 Karine Tremblay
Karine Tremblay Camélia Dubois-Bouchard
Camélia Dubois-Bouchard Diane Brisson
Diane Brisson Daniel Gaudet
Daniel Gaudet- 1Department of Medicine, Université de Montréal, Montreal, Canada
- 2ECOGENE-21 Clinical Research Center, Saguenay, QC, Canada
Background: There are important inter-individual variations in the incidence and severity of acute pancreatitis in patients with severe hypertriglyceridemia. Several genes involved in triglyceride-rich lipoprotein metabolism or serine proteases pathways are known to influence the risk of pancreatitis.
Aim: To evaluate the association between genes regulating serine proteases, chymotrypsin C (CTRC) and serine peptidase inhibitor kazal type1 (SPINK1), and recurrence of hospitalizations for acute pancreatitis or severe abdominal pain in patients with Lipoprotein Lipase Deficiency (LPLD), a rare and extreme monogenic model of severe hypertriglyceridemia and pancreatitis.
Method: The CTRC and SPINK1 genes promoter and coding regions sequencing has been performed in a sample of 38 LPLD adults (22 men and 16 women) and 100 controls (53 men and 47 women). Estimation of the association of CTRC and SPINK1 gene variants or combinations of variants with history of hospitalizations for pancreatitis or acute abdominal pain in LPLD was investigated using non-parametric analyses with correction for multiple testing and logistic regression models controlling for age, gender, family history, and life habits.
Results: Gene sequencing followed by genotype-stratified analyses of the CTRC and SPINK1 genes in LPLD and controls revealed a positive association between recurrence of hospitalizations and the rs545634 (CTRC)—rs11319 (SPINK1) combination [OR = 41.4 (CI: 2.0–848.0); p = 0.016]. In all models, a positive family history of pancreatitis was a significant predictor of recurrent hospitalizations independently of the contribution of SPINK1 or CTRC (p < 0.001).
Conclusion: These results suggest that a positive family history of pancreatitis and genetic markers in the serine protease pathways could be associated with a risk of recurrent hospitalization for acute pancreatitis in severe hypertriglyceridemia due to LPLD.
Introduction
Very severe hypertriglyceridemia (defined as fasting plasma TG concentration >10 mmol/L or 900 mg/dl) increases the risk of acute pancreatitis and has a prevalence of approximately 1/600 in North America (Johansen et al., 2011). Both severe hypertriglyceridemia and acute pancreatitis are associated with important clinical and socio-economic burden (Gaudet et al., 2013). However, there are important inter-individual variations in the incidence and severity of acute pancreatitis in patients with severe hypertriglyceridemia. Although rare, some extreme forms of severe hypertriglyceridemia and pancreatitis risk exist. This is the case of Lipoprotein Lipase Deficiency (LPLD) [MIM: 238600—Familial hyperchylomicronemia]. LPLD is a rare monogenic disease transmitted on an autosomal recessive mode (Monsalve et al., 1990; Ma et al., 1991; Mattei et al., 1993; Brunzell and Deeb, 2001). It is associated with recurrent, severe abdominal pain, increased risk of acute pancreatitis and other morbidities such as pulmonary embolism-like syndrome, coronary heart disease with or without atherosclerosis, and metabolic consequences of pancreatic insufficiency, including insulinopenic diabetes (Brunzell and Deeb, 2001; Tremblay et al., 2011). Although rare, LPLD is the most common form of the Familial Chylomicronemia Syndrome (FCS). The estimated worldwide prevalence of LPLD is 1–2:1,000,000 (Fredrickson et al., 1978). Numerous loss-of-function lipoprotein lipase gene (LPL) [MIM:609708] mutations were identified, but only a small number are null alleles, recognized to be responsible to the markedly reduced or absent lipoprotein lipase activity observed in LPLD (Brunzell and Deeb, 2001). LPLD results in chylomicrons accumulation and severe hypertriglyceridemia (>10 mmol/L) in the fasting state, and is associated with characteristic clinical signs such as eruptive xanthomas, lipaemia retinalis, and hepatosplenomegaly (Brunzell and Deeb, 2001). Other causes of FCS are documented, such as apolipoprotein C-II (APOC2), glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1), lipase maturation factor 1 (LMF1) or apolipoprotein A-V (APOA5) gene deficiencies, which all result in functional LPLD (Surendran et al., 2012). Currently, no available lipid-lowering drug therapy is effective to control the risk associated with LPLD, and affected patients typically do not respond to fibrates or niacin (Brisson et al., 2010; Zhou and Sahin-Toth, 2011; Beer et al., 2012). LPLD, like all other causes of FCS, is predominantly treated by severe dietary fat restriction and the use of medium-chain triglycerides (MCT) (Brunzell and Deeb, 2001), which does not fully eliminate the risk of pancreatitis or disease progression, while interfering with patient's quality of life on a daily basis. Recently, LPL gene replacement therapy (Gaudet et al., 2010, 2012) has been approved in Europe for severely affected LPLD patients. New emerging classes of treatments are in clinical development for FCS, including (among others) DGAT-1 inhibitors (Meyers et al., 2012), apoC3 antisense therapy, MTP inhibitors (Sacks et al., 2013) and peptide linker technologies.
As in many Mendelian diseases, important variations are noted in the clinical expression of LPLD. In particular, the incidence and the severity of abdominal pain and acute pancreatitis vary importantly across an individual's lifespan and between affected individuals, even in the same family. Although the pancreatitis risk is extremely high in LPLD (Tremblay et al., 2011), some patients do not experience a crisis before advanced age while others were frequently hospitalized since young age or eventually die from complications. Such heterogeneity in LPLD phenotypic expression complicates the individualization of risk evaluation and stratification.
The genetic basis of pancreatitis is now well documented and numerous candidate genes have been reported (Chen and Ferec, 2009; Whitcomb, 2010, 2013). All these genes could theoritically influence the trajectory of pancreatitis risk in LPLD. The variability of pancreatitis expression in LPLD is obviously complex and can also be affected by several environmental factors and by epigenetic regulatory mechanisms such as methylation, and/or microRNA (Jiang et al., 2004; Chahwan et al., 2011). In this study, we used a candidate gene approach to evaluate the association between the recurrence of hospitalization for acute pancreatitis or severe abdominal pain in LPLD and genes known to contribute to pancreatitis susceptibility. Specifically, we have selected the chymotrypsinogen C (CTRC) and serine protease inhibitor Kazal-type 1 (SPINK1) genes based on their important physiological role in serine proteases regulation and because they might have a functional relation with LPLD through the high density lipoprotein (HDL) particle which is significantly affected in this disease. In particular, the HDL cholesterol concentration, and the number of HDL particles are significantly decreased in LPLD. HDL is an efficient carrier of alpha(1)-antitrypsin and has been reported to play a significant role in proteases inhibition.
Materials and Methods
Subjects
A total of 138 Caucasian adults have participated in this study: 100 normolipemic controls and 38 genetically confirmed patients with LPLD. All LPLD patients were identical-by-descent (IBD) homozygotes or compound heterozygous for null alleles in the LPL gene and presented fasting plasma TG values >10 mmol/L and clinical characteristics of FCS (Gaudet et al., 1998; Gagné and Gaudet, 2007; Tremblay et al., 2011). Null LPL alleles are associated with <5% normal LPL activity. Fasting plasma TG, cholesterol, non-esterified fatty acids, and apolipoprotein B (apo B) were measured as previously described (St-Pierre et al., 2001). Among the 38 LPLD participants, 18 had a history of recurrent (≥5) hospitalizations for severe abdominal pain or acute pancreatitis whereas 8 had not yet been hospitalized for such condition. Overall, approximately 80% of LPLD patients had already suffered from at least one episode of pancreatitis or abdominal pain requiring hospitalization and the number of hospitalizations per individual ranged between 0 and 96. Calculation of the number of hospitalizations for pancreatitis or severe abdominal pain was performed by questionnaire and by reviewing patient's medical charts. Applying the criteria of the Atlanta classification for pancreatitis (Banks et al., 2013), the hospitalizations having been considered in this study group all definite, probable and suspected pancreatitis episodes. Hospitalizations for conditions clearly not suspected to be related to FCS (eg., appendicectomy, parietal pain) were not considered in the analyses. The family history was considered “positive” when at least one known relative (available in our database) has already been hospitalized for acute pancreatitis. All LPLD patients have been followed in a Lipid Clinic, received nutritional counseling and were prescribed a strict type-1 adapted low fat diet. This study has been approved by the Local Ethics Review Board in accordance with the Declaration of Helsinki and all subjects gave informed consent.
CTRC and SPINK1 Sequencing
DNA has been extracted from blood leukocytes of all participants using Qiagen kit and following manufacturer's instruction (Qiagen Inc., Valencia, CA, USA). CTRC [1p36.21—GeneID: 11330] and SPINK1 [5q32—GeneID: 6690] sequences information were obtained from UCSC Genome Browser (http://genome.ucsc.edu/, University of California, Santa Cruz, CA, USA). For CTRC, the PCR was divided into seven regions that spanned 3.1 kb, covering all eight coding and promoter sequences (Figure 1). For SPINK1, the PCR was divided into four regions that spanned 2.2 kb, covering all four coding and promoter regions (Figure 1). Analysis of sequences were performed with CodonCode Aligner (http://www.codoncode.com/aligner/, CodonCode Corporation Dedham, MA, USA). Oligonucleotide sequences as well as PCR amplification specific annealing temperatures are listed in the Supplementary Table 1. The PCR reactions were done in 25 μL volume containing 0–1 mM of Magnesium Chloride, 1X of Qiagen HotStart Taq PCR Buffer or 1X of NEB Taq PCR Buffer, 0.25 mM of dNTPs, 1 U/reaction of HotStart Taq DNA polymerase (Qiagen, Valencia, CA, USA or New England Biolabs Inc., Ipswich, Ma, USA), 0.2 μM of each primer and 20 ng/μl human genomic DNA. The PCR amplification was performed at 95°C for 10 min, 35 cycles of: first, 94°C during 30 s, secondly, annealing temperature for 30 s and thirdly, 72°C for 30 s. Finally, one cycle at 72°C for 7 min. Amplification products were purified with multiscreen PCR plates (MSNU 030 PCR, Millipore Corporation, Bedford, Ma, USA). Sequencing reactions were done in 10 μL volume containing 2 μL of purified and diluted PCR product, 0.3 μL of BigDye terminator v1.1 (Applied Biosystem Inc., Foster City, CA, USA), 1.75 μL of BigDye Terminator Sequencing Buffer 5X (Applied Biosystem Inc., Foster City, CA, USA) and 0.5 μL of the forward or reverse PCR primer (Supplementary Table 1). The sequencing reaction cycles were done following manufacturer's instructions (BigDye® Terminator v1.1 Cycle Sequencing Kit). Reaction sequences were precipitated in 3 M of sodium acetate (Ambion®, Foster City, CA, USA) and 95% ethanol, resuspended in 10 μL of Hi-Di™ Formamide (Applied Biosystem Inc., Foster City, CA, USA) and analyzed on ABI PRISM 3100 Genetic Analyzer (Applied Biosystem Inc.).
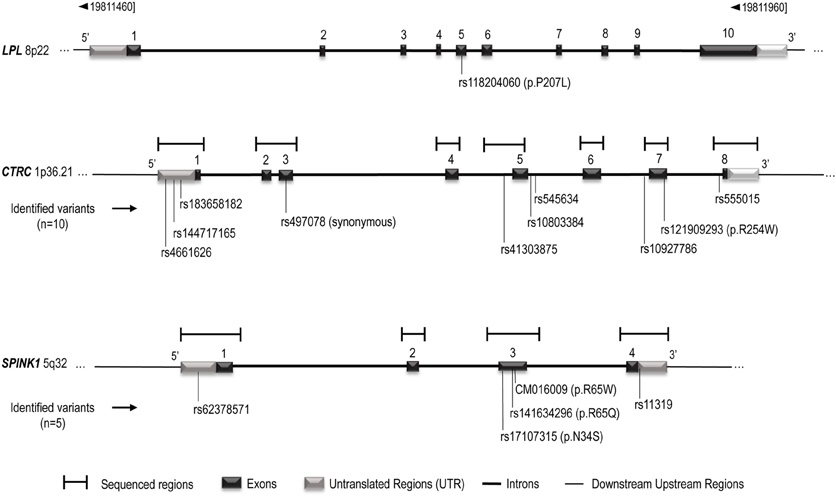
Figure 1. Schematic representation of the LPL, CTRC, and SPINK1 genes structure. The gene representation is based on UCSC Genome Browser (http://genome.ucsc.edu/, February 2009, Santa Cruz, CA, USA) and Ensembl (http://uswest.ensembl.org/index.html, september 2013, Cambridge, United Kingdom), and is not scaled. CTRC and SPINK1 sequenced regions and identified variants are indicated. Complementary informations on sequenced regions are avaible in Supplementary Table 1.
Statistical Analyses
All statistical analyses were performed using SPSS software (IBM Corporation, SPSS Statistics for Windows, Versions 11.5.0, and 21.0, Armonk, NY, USA). Continuous variable comparisons between the different phenotypes were done using Kruskal-Wallis non-parametric tests combined to Mann-Whitney tests to perform inter-group comparisons. P-values are reported after Bonferroni correction for multiple testing. Chi-Square tests were used to compare dichotomic variables as well as minor allele frequencies and genotypes distribution between studied groups, and z-test was performed to inter-group comparisons. P-values were also adjusted using the Bonferroni method. For interaction analyses, significance level was fixed at p ≤ 0.005 since we have tested 10 SNPs combinations. Estimation of the association of CTRC and SPINK1 variants or combinations with recurrent (≥5) hospitalizations for pancreatitis or acute abdominal pain in LPLD was also investigated using multinominal logistic regression models controlling for age, gender, and smoking status. Subjects with known alcohol abuse or known to be non-compliant to the diet were excluded from the study. In regression models, comparisons were made using normolipidemic controls as the reference. Results are reported for the groups of LPLD patients with ≥5 or <5 hospitalizations as odds ratio (OR) and 95% confidence interval (CI) at a significance level fixed at p < 0.05.
Results
The characteristics of the two LPLD groups (defined on the basis of the number of hospitalizations for pancreatitis or abdominal pain) and controls are presented in Table 1. LPLD patients had significantly higher total triglyceride and cholesterol levels, and lower LDL-C and HDL-C than controls. Both LPLD groups were comparable for other lipid-lipoprotein variables, including total cholesterol, HDL-C, non-esterified fatty acids and total apolipoprotein B. LPLD subjects with recurrent hospitalizations were older, in addition to present a higher proportion of smokers than controls. They also presented a greater proportion of positive family history of pancreatitis than controls and LPLD subjects with <5 hospitalizations. All groups were comparable for body mass index.
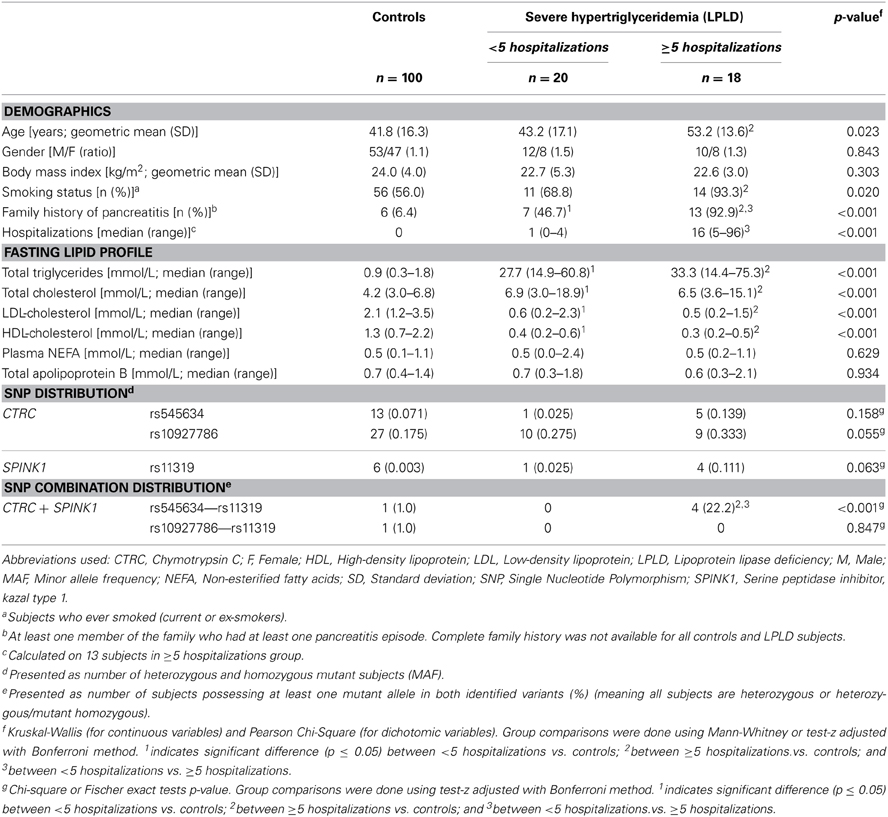
Table 1. Characteristics of the Participants.
Table 1 also presents the compared frequency of the CTRC and SPINK1 variants showing an association with recurrent hospitalizations for pancreatitis or acute abdominal pain while Table 2 presents the characteristics of all CTRC and SPINK1 variants (n = 15) identified through gene sequencing in the different groups. All identified variants are single nucleotide polymorphisms (SNP) located in coding, non-coding or untranslated regions (Table 2 and Figure 1). The Supplementary Table 2 presents the associations in more details. Minor allele frequency comparisons between the studied groups revealed a trend of association with recurrent pancreatitis for only three SNPs (p-values < 0.2) (Table 1). Of note, among the eight LPLD individuals who had never been hospitalized for pancreatitis, the rs545634 was not observed while three heterozygous were observed for rs10927786 and one heterozygous for rs11319. We then performed two-by-two interaction analyses among the associated SNPs (Table 1). At a significance level of 0.005, only one SNP combination, CTRC (rs545634) and SPINK1 (rs11319), showed a positive association after correction for multiple testing (p < 0.001). Genotype-stratified analyses confirmed that all subjects carrying this combination were heterozygotes (p < 0.001). We have then evaluated the association of this CTRC (rs545634) and SPINK1 (rs11319) combination with recurrent pancreatitis in logistic regression analysis, including age, gender and smoking status as covariates (Table 3). Results of multivariate analyses confirmed the association of the rs545634-rs11319 SNPs combination with an increased odd of recurrent hospitalizations for acute pancreatitis or severe abdominal pain (OR = 41.4, p = 0.016). A positive family history of pancreatitis was also a significant covariate of recurrent hospitalizations in all groups (p < 0.001). When the family history was included in the regression models, the contribution of SPINK1-CTRC combination to the risk of hospitalization for pancreatitis or abdominal pain remained significant (p = 0.032).
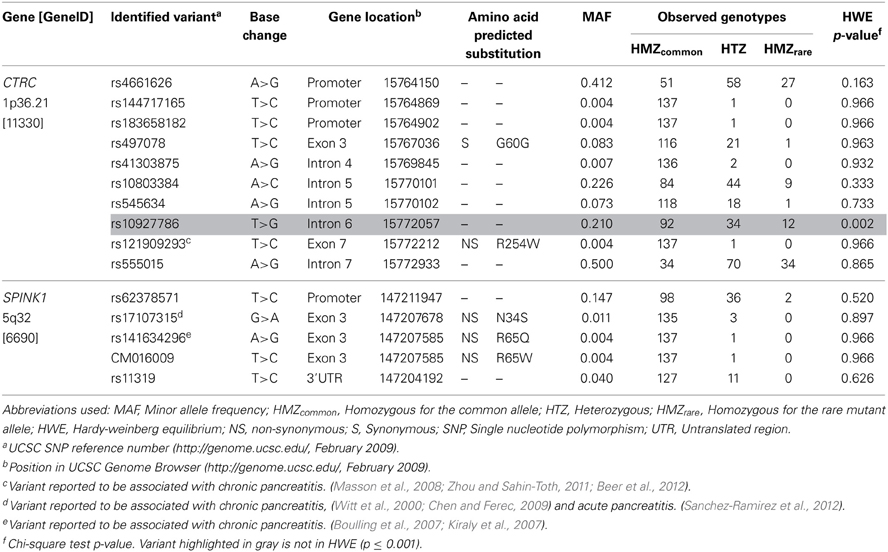
Table 2. Genetic Characteristics of the CTRC and SPINK1 Variants Identified.
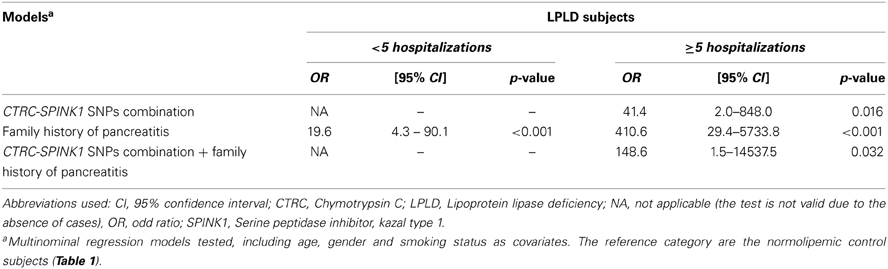
Table 3. Association of CTRC and SPINK1 SNPs Combination and Family History with Recurrence (≥5) of Hospitalizations for Pancreatitis in Severe Hypertriglyceridemia due to LPLD.
Discussion
Both pancreatitis and hypertriglyceridemia are complex traits and inter-individual variations are regularly observed in the expression of pancreatitis in patients with severe hypertriglyceridemia. Such differences are noticed even in a homogeneous group of individuals affected by LPLD, a disease characterized by extreme plasma triglyceride values and high risk of pancreatitis (Brunzell and Deeb, 2001; Tremblay et al., 2011). LPLD is a very rare disease and we thus report data on one of the largest sample of LPLD patients having been ever published. In this sample, we observed important variations in term of pancreatitis morbidity (assessed by the recurrence of hospitalizations), despite the fact that all patients were carrying the same LPL gene defect. Indeed, approximately 80% of LPLD adults in this study had been hospitalized at least once for acute pancreatitis (range: 1–96) but 20% had never suffered from pancreatitis, even at age >50. The results presented herein suggest that genetic markers in the serine protease pathways could influence the risk of recurrent hospitalizations for acute pancreatitis or acute abdominal pain in LPLD.
Clinically, the diagnosis of acute pancreatitis requires two of the following criteria: abdominal pain suggestive of acute pancreatitis, serum amylase and/or lipase activity at least three times greater than the upper limit of normal, and characteristic findings of acute pancreatitis on abdominal ultrasonography or on contrast-enhanced computed tomography scanning or magnetic resonance imaging (Frossard et al., 2008; Banks et al., 2013). However, due to severe chylomicronemia, FCS is by nature associated with spurious estimation of several laboratory values and patients can present with pancreatitis and normal amylase or lipase values. Furthermore, imaging results were not available in all patients medical files. This might lead to underestimating the number of definite pancreatitis episodes in these patients. To overwhelm this issue, we considered all cases of hospitalizations for acute abdominal pain which were suggestive of pancreatitis. This includes all definite, probable and suspected cases of pancreatitis.
Family history is an important predictor of risk of several lipid-related diseases, including pancreatitis. In this study, LPLD patients with ≥5 hospitalizations for pancreatitis or severe abdominal pain presented at least twice the proportion of positive family history of pancreatitis than that observed in patients with less hospitalizations and controls. The family history is obviously not reflecting the genetic background only and takes into account shared life habits and the environemental background.
Although hereditary forms of pancreatitis exist (LaRusch et al., 2012), the majority of genes having been associated with pancreatitis explain the predisposition, rather than the cause, of acute pancreatitis. All FCS causing genes, including LPL are well-documented pancreatitis susceptibility genes. Pancreatitis is a complex disease and the mechanisms explaining the pancreatitis risk in LPLD are not clearly understood. One hypothesis is that large chylomicrons lodged in pancreatic capillaries expose them to pancreatic lipase, with the subsequent release of free fatty acids through the hydrolysis of chylomicron-associated triglycerides. Local high concentrations of free fatty acids are thought to damage pancreatic cells leading to pancreatitis (Ross et al., 2004, 2006; Rip et al., 2005, 2006; Gaudet et al., 2010). Another hypothesis is that phospholipids and oxidized phospholipids at the surface of buyant-chylomicrons once hydrolyzed by phospholipase serve as triggers for proteases. In all cases, if enough proteases become activated intracellularly, they can overwhelm the first line of defense (pancreatic secretory trypsin inhibitor) and resist backup defenses (proteolytic degradation). Activated cationic trypsin can then trigger the entire zymogen activation cascade (Frossard et al., 2008). It is possible that mutations in CTRC and SPINK1 genes contribute to this process and further increase the pancreatitis risk conferred by LPLD by either increasing the level of activated proteases, blocking active site of trypsin or decreasing proteases degradation. Indeed, mutant variants in CTRC and SPINK1 genes are known to contribute to pancreatitis susceptibility (Chen and Ferec, 2009; Whitcomb, 2010). CTRC encodes the protease chymotrypsin C, a protease produced in small quantities by the acinar cells and found in an inactive form in the zymogen granules (Szmola and Sahin-Toth, 2007), which degrades trypsin when trypsinogen is prematurely activated in the pancreas (Szmola and Sahin-Toth, 2010). Genetic variations in CTRC can predispose to chronic pancreatitis by diminishing its protective trypsin-degrading activity (Szmola and Sahin-Toth, 2007; Rosendahl et al., 2008; Whitcomb, 2010). SPINK1 encodes pancreatic secretory trypsin inhibitor, which prevents trypsin-catalyzed premature activation of zymogens within the pancreas and the pancreatic duct (Chen and Ferec, 2009; Ohmuraya and Yamamura, 2011; Kume et al., 2012). Mutations in this gene have been associated with hereditary pancreatitis (Ohmuraya and Yamamura, 2011; Ohmuraya et al., 2012), and it is thought to function in the prevention of trypsin-catalyzed premature activation of zymogens within the pancreas. CTRC and SPINK1 have pleiotropic functions but their role in serine proteases (trypsin and chymotrypsin) physiology is shared in common. This might explain why the effect of the combination of variants in these two genes on risk of pancreatitis in LPLD was more pronounced than single variants effect in the present study: CTRC-rs545634, located in the 5th intron, could be responsible of an alternate splicing site which would affect this enzyme's integrity and its inhibitory effect, combined to SPINK1-rs11319, located in the 5'UTR, that could affect mRNA stability and be responsible of a lesser production of this trypsin inhibitor, may be compatible with an increased pancreatitis risk. Further functional studies on these variants are needed to elucidate the role of their mutant allele in this hypothetic loss of inhibitory function responsible of their associated pancreatitis risk in LPLD.
Trypsin and chymotrypsin are two structurally similar serine proteases synthesized in the pancreas as inactive zymogen precursors (trypsinogen and chymotrypsinogen) secreted in the duodenum via the pancreatic duct and converted to the mature, active enzyme by proteolysis (Frossard et al., 2008; Whitcomb, 2010). They recognize different protein substrates. Trypsinogen and chymotrypsinogen also enter the bloodstream, where they can be detected in serum (Artigas et al., 1981). In blood, serine proteases of the trypsin-like family have long been recognized to be critical effectors of biological processes as diverse as blood coagulation, fibrinolysis, and immunity (Antalis et al., 2011). Serum trypsin levels have been shown to be significantly higher in acute pancreatitis patients than in controls (Artigas et al., 1981). Mass spectrometry and western blotting have shown that HDL are efficient carriers of alpha(1)-antitrypsin (Ortiz-Munoz et al., 2009). HDL-associated alpha(1)-antitrypsin appears to be able to inhibit trypsin activity, extracellular matrix degradation, cell detachment, and apoptosis induced by proteases in human vascular smooth muscle cells. The number of HDL particles and their cholesterol content is in general very low in presence of severe hypertriglyceridemia, a feature systematically observed in LPLD. It is possible that low HDL-associated alpha(1)-antitrypsin is a factor contributing to LPLD morbidity, including hepatomegaly and deleterious liver effects, in presence of mutant alleles in protease-regulating genes, such as CTRC and SPINK1. Clinically, serum serine proteases have been directly associated with triglycerides metabolism, including GPIHPB1-related factors and glycosylphosphatidylinositol linkage (Deeg and Bowen, 2002). Severe hypertriglyceridemia induced by PEG-L-Asparaginase in patients treated for acute lymphocytic leukemia can induced acute pancreatitis in patients undergoing chemotherapy (Konig and Malek, 2012). This has been associated with increased levels of serum trypsin and elastase 10 and 20 days after beginning L-asparaginase therapy (Shimizu et al., 1998). Alcohol abuse is associated with both hypertriglycridemia, and proteases activation (Joly et al., 1992). It is one of the most common cause of acute pancreatitis (Thrower et al., 2008). In this study, we excluded all known cases of alcohol abuse, although it is almost not viable long term in FCS due to nature of the disease and the huge risk of heavy alcohol consumption for these patients. The links between the proteases networks and lipid metabolism are complex and the connection with pancreatitis risk and other morbidities in LPLD remain to be documented.
This study has limitations. LPLD is an ultra-rare, and extreme cause of hypertriglyceridemia and is thus not representative of all causes of severe hypertriglyceridemia. The rarity of LPLD, implies that we have to deal with small sample size which also limits the power of association studies. Despite this, the results suggest that genetic markers in the protease cascade could be associated with the expression of pancreatitis in LPLD. However, functional analyses were not performed in this study and we cannot infer at this point that there is a physiological association between protease regulators and the recurrence of pancreatitis in presence of severe hypertriglyceridemia. Our findings need to be validated and replicated in larger LPLD and severe hypertriglyceridemia cohorts and the functional effect of identified variants needs to be characterized before this SNP-combination could be considered as a clinical biomarker of recurrence of pancreatitis in LPLD. Since other genes are known to confer pancreatitis susceptibility, additional analyses are needed to understand the complex genetic architecture of pancreatitis in LPLD. Such studies, including whole genome sequencing, resequencing of extremes, functional gene expression analyses, physiologicial and metabolic studies, are actually underway.
Author Contributions
Karine Tremblay co-designed the study, co-supervised the CTRC and SPINK1 genes sequencing, performed the statistical analyses and drafted the manuscript. Camélia Dubois-Bouchard participated in the review of the literature, the selection of candidate genes and in CTRC and SPINK1 genes sequencing activities under Karine Tremblay supervision. She also participated in the statistical analyses and to the writing of the manuscript. Diane Brisson supervised LPL genotyping, participated in statistical analyses and revised the manuscript. Daniel Gaudet co-designed and supervised the study, coordinated the clinical tasks, revised the manuscript and approved the version to be published. During the study, Camélia Dubois-Bouchard was a Université de Montréal M Sc. student and Karine Tremblay a Université de Montréal postdoctoral fellow, both under Daniel Gaudet supervision.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors are thankful to all participants and to the staff of the ECOGENE-21 Clinical Research Center. Karine Tremblay is a Université de Montréal postdoctoral and CCRP fellow, and was the recipient of bursary of excellence from the Canadian Heart and Stroke Foundation and from the “Fonds de la Recherche en Santé du Québec.” Camélia Dubois-Bouchard is a master student who received a graduate bursary from the Department of Medicine of the Université de Montréal. Daniel Gaudet had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. This study was supported by a Canadian Institutes of Health Research team grant (# CTP-82941).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fgene.2014.00090/abstract
References
Antalis, T. M., Bugge, T. H., and Wu, Q. (2011). Membrane-anchored serine proteases in health and disease. Prog. Mol. Biol. Transl. Sci. 99, 1–50. doi: 10.1016/B978-0-12-385504-6.00001-4
Artigas, J. M., Garcia, M. E., Faure, M. R., and Gimeno, A. M. (1981). Serum trypsin levels in acute pancreatic and non-pancreatic abdominal conditions. Postgrad. Med. J. 57, 219–222. doi: 10.1136/pgmj.57.666.219
Banks, P. A., Bollen, T. L., Dervenis, C., Gooszen, H. G., Johnson, C. D., Sarr, M. G., et al. (2013). Classification of acute pancreatitis–2012: revision of the Atlanta classification and definitions by international consensus. Gut 62, 102–111. doi: 10.1136/gutjnl-2012-302779
Beer, S., Zhou, J., Szabo, A., Keiles, S., Chandak, G. R., Witt, H., et al. (2012). Comprehensive functional analysis of chymotrypsin C (CTRC) variants reveals distinct loss-of-function mechanisms associated with pancreatitis risk. Gut 62, 1616–1624. doi: 10.1136/gutjnl-2012-303090
Boulling, A., Le Marechal, C.Trouve, P., Raguenes, O., Chen, J. M., and Ferec, C. (2007). Functional analysis of pancreatitis-associated missense mutations in the pancreatic secretory trypsin inhibitor (SPINK1) gene. Eur. J. Hum. Genet. 15, 936–942. doi: 10.1038/sj.ejhg.5201873
Brisson, D., Tremblay, M., Methot, J., Tremblay, K., and Gaudet, D. (2010). Comparison of the efficacy of fibrates on hypertriglyceridemic phenotypes with different genetic and clinical characteristics. Pharmacogenet. Genomics 20, 742–747. doi: 10.1097/FPC.0b013e328340095e
Brunzell, J., and Deeb, S. (2001). “Familial lipoprotein lipase deficiency, apo CII deficiency and hepatic lipase deficiency,” in The Metabolic and Molecular Bases of Inherited Disease, 8 Edn, eds C. R. Scriver, A. L. Beaudet, W. S. Sly, and D. Valle (New York, NY: McGraw-Hill), 2789–2816.
Chahwan, R., Wontakal, S. N., and Roa, S. (2011). The multidimensional nature of epigenetic information and its role in disease. Discov. Med. 11, 233–243.
Chen, J. M., and Ferec, C. (2009). Chronic pancreatitis: genetics and pathogenesis. Annu. Rev. Genomics Hum. Genet. 10, 63–87. doi: 10.1146/annurev-genom-082908-150009
Deeg, M. A., and Bowen, R. F. (2002). Phosphorylation decreases trypsin activation and apolipoprotein al binding to glycosylphosphatidylinositol-specific phospholipase D. Biochem. Cell Biol. 80, 253–260. doi: 10.1139/o02-004
Fredrickson, D. S., Goldstein, J. L., and Brown, M. S. (1978). “The familial hyperlipoproteinemias,” in The Metabolic Basis of Inherited Disease, 4th Edn, eds J. B. Stanbury, J. B. Wyngaarden, and D. S. Fredrickson (New York, NY: McGraw-Hill), 604.
Frossard, J. L., Steer, M. L., and Pastor, C. M. (2008). Acute pancreatitis. Lancet 371, 143–152. doi: 10.1016/S0140-6736(08)60107-5
Gagné, C., and Gaudet, D. (2007). Les dyslipoprotéinémies: l'approche Clinique. Quebec: Lipimed Communications.
Gaudet, D., de Wal, J., Tremblay, K., Dery, S., van Deventer, S., Freidig, A., et al. (2010). Review of the clinical development of alipogene tiparvovec gene therapy for lipoprotein lipase deficiency. Atheroscler. Suppl. 11, 55–60. doi: 10.1016/j.atherosclerosissup.2010.03.004
Gaudet, D., Methot, J., and Kastelein, J. (2012). Gene therapy for lipoprotein lipase deficiency. Curr. Opin. Lipidol. 23, 310–320. doi: 10.1097/MOL.0b013e3283555a7e
Gaudet, D., Signorovitch, J., Swallow, E., Fan, L., Tremblay, K., Brisson, D., et al. (2013). Medical resource use and costs associated with chylomicronemia. J. Med. Econ. 16, 657–666. doi: 10.3111/13696998.2013.779277
Gaudet, D., Vohl, M. C., Julien, P., Tremblay, G., Perron, P., Gagne, C., et al. (1998). Relative contribution of low-density lipoprotein receptor and lipoprotein lipase gene mutations to angiographically assessed coronary artery disease among French Canadians. Am. J. Cardiol. 82, 299–305. doi: 10.1016/S0002-9149(98)00328-2
Jiang, Y. H., Bressler, J., and Beaudet, A. L. (2004). Epigenetics and human disease. Annu. Rev. Genomics Hum. Genet. 5, 479–510. doi: 10.1146/annurev.genom.5.061903.180014
Johansen, C. T., Kathiresan, S., and Hegele, R. A. (2011). Genetic determinants of plasma triglycerides. J. Lipid. Res. 52, 189–206. doi: 10.1194/jlr.R009720
Joly, J. P., Sesboue, R., Hillemand, B., and Martin, J. P. (1992). Serum trypsin-like activity in chronic alcoholized men: possible relationship with lipids, apoA-1 and apoB lipoproteins. Alcohol. Alcohol. 27, 563–569.
Kiraly, O., Boulling, A., Witt, H., Le Marechal, C., Chen, J. M., Rosendahl, J., et al. (2007). Signal peptide variants that impair secretion of pancreatic secretory trypsin inhibitor (SPINK1) cause autosomal dominant hereditary pancreatitis. Hum. Mutat. 28, 469–476. doi: 10.1002/humu.20471
Konig, M., and Malek, R. (2012). PEG-L-Asparaginase induced hypertriglyceridemia. Endocr. Rev. 33 (03_MeetingAbstracts), SUN-248. doi: 10.1210/endo-meetings.2012.33.03_MeetingAbstracts.SUN-248
Kume, K., Masamune, A., Ariga, H., Hayashi, S., Takikawa, T., Miura, S., et al. (2012). Do genetic variants in the SPINK1 gene affect the level of serum PSTI? J. Gastroenterol. 47, 1267–1274. doi: 10.1007/s00535-012-0590-3
LaRusch, J., Barmada, M. M., Solomon, S., and Whitcomb, D. C. (2012). Whole exome sequencing identifies multiple, complex etiologies in an idiopathic hereditary pancreatitis kindred. JOP 13, 258–262.
Ma, Y., Henderson, H. E., Murthy, V., Roederer, G., Monsalve, M. V., Clarke, L. A., et al. (1991). A mutation in the human lipoprotein lipase gene as the most common cause of familial chylomicronemia in French Canadians. N. Engl. J. Med. 324, 1761–1766. doi: 10.1056/NEJM199106203242502
Masson, E., Chen, J. M., Scotet, V., Le Marechal, C., and Ferec, C. (2008). Association of rare chymotrypsinogen C (CTRC) gene variations in patients with idiopathic chronic pancreatitis. Hum. Genet. 123, 83–91. doi: 10.1007/s00439-007-0459-3
Mattei, M. G., Etienne, J., Chuat, J. C., Nguyen, V. C., Brault, D., Bernheim, A., et al. (1993). Assignment of the human lipoprotein lipase (LPL) gene to chromosome band 8p22. Cytogenet. Cell Genet. 63, 45–46. doi: 10.1159/000133499
Meyers, C., Gaudet, D., Tremblay, K., Amer, A., Chen, J., and Aimin, F. (2012). The DGAT1 Inhibitor LCQ908 decreases triglyceride levels in patients with the familial chylomicronemia syndrome. J. Clin. Lipidol. 6, 266–267. doi: 10.1016/j.jacl.2012.04.034
Monsalve, M. V., Henderson, H., Roederer, G., Julien, P., Deeb, S., Kastelein, J. J., et al. (1990). A missense mutation at codon 188 of the human lipoprotein lipase gene is a frequent cause of lipoprotein lipase deficiency in persons of different ancestries. J. Clin. Invest. 86, 728–734. doi: 10.1172/JCI114769
Ohmuraya, M., Sugano, A., Hirota, M., Takaoka, Y., and Yamamura, K. (2012). Role of Intrapancreatic SPINK1/Spink3 Expression in the Development of Pancreatitis. Front. Physiol. 3:126. doi: 10.3389/fphys.2012.00126
Ohmuraya, M., and Yamamura, K. (2011). Roles of serine protease inhibitor Kazal type 1 (SPINK1) in pancreatic diseases. Exp. Anim. 60, 433–444. doi: 10.1538/expanim.60.433
Ortiz-Munoz, G., Houard, X., Martin-Ventura, J. L., Ishida, B. Y., Loyau, S., Rossignol, P., et al. (2009). HDL antielastase activity prevents smooth muscle cell anoikis, a potential new antiatherogenic property. FASEB J. 23, 3129–3139. doi: 10.1096/fj.08-127928
Rip, J., Nierman, M. C., Ross, C. J., Jukema, J. W., Hayden, M. R., Kastelein, J. J., et al. (2006). Lipoprotein lipase S447X: a naturally occurring gain-of-function mutation. Arterioscler. Thromb. Vasc. Biol. 26, 1236–1245. doi: 10.1161/01.ATV.0000219283.10832.43
Rip, J., Nierman, M. C., Sierts, J. A., Petersen, W., Van den Oever, K., Van Raalte, D., et al. (2005). Gene therapy for lipoprotein lipase deficiency: working toward clinical application. Hum. Gene Ther. 16, 1276–1286. doi: 10.1089/hum.2005.16.1276
Rosendahl, J., Witt, H., Szmola, R., Bhatia, E., Ozsvari, B., Landt, O., et al. (2008). Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat. Genet. 40, 78–82. doi: 10.1038/ng.2007.44
Ross, C. J., Twisk, J., Bakker, A. C., Miao, F., Verbart, D., Rip, J., et al. (2006). Correction of feline lipoprotein lipase deficiency with adeno-associated virus serotype 1-mediated gene transfer of the lipoprotein lipase S447X beneficial mutation. Hum. Gene Ther. 17, 487–499. doi: 10.1089/hum.2006.17.487
Ross, C. J., Twisk, J., Meulenberg, J. M., Liu, G., van den Oever, K., Moraal, E., et al. (2004). Long-term correction of murine lipoprotein lipase deficiency with AAV1-mediated gene transfer of the naturally occurring LPL(S447X) beneficial mutation. Hum. Gene Ther. 15, 906–919. doi: 10.1089/hum.2004.15.906
Sacks, F. M., Stanesa, M., and Hegele, R. A. (2013). Severe hypertriglyceridemia with pancreatitis: thirteen years' treatment with lomitapide. JAMA Intern. Med. 174, 443–447. doi: 10.1001/jamainternmed.2013.13309
Sanchez-Ramirez, C. A., Flores-Martinez, S. E., Garcia-Zapien, A. G., Montero-Cruz, S. A., Larrosa-Haro, A., and Sanchez-Corona, J. (2012). Screening of R122H and N29I mutations in the PRSS1 gene and N34S mutation in the SPINK1 gene in Mexican pediatric patients with acute and recurrent pancreatitis. Pancreas 41, 707–711. doi: 10.1097/MPA.0b013e31823cd873
Shimizu, T., Yamashiro, Y., Igarashi, J., Fujita, H., and Ishimoto, K. (1998). Increased serum trypsin and elastase-1 levels in patients undergoing L-asparaginase therapy. Eur. J. Pediatr. 157, 561–563. doi: 10.1007/s004310050878
St-Pierre, J., Vohl, M. C., Brisson, D., Perron, P., Despres, J. P., Hudson, T. J., et al. (2001). A sequence variation in the mitochondrial glycerol-3-phosphate dehydrogenase gene is associated with increased plasma glycerol and free fatty acid concentrations among French Canadians. Mol. Genet. Metab. 72, 209–217. doi: 10.1006/mgme.2000.3144
Surendran, R. P., Visser, M. E., Heemelaar, S., Wang, J., Peter, J., Defesche, J. C., et al. (2012). Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J. Intern. Med. 20, 185–196. doi: 10.1111/j.1365-2796.2012.02516.x
Szmola, R., and Sahin-Toth, M. (2007). Chymotrypsin C (caldecrin) promotes degradation of human cationic trypsin: identity with Rinderknecht's enzyme Y. Proc. Natl. Acad. Sci. U.S.A. 104, 11227–11232. doi: 10.1073/pnas.0703714104
Szmola, R., and Sahin-Toth, M. (2010). Pancreatitis-associated chymotrypsinogen C (CTRC) mutant elicits endoplasmic reticulum stress in pancreatic acinar cells. Gut 59, 365–372. doi: 10.1136/gut.2009.198903
Thrower, E., Husain, S., and Gorelick, F. (2008). Molecular basis for pancreatitis. Curr. Opin. Gastroenterol. 24, 580–585. doi: 10.1097/MOG.0b013e32830b10e6
Tremblay, K., Methot, J., Brisson, D., and Gaudet, D. (2011). Etiology and risk of lactescent plasma and severe hypertriglyceridemia. J. Clin. Lipidol. 5, 37–44. doi: 10.1016/j.jacl.2010.11.004
Whitcomb, D. C. (2010). Genetic aspects of pancreatitis. Annu. Rev. Med. 61, 413–424. doi: 10.1146/annurev.med.041608.121416
Whitcomb, D. C. (2013). Genetic risk factors for pancreatic disorders. Gastroenterology 144, 1292–1302. doi: 10.1053/j.gastro.2013.01.069
Witt, H., Luck, W., Hennies, H. C., Classen, M., Kage, A., Lass, U., et al. (2000). Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat. Genet. 25, 213–216. doi: 10.1038/76088
Keywords: severe hypertriglyceridemia, lipoprotein lipase deficiency (LPLD), pancreatitis, CTRC gene, SPINK1 gene, LPL gene
Citation: Tremblay K, Dubois-Bouchard C, Brisson D and Gaudet D (2014) Association of CTRC and SPINK1 gene variants with recurrent hospitalizations for pancreatitis or acute abdominal pain in lipoprotein lipase deficiency. Front. Genet. 5:90. doi: 10.3389/fgene.2014.00090
Received: 12 February 2014; Accepted: 02 April 2014;
Published online: 22 April 2014.
Edited by:
M. Z. A. Bhuiyan, University Hospital Lausanne (CHUV), SwitzerlandReviewed by:
Bruna De Felice, University of Naples II, ItalyXusheng Wang, St. Jude Children's Research Hospital, USA
Copyright © 2014 Tremblay, Dubois-Bouchard, Brisson and Gaudet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Gaudet, Department of Medicine, ECOGENE-21 Research Center, Université de Montréal, Pavillon des Augustines, 5th floor, 305 St-Vallier street, Chicoutimi, QC G7H 5H6, Canada e-mail: daniel.gaudet@umontreal.ca
†These authors have contributed equally to this work.