 Ilias Gkikas
Ilias Gkikas Dionysia Petratou
Dionysia Petratou Nektarios Tavernarakis
Nektarios Tavernarakis- 1Institute of Molecular Biology and Biotechnology, Foundation for Research and Technology-Hellas, Heraklion, Crete, Greece
- 2Department of Basic Sciences, Faculty of Medicine, University of Crete, Heraklion, Crete, Greece
The aging process has been associated with numerous pathologies at the cellular, tissue, and organ level. Decline or loss of brain functions, including learning and memory, is one of the most devastating and feared aspects of aging. Learning and memory are fundamental processes by which animals adjust to environmental changes, evaluate various sensory signals based on context and experience, and make decisions to generate adaptive behaviors. Age-related memory impairment is an important phenotype of brain aging. Understanding the molecular mechanisms underlying age-related memory impairment is crucial for the development of therapeutic strategies that may eventually lead to the development of drugs to combat memory loss. Studies in invertebrate animal models have taught us much about the physiology of aging and its effects on learning and memory. In this review we survey recent progress relevant to conserved molecular pathways implicated in both aging and memory formation and consolidation.
Introduction
During the past century, age-related memory impairments have emerged as one of the top public health threats. Both psychiatric and neurodegenerative disorders comprising schizophrenia, depression, Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) are associated with age-related memory impairment. In humans, cognitive decline starts in mid-life and deepens with advancing age suggesting that the greatest risk factor is age itself. Thus, ultimately, prevention of these pathologies necessitates thorough understanding of the molecular mechanisms underlying their links with the aging process (Bishop et al., 2010).
Our knowledge of the molecular regulatory mechanisms of aging impinging on cognitive capacity is steadily increasing in recent years. Interestingly, analyses of vertebrate and invertebrate model systems suggest that molecular and genetic pathways regulating cognitive aging are highly conserved in yeast, flies, worms, and mammals (Barco et al., 2006; Ardiel and Rankin, 2010; Bishop et al., 2010; Kauffman et al., 2010). Accumulating evidence from these models suggest a dynamic association between cognitive functions and aging. Similarly to several phenotypes and biomarkers of aging, which can vary substantially among individuals, cognitive decline displays significant severity fluctuation within a population. Consequently, it is important to identify key regulators of both cognitive impairment and longevity pathways. A plethora of molecular and cellular studies indicate a strong entanglement between lifespan regulation pathways and cognitive decline or neurodegeneration. In this review, we survey the molecular mechanisms and genes associated with longevity that have also been implicated in cognitive aging (summarized in Table 1). We further focus on recent work in invertebrate model organisms linking learning and memory impairment with age.
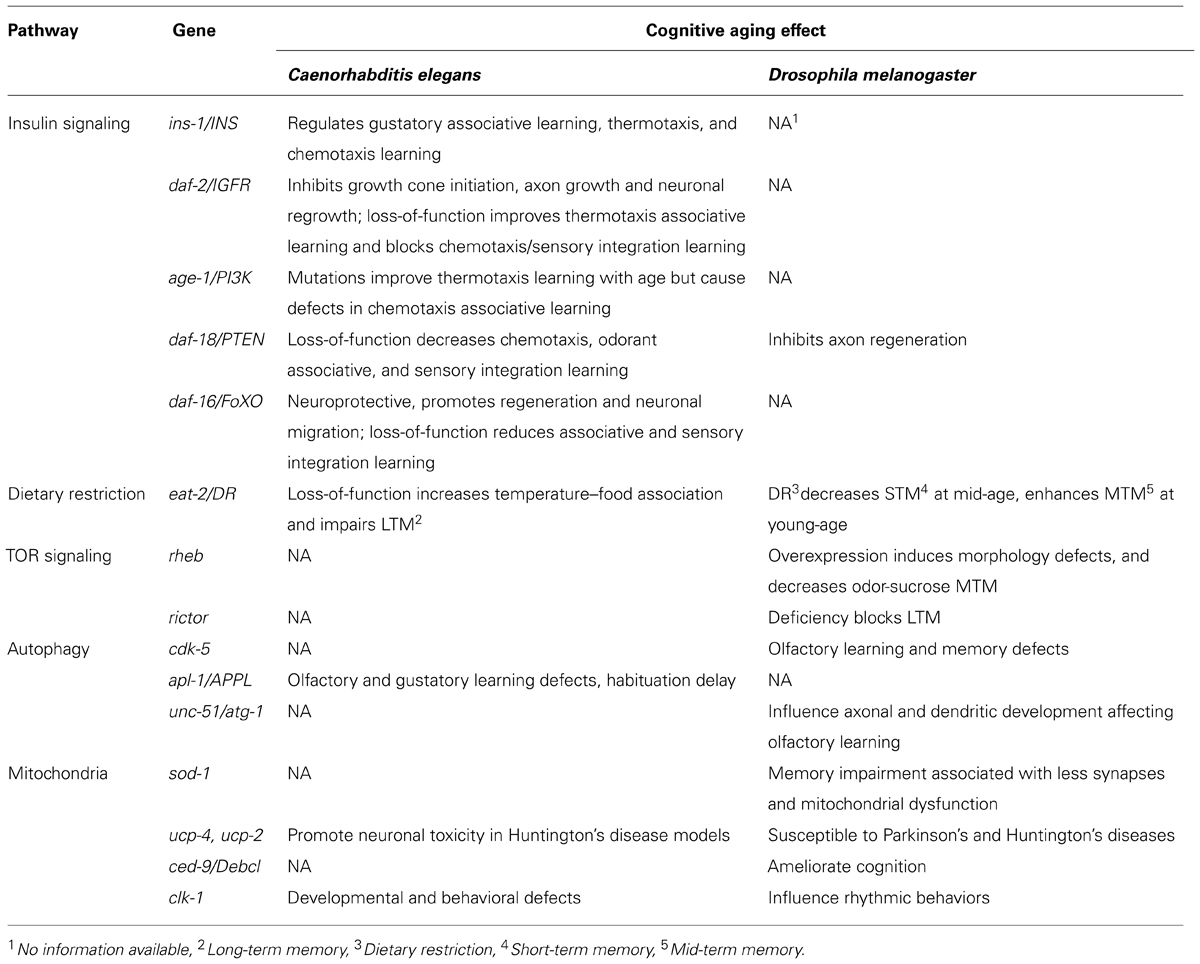
TABLE 1. Conserved signaling pathway genes and cognitive aging in worms and flies.
Reduced Insulin/IGF-1 Signaling Promotes Learning Ability During Aging
The insulin/IGF-1 (IIS) signal transduction pathway and its downstream effectors have been found to influence lifespan in a wide range of diverse organisms, suggesting a tightly conserved role of these mechanisms in aging. Reduction of IIS signaling promotes longevity in Caenorhabditis elegans and flies (Kenyon et al., 1993; Kenyon, 2010; Partridge, 2010). Whether this function is conserved in mice and humans remains unclear (Clancy et al., 2001; Tatar et al., 2001; Bluher et al., 2003; Suh et al., 2008; Bokov et al., 2011). Main components of insulin signaling in C. elegans are the insulin homolog INS-1, its receptor DAF-2, and the PIP3-kinase (phosphatidylinositol-triphosphate kinase) homolog AGE-1. Insulin signaling has been implicated in learning and memory, and in neuronal aging. Reduction of IIS attenuates protein aggregation and insolubility, and prevents amyloid-beta toxic effects. These processes are tightly associated with impaired nervous system function and age-related neurodegenerative diseases (Florez-McClure et al., 2007; David et al., 2010; Keowkase et al., 2010; Zhang et al., 2011; Tamura et al., 2013). In mouse models of AD, reduced IGF1 signaling protects from disease-associated neuronal loss and behavioral impairment, allthough IGF1R haploinsufficiency does not necessarily extent lifespan in mice (Cohen et al., 2009; Bokov et al., 2011).
In C. elegans, IIS has been shown to influence thermotaxis learning (Kodama et al., 2006) and salt chemotaxis learning (Tomioka et al., 2006). Moreover, long-lived IIS mutants show improved ability to associate temperature with food at both young and old age (Murakami, 2007). By contrast, some of these mutants are impaired, at young age, in their ability to associate NaCl with the absence of food (Vellai et al., 2006), or to intergrate sensory stimuli, such as Cu2+ and diacetyl perception, towards decision-making (Jiu et al., 2010). Moreover, long-lived age-1 mutant animals display delayed age-related decline of isothermal tracking and locomotion. Similarly, age-1 and daf-2 mutants associate temperature and starvation more efficiently compared to wild type controls, while young adults of these muants show increased temperature–food association. The enhanced association capacity of daf-2 mutants is dependent on the neuronal Ca2+-sensor NCS-1, which modulates isothermal tracking in the amphid interneurons, a key component of the thermosensory circuit (Murakami et al., 2005). AGE-1 also acts in the benzaldehyde-sensing amphid wing C (AWC) sensory neurons to direct benzaldehyde–starvation associative plasticity (Lin et al., 2010). While, mutations in the daf-2 IIS receptor improve memory performance in C. elegans early in adulthood, maintaining learning ability with age, no extension in long-term memory (LTM) during aging is evident. Reduced insulin signaling does not alter neuronal plasticity but rather establishes an association more rapidly and prolongs the duration of this association early in adulthood (Kauffman et al., 2010).
Neuronal cells not only degenerate with age but the nervous system also loses the ability to regenerate after injury. Genetic experiments indicate that axon regeneration in aging C. elegans motor neurons is repressed by elevated IIS, which inhibits both growth cone initiation and axon growth (but not axon guidance) in aged animals. IIS impairs regeneration by blocking the function of DAF-16, a FOXO transcription factor and downstream effector of IIS. DAF-16/FOXO is necessary and sufficient to promote neuronal regeneration in a cell-autonomous manner. (Byrne et al., 2014). DAF-16 has also been shown to promote developmental neuronal migration and to affect aspects of neuronal cell morphology, such as neurite outgrowth (Christensen et al., 2011; Kennedy et al., 2013). DLK-1, a mitogen activated kinase kinase kinase (MAPKKK) that regulates presynaptic development is downregulated by IIS. DAF-16 upregulates expression of dlk-1 in a neuron-specific manner, to promote neuronal regeneration indipendently of lifespan (Byrne et al., 2014). In addition, DAF-16 and HSF-1, the C. elegans heat shock transcription factor ortholog, show neuroprotective characteristics since their activation can defer the morphological and functional defects that emerge on the synapses of touch receptor neurons with physiological aging (Toth et al., 2012).
The totality of these findings suggest that in addition to extending lifespan, reduced IIS signaling also promotes learning ability with age. However, this effect is not accompanied by maintenance or extension of long term-memory during aging. Instead, IIS signaling appears to play a more significant role in the retrieval rather than acquisition of memory.
Dietary Restriction and Long-Term Memory
Dietary restriction (DR), a reduction in total food intake, has been shown to increase lifespan and reduce fecundity in a wide range of organisms such as yeast, nematodes, flies, and rodents (Masoro, 2005; Mair and Dillin, 2008; Piper and Bartke, 2008). Recent studies in primates indicate that DR prevents from aging-related pathologies like brain atrophy,but it is still under debate wether it extends lifespan (Colman et al., 2009; Mattison et al., 2012; Cava and Fontana, 2013; Colman et al., 2014). Little is known about the genes mediating these effects of DR. In C. elegans, knock-down of mekk-3 a homolog of the mammalian mitogen-activated MEKK3-like kinase, recapitulates DR and extends lifespan. MEKK-3 deficiency leads to reprograming of fatty acid metabolism and lowering reactive oxygen species (ROS) generation, through the nuclear hormone receptor NHR-49 and DAF-22, an ortholog of human sterol carrier protein SCP2 (Chamoli et al., 2014).
The C. elegans feeding-defective mutant eat-2 has been utilized as a model of DR. eat-2 mutants ingest food poorly and, as a consequence, are long-lived. Lifespan extension by eat-2 mutations is at least in part mediated through a daf-16-independent pathway (Avery, 1993; Raizen et al., 1995; Lakowski and Hekimi, 1998; Panowski et al., 2007). DR has also been suggested to attenuate age-related cognitive decline in rats (Adams et al., 2008). In C. elegans, young adult eat-2 mutants show increased consistency of isothermal tracking (temperature–food association; Murakami et al., 2005). Contrary to daf-2, eat-2 mutants exhibit significantly impaired LTM during young adulthood, but maintain memory capacity longer with age. Although young eat-2 mutants display normal benzaldehyde chemotaxis, they require more training to form long-term memories. The duration of short-term memory in eat-2 animals is similar to wild type, contrary to significant short term associative memory extension observed in daf-2 mutants (Kauffman et al., 2010).
Dietary restriction also affects learning performance during aging in Drosophila melanogaster. The performance of young and old flies in an aversive learning test, where an odor is associated with a noxius mechanical shock, has been examined. These experiments showed that dietary-restricted flies, that live on average 14% longer than rich-diet fed flies, appear to have a better learning ability, even at old age. Young, dietary restricted flies show enhanced mid-term memory but their short-term memory is not affected. By contrast, short-term memory of mid-aged flies is poorer, compared with flies that grew on rich diet. Mid-term memory performance of mid-aged and old flies is not improved (Burger et al., 2010). These results are consistent with findings in C. elegans, in that only long term-memory is affected by DR during aging (Kauffman et al., 2010). While DR and reduced IIS signaling both increase longevity, the two pathways influence cognitive ability of young adults in an opposing manner.The differential effects of IIS and DR on learning and memory decline with age are likely due to their differential regulation of expression levels and activity of CRH-1, the cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) transcription factor homolog in C. elegans (Kauffman et al., 2010).
Mitochondrial Function and Cognitive Aging
Mitochondria play pivotal role in adenosine triphosphate (ATP) production, calcium homeostasis, and apoptosis regulation, and are the main source of endogenous ROS. The functionality of these organelles influences aging through multiple pathways that may be directly or indirectly relevant to cognitive decline. The link between mitochondrial dysfunction, neurodegeneration, and cognition has been a subject of intensive study in many metazoans, ranging from C. elegans to humans (Bishop et al., 2010; Aksenov et al., 2013). A growing body of evidence suggests that neuronal structure and function are particularly vulnerable to mitochondrial function impairment (Stein and Murphy, 2012). However, the contribution of mitochondria to selective neurodegeneration in a variety of neurodegenerative pathologies associated with cognitive decline remains a matter of debate.
Aging studies in invertebrate model organisms provide a common ground for mitopathology and cognitive research. Several conserved groups of genes influencing mitochondrial metabolism, neural plasticity and synaptic function show expression changes during aging. In C. elegans, loss of α-tubulin acetyltransferase gene mec-17 causes axon degeneration, thereby leading to neuronal dysfunction. Axons lacking MEC-17 contain less mitochondria, display transport defects, and loss of synaptic integrity (Neumann and Hilliard, 2014). Atat1, the mouse homolog of MEC-17 is associated with the formation of dentate gyrus, which is essential for learning and memory (Kim et al., 2013). Moreover, studies in C. elegans ric-7 mutants, where axonal mitochondria trafficking is impaired, suggest that mitochondria are important for protection of axons against degeneration (Rawson et al., 2014). During physiological aging, nematode touch receptor neurons display morphological and functional abnormalities, such as neurite outgrowth defects and reduced number of synapses. Positioning of mitochondria in branches required for neurite outgrowth and the accumulation of vesicles in neuronal processes suggests that trafficking deficiency underlies these age-related abnormalities (Toth et al., 2012). These morphological changes of neurons have been associated with a decline in cognition, learning, and memory during aging (Vohra et al., 2010; Kimata et al., 2012; Kim et al., 2013; Wang et al., 2013).
The nematode genome encodes five superoxide dismutases (SODs) that function in cytoplasm, mitochondria, and extracellularly. Specifically, SOD-1 regulates detoxification of syperoxide radicals in mitochondria and guards from accumulation of oxidative damage during aging (Harman, 1968; McCord and Fridovich, 1969; Yanase et al., 2009; Back et al., 2010). Nonetheless, lifespan extension in mutants overexpressing sod-1 is not related with reduction of oxidative damage (Cabreiro et al., 2011). Transgenic C. elegans expressing the human G93A SOD1 variant, associated with familial amyotrophic lateral sclerosis (ALS), in motor neurons show motor defects and increased autophagy in an age-dependent manner (Aksenov et al., 2013; Li et al., 2013). SOD-1 overexpression has also been associated with mitochondrial swelling, and learning and memory impairment in flies, mice, and humans (Shin et al., 2004; Perluigi and Butterfield, 2012; Haddadi et al., 2014). For example, transgenic flies expressing a zinc-deficient SOD1 mutant display behavioral defects, including impairment of locomotion, associated with mitochondrial respiratory chain deficiency and matrix vacuolization, that is not accompanied by shortening of lifespan (Bahadorani et al., 2013). Moreover, SOD-1 activity and expression levels decline during normal aging of Drosophila. At the same time, knock-down of sod-1 in the mushroom bodies deteriorates mid-term memory and LTM. These memory defects associate with reduced synapse formation and mitochondrial damage during Drosophila aging (Haddadi et al., 2014).
Converging evidence implicates members of the antiapoptotic BCL-2 family of proteins in neuronal injury and synapse deformation, through impairment of mitochondrial dynamics (Berman et al., 2009). CED-9, the C. elegans homolog of BCL-2, interacts with the mitofusin FZO-1 and the dymanin related protein EAT-3 to promote mitochondrial fusion under specific conditions. The C. elegans eat-3 encodes a homolog of human OPA-1 which is associated with Dominant Optic Atrophy disorder (Breckenridge et al., 2009; Rolland et al., 2009). In Drosophila, the BCL-2 homologous proteins, Buffy and Debcl are involved in the permeabilization of mitochondria to cytochrome-c that is mediated by pro-death mitochondrial proteins including Reaper and Hid (Abdelwahid et al., 2011). Unlike in worms, Buffy inhibition results in normal flies, while knockdown of Debcl protects against polyglutamine (polyQ)-induced neurodegeneration through maintaining mitochondrial homeostasis. The Debcl ortholog in mice, Bax/Bak, was found to regulate neurogenesis in adult brain regions such as hippocampus and cerebellum and promote discrimination learning without affecting significantly spatial memory and learning (Senoo-Matsuda et al., 2005; Galindo et al., 2009; Sahay et al., 2011; Hardwick and Soane, 2013).
Neurons are particularly vulnerable to mitochondrial dysfunction. Interestingly, expression of the human mitochondrial uncoupling protein (UCP) ucp2 in Drosophila dopaminergic neurons increases ATP production and locomotion activity, and results in neuroprotection against pathogenic stress associated with PD (Islam et al., 2012). Beyond neurons, enhanced expression of mitochondria UCPs in flies ameliorates HD phenotypes in glia cells by moderating ROS and ATP production (Besson et al., 2010). In C. elegans, depletion of UCP-4 exacerbates neuronal toxicity in animals expressing an expanded polyQ repeat protein in touch neurons, suggesting that similarly to flies, under normal conditions UCP-4 protects from neuronal injuries in worms (Parker et al., 2012). However, overexpression of ucp-4 in worms does not extend lifespan (Sagi and Kim, 2012). Alterations in the expression of mitochondrial respiratory chain genes result in similar effects. For example, mutations in the mev-1 and gas-1 genes, encoding subunits of complex II and I of the respiratory chain, respectively, increase ROS production, shorten lifespan, and retard behavioral rates (Kayser et al., 2004). In another example, animals carrying mutations in mitochondria complex IV sft-1 gene, show increased lifespan that is dependent on DAF-16 (Maxwell et al., 2013). Depletion of SURF1, the mouse ortholog of sft-1, also increases lifespan and improves cognitive function in mice (Lin et al., 2013a). Knockdown of clk-1, a gene required for ubiquinone biosynthesis reduces respiration rates and increases C. elegans lifespan, also delaying behavioral rates (Rea et al., 2007). Loss-of-function mutations in clk-1 extend lifespan and slow development and behavioral rates (Takahashi et al., 2012). Similarly, knockdown of the mouse clk-1 ortholog causes mild mitochondrial dysfunction and extends lifespan (Lapointe and Hekimi, 2008; Deepa et al., 2013). In Drosophila, reduced expression of complex I and IV genes specifically in adult neurons is sufficient to extend lifespan (Copeland et al., 2009). Furthermore, observations in clk-1 mutants indicate that neurite outgrowth is inhibited in aged worms (Tank et al., 2011). The association between neuronal morphology and behavioral effects suggests that mitochondria dysfunction may, in part, underlie memory and learning decline during aging (Ardiel and Rankin, 2010; Kimata et al., 2012; Stein and Murphy, 2012). However, little is known about the molecular mechanisms that mediate the effects of alterations in mitochondrial metabolism on both cognitive capacity and longevity.
Autophagy and Protein Homeostasis in Learning and Memory
The autophagic pathway has also been implicated in aging and cognitive decline. Autophagic activity decreases during the course of aging and genes that control this process are strongly associated with lifespan regulation in flies and worms (Lionaki et al., 2013). In Drosophila, overexpression of autophagy-related genes in neurons enhances longevity, while their repression causes neuronal defects and shortening of lifespan (Simonsen et al., 2008). Similarly, increasing autophagy mediates lifespan extension in worms (Hansen et al., 2008). UNC-51, a nematode autophagy regulator also controls axonal and dendritic development and its homolog affects olfactory learning in flies (Mochizuki et al., 2011). Worms lacking UNC-51 display axonal membrane defects, indicating a role of autophagy in synaptic plasticity, which indirectly interferes with learning and memory (Sigmond et al., 2008; Ragagnin et al., 2013). In Drosophila, inhibition of the cyclin-dependent kinase 5 (cdk5) kinase ortholog decreases autophagy, shortens lifespan and causes structural defects in central brain regions associated with olfactory learning and memory (Trunova and Giniger, 2012). In both flies and worms, autophagy deficiency leads to abnormal accumulation of protein aggregates thus promoting pathological mechanisms associated with neurodegenerative disorders, such as HD and AD (Ling et al., 2009; Low et al., 2013). For example, accumulation of intracellular APL-1, a β-amyloid precursor protein, upon autophagy impairment, causes behavioral deficiencies, including olfactory and gustatory learning defects, and habituation delay in C. elegans (Ewald et al., 2012; Ewald and Li, 2012; Chen et al., 2013). Accumulation of APL-1 also occurs during normal aging and can reach pathological levels contributing to the pathogenesis of AD (Nilsson et al., 2013).
Age-induced memory impairment studies in Drosophila suggest that cognitive aging is strongly associated with the autophagic pathway. Indeed, spermidine-induced autophagy reduces aggregation of ubiquitinated proteins and protects from age-related memory impairment, in the aged Drosophila brain (Gupta et al., 2013). Spermidine activates autophagy to also promote longevity in different metazoans ranging from C. elegans to mice (Eisenberg et al., 2009; Wang et al., 2012). Other studies suggest that spermidine may not act directly through autophagy to facilitate neuroprotection and memory during aging. Instead, spermidine administration may influence histone acetyltransferase activity to modulate autophagy (Simonsen and Tooze, 2009; Davis, 2013; Graff and Tsai, 2013). These findings indicate that although the protective effect of spermidine does require activation of the autophagy pathway, the involvement of additional regulatory pathways remains to be elucidated. In conclusion, the exact mechanism by which autophagy controls cognitive aging is multifaceted and remains poorly understood. Additional studies are required to elucidate the contribution of autophagy in both longevity and cognitive capacity maintenance during aging.
Tor Signaling and Long-Term Memory
Reduced signaling through the target of rapamycin (TOR) kinase has been shown to extend lifespan in diverse organisms (Vellai et al., 2003; Jia et al., 2004; Kapahi et al., 2004; Kaeberlein et al., 2005; Powers et al., 2006; Kenyon, 2010). The evolutionarily conserved mTOR functions in two complexes, mTORC1 and mTORC2 (Hay and Sonenberg, 2004; Wullschleger et al., 2006; Guertin and Sabatini, 2007). Tight regulation of the upstream components of the TOR pathway is important for proper neural growth and function throughout development and adulthood in C. elegans (Goldsmith et al., 2010). Overexpression of the small GTPase RAS homolog enriched in brain (Rheb), an upstream activator of TOR, in Drosophila photoreceptor cells downregulates autophagy, causes axon guidance defects and induces cell death (Knox et al., 2007; Wang et al., 2009). Selective overexpression of Rheb in distinct subsets of central brain neurons results in enlarged cell bodies and projections. In addition, Rheb overexpression in the mushroom bodies decreases mid-term odor-sucrose memory (Brown et al., 2012).
In the PIP3/PTEN/Akt/TOR pathway phosphorylated Akt activates TOR to regulate cell cycle and protein synthesis. In flies, the PTEN/Akt pathway is implicated in axon regeneration (Song et al., 2012). Similarly, axon regeneration is evident after the loss of DAF-18/PTEN in young adult worms (Byrne et al., 2014). Reduced TORC2 activity causes LTM deterioration in fruit flies (Huang et al., 2013). Rapamycin, a protein synthesis inhibitor that acts through the TOR pathway (mainly mTORC1), blocks long-term facilitation (LTF) in Aplysia californica (Hu et al., 2006). Moreover, rapamycin completely disrupts pre-existing long-term synaptic plasticity in Aplysia (Hu et al., 2011). While rapamycin extends lifespan mainly by blocking the TOR pathway, it may exert its effects on cognition through a different mechanism (Neff et al., 2013).
The TOR pathway controls translation of 5′TOP mRNAs containing a 5′ terminal oligopyrimidine tract. 5′TOP mRNAs encode proteins of the translational machinery. Under physiological conditions, 5′TOP mRNAs are largely repressed. Serotonin, which activates the TOR pathway, aleviates this repression, in a rapamycin-sensitive manner (Garelick and Kennedy, 2011; Labban and Sossin, 2011). eEF2 (eukaryotic elongation factor 2) is implicated in LTF in Aplysia, but is differentially regulated by eEF2 kinase in the neurites and the soma of sensory neurons involved in LTF (Weatherill et al., 2011). TORC1 mediates regulation of phosphorylation of eEF2 through the eEF2K (Carroll et al., 2004). Both in Aplysia, and in rodents, eEF2K function is associated with increased memory processing, through enhancing expression of genes implicated in the regulation of syntaptic strength (Weatherill et al., 2011).
Similarly, long term administration of rapamycin eliminates neuronal demyelination and neurodegeneration observed during aging in senescence-accelerated OXYS rats, a strain characterized by overproduction of free radicals (Kolosova et al., 2013). In mouse models of AD, rapamycin administrated either prior or after the onset of AD symptoms, improves animal cognition, probably through the preservation of brain vascular integrity and function (Lin et al., 2013b). Moreover, chronic treatment with rapamycin enhances spatial learning and memory with age, as well as the ability to recall a memory, even when the administration takes place late in life (Halloran et al., 2012). However, short-term administration following the emergence of learning and memory defects with aging, is not accompanied by such positive effects. The improvement of cognitive ability with rapamycin is mediated through reduction of TOR signaling and of IL-1β levels in the hippocampus, the facilitation of NMDA signaling, and increased CREB phosphorylation (Majumder et al., 2012). Furthermore, increased phosphorylation of S6, a target of TOR, is observed in the prefrontal cortex, after the administration of rapamycin, in OXYS rats (Kolosova et al., 2013).
Conclusion
Understanding how neuronal aging and cognitive impairment are influenced by mechanisms that modulate lifespan is an ongoing challenge. Such well-studied mechanisms include the IIS signaling pathway, DR, mitochondrial dysfunction, autophagy, and the TOR signaling pathway. Accumulating evidence indicates that these pathways also impinge on age-related neuronal dysfunction and memory-impairment. Indeed, manipulation of these pathways in a variety of metazoans affects neuronal structure and function and consequently promotes age-related memory impairment. It is likely that the decline in different forms of memory is independently mediated by distinct aging mechanisms (Figure 1). Decreased IIS signaling promotes decision making and associative learning. However this is not a general rule and, instead, appears to be dependent on different types of association. Nonetheless, DAF-16 activation likely delays morphological changes that occur with aging and promotes neuronal regeneration. DR exerts negative effects on LTM but enhances association making and memory. DR effects on short-term and mid-term memory appear to be age dependent.
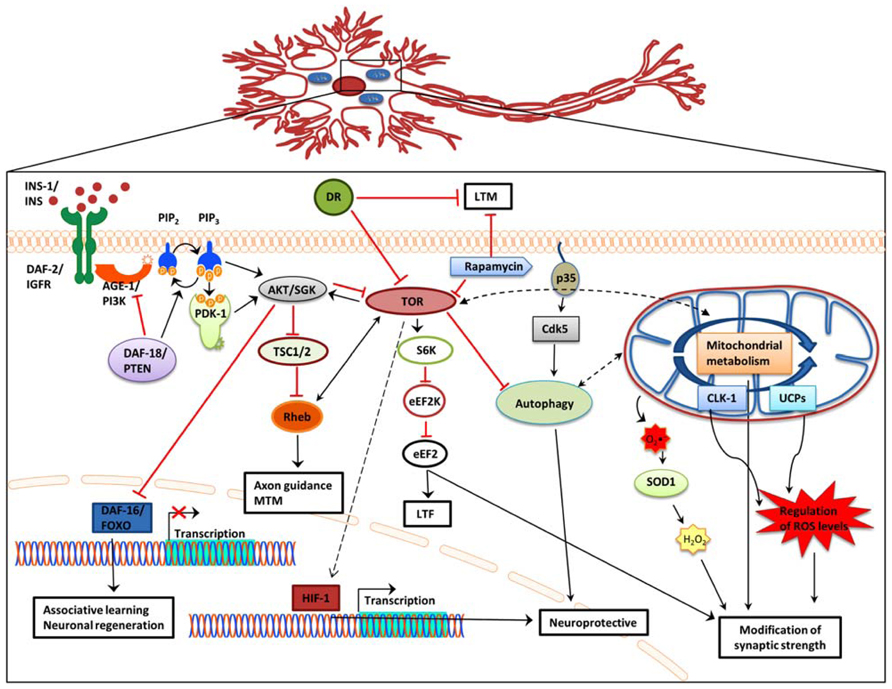
FIGURE 1. Effects of IIS, DR, and TOR signaling, autophagy, and mitochondrial function on memory formation. In addition to extending lifespan, attenuation of IIS signaling, and subsequent de-repression of DAF-16 also reinforces associative learning and promotes axon regeneration and neuronal migration. Dietary restriction significantly impairs long-term memory, while it does not affect short-term memory. Blocking TOR signaling causes long-term-facilitation defects, while Rheb overexpression decreases mid-term memory and causes axon guidance defects. Regulation of autophagy through Cdk-5 affects lifespan and has also been associated with central nervous system structure defects, including olfactory learning and memory. Several mitochondrial genes promote neuroprotection. Impairment of mitochondrial function causes over production of ROS and concomitant synaptic deficiency. AGE-1/PI3K,phosphoinositide 3-kinase; AKT, protein kinase B; Cdk5, cyclin-dependent kinase 5; CLK-1, clock 1; DAF-2/IGFR, insulin-like growth factor receptor; DAF-16/FOXO, forkhead box O; DAF-18/PTEN, phosphatase and ensin homolog; DR, dietary restriction; eEF2, eukaryotic elongation factor 2; eEF2K, eukaryotic elongation factor 2 kinase; HIF-1, hypoxia-induced factor 1; INS, insulin; LTF, long-term facilitation; LTM, long-term memory; MTM, mid-term memory; PDK-1, 3-phosphoinositide-dependent kinase 1; PIP2, phosphatidylinositol (4,5)-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; Rheb, Ras homolog enriched in brain; S6K, ribosomal protein S6 kinase; SGK, serum- and glucocorticoid-inducible kinase; SOD1, superoxide dismutase 1; TSC1/2, tuberous sclerosis 1/2; TOR, target of rapamycin; UCPs, uncoupling proteins; Black arrow, direct stimulation; black dashed arrow, indirect stimulation; black dashed double head arrow, interplay; red arrow, inhibition.
The TOR pathway influences cognition by controlling protein biosynthesis, cell cycle, and metabolism (Garelick and Kennedy, 2011; Santos et al., 2011). Studies in invertebrates suggest that increased TOR signaling downregulates autophagy and causes axon guidance defects, while also promoting memory processing and synapse integrity. Conversely, in addition to increasing lifespan, downregulation of TOR signaling, mainly through rapamycin treatment, blocks LTM and LTF but also causes axon regeneration. In rodents rapamycin administration offsets the negative impact of aging on spatial learning and memory, increases memory recall ability, and enhances the vascular integrity of the brain. Moreover, morphological signs of AD and aging, such as neuronal demyelination and neurodegeneration are ameliorated. Activation of mTORC1 promotes mRNA translation, which likely enhances synapse formation. These distinct, but not necessarily conflicting results of manipulating TOR signaling could reflect a dose or compartment dependent regulation of cognition through TOR signaling.
In both worms and flies, activation of autophagy appears to promote lifespan and cognitive function. In mammals, little it is known about the role of autophagy during aging. However, recent studies in mice suggest that overexpression of autophagy-related gene Atg5 also increases lifespan (Pyo et al., 2013). Manipulation of mitochondria function during aging causes similar effects, either causing axon degeneration, or promoting axon integrity and stability (Keller et al., 2011; Fang et al., 2012). These ostensibly contradictory observations may suggest a neuron-specific function of mitochondria in aging. Despite recent progress and findings, several open questions need to be addressed. The involvement of epigenetic mechanisms and environmental conditions on nervous system aging is largely unknown. In addition, whether aging differentially affects subpopulations of neurons or different brain areas and to what extent remains unclear. While significant progress has been achieved towards deciphering the link between pathways that modulate both lifespan and aspects of neuronal function in invertebrate models, the relevance of these findings to neuronal aging and pathophysiology in higher organisms including humans has not been evaluated yet. Addressing these key issues will contribute towards developing informed strategies and therapeutic approaches aiming to battle age-related decline of nervous system performance and numerous neurodegenerative conditions associated with aging.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Maria Markaki for discussions and comments on the manuscript. Work in the authors’ laboratory is funded by grants from the European Research Council (ERC), the European Commission Framework Programmes, and the Greek Ministry of Education. Ilias Gkikas is supported by the European Union Seventh Framework program through the Marie Curie Initial Training fellowship (Ageing Network – MarriAge). Dionysia Petratou is supported jointly by the European Union (European Social Fund – ESF) and Greek national program “THALES-MINOS” of the National Strategic Reference Framework (NSRF).
References
Abdelwahid, E., Rolland, S., Teng, X., Conradt, B., Hardwick, J. M., and White, K. (2011). Mitochondrial involvement in cell death of non-mammalian eukaryotes. Biochim. Biophys. Acta 1813, 597–607. doi: 10.1016/j.bbamcr.2010.10.008
Adams, M. M., Shi, L., Linville, M. C., Forbes, M. E., Long, A. B., Bennett, C., et al. (2008). Caloric restriction and age affect synaptic proteins in hippocampal CA3 and spatial learning ability. Exp. Neurol. 211, 141–149. doi: 10.1016/j.expneurol.2008.01.016
Aksenov, V., Long, J., Liu, J., Szechtman, H., Khanna, P., Matravadia, S., et al. (2013). A complex dietary supplement augments spatial learning, brain mass, and mitochondrial electron transport chain activity in aging mice. Age (Dordr.) 35, 23–33. doi: 10.1007/s11357-011-9325-2
Ardiel, E. L., and Rankin, C. H. (2010). An elegant mind: learning and memory in Caenorhabditis elegans. Learn. Mem. 17, 191–201. doi: 10.1101/lm.960510
Back, P., Matthijssens, F., Vlaeminck, C., Braeckman, B. P., and Vanfleteren, J. R. (2010). Effects of sod gene overexpression and deletion mutation on the expression profiles of reporter genes of major detoxification pathways in Caenorhabditis elegans. Exp. Gerontol. 45, 603–610. doi: 10.1016/j.exger.2010.01.014
Bahadorani, S., Mukai, S. T., Rabie, J., Beckman, J. S., Phillips, J. P., and Hilliker, A. J. (2013). Expression of zinc-deficient human superoxide dismutase in Drosophila neurons produces a locomotor defect linked to mitochondrial dysfunction. Neurobiol. Aging 34, 2322–2330. doi: 10.1016/j.neurobiolaging.2013.03.024
Barco, A., Bailey, C. H., and Kandel, E. R. (2006). Common molecular mechanisms in explicit and implicit memory. J. Neurochem. 97, 1520–1533. doi: 10.1111/j.1471-4159.2006.03870.x
Berman, S. B., Chen, Y. B., Qi, B., McCaffery, J. M., Rucker, E. B. III, Goebbels, S., et al. (2009). Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J. Cell Biol. 184, 707–719. doi: 10.1083/jcb.200809060
Besson, M. T., Dupont, P., Fridell, Y. W., and Lievens, J. C. (2010). Increased energy metabolism rescues glia-induced pathology in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 19, 3372–3382. doi: 10.1093/hmg/ddq249
Bishop, N. A., Lu, T., and Yankner, B. A. (2010). Neural mechanisms of ageing and cognitive decline. Nature 464, 529–535. doi: 10.1038/nature08983
Bluher, M., Kahn, B. B., and Kahn, C. R. (2003). Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 299, 572–574. doi: 10.1126/science.1078223
Bokov, A. F., Garg, N., Ikeno, Y., Thakur, S., Musi, N., DeFronzo, R. A., et al. (2011). Does reduced IGF-1R signaling in Igf1r+/– mice alter aging? PLoS ONE 6:e26891. doi: 10.1371/journal.pone.0026891
Breckenridge, D. G., Kang, B. H., and Xue, D. (2009). Bcl-2 proteins EGL-1 and CED-9 do not regulate mitochondrial fission or fusion in Caenorhabditis elegans. Curr. Biol. 19, 768–773. doi: 10.1016/j.cub.2009.03.022
Brown, H. L., Kaun, K. R., and Edgar, B. A. (2012). The small GTPase Rheb affects central brain neuronal morphology and memory formation in Drosophila. PLoS ONE 7:e44888. doi: 10.1371/journal.pone.0044888
Burger, J. M., Buechel, S. D., and Kawecki, T. J. (2010). Dietary restriction affects lifespan but not cognitive aging in Drosophila melanogaster. Aging Cell 9, 327–335. doi: 10.1111/j.1474-9726.2010.00560.x
Byrne, A. B., Walradt, T., Gardner, K. E., Hubbert, A., Reinke, V., and Hammarlund, M. (2014). Insulin/IGF1 signaling inhibits age-dependent axon regeneration. Neuron 81, 561–573. doi: 10.1016/j.neuron.2013.11.019
Cabreiro, F., Ackerman, D., Doonan, R., Araiz, C., Back, P., Papp, D., et al. (2011). Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radic. Biol. Med. 51, 1575–1582. doi: 10.1016/j.freeradbiomed.2011.07.020
Carroll, M., Warren, O., Fan, X., and Sossin, W. S. (2004). 5-HT stimulates eEF2 dephosphorylation in a rapamycin-sensitive manner in Aplysia neurites. J. Neurochem. 90, 1464–1476. doi: 10.1111/j.1471-4159.2004.02634.x
Cava, E., and Fontana, L. (2013). Will calorie restriction work in humans? Aging (Albany NY) 5, 507–514.
Chamoli, M., Singh, A., Malik, Y., and Mukhopadhyay, A. (2014). A novel kinase regulates dietary restriction-mediated longevity in Caenorhabditis elegans. Aging Cell. doi: 10.1111/acel.12218 [Epub ahead of print].
Chen, C. H., Chen, Y. C., Jiang, H. C., Chen, C. K., and Pan, C. L. (2013). Neuronal aging: learning from C. elegans. J. Mol. Signal. 8, 14. doi: 10.1186/1750-2187-8-14
Christensen, R., de la Torre-Ubieta, L., Bonni, A., and Colon-Ramos, D. A. (2011). A conserved PTEN/FOXO pathway regulates neuronal morphology during C. elegans development. Development 138, 5257–5267. doi: 10.1242/dev.069062
Clancy, D. J., Gems, D., Harshman, L. G., Oldham, S., Stocker, H., Hafen, E., et al. (2001). Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 292, 104–106. doi: 10.1126/science.1057991
Cohen, E., Paulsson, J. F., Blinder, P., Burstyn-Cohen, T., Du, D., Estepa, G., et al. (2009). Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 139, 1157–1169. doi: 10.1016/j.cell.2009.11.014
Colman, R. J., Anderson, R. M., Johnson, S. C., Kastman, E. K., Kosmatka, K. J., Beasley, T. M., et al. (2009). Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 325, 201–204. doi: 10.1126/science.1173635
Colman, R. J., Beasley, T. M., Kemnitz, J. W., Johnson, S. C., Weindruch, R., and Anderson, R. M. (2014). Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 5, 3557. doi: 10.1038/ncomms4557
Copeland, J. M., Cho, J., Lo, T., Hur, J. H., Bahadorani, S., Arabyan, T., et al. (2009). Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 19, 1591–1598. doi: 10.1016/j.cub.2009.08.016
David, D. C., Ollikainen, N., Trinidad, J. C., Cary, M. P., Burlingame, A. L., and Kenyon, C. (2010). Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 8:e1000450. doi: 10.1371/journal.pbio.1000450
Davis, R. L. (2013). Spermidine cures flies of senior moments. Nat. Neurosci. 16, 1363–1364. doi: 10.1038/nn.3518
Deepa, S. S., Pulliam, D., Hill, S., Shi, Y., Walsh, M. E., Salmon, A., et al. (2013). Improved insulin sensitivity associated with reduced mitochondrial complex IV assembly and activity. FASEB J. 27, 1371–1380. doi: 10.1096/fj.12-221879
Eisenberg, T., Knauer, H., Schauer, A., Buttner, S., Ruckenstuhl, C., Carmona-Gutierrez, D., et al. (2009). Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314. doi: 10.1038/ncb1975
Ewald, C. Y., Cheng, R., Tolen, L., Shah, V., Gillani, A., Nasrin, A., et al. (2012). Pan-neuronal expression of APL-1, an APP-related protein, disrupts olfactory, gustatory, and touch plasticity in Caenorhabditis elegans. J. Neurosci. 32, 10156–10169. doi: 10.1523/JNEUROSCI.0495-12.2012
Ewald, C. Y., and Li, C. (2012). Caenorhabditis elegans as a model organism to study APP function. Exp. Brain Res. 217, 397–411. doi: 10.1007/s00221-011-2905-7
Fang, Y., Soares, L., Teng, X., Geary, M., and Bonini, N. M. (2012). A novel Drosophila model of nerve injury reveals an essential role of Nmnat in maintaining axonal integrity. Curr. Biol. 22, 590–595. doi: 10.1016/j.cub.2012.01.065
Florez-McClure, M. L., Hohsfield, L. A., Fonte, G., Bealor, M. T., and Link, C. D. (2007). Decreased insulin-receptor signaling promotes the autophagic degradation of beta-amyloid peptide in C. elegans. Autophagy 3, 569–580.
Galindo, K. A., Lu, W. J., Park, J. H., and Abrams, J. M. (2009). The Bax/Bak ortholog in Drosophila, Debcl, exerts limited control over programmed cell death. Development 136, 275–283. doi: 10.1242/dev.019042
Garelick, M. G., and Kennedy, B. K. (2011). TOR on the brain. Exp. Gerontol. 46, 155–163. doi: 10.1016/j.exger.2010.08.030
Goldsmith, A. D., Sarin, S., Lockery, S., and Hobert, O. (2010). Developmental control of lateralized neuron size in the nematode Caenorhabditis elegans. Neural Dev. 5, 33. doi: 10.1186/1749-8104-5-33
Graff, J., and Tsai, L. H. (2013). Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 14, 97–111. doi: 10.1038/nrn3427
Guertin, D. A., and Sabatini, D. M. (2007). Defining the role of mTOR in cancer. Cancer Cell 12, 9–22. doi: 10.1016/j.ccr.2007.05.008
Gupta, V. K., Scheunemann, L., Eisenberg, T., Mertel, S., Bhukel, A., Koemans, T. S., et al. (2013). Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat. Neurosci. 16, 1453–1460. doi: 10.1038/nn.3512
Haddadi, M., Jahromi, S. R., Sagar, B. K., Patil, R. K., Shivanandappa, T., and Ramesh, S. R. (2014). Brain aging, memory impairment and oxidative stress: a study in Drosophila melanogaster. Behav. Brain Res. 259, 60–69. doi: 10.1016/j.bbr.2013.10.036
Halloran, J., Hussong, S. A., Burbank, R., Podlutskaya, N., Fischer, K. E., Sloane, L. B., et al. (2012). Chronic inhibition of mammalian target of rapamycin by rapamycin modulates cognitive and non-cognitive components of behavior throughout lifespan in mice. Neuroscience 223, 102–113. doi: 10.1016/j.neuroscience.2012.06.054
Hansen, M., Chandra, A., Mitic, L. L., Onken, B., Driscoll, M., and Kenyon, C. (2008). A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 4:e24. doi: 10.1371/journal.pgen.0040024
Hardwick, J. M., and Soane, L. (2013). Multiple functions of BCL-2 family proteins. Cold Spring Harb. Perspect. Biol. 5. doi: 10.1101/cshperspect.a008722
Harman, D. (1968). Free radical theory of aging: effect of free radical reaction inhibitors on the mortality rate of male LAF mice. J. Gerontol. 23, 476–482. doi: 10.1093/geronj/23.4.476
Hay, N., and Sonenberg, N. (2004). Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945. doi: 10.1101/gad.1212704
Hu, J. Y., Baussi, O., Levine, A., Chen, Y., and Schacher, S. (2011). Persistent long-term synaptic plasticity requires activation of a new signaling pathway by additional stimuli. J. Neurosci. 31, 8841–8850. doi: 10.1523/JNEUROSCI.1358-11.2011
Hu, J. Y., Wu, F., and Schacher, S. (2006). Two signaling pathways regulate the expression and secretion of a neuropeptide required for long-term facilitation in Aplysia. J. Neurosci. 26, 1026–1035. doi: 10.1523/JNEUROSCI.4258-05.2006
Huang, W., Zhu, P. J., Zhang, S., Zhou, H., Stoica, L., Galiano, M., et al. (2013). mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat. Neurosci. 16, 441–448. doi: 10.1038/nn.3351
Islam, R., Yang, L., Sah, M., Kannan, K., Anamani, D., Vijayan, C., et al. (2012). A neuroprotective role of the human uncoupling protein 2 (hUCP2) in a Drosophila Parkinson’s disease model. Neurobiol. Dis. 46, 137–146. doi: 10.1016/j.nbd.2011.12.055
Jia, K., Chen, D., and Riddle, D. L. (2004). The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 131, 3897–3906. doi: 10.1242/dev.01255
Jiu, Y. M., Yue, Y., Yang, S., Liu, L., Yu, J. W., Wu, Z. X., et al. (2010). Insulin-like signaling pathway functions in integrative response to an olfactory and a gustatory stimuli in Caenorhabditis elegans. Protein Cell 1, 75–81. doi: 10.1007/s13238-010-0003-4
Kaeberlein, M., Powers, R. W. III, Steffen, K. K., Westman, E. A., Hu, D., Dang, N., et al. (2005). Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310, 1193–1196. doi: 10.1126/science.1115535
Kapahi, P., Zid, B. M., Harper, T., Koslover, D., Sapin, V., and Benzer, S. (2004). Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 14, 885–890. doi: 10.1016/j.cub.2004.03.059
Kauffman, A. L., Ashraf, J. M., Corces-Zimmerman, M. R., Landis, J. N., and Murphy, C. T. (2010). Insulin signaling and dietary restriction differentially influence the decline of learning and memory with age. PLoS Biol. 8:e1000372. doi: 10.1371/journal.pbio.1000372
Kayser, E. B., Sedensky, M. M., Morgan, P. G., and Hoppel, C. L. (2004). Mitochondrial oxidative phosphorylation is defective in the long-lived mutant clk-1. J. Biol. Chem. 279, 54479–54486. doi: 10.1074/jbc.M403066200
Keller, L. C., Cheng, L., Locke, C. J., Muller, M., Fetter, R. D., and Davis, G. W. (2011). Glial-derived prodegenerative signaling in the Drosophila neuromuscular system. Neuron 72, 760–775. doi: 10.1016/j.neuron.2011.09.031
Kennedy, L. M., Pham, S. C., and Grishok, A. (2013). Nonautonomous regulation of neuronal migration by insulin signaling, DAF-16/FOXO, and PAK-1. Cell Rep. 4, 996–1009. doi: 10.1016/j.celrep.2013.07.045
Kenyon, C., Chang, J., Gensch, E., Rudner, A., and Tabtiang, R. (1993). A C. elegans mutant that lives twice as long as wild type. Nature 366, 461–464. doi: 10.1038/366461a0
Keowkase, R., Aboukhatwa, M., and Luo, Y. (2010). Fluoxetine protects against amyloid-beta toxicity, in part via daf-16 mediated cell signaling pathway, in Caenorhabditis elegans. Neuropharmacology 59, 358–365. doi: 10.1016/j.neuropharm.2010.04.008
Kim, G. W., Li, L., Gorbani, M., You, L., and Yang, X. J. (2013). Mice lacking alpha-tubulin acetyltransferase 1 are viable but display alpha-tubulin acetylation deficiency and dentate gyrus distortion. J. Biol. Chem. 288, 20334–20350. doi: 10.1074/jbc.M113.464792
Kimata, T., Sasakura, H., Ohnishi, N., Nishio, N., and Mori, I. (2012). Thermotaxis of C. elegans as a model for temperature perception, neural information processing and neural plasticity. Worm 1, 31–41. doi: 10.4161/worm.19504
Knox, S., Ge, H., Dimitroff, B. D., Ren, Y., Howe, K. A., Arsham, A. M., et al. (2007). Mechanisms of TSC-mediated control of synapse assembly and axon guidance. PLoS ONE 2:e375. doi: 10.1371/journal.pone.0000375
Kodama, E., Kuhara, A., Mohri-Shiomi, A., Kimura, K. D., Okumura, M., Tomioka, M., et al. (2006). Insulin-like signaling and the neural circuit for integrative behavior in C. elegans. Genes Dev. 20, 2955–2960. doi: 10.1101/gad.1479906
Kolosova, N. G., Vitovtov, A. O., Muraleva, N. A., Akulov, A. E., Stefanova, N. A., and Blagosklonny, M. V. (2013). Rapamycin suppresses brain aging in senescence-accelerated OXYS rats. Aging (Albany NY) 5, 474–484.
Labban, M., and Sossin, W. S. (2011). Translation of 5′ terminal oligopyrimidine tract (5′TOP) mRNAs in Aplysia californica is regulated by the target of rapamycin (TOR). Biochem. Biophys. Res. Commun. 404, 816–821. doi: 10.1016/j.bbrc.2010.12.066
Lakowski, B., and Hekimi, S. (1998). The genetics of caloric restriction in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 95, 13091–13096. doi: 10.1073/pnas.95.22.13091
Lapointe, J., and Hekimi, S. (2008). Early mitochondrial dysfunction in long-lived Mclk1+/– mice. J. Biol. Chem. 283, 26217–26227. doi: 10.1074/jbc.M803287200
Li, J., Huang, K. X., and Le, W. D. (2013). Establishing a novel C. elegans model to investigate the role of autophagy in amyotrophic lateral sclerosis. Acta pharmacol. Sin. 34, 644–650. doi: 10.1038/aps.2012.190
Lin, A. L., Pulliam, D. A., Deepa, S. S., Halloran, J. J., Hussong, S. A., Burbank, R. R., et al. (2013a). Decreased in vitro mitochondrial function is associated with enhanced brain metabolism, blood flow, and memory in Surf1-deficient mice. J. Cereb. Blood Flow Metab. 33, 1605–1611. doi: 10.1038/jcbfm.2013.116
Lin, A. L., Zheng, W., Halloran, J. J., Burbank, R. R., Hussong, S. A., Hart, M. J., et al. (2013b). Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J. Cereb. Blood Flow Metab. 33, 1412–1421. doi: 10.1038/jcbfm.2013.82
Lin, C. H., Tomioka, M., Pereira, S., Sellings, L., Iino, Y., and van der Kooy, D. (2010). Insulin signaling plays a dual role in Caenorhabditis elegans memory acquisition and memory retrieval. J. Neurosci. 30, 8001–8011. doi: 10.1523/JNEUROSCI.4636-09.2010
Ling, D., Song, H. J., Garza, D., Neufeld, T. P., and Salvaterra, P. M. (2009). Abeta42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS ONE 4:e4201. doi: 10.1371/journal.pone.0004201
Lionaki, E., Markaki, M., and Tavernarakis, N. (2013). Autophagy and ageing: insights from invertebrate model organisms. Ageing Res. Rev. 12, 413–428. doi: 10.1016/j.arr.2012.05.001
Low, P., Varga, A., Pircs, K., Nagy, P., Szatmari, Z., Sass, M., et al. (2013). Impaired proteasomal degradation enhances autophagy via hypoxia signaling in Drosophila. BMC Cell Biol. 14, 29. doi: 10.1186/1471-2121-14-29
Mair, W., and Dillin, A. (2008). Aging and survival: the genetics of life span extension by dietary restriction. Annu. Rev. Biochem. 77, 727–754. doi: 10.1146/annurev.biochem.77.061206.171059
Majumder, S., Caccamo, A., Medina, D. X., Benavides, A. D., Javors, M. A., Kraig, E., et al. (2012). Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1beta and enhancing NMDA signaling. Aging Cell 11, 326–335. doi: 10.1111/j.1474-9726.2011.00791.x
Masoro, E. J. (2005). Overview of caloric restriction and ageing. Mech. Ageing Dev. 126, 913–922. doi: 10.1016/j.mad.2005.03.012
Mattison, J. A., Roth, G. S., Beasley, T. M., Tilmont, E. M., Handy, A. M., Herbert, R. L., et al. (2012). Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 489, 318–321. doi: 10.1038/nature11432
Maxwell, S., Harding, J., Brabin, C., Appleford, P. J., Brown, R., Delaney, C., et al. (2013). The SFT-1 and OXA-1 respiratory chain complex assembly factors influence lifespan by distinct mechanisms in C. elegans. Longev. Healthspan 2, 9. doi: 10.1186/2046-2395-2-9
McCord, J. M., and Fridovich, I. (1969). Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244, 6049–6055.
Mochizuki, H., Toda, H., Ando, M., Kurusu, M., Tomoda, T., and Furukubo-Tokunaga, K. (2011). Unc-51/ATG1 controls axonal and dendritic development via kinesin-mediated vesicle transport in the Drosophila brain. PLoS ONE 6:e19632. doi: 10.1371/journal.pone.0019632
Murakami, H., Bessinger, K., Hellmann, J., and Murakami, S. (2005). Aging-dependent and -independent modulation of associative learning behavior by insulin/insulin-like growth factor-1 signal in Caenorhabditis elegans. J. Neurosci. 25, 10894–10904. doi: 10.1523/JNEUROSCI.3600-04.2005
Murakami, S. (2007). Caenorhabditis elegans as a model system to study aging of learning and memory. Mol. Neurobiol. 35, 85–94. doi: 10.1007/BF02700625
Neff, F., Flores-Dominguez, D., Ryan, D. P., Horsch, M., Schroder, S., Adler, T., et al. (2013). Rapamycin extends murine lifespan but has limited effects on aging. J. Clin. Invest. 123, 3272–3291. doi: 10.1172/JCI67674
Neumann, B., and Hilliard, M. A. (2014). Loss of MEC-17 Leads to microtubule instability and axonal degeneration. Cell Rep. 6, 93–103. doi: 10.1016/j.celrep.2013.12.004
Nilsson, P., Loganathan, K., Sekiguchi, M., Matsuba, Y., Hui, K., Tsubuki, S., et al. (2013). Abeta secretion and plaque formation depend on autophagy. Cell Rep. 5, 61–69. doi: 10.1016/j.celrep.2013.08.042
Panowski, S. H., Wolff, S., Aguilaniu, H., Durieux, J., and Dillin, A. (2007). PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature 447, 550–555. doi: 10.1038/nature05837
Parker, J. A., Vazquez-Manrique, R. P., Tourette, C., Farina, F., Offner, N., Mukhopadhyay, A., et al. (2012). Integration of beta-catenin, sirtuin, and FOXO signaling protects from mutant huntingtin toxicity. J. Neurosci. 32, 12630–12640. doi: 10.1523/JNEUROSCI.0277-12.2012
Partridge, L. (2010). The new biology of ageing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 147–154. doi: 10.1098/rstb.2009.0222
Perluigi, M., and Butterfield, D. A. (2012). Oxidative stress and Down syndrome: a route toward Alzheimer-like dementia. Curr. Gerontol. Geriatr. Res. 2012, 724904. doi: 10.1155/2012/724904
Piper, M. D., and Bartke, A. (2008). Diet and aging. Cell Metab. 8, 99–104. doi: 10.1016/j.cmet.2008.06.012
Powers, R. W. III, Kaeberlein, M., Caldwell, S. D., Kennedy, B. K., and Fields, S. (2006). Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 20, 174–184. doi: 10.1101/gad.1381406
Pyo, J. O., Yoo, S. M., Ahn, H. H., Nah, J., Hong, S. H., Kam, T. I., et al. (2013). Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 4, 2300. doi: 10.1038/ncomms3300
Ragagnin, A., Guillemain, A., Grant, N. J., and Bailly, Y. J. R. (2013). “Neuronal autophagy and prion proteins,” in Autophagy - A Double-Edged Sword - Cell Survival or Death?, ed. Y. Bailly (Croatia: InTech), 377–419.
Raizen, D. M., Lee, R. Y., and Avery, L. (1995). Interacting genes required for pharyngeal excitation by motor neuron MC in Caenorhabditis elegans. Genetics 141, 1365–1382.
Rawson, R. L., Yam, L., Weimer, R. M., Bend, E. G., Hartwieg, E., Horvitz, H. R., et al. (2014). Axons degenerate in the absence of mitochondria in C. elegans. Curr. Biol. 24, 760–765. doi: 10.1016/j.cub.2014.02.025
Rea, S. L., Ventura, N., and Johnson, T. E. (2007). Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 5:e259. doi: 10.1371/journal.pbio.0050259
Rolland, S. G., Lu, Y., David, C. N., and Conradt, B. (2009). The BCL-2-like protein CED-9 of C. elegans promotes FZO-1/Mfn1,2- and EAT-3/Opa1-dependent mitochondrial fusion. J. Cell Biol. 186, 525–540. doi: 10.1083/jcb.200905070
Sagi, D., and Kim, S. K. (2012). An engineering approach to extending lifespan in C. elegans. PLoS Genet. 8:e1002780. doi: 10.1371/journal.pgen.1002780
Sahay, A., Scobie, K. N., Hill, A. S., O’Carroll, C. M., Kheirbek, M. A., Burghardt, N. S., et al. (2011). Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature 472, 466–470. doi: 10.1038/nature09817
Santos, R. X., Correia, S. C., Cardoso, S., Carvalho, C., Santos, M. S., and Moreira, P. I. (2011). Effects of rapamycin and TOR on aging and memory: implications for Alzheimer’s disease. J. Neurochem. 117, 927–936. doi: 10.1111/j.1471-4159.2011.07262.x
Senoo-Matsuda, N., Igaki, T., and Miura, M. (2005). Bax-like protein Drob-1 protects neurons from expanded polyglutamine-induced toxicity in Drosophila. EMBO J. 24, 2700–2713. doi: 10.1038/sj.emboj.7600721
Shin, J. H., London, J., Le Pecheur, M., Hoger, H., Pollak, D., and Lubec, G. (2004). Aberrant neuronal and mitochondrial proteins in hippocampus of transgenic mice overexpressing human Cu/Zn superoxide dismutase 1. Free Radic. Biol. Med. 37, 643–653. doi: 10.1016/j.freeradbiomed.2004.05.019
Sigmond, T., Feher, J., Baksa, A., Pasti, G., Palfia, Z., Takacs-Vellai, K., et al. (2008). Qualitative and quantitative characterization of autophagy in Caenorhabditis elegans by electron microscopy. Methods Enzymol. 451, 467–491. doi: 10.1016/S0076-6879(08)03228-X
Simonsen, A., Cumming, R. C., Brech, A., Isakson, P., Schubert, D. R., and Finley, K. D. (2008). Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4, 176–184.
Simonsen, A., and Tooze, S. A. (2009). Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell Biol. 186, 773–782. doi: 10.1083/jcb.200907014
Song, Y., Ori-McKenney, K. M., Zheng, Y., Han, C., Jan, L. Y., and Jan, Y. N. (2012). Regeneration of Drosophila sensory neuron axons and dendrites is regulated by the Akt pathway involving Pten and microRNA bantam. Genes Dev. 26, 1612–1625. doi: 10.1101/gad.193243.112
Stein, G. M., and Murphy, C. T. (2012). The intersection of aging, longevity pathways, and learning and memory in C. elegans. Front. Genet. 3:259. doi: 10.3389/fgene.2012.00259
Suh, Y., Atzmon, G., Cho, M. O., Hwang, D., Liu, B., Leahy, D. J., et al. (2008). Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc. Natl. Acad. Sci. U.S.A. 105, 3438–3442. doi: 10.1073/pnas.0705467105
Takahashi, M., Ogawara, M., Shimizu, T., and Shirasawa, T. (2012). Restoration of the behavioral rates and lifespan in clk-1 mutant nematodes in response to exogenous coenzyme Q(10). Exp. Gerontol. 47, 276–279. doi: 10.1016/j.exger.2011.12.012
Tamura, T., Sone, M., Nakamura, Y., Shimamura, T., Imoto, S., Miyano, S., et al. (2013). A restricted level of PQBP1 is needed for the best longevity of Drosophila. Neurobiol. Aging 34, 356.e311–356.e320. doi: 10.1016/j.neurobiolaging.2012.07.015
Tank, E. M. H., Rodgers, K. E., and Kenyon, C. (2011). Spontaneous age-related neurite branching in Caenorhabditis elegans. J. Neurosci. 31, 9279–9288. doi: 10.1523/JNEUROSCI.6606-10.2011
Tatar, M., Kopelman, A., Epstein, D., Tu, M. P., Yin, C. M., and Garofalo, R. S. (2001). A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 292, 107–110. doi: 10.1126/science.1057987
Tomioka, M., Adachi, T., Suzuki, H., Kunitomo, H., Schafer, W. R., and Iino, Y. (2006). The insulin/PI 3-kinase pathway regulates salt chemotaxis learning in Caenorhabditis elegans. Neuron 51, 613–625. doi: 10.1016/j.neuron.2006.07.024
Toth, M. L., Melentijevic, I., Shah, L., Bhatia, A., Lu, K., Talwar, A., et al. (2012). Neurite sprouting and synapse deterioration in the aging Caenorhabditis elegans nervous system. J. Neurosci. 32, 8778–8790. doi: 10.1523/JNEUROSCI.1494-11.2012
Trunova, S., and Giniger, E. (2012). Absence of the Cdk5 activator p35 causes adult-onset neurodegeneration in the central brain of Drosophila. Dis. Model. Mech. 5, 210–219. doi: 10.1242/dmm.008847
Vellai, T., McCulloch, D., Gems, D., and Kovacs, A. L. (2006). Effects of sex and insulin/insulin-like growth factor-1 signaling on performance in an associative learning paradigm in Caenorhabditis elegans. Genetics 174, 309–316. doi: 10.1534/genetics.106.061499
Vellai, T., Takacs-Vellai, K., Zhang, Y., Kovacs, A. L., Orosz, L., and Muller, F. (2003). Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 426, 620. doi: 10.1038/426620a
Vohra, B. P., Sasaki, Y., Miller, B. R., Chang, J., DiAntonio, A., and Milbrandt, J. (2010). Amyloid precursor protein cleavage-dependent and -independent axonal degeneration programs share a common nicotinamide mononucleotide adenylyltransferase 1-sensitive pathway. J. Neurosci. 30, 13729–13738. doi: 10.1523/JNEUROSCI.2939-10.2010
Wang, I. F., Guo, B. S., Liu, Y. C., Wu, C. C., Yang, C. H., Tsai, K. J., et al. (2012). Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. U.S.A. 109, 15024–15029. doi: 10.1073/pnas.1206362109
Wang, T., Lao, U., and Edgar, B. A. (2009). TOR-mediated autophagy regulates cell death in Drosophila neurodegenerative disease. J. Cell Biol. 186, 703–711. doi: 10.1083/jcb.200904090
Wang, Y., Yin, H., Li, J., Zhang, Y., Han, B., Zeng, Z., et al. (2013). Amelioration of beta-amyloid-induced cognitive dysfunction and hippocampal axon degeneration by curcumin is associated with suppression of CRMP-2 hyperphosphorylation. Neurosci. Lett. 557(Pt B), 112–117. doi: 10.1016/j.neulet.2013.10.024
Weatherill, D. B., McCamphill, P. K., Pethoukov, E., Dunn, T. W., Fan, X., and Sossin, W. S. (2011). Compartment-specific, differential regulation of eukaryotic elongation factor 2 and its kinase within Aplysia sensory neurons. J. Neurochem. 117, 841–855. doi: 10.1111/j.1471-4159.2011.07251.x
Wullschleger, S., Loewith, R., and Hall, M. N. (2006). TOR signaling in growth and metabolism. Cell 124, 471–484. doi: 10.1016/j.cell.2006.01.016
Yanase, S., Onodera, A., Tedesco, P., Johnson, T. E., and Ishii, N. (2009). SOD-1 deletions in Caenorhabditis elegans alter the localization of intracellular reactive oxygen species and show molecular compensation. J. Gerontol. A Biol. Sci. Med. Sci. 64, 530–539. doi: 10.1093/gerona/glp020
Keywords: Alzheimer’s disease, autophagy, dietary restriction, insulin/IGF-1 signaling, learning, mitochondria, neurodegeneration, TOR signaling
Citation: Gkikas I, Petratou D and Tavernarakis N (2014) Longevity pathways and memory aging. Front. Genet. 5:155. doi: 10.3389/fgene.2014.00155
Received: 27 March 2014; paper pending published: 22 April 2014;
Accepted: 10 May 2014; Published online: 04 June 2014.
Edited by:
Shin Murakami, Touro University-California, USAReviewed by:
Douglas Gray, Ottawa Hospital Research Institute, CanadaDaniele Bano, Deutsches Zentrum für Neurodegenerative Erkrankungen, Germany
Marta Artal Sanz, University Pablo de Olavide, Spain
Copyright © 2014 Gkikas, Petratou and Tavernarakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nektarios Tavernarakis, Institute of Molecular Biology and Biotechnology, Foundation for Research and Technology-Hellas, N. Plastira 100, Vassilika Vouton, P.O. Box 1385, Heraklion 71110, Crete, Greece e-mail: tavernarakis@imbb.forth.gr