension reduction of such data sets (Izenman, 2008; Clarke et al., 2009).
In Figure 1 we show a visualization of the general problem. If the measurement is limited to genotype data only, it is necessary to catalog all equivalent genetic, environmental and epigenetic variations because otherwise they may be mistakenly considered as different from each other and one would expect them leading to different phenotypes.
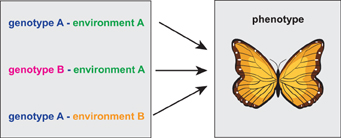
Figure 1. Schematic visualization of a situation when three different genotype-environment constellations lead to the same phenotype. Here it is important to note that the similarity of the three genotype-environment configurations can only be judged on the phenotype level of the organism.
4. Omics High-Throughput Data that Provide Dynamic Information
In order to obtain information about the activity of molecular and cellular programs as encoded in the DNA of an organism, it is necessary to measure entities that reflect these activity states appropriately. In this respect, the expression levels of genes or proteins provide valuable information to close this gap (Speed, 2003). For example, by using DNA microarray or next-generation sequencing technologies, gene expression and RNA-seq data can be obtained representing the abundance of mRNAs in a given sample (Wang et al., 2009). By comparison with different samples, e.g., taken from a normal or a control group of patients, it is possible to infer which genes are (statistically significant) expressed or not expressed (Ge et al., 2003; Storey and Tibshirani, 2003). This information can be a valuable surrogate for the activity of these genes in their underlying physiological conditions, provided by the samples. Such a comparison is not limited to individual genes, but can also be conducted for gene-sets or groups of genes that correspond for example to biological pathways; either defined by expert knowledge or databases like Gene Ontology or KEGG (Subramanian et al., 2005; Abatangelo et al., 2009; Emmert-Streib and Glazko, 2011; Tripathi and Emmert-Streib, 2012). In this way it is possible to enable a systems approach to medicine, acknowledging the fact that genes do not operate in isolation but function collectively in a variable manner (Ahn et al., 2006; Emmert-Streib et al., 2012b).
In order to distinguish “dynamic” from “static” Omics data that allow capturing dynamic aspects of the samples from patients, we suggest the following terminology.
DynOmics Data: Omics data that represent dynamic aspects of a molecular and cellular system by reflecting the activity level of genes and gene products.
Particular examples for dynOmics data are transcriptomics, proteomics and metabolomics data.
In Figure 2 we provide a summary of the connection between the genetic, genomic and phenotype level, as described in the previous sections. A direct mapping from the genotype to the phenotype, as indicated by the red arrow, could principally provide a shortcut in explaining for instance clinical patient characteristics. However, the danger is that this incurs problems by neglecting valuable information about the dynamic activity state of the cells, as represented, e.g., by the expression levels of genes or proteins. In other words, due to the static nature of genotype data this information should be seen as potential functional information about a patient, because information about the activity or usage of the diverse genetic programs is not captured by such data at all. Here by potential functional information we mean that the DNA is just a storage or a database of information (Noble, 2008) and this information is not indicative of the activity of the stored entities. For example, despite the fact that the CNV or the methylation of the DNA can change over the time of the evolution of a tumor, this does not say anything definite about the actual expression of the genes and, hence, their activation.
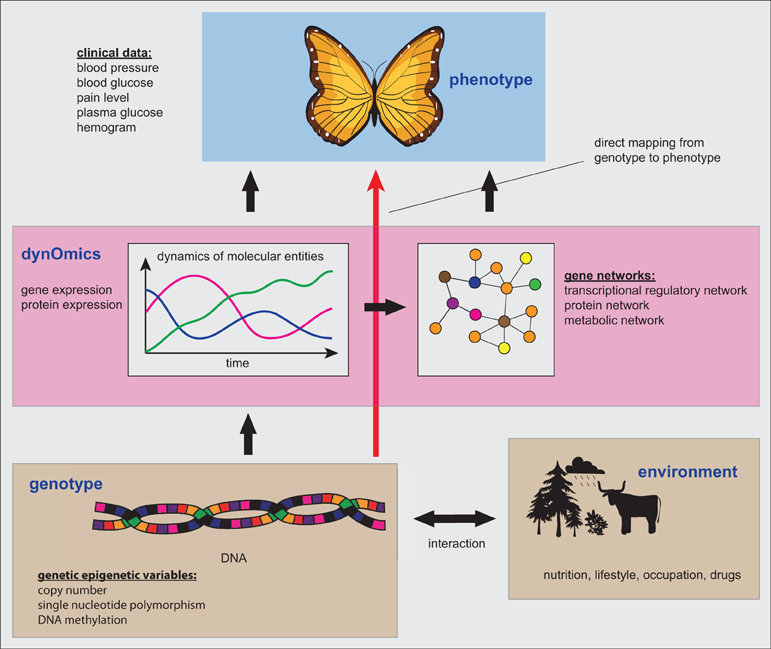
Figure 2. The connection between the genetic, genomic and phenotype level. Gene networks form one particular type of information that can be inferred from dynOmics data.
We would like to note that the consideration of dynamic high-throughput data, e.g., in the form of gene expression or proteomics data, does not only allow to identify differentially expressed genes or gene sets, but for sufficiently large samples sizes and variable sample conditions such data allow also to infer gene regulatory or protein networks (Belcastro et al., 2011; Emmert-Streib et al., 2012a; Emmert-Streib, 2013). These networks have the additional advantage of holding expedient clues for the molecular causes of the observed phenotypes (Emmert-Streib and Dehmer, 2011) that can be explored, e.g., by triggering follow-up experiments in the biomedical sciences. The difference to studies, e.g., utilizing DNA biomarkers to estimate the patient's disease risk (Ng et al., 2009) is that, e.g., regulatory networks provide direct insights into the molecular interaction structure of gene products (de Matos Simoes et al., 2013) and, hence, biological disease mechanisms on a level of detail that is absent in biomarker studies that are merely aiming to predict a phenotypic outcome. Furthermore, such networks can be utilized in identifying drug targets or drug mechanisms to extend traditional pharmacogenomics and pharmacodynamics approaches (Hopkins, 2008; Arrell and Terzic, 2010; Ghosh and Basu, 2012; Leung et al., 2012; Madhamshettiwar et al., 2012).
Finally, we just want to briefly mention that, strictly, there are two different types of dynOmics data that can be distinguish. The first type of dynOmics data contains explicit information about the temporal behavior of molecular entities as, e.g., provided by time series data of the concentration of mRNAs. In contrast, the second type of dynOmics data contains implicit information about the temporal behavior. An example for such dynOmics data are condition specific samples, e.g., from treatment and control patients. In the latter case, no time series (or longitudinal) data are available, yet, the data provide information about the activity level of genes in the form: “Is gene X active (expressed) or not.” For reasons of simplicity, we termed both types of dynamic information dynOmics data. However, as the above discussion indicates, a more refined subdivision is possible and, depending on the context, sensible.
5. Practical Exploitation of Dynomics Data
Finally, we discuss a couple of particular examples of approaches where dynOmics data sets have been utilized in disease diagnoses and personalized medicine.
In Huang et al. (2010) data from the public gene expression repository GENE EXPRESSION OMNIBUS (GEO), provided by the NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION (NCBI), have been utilized to construct a classifier for query expression profiles. Specifically, expression data from over 9000 microarray experiments have been gathered for 110 different disorders. These data sets have been used in combination with a Bayesian approach to learn a classifier for these 110 disease classes. This resulted in a method that allows to make (probabilistic) predictions about an unknown disease state as represented by a query expression profile that could be, e.g., obtained from a patient. Overall, the presented method has the capability to transform biological knowledge, as provided by the GEO database, into novel discoveries by means of the developed diagnoses tool.
For data providing information about the molecular interactions of proteins, as represented by protein interaction networks, similar approaches have been developed (Oti et al., 2006; Yang et al., 2011). For instance, in Wu et al. (2008) a method called CIPHER has been introduced that assumes that diseases with a similar phenotype are the effect of functionally related proteins that are close in a protein interaction network. In order to predict potential disease-genes based on phenotypic information about the disorder, CIPHER integrates two different types of data to define three different, connected parts. First, the ONLINE MENDELIAN INHERITANCE IN MAN (OMIM) database is used to estimate the similarity between disease phenotypes by using text mining tools. Further, OMIM is also used to obtain information about gene-phenotype associations. Second, in order to assess the functional similarity between proteins the human protein interaction network is used. Here it is important to emphasize that a protein interaction network provides information about the activity of proteins in the form of their interactions and, hence, represents a type of dynOmics data.
A seminal study that demonstrates impressively the advantages of using dynOmics data has been conducted in Chen et al. (2012). This study analyzed Omics profiles, comprising genetic, transcriptomic, proteomic, metabolomic and autoantibody profiles from a single individual measured over a period of 14 months. As a result, it has been particularly highlighted that the measurement of dynamic entities is crucial if one wants to make predictions about a patient that go beyond potential effects.
6. Conclusions
Genome-wide high-throughput technologies provide an unprecedented opportunity for systems medicine. The major purpose of this paper has been to advocate high-throughput data that provide dynamic information about cellular states, which we termed dynOmics data. However, we would like to emphasize that this does not mean that genotype data should not be used for systems and personalized medicine. Instead, the concern of the present paper is to balance the current trend in this field that might give the misleading impression that the usage of next-generation sequencing technologies to generate, e.g., DNA-seq data is the only way to achieve the translation from basic research to medical practice. Instead, we hypothesized that dynOmics data provide an indispensable source of information representing dynamic patient signatures that should be utilized for the complementation of genotype data.
Due to the fact that gene expression data and proteomics data contain a wealth of dynamic information that is per se not contained in genotype data, there are inherent limitations of approaches that are solely based on such data. Furthermore, potentially, dynOmics data may represent denoised information compared to sequence information, because the plurality of the genetic information is decided on the functional cellular and the phenotype level. Here it is important to distinguish between “data” and “information” to comprehend the meaning of denoised information. Whereas “data” refer only to the measured numbers, “information” implies a semantic biological content. For this reason the fact that DNA sequencing can be performed with a higher accuracy than, e.g., the measurement of the mRNA expression, does not contradict the observation that the uncertainly in the interpretation of the functional meaning of these numbers is generally reduced from the DNA to the mRNA and the protein level.
Another important point for future developments in personalized medicine would be the integration of different types of genotype and dynOmics data, e.g., from transcriptomics, proteomics, metabolomics and epigenomics experiments. However, in medical practise, we are far away from such a reality (Romero et al., 2006; Ostrowski and Wyrwicz, 2009; Chan and Ginsburg, 2011) and much more basic research is necessary before we can begin translating these results into practical patient care.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Ricardo de Matos Simoes, Shailesh Tripathi, Paul Mullan and Manuel Salto-Tellez for fruitful discussions. Funding: Matthias Dehmer thanks the Austrian Science Funds for supporting this work (project P22029-N13).
References
Abatangelo, L., Maglietta, R., Distaso, A., D'Addabbo, A., Creanza, T., Mukherjee, S., et al. (2009). Comparative study of gene set enrichment methods. BMC Bioinformatics 10:275. doi: 10.1186/1471-2105-10-275
Ahn, A. C., Tewari, M., Poon, C.-S., and Phillips, R. S. (2006). The limits of reductionism in medicine: could systems biology offer an alternative? PLoS Med. 3:e208. doi: 10.1371/journal.pmed.0030208
Alkan, C., Kidd, J. M., Marques-Bonet, T., Aksay, G., Antonacci, F., Hormozdiari, F., et al. (2009). Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 41, 1061–1067. doi: 10.1038/ng.437
Ansorge, W. (2009). Next-generation dna sequencing techniques. New Biotechnol. 25, 195–203. doi: 10.1016/j.nbt.2008.12.009
Arrell, D. K., and Terzic, A. (2010). Network systems biology for drug discovery. Clin. Pharmacol. Ther. 88, 120–125. doi: 10.1038/clpt.2010.91
Ballestar, E., and Esteller, M. (2008). “Chapter 9 epigenetic gene regulation in cancer,” in Long-Range Control of Gene Expression, Vol. 61 of Advances in Genetics, eds V. van Heyningen and R. E. Hill (Boston, MA: Academic Press), 247–267.
Baylin, S. B. (2005). Dna methylation and gene silencing in cancer. Nat. Clin. Pract. Oncol. 2(Suppl. 1), S4–S11. doi: 10.1038/ncponc0354
Belcastro, V., Siciliano, V., Gregoretti, F., Mithbaokar, P., Dharmalingam, G., Berlingieri, S. et al. (2011). Transcriptional gene network inference from a massive dataset elucidates transcriptome organization and gene function. Nucleic Acids Res. 39, 8677–8688. doi: 10.1093/nar/gkr593
Beroukhim, R., Mermel, C. H., Porter, D., Wei, G., Raychaudhuri, S., Donovan, J., et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. doi: 10.1038/nature08822
Butte, A. J. (2008). Translational bioinformatics: coming of age. J. Am. Med. Inf. Assoc. 15, 709–714. doi: 10.1197/jamia.M2824
Chan, I. S., and Ginsburg, G. S. (2011). Personalized medicine: progress and promise. Annu. Rev. Genomics Hum. Genet. 12, 217–244. doi: 10.1146/annurev-genom-082410-101446
Chen, R., Mias, G. I., Li-Pook-Than, J., Jiang, L., Lam, H. Y., Chen, R., et al. (2012). Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 148, 1293–1307. doi: 10.1016/j.cell.2012.02.009
Chin, L., Andersen, J. N., and Futreal, P. A. (2011). Cancer genomics: from discovery science to personalized medicine. Nat. Med. 17, 297–303. doi: 10.1038/nm.2323
Clarke, B., Fokoue, E., and Zhang, H. H. (2009). Principles and Theory for Data Mining and Machine Learning. Dordrecht; New York: Springer. doi: 10.1007/978-0-387-98135-2
Collins, F. S., Guyer, M. S., and Chakravarti, A. (1997). Variations on a theme: cataloging human dna sequence variation. Science 278, 1580–1581. doi: 10.1126/science.278.5343.1580
Consortium, International Human Genome Sequencing (2004). Finishing the euchromatic sequence of the human genome. Nature 431, 931–945. doi: 10.1038/nature03001
Dawson, M. A., and Kouzarides, T. (2012). Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27. doi: 10.1016/j.cell.2012.06.013
de Matos Simoes, R., Dehmer, M., and Emmert-Streib, F. (2013). Interfacing cellular networks of S. cerevisiae and E. coli: connecting dynamic and genetic information. BMC Genomics 14:324. doi: 10.1186/1471-2164-14-324
Djupedal, I., and Ekwall, K. (2009). Epigenetics : heterochromatin meets RNAi. Cell Res. 19, 282–295. doi: 10.1038/cr.2009.13
Egger, G., Liang, G., Aparicio, A., and Jones, P. A. (2004). Epigenetics in human disease and prospects for epigenetic therapy. Nature 429, 457–463. doi: 10.1038/nature02625
Ehrlich, M., Gama-Sosa, M. A., Huang, L.-H., Midgett, R. M., Kuo, K. C., McCune, R. A., et al. (1982). Amount and distribution of 5-methylcytosine in human dna from different types of tissues or cells. Nucleic Acids Res. 10, 2709–2721. doi: 10.1093/nar/10.8.2709
Emmert-Streib, F. (2013). Influence of the experimental design of gene expression studies on the inference of gene regulatory networks: environmental factors. PeerJ 1, e10. doi: 10.7717/peerj.10
Emmert-Streib, F., and Dehmer, M. (2011). Networks for systems biology: conceptual connection of data and function. IET Syst. Biol. 5, 185. doi: 10.1049/iet-syb.2010.0025
Emmert-Streib, F., and Glazko, G. (2011). Pathway analysis of expression data: deciphering functional building blocks of complex diseases. PLoS Comput. Biol. 7:e1002053. doi: 10.1371/journal.pcbi.1002053
Emmert-Streib, F., Glazko, G., Altay, G., and de Matos Simoes, R. (2012a). Statistical inference and reverse engineering of gene regulatory networks from observational expression data. Front. Genet. 3:8. doi: 10.3389/fgene.2012.00008
Emmert-Streib, F., Tripathi, S., and de Matos Simoes, R. (2012b). Harnessing the complexity of gene expression data from cancer: from single gene to structural pathway methods. Biol. Dir. 7, 44. doi: 10.1186/1745-6150-7-44
Falconer, D. S., and Mackay, T. F. C. (1996). Introduction to Quantitative Genetics. Harlow: Longmans Green.
Fernald, G. H., Capriotti, E., Daneshjou, R., Karczewski, K. J., and Altman, R. B. (2011). Bioinformatics challenges for personalized medicine. Bioinformatics 27, 1741–1748. doi: 10.1093/bioinformatics/btr295
Förster, J., Famili, I., Fu, P., Palsson, B., and Nielsen, J. (2003). Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res. 13, 244–253. doi: 10.1101/gr.234503
Freeman, J. L., Perry, G. H., Feuk, L., Redon, R., McCarroll, S. A., Altshuler, D. M., et al. (2006). Copy number variation: new insights in genome diversity. Genome Res. 16, 949–961. doi: 10.1101/gr.3677206
Garbett, K., Ebert, P. J., Mitchell, A., Lintas, C., Manzi, B., Mirnics, K., et al. (2008). Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobio. Dis. 30, 303–311. doi: 10.1016/j.nbd.2008.01.012
Ge, Y., Dudoit, S., and Speed, T. (2003). Resampling-based multiple testing for microarray data analysis. TEST 12, 1–77. doi: 10.1007/BF02595811
Ghosh, D., and Poisson, L. M. (2009). ‘Omics’ data and levels of evidence for biomarker discovery. Genomics 93, 13–16. doi: 10.1016/j.ygeno.2008.07.006
Ghosh, S., and Basu, A. (2012). Network medicine in drug design: implications for neuroinflammation. Drug Discov. Today 17, 600–607. doi: 10.1016/j.drudis.2012.01.018
Hecht, S. S. (2003). Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat. Rev. Cancer 3, 733–744. doi: 10.1038/nrc1190
Highnam, G., and Mittelman, D. (2012). Personal genomes and precision medicine. Genome Biol. 13, 324. doi: 10.1186/gb-2012-13-12-324
Hopkins, A. L. (2008). Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 4, 682–690. doi: 10.1038/nchembio.118
Huang, H., Liu, C.-C., and Zhou, X. J. (2010). Bayesian approach to transforming public gene expression repositories into disease diagnosis databases. Proc. Natl. Acad. Sci. U.S.A. 107, 6823–6828. doi: 10.1073/pnas.0912043107
Izenman, A. J. (2008). Modern Multivariate Statistical Techniques. New York, NY: Springer. doi: 10.1007/978-0-387-78189-1
Kresse, S. H., Ohnstad, H. O., Bjerkehagen, B., Myklebost, O., and Meza-Zepeda, L. A. (2010). DNA copy number changes in human malignant fibrous histiocytomas by array comparative genomic hybridisation. PLoS ONE 5:e15378. doi: 10.1371/journal.pone.0015378
LaFramboise, T. (2009). Single nucleotide polymorphism arrays: a decade of biological, computational and technological advances. Nucleic Acids Res. 37, 4181–4193. doi: 10.1093/nar/gkp552
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. doi: 10.1038/35057062
Law, J. A., and Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220. doi: 10.1038/nrg2719
Lee, T. I., Rinaldi, N. J., Robert, F., Odom, D. T., Bar-Joseph, Z., Gerber, G. K., et al. (2002). Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298, 799–804. doi: 10.1126/science.1075090
Leung, E. L., Cao, Z.-W., Jiang, Z.-H., Zhou, H., and Liu, L. (2012). Network-based drug discovery by integrating systems biology and computational technologies. Brief. Bioinform. 14, 491–505. doi: 10.1093/bib/bbs043
Lussier, Y. A., Butte, A. J., and Hunter, L. (2010). Guest editorial: current methodologies for translational bioinformatics. J. Biomed. Inform. 43, 355–357. doi: 10.1016/j.jbi.2010.05.002
Madhamshettiwar, P., Maetschke, S., Davis, M., Reverter, A., and Ragan, M. (2012). Gene regulatory network inference: evaluation and application to ovarian cancer allows the prioritization of drug targets. Genome Med. 4, 41. doi: 10.1186/gm340
Manolio, T. A. (2010). Genomewide association studies and assessment of the risk of disease. New Eng. J. Med. 363, 166–176. doi: 10.1056/NEJMra0905980
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/nature08494
Mardis, E. R. (2008). Next-generation dna sequencing methods. Annu. Rev. Genomics Hum. Genet. 9, 387–402. doi: 10.1146/annurev.genom.9.081307.164359
McCarroll, S. A., and Altshuler, D. M. (2007). Copy-number variation and association studies of human disease. Nat. Genet. 39(Suppl. 7), S37–S42. doi: 10.1038/ng2080
McCarthy, M., Abecasis, G., Cardon, L., Goldstein, D., Little, J., Ioannidis, J., et al. (2008). Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 9, 356–369. doi: 10.1038/nrg2344
Metzker, M. (2009). Sequencing technologies-the next generation. Nat. Rev. Genet. 11, 31–46. doi: 10.1038/nrg2626
Moreno-Risueno, M. A., Busch, W., and Benfey, P. N. (2010). Omics meet networks - using systems approaches to infer regulatory networks in plants, Curr. Opin. Plant Biol. 13, 126–131. doi: 10.1016/j.pbi.2009.11.005
Moss, T. J., and Wallrath, L. L. (2007). Connections between epigenetic gene silencing and human disease. Mutat. Res. Fundam. Mol. Mech. Mutagen. 618, 163–174. doi: 10.1016/j.mrfmmm.2006.05.038
Murthy, S. S., and Testa, J. R. (1999). Asbestos, chromosomal deletions, and tumor suppressor gene alterations in human malignant mesothelioma. J. Cell. Physiol. 180, 150–157. doi: 10.1002/(SICI)1097-4652(199908)180:2<150::AID-JCP2>3.0.CO;2-H
Ng, P. C., Murray, S. S., Levy, S., and Venter, J. C. (2009). An agenda for personalized medicine. Nature 461, 724–726. doi: 10.1038/461724a
Noble, D. (2008). Genes and causation. Phil. Trans. R. Soc. A 366, 3001–3015. doi: 10.1098/rsta.2008.0086
Ostrowski, J., and Wyrwicz, L. S. (2009). Integrating genomics, proteomics and bioinformatics in translational studies of molecular medicine. Exp. Rev. Mol. Diagn. 9, 623–630. doi: 10.1586/erm.09.41
Oti, M., Snel, B., Huynen, M., and Brunner, H. (2006). Predicting disease genes using protein–protein interactions. J. Med. Genet. 43, 691–698. doi: 10.1136/jmg.2006.041376
Palsson, B. (2006). Systems Biology, Cambridge; New York: Cambridge University Press. doi: 10.1017/CBO9780511790515
Pearson, T., and Manolio, T. (2008). How to interpret a genome-wide association study. JAMA 299, 1335–1344. doi: 10.1001/jama.299.11.1335
Pinto, D., Darvishi, K., Shi, X., Rajan, D., Rigler, D., Fitzgerald, T., et al. (2011). Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat. Biotech. 29, 512–520. doi: 10.1038/nbt.1852
Richardson, B. C. (2002). Role of dna methylation in the regulation of cell function: autoimmunity, aging and cancer. J. Nutr. 132, 2401S–2405S.
Robertson, K. D. (2005). DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610. doi: 10.1038/nrg1655
Romero, R., Espinoza, J., Gotsch, F., Kusanovic, J., Friel, L., Erez, O., et al. (2006). The use of high-dimensional biology (genomics, transcriptomics, proteomics, and metabolomics) to understand the preterm parturition syndrome. BJOG Int. J. Obstet. Gynaecol. 113, 118–135. doi: 10.1111/j.1471-0528.2006.01150.x
Rual, J.-F., Venkatesan, K., Hao, T., and Hirozane-Kishikawa, T. (2005). Towards a proteome-scale map of the human protein-protein interaction network. Nature 437, 1173–1178. doi: 10.1038/nature04209
Sachidanandam, R., Weissman, D., Schmidt, S. C., Kakol, J. M., Stein, L. D., Marth, G., et al. (2001). A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409, 928–933. doi: 10.1038/35057149
Sassone-Corsi, P., Imhof, A., and Katada, S. (2012). Connecting threads: epigenetics and metabolism. Cell 148, 24–28. doi: 10.1016/j.cell.2012.01.001
Sechi, S., (ed). (2007). Quantitative Proteomics by Mass Spectrometry. (Totowa, NJ: Humana Press). doi: 10.1007/978-1-59745-255-7
Shendure, J., and Ji, H. (2008). Next-generation dna sequencing. Nat. Biotechnol. 26, 1135–1145. doi: 10.1038/nbt1486
Sladek, R., Rocheleau, G., Rung, J., Dina, C., Shen, L., Serre, D., et al. (2007). A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445, 881–885. doi: 10.1038/nature05616
Soffritti, M., Maltoni, C., Maffei, F., and Biagi, R. (1989). Formaldehyde: an experimental multipotential carcinogen. Toxicol. Indust. Health 5, 699–730. doi: 10.1177/074823378900500510
Speed, T. (2003). Statistical Analysis of Gene Expression Microarray Data. Boca Raton, FL: Chapman and Hall/CRC. doi: 10.1201/9780203011232
Stelzl, U., Worm, U., Lalowski, M., Haenig, C., Brembeck, F., Goehler, H., et al. (2005). A human protein-protein interaction network: a resource for annotating the proteome. Cell 122, 957–968. doi: 10.1016/j.cell.2005.08.029
Stepanov, V. A. (2010). Genomes, populations and diseases: ethnic genomics and personalized medicine. Acta Nat. 2, 15–30. Available online at: http://www.ncbi.nlm.nih.gov/pubmed/?term=%22Genomes%2C+populations+and+diseases%3A+ethnic+genomics+and+653+personalized+medicine%22
Stephens, P. J., McBride, D. J., Lin, M.-L., Varela, I., Pleasance, E. D., Simpson, J. T., et al. (2009). Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 462, 1005–1010. doi: 10.1038/nature08645
Storey, J., and Tibshirani, R. (2003). Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 100, 9440–9445. doi: 10.1073/pnas.1530509100
Subramanian, A., Tamayo, P., Mootha, V., Mukherjee, S., Ebert, B., Gillette, M., et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550. doi: 10.1073/pnas.0506580102
The ENCODE Project Consortium (2011). A user's guide to the encyclopedia of DNA elements (eNCODE). PLoS Biol. 9:e1001046. doi: 10.1371/journal.pbio.1001046
Tripathi, S., and Emmert-Streib, F. (2012). Assessment method for a power analysis to identify differentially expressed pathways. PLoS ONE 7:e37510. doi: 10.1371/journal.pone.0037510
Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G., et al. (2001). The sequence of the human genome. Science 291, 1304–1351. doi: 10.1126/science.1058040
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Welch, B. M., and Kawamoto, K. (2012). Clinical decision support for genetically guided personalized medicine: a systematic review. J. Am. Med. Inform. Assoc. 20, 388–400. doi: 10.1136/amiajnl-2012-000892
Werner, T. (2010). Next generation sequencing in functional genomics. Brief. Bioinformat. 11, 499–511. doi: 10.1093/bib/bbq018
Wheeler, D. L., Barrett, T., Benson, D. A., Bryant, S. H., Canese, K., Chetvernin, V., et al. (2007). Database resources of the national center for biotechnology information. Nucleic Acids Res. 35(Suppl. 1), D5–D12. doi: 10.1093/nar/gkl1031
Wu, X., Jiang, R., Zhang, M. Q., and Li, S. (2008). Network-based global inference of human disease genes. Mol. Syst. Biol. 4, 189. doi: 10.1038/msb.2008.27
Yang, P., Li, X., Wu, M., Kwoh, C.-K., and Ng, S.-K. (2011). Inferring gene-phenotype associations via global protein complex network propagation, PLoS ONE 6:e21502. doi: 10.1371/journal.pone.0021502
Yeager, M., Orr, N., Hayes, R. B., Jacobs, K. B., Kraft, P., Wacholder, S., et al. (2007). Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat. Genet. 39, 645–649. doi: 10.1038/ng2022
Yu, H., Braun, P., Yildirim, M. A., Lemmens, I., Venkatesan, K., Sahalie, J., et al. (2008). High-quality binary protein interaction map of the yeast interactome network. Science 322, 104–110. doi: 10.1126/science.1158684
Zhang, J., Chiodini, R., Badr, A., and Zhang, G. (2011). The impact of next-generation sequencing on genomics. J. Genet. Genomics 38, 95–109. doi: 10.1016/j.jgg.2011.02.003
Keywords: genome medicine, personalized medicine, next-generation sequencing data, dynOmics data, high-throughput data
Citation: Emmert-Streib F and Dehmer M (2013) Enhancing systems medicine beyond genotype data by dynamic patient signatures: having information and using it too. Front. Genet. 4:241. doi: 10.3389/fgene.2013.00241
Received: 11 September 2013; Accepted: 24 October 2013;
Published online: 19 November 2013.
Edited by:
Galina Glazko, University of Arkansas for Medical Sciences, USAReviewed by:
Galina Glazko, University of Arkansas for Medical Sciences, USADov J. Stekel, University of Nottingham, UK
Copyright © 2013 Emmert-Streib and Dehmer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank Emmert-Streib, Computational Biology and Machine Learning Laboratory, Faculty of Medicine, Health and Life Sciences, Center for Cancer Research and Cell Biology, School of Medicine, Dentistry and Biomedical Sciences, Queen's University Belfast, 97 Lisburn Road, Belfast, BT9 7BL, UK e-mail: v@bio-complexity.com