 Victoria P. Belancio1,2*
Victoria P. Belancio1,2* David E. Blask1,2
David E. Blask1,2 Prescott Deininger2,3
Prescott Deininger2,3 Steven M. Hill1,2
Steven M. Hill1,2 S. Michal Jazwinski2,4
S. Michal Jazwinski2,4- 1Department of Structural and Cellular Biology, Tulane School of Medicine, Tulane University, New Orleans, LA, USA
- 2Tulane Cancer Center, Tulane Center for Aging, and Tulane Center for Circadian Biology, New Orleans, LA, USA
- 3Department of Epidemiology, Tulane University, New Orleans, LA, USA
- 4Department of Medicine, Tulane University, New Orleans, LA, USA
It is widely accepted that aging is characterized by a gradual decline in the efficiency and accuracy of biological processes, leading to deterioration of physiological functions and development of age-associated diseases. Age-dependent accumulation of genomic instability and development of metabolic syndrome are well-recognized components of the aging phenotype, both of which have been extensively studied. Existing findings strongly support the view that the integrity of the cellular genome and metabolic function can be influenced by light at night (LAN) and associated suppression of circadian melatonin production. While LAN is reported to accelerate aging by promoting age-associated carcinogenesis in several animal models, the specific molecular mechanism(s) of its action are not fully understood. Here, we review literature supporting a connection between LAN-induced central circadian disruption of peripheral circadian rhythms and clock function, LINE-1 retrotransposon-associated genomic instability, metabolic deregulation, and aging. We propose that aging is a progressive decline in the stability, continuity, and synchronization of multi-frequency oscillations in biological processes to a temporally disorganized state. By extension, healthy aging is the ability to maintain the most consistent, stable, and entrainable rhythmicity and coordination of these oscillations, at the molecular, cellular, and systemic levels.
Genomic Instability, its Sources, and Impact on Aging
Genomic instability is a hallmark of many human diseases with cancer and progeroid syndromes representing the most common outcomes associated with the loss of genome integrity (Anisimov, 2003; Campisi, 2005; Coppede and Migliore, 2010; Vijg and Suh, 2013). The link between genomic instability, cancer, and aging is not surprising as accumulation of mutations resulting in clinically relevant tumors takes time. Further evidence for the importance of cancer prevention for extended longevity resides in the resistance of long-lived rodents to spontaneous and induced tumorigenesis. These exceptional animals exemplify unique evolutionary adaptations preventing cancer development (Gorbunova et al., 2012; Tian et al., 2013). Humans also possess genes positively associated with longevity (Jazwinski et al., 2010; Kim et al., 2012), and exceptionally long-lived individuals typically do not develop cancer, often despite practicing unhealthy lifestyles. This is probably achieved by assuring the fidelity of DNA damage repair, which normally declines with age (Gorbunova et al., 2007).
Genomic instability arises from either exogenous or endogenous sources. Numerous exogenous carcinogenic agents (IR, UV, heavy metals, cigarette smoke, etc.) are well recognized. Artificial LAN represents a recent and unique addition to the list of genome offenders. Night shift work, which is regarded in epidemiological studies as a surrogate for LAN, involving circadian disruption has been recognized as a probable carcinogen (class 2a) by the World Health Organization (Bonde et al., 2012; Stevens et al., 2013, 2014). While the mechanistic relationship between this environmental factor and genomic instability is not well understood, its negative effect on genome integrity is substantiated by the fact that LAN increases cancer risk in humans and promotes aging and cancer growth in animal models (Davis et al., 2001; Schernhammer et al., 2001; Anisimov et al., 2004; Megdal et al., 2005; Vinogradova et al., 2009; Zhu et al., 2009; Wu et al., 2011).
Among established endogenous sources of DNA damage are reactive oxygen species (ROS), stalled replication forks, replication errors, and mitochondrial dysfunction. Much research has been dedicated to understanding their origin and their contribution to aging (Vijg and Suh, 2013). Another, frequently overlooked, source of endogenous genomic instability are transposable elements. These entities, which are present in most analyzed genomes, can rearrange the genetic material of their hosts in the process of their mobilization (reviewed in Belancio et al., 2010a). While their role in aging and cancer has long been debated, the empirical evidence for their actual involvement in these processes has only recently begun to accumulate (Gasior et al., 2006; Belancio et al., 2010b; Evrony et al., 2012; Lee et al., 2012; Solyom et al., 2012; De Cecco et al., 2013).
Genomic instability manifests itself in different ways. Single base-pair substitutions or deletions are the smallest genetic changes that can completely abolish gene function when they occur at positions critical for gene expression or activity. Another type of mutation is large genomic rearrangements such as deletions, insertions, inversions, and translocations, often referred to as chromosomal instability. They commonly result from the misrepair of DNA double-strand breaks (DSBs), which can be caused by stalled replication forks or external and endogenous DNA damaging agents. While all types of mutations are known to contribute to tumorigenesis, the rate and the spectrum of their accumulation with age demonstrates significant variation and tissue-specificity (Vijg and Dolle, 2002). Large genomic deletions, rather than point mutations, are believed to contribute to the aging phenotype as they are more likely to perturb regulation of gene expression, leading to accumulation of dysfunctional mosaic cells in aging tissues (Vijg and Dolle, 2002; Hsieh et al., 2013).
The spectrum and rate of accumulation of mutations can be greatly affected by genotype and environmental exposures with many, seemingly independent, cellular processes, and external factors influencing genome stability. Among the relevant genes are those involved in DNA repair, circadian regulation, and metabolism (Fu et al., 2002; Grimaldi et al., 2010; Kang et al., 2011; Gotoh et al., 2014). The pathways they specify can be disrupted or altered by various environmental cues such as LAN and diet. Not surprisingly, mutations abrogating these pathways lead to increased genomic instability and age-associated diseases. The majority of proteins involved in DNA repair, metabolism, and circadian pathways are highly conserved among evolutionarily distant organisms, further underscoring the fundamental importance of maintaining DNA integrity. Thus, the genetic, metabolic, and environmental effects on aging can be considered in the context of interconnected entities of the same system, synchronized with its environment, rather than individual, autonomous pathways. Here, we discuss emerging connections between genomic instability, transposable elements, circadian regulation, and metabolism.
Retroelements and Aging
Retroelements are mobile genetic entities that are a universal feature of many evolutionarily diverse organisms (reviewed in Belancio et al., 2008). Only 25% of the genome of the naked mole rat, a long-lived rodent, is occupied by transposon-derived repeats compared to 40% in human, 37% in mouse, and 35% in rat (Keane et al., 2014). Retroelements belong to two evolutionarily related groups of LTR (long terminal repeat) and non-LTR retrotransposons. In mammals, they are represented by endogenous retroviruses and Long and Short Interspersed Elements (LINEs and SINEs) and SVA elements, respectively (reviewed in Belancio et al., 2008). Non-LTR retroelements amplify through a “copy-and-paste” mechanism, which has allowed them to amass to over 500,000 copies per genome (Lander et al., 2001; Bibillo and Eickbush, 2002). LINEs, SINEs, and SVA are the only retrotransposons currently active in the human genome (Lander et al., 2001).
L1 elements can contribute to genomic instability through the retrotransposition of themselves and their parasites Alu and SVA (Moran et al., 1996; Dewannieux et al., 2003; Hancks et al., 2011; Raiz et al., 2012), as well as by induction of DSBs (Gasior et al., 2006; Belancio et al., 2010b; Kines et al., 2014). Both types of damage rely on the function of the endonuclease domain (EN) of the L1 ORF2 protein (Feng et al., 1996). EN is responsible for breaking genomic DNA to initiate de novo integration. The L1 ORF2p also possesses a reverse transcriptase (RT) domain, which functionally connects L1 to all RT-using entities (Mathias et al., 1991).
Historically, L1 activity was believed to be restricted to the germ line, early embryogenesis, and transformed somatic cells. The discovery of endogenous L1 mRNA expression in normal human tissues opened the possibility of L1 involvement in aging (Belancio et al., 2010b), and the report of endogenous L1 mobilization within human cortex and caudate neurons (Evrony et al., 2012) provided the first direct evidence of L1 activity in normal cells. Next generation sequencing (NGS) conducted at the single cell level showed a rate of 0.04–0.07 somatic L1 inserts per neuron. With an estimated 100 billion neurons per human brain and 50 trillion cells per human body, this finding suggests that there are about 4 billion neurons containing somatic L1 inserts in an average human brain and millions of de novo L1 integration events in every normal individual.
While some understanding of the rate of L1 retrotransposition in vivo is emerging, the amount of damage associated with L1-induced DSBs remains unknown. Some evidence exists that DSBs associated with L1 activity are 10–100 times more frequent than de novo L1 integrations (Gasior et al., 2006), suggesting that L1 may be responsible for 0.4–7 DSBs per cell. DSBs are one of the most harmful lesions in mammalian cells, because they are typically mutagenic when misrepaired by the NHEJ repair pathway (Gorbunova and Seluanov, 2005). DSB-induced mutations and unrepaired DSBs are known to accumulate with age (Vijg and Dolle, 2002; Sedelnikova et al., 2004). DSBs can be toxic to mammalian cells when unrepaired. Consistent with this notion, transient L1 overexpression in primary normal human cells and stem cells leads to apoptosis or senescence (Belancio et al., 2010b). This could potentially be one of the reasons for detection of low L1 retrotransposition in vivo, as normal cells supporting high L1 activity may be efficiently eliminated. L1-induced senescence of adult stem cells could contribute to their depletion with age. All of the above suggest that L1 may be responsible for the generation of mutations reported to accumulate with age as well as for promoting cellular senescence which is reported to increase with age (Jeyapalan et al., 2007).
The estimated L1 insertion frequency reflects retrotransposition in a cellular environment with all mechanisms in place to suppress these elements. There is a continually growing list of mammalian genes that negatively regulate different steps of the L1 replication cycle (reviewed in Belancio et al., 2008). Even though most of these have not yet been validated in vivo, their increasing number and the diversity of the pathways reported to control L1 activity underscore the necessity of their efficient suppression. It has been hypothesized that genomes deficient in cellular functions critical for L1 downregulation are burdened with higher rates of L1-induced genomic instability. For example, most human cancers support higher L1 expression than the normal tissues from which they have originated (Bratthauer and Fanning, 1992; Bratthauer et al., 1994). Recent NGS studies of L1 retrotransposition in human cancers provide experimental support for higher de novo L1 mobilization in human tumors relative to normal somatic tissues (Lee et al., 2012; Solyom et al., 2012; Tubio et al., 2014). As most human cancers harbor defects in many DNA repair or DDR pathways, these findings suggest that the rate of de novo L1 retrotransposition in normal tissues may increase with acquired elimination or age-associated decline of negative regulators of the L1 replication cycle. The significant redundancy of pathways suppressing L1 activity suggests that increased L1 mobilization may occur with sequential inactivation of multiple pathways or through circadian disruption of a “master regulator” controlling multiple cellular processes.
There are several tangible connections between L1 activity and the host circadian system (Figure 1). One of the recently discovered factors suppressing L1 is melatonin signaling (Deharo et al., 2014). Activation of melatonin receptor 1 (MT1) suppresses L1 expression in an in vivo cancer model and dramatically decreases L1 retrotransposition in cultured cells. This connects the activity of an endogenous DNA damaging entity with a component of the host circadian system and with the periodicity of environmental light exposure. In addition to its direct effect on L1 through downregulation of L1 ORF1 protein, melatonin signaling is also involved in the synchronization of uniquely timed biochemical functions in peripheral tissues and their associated cellular clock mechanisms which include the DDR. This suggests that circadian disruption may also indirectly increase L1 activity via deregulation of pathways important for the suppression of these elements. The best example is the reported circadian regulation of the nucleotide excision repair (NER) pathway activity in mice (Kang et al., 2009), which is also a suppressor of L1 retrotransposition in cultured cells (Gasior et al., 2008).
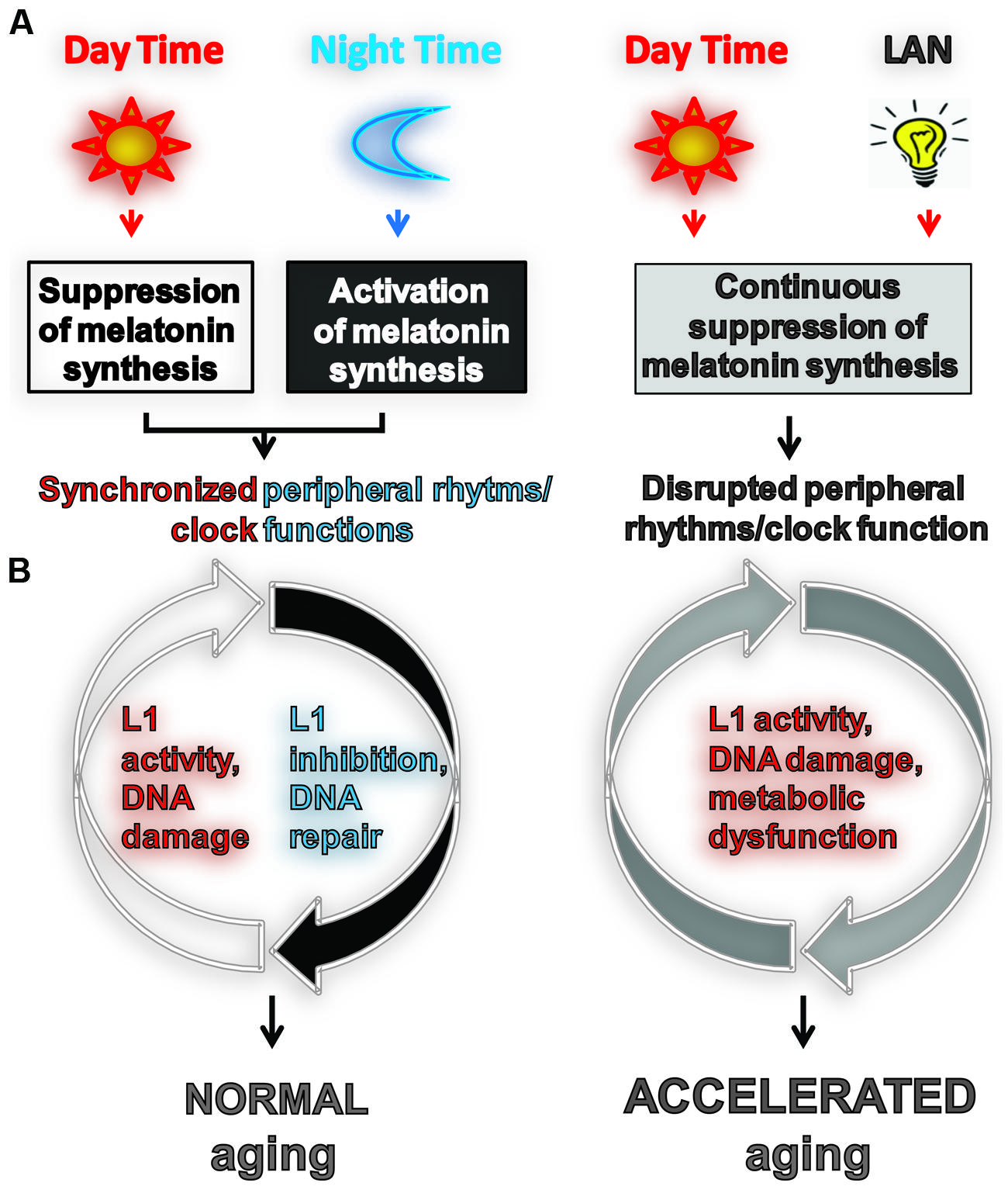
FIGURE 1. Light exposure at night accelerates aging by impeding or enhancing processes associated with aging. (A) Usually aging involves normal light exposure that is characterized by alternating intervals of light and dark over a 24-h period, which result in circadian production of nocturnal melatonin. This leads to the synchronization of peripheral clock (PC) function controlling many biochemical processes in cells including L1 expression and activity (Deharo et al., 2014) and the DNA damage response (DDR). (B) Exposure to light at night (LAN) is reported to accelerate aging. LAN blocks nocturnal melatonin production which prevents synchronization of PCs, leading to the disruption of timely function of many biochemical processes in cells including L1 expression and activity, DDR, and metabolism.
Circadian Connection Between Aging, Metabolism, and Genome Stability
The functions performed by individual cells are coordinated with the activity of their neighboring and distant cells by the circadian system (Dibner et al., 2010). The circadian system typically contains three major components: a central clock (CC), an entrainment pathway(s), and CC-responsive peripheral tissues and their associated peripheral clocks (PCs). The CC is located in the hypothalamic suprachiasmatic nucleus (SCN) of the brain and it is often referred to as the “master clock” because of its autonomous nature. The autoregulatory activity of the CC provides temporal organization of rhythmic function of PCs and thus many molecular processes in somatic tissues. While self-sufficient, the CC is entrained (synchronized) by external stimuli which is essential for adaptation of various functions within organisms in anticipation of daily changes in their environment.
The most potent external stimulus influencing the activity of the CC is environmental light/dark cycle. Daily periodicity of the light/dark cycle synchronizes the CC-driven oscillation of melatonin production in the pineal gland. Melatonin is a neurohormone that is produced during the dark phase of the 24 h light/dark cycle. Melatonin is an ancient and evolutionarily conserved molecule that is found in animals, plants, and microbes. Its main role in mammals is to inform the CC and all peripheral cells (including their endogenous clockworks) of the onset of nighttime (darkness) and to initiate actions associated with the nighttime of the daily cycle (Pfeffer et al., 2012). Like light during the daytime, melatonin during the nighttime helps to reset the CC in mammals. Melatonin functions through its G-protein coupled receptors MT1 and MT2 both of which are expressed in the CNS and peripheral tissues (Masana and Dubocovich, 2001; Poirel et al., 2003). Interestingly, the nocturnal mode of melatonin production is the same in both nocturnal and diurnal animals even though they exhibit inverse times of their sleep/wake activity (Dauchy et al., 2010). Despite these behavioral differences, the disruption of the circadian melatonin signal in both leads to the same negative effects on their health (Vinogradova et al., 2009), supporting an important, sleep-independent role of melatonin on health.
Melatonin production can be easily disrupted by LAN which commonly occurs in shift workers (Lewy et al., 1980; Blask et al., 2011). Melatonin synthesis also declines with age (Reiter et al., 2002; Hill et al., 2010; Zhdanova et al., 2011; Baba et al., 2012). LAN-induced melatonin suppression is associated with metabolic dysfunction, obesity, and an increased risk of several malignancies (Anisimov et al., 2004; Blask et al., 2005, 2011, 2014; Lai et al., 2009; Vinogradova et al., 2009; Wu et al., 2011). While the effect of LAN on human aging is not known, the LAN-induced shortening of life span in rodent models supports the existence of a biological connection between CC and PC functions, genomic instability, metabolism, and aging (Reiter et al., 2002; Wood et al., 2009; Figure 1). Recent findings have demonstrated a direct relationship between the biological clock, aging, and sphingolipid metabolism, arguing for a conserved circadian-based mechanism of aging from fungi to humans (Case et al., 2014).
Metabolic cycles are tightly coupled with both CC and PCs allowing both diurnal and nocturnal mammalian species to coordinate nutrient use and storage with light/dark entrained sleep/wake cycles in the overall regulation of organismal bioenergetics (Bass, 2012). Metabolically active tissues (e.g., liver, adipose tissue, skeletal muscle) are highly responsive to circadian oscillations in circulating glucose, fatty acids, triglycerides, and metabolic hormones (Bellet and Sassone-Corsi, 2010). Circadian clocks control several critical metabolic pathways and, conversely, metabolic processes exert important feedback effects on the molecular clock machinery. A significant player in the bidirectional interactions between circadian signaling and metabolic activities is thought to be nicotinamide adenine dinucleotide (NAD+) which functions as an electron shuttle in oxidoreductase reactions. Additionally, NAD+ is a critical cofactor for sirtuins, most notably SIRT1, which regulate metabolism in response to caloric restriction and as modulators of oxidative damage and DNA repair processes that appear to be critical for lifespan. The circadian regulation of NAD+-dependent sirtuin activity may have implications for healthy aging and oxidative metabolism that is particularly relevant to the association of circadian period length and longevity (Bass, 2012; Masri and Sassone-Corsi, 2014). Further support for the presence and conservation of this mechanism comes from the discovery that human Sirt1 and six modulate the function of circadian clock genes (Asher et al., 2008; Jung-Hynes et al., 2010; Chang and Guarente, 2013; Masri et al., 2014) and the expression of Sirt1 and some clock genes in normal tissues is enhanced by melatonin (Chang et al., 2009; Yu et al., 2014).
Melatonin is a powerful antioxidant that suppresses ROS (Reiter et al., 2010) and through its receptor downregulates L1 retrotransposons (Deharo et al., 2014), both of which may elicit DNA damage in the absence of melatonin. Suppression of melatonin signaling is also known to impair cell signaling pathways important for DNA repair, apoptosis, and cellular differentiation (Xiang et al., 2008; Hill et al., 2009; Blask et al., 2011). There is growing support for a connection between systemic DDR, which could be caused by L1 activity, and attenuation of p53 function and metabolic changes, consistent with the age-dependent and LAN-induced decline in DDR efficiency and development of metabolic syndrome caused by decline in circadian clock activity (Erol, 2010; Feng et al., 2012). Furthermore, caloric restriction delays age-associated decline in melatonin production in rhesus monkeys (Roth et al., 2001) and resets circadian rhythms in mice (Froy et al., 2008). In contrast, high fat diet disrupts the normal circadian cycle (Eckel-Mahan et al., 2013). Interestingly, DNA damage itself can reset the circadian clock (Gamsby et al., 2009) suggesting the possibility that controlled periodicity of intrinsic DNA damage in tissues with a synchronized clock may facilitate its maintenance (Figure 2). On the other hand, deregulation of intrinsic DNA damage response caused by LAN may further promote desynchronization of PCs. Thus, melatonin signaling is positively associated with molecular functions that activate cellular pathways involved in the maintenance of genome stability and metabolism that are central to healthy aging. Collectively, current data suggest that LAN-induced disruption of the CC and the associated disruption of the PC activate L1-associated DNA damage and metabolic changes in normal tissues, which may contribute to the LAN-induced acceleration of aging manifested in age-associated changes and diseases.
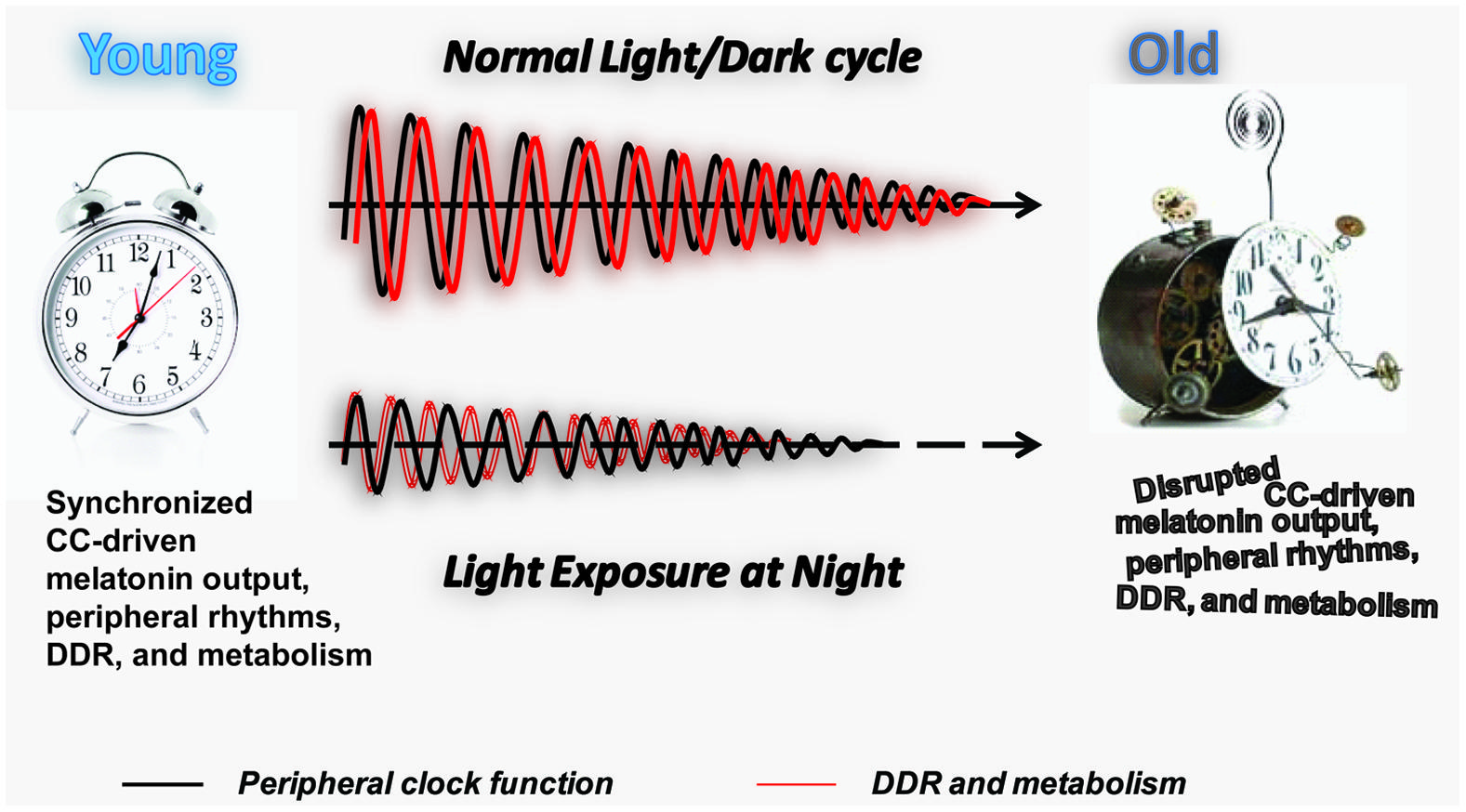
FIGURE 2. Longitudinal effect of LAN on PC function, DDR, and metabolism. Schematic representation of the effect of normal light exposure versus LAN on the age-associated deterioration of PCs. The maintenance of the normal light/dark cycle promotes circadian melatonin output and synchronization of the PC (black line) with DDR and metabolic function (red line). An age-associated decline in melatonin production and melatonin receptor expression (Hill et al., 2010) leads to the gradual decline in the amplitude of the peripheral rhythms and potentially their synchronization with DDR and metabolic function. We hypothesize that LAN accelerates aging by promoting age-associated decline in the amplitude of the peripheral rhythms and their synchronization with DDR and metabolic function at early age. Individual genomes may provide molecular machinery to resist adverse effects of LAN, explaining the variation in lifespan observed in the human population.
Concluding Remarks
Aging has largely been discussed as a complex, but for the most part, linear progression from the beginning to the end of life. The multitude of differences between convenient experimental and simplistic approaches and the actual complexity of life as we age in a continuously changing environment (Chang et al., 2009; Yu et al., 2014) forces us to refine this view. We propose that during aging there is a progressive loss of synchronized oscillation of biological processes along the axis of life accompanied by a continuous decline in their amplitude (Figure 2). By extension, healthy aging is the ability to maintain the most consistent light/dark entrainable rhythmicity and coordination at the molecular, cellular, and systemic levels throughout the lifespan, originating in the genetically programmed resistance to environmental cues and stress capable of disrupting this balanced progression. This definition has the potential to explain the puzzling coexistence of an unhealthy lifestyle with an exceptionally long survival.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to the many investigators whose work could not be cited adequately due to space restrictions. The authors’ research is supported by grants from the National Institutes of Health: NIH R37 AG006168 (S. Michal Jazwinski) and NIH R01 GM45668 (Prescott Deininger) and from Life Extension Foundation and Kay Yaw Cancer Fund (Victoria P. Belancio).
References
Anisimov, V. N. (2003). The relationship between aging and carcinogenesis: a critical appraisal. Crit. Rev. Oncol. Hematol. 45, 277–304. doi: 10.1016/S1040-8428(02)00121-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Anisimov, V. N., Baturin, D. A., Popovich, I. G., Zabezhinski, M. A., Manton, K. G., Semenchenko, A. V.,et al. (2004). Effect of exposure to light-at-night on life span and spontaneous carcinogenesis in female CBA mice. Int. J. Cancer 111, 475–479. doi: 10.1002/ijc.20298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Asher, G., Gatfield, D., Stratmann, M., Reinke, H., Dibner, C., Kreppel, F.,et al. (2008). SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134, 317–328. doi: 10.1016/j.cell.2008.06.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baba, K., Mazzoni, F., Owino, S., Contreras-Alcantara, S., Strettoi, E., Tosini, G.,et al. (2012). Age-related changes in the daily rhythm of photoreceptor functioning and circuitry in a melatonin-proficient mouse strain. PLoS ONE 7:e37799. doi: 10.1371/journal.pone.0037799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bass, J. (2012). Circadian topology of metabolism. Nature 491, 348–356. doi: 10.1038/nature11704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belancio, V. P., Hedges, D. J., and Deininger, P. (2008). Mammalian non-LTR retrotransposons: for better or worse, in sickness and in health. Genome Res. 18, 343–358. doi: 10.1101/gr.5558208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belancio, V. P., Roy-Engel, A. M., and Deininger, P. L. (2010a). All y’all need to know ‘bout retroelements in cancer. Semin. Cancer Biol. 20, 200–210. doi: 10.1016/j.semcancer.2010.06.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belancio, V. P., Roy-Engel, A. M., Pochampally, R. R., and Deininger, P. (2010b). Somatic expression of LINE-1 elements in human tissues. Nucleic Acids Res. 38, 3909–3922. doi: 10.1093/nar/gkq132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bellet, M. M., and Sassone-Corsi, P. (2010). Mammalian circadian clock and metabolism - the epigenetic link. J. Cell Sci. 123, 3837–3848. doi: 10.1242/jcs.051649
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bibillo, A., and Eickbush, T. H. (2002). The reverse transcriptase of the R2 non-LTR retrotransposon: continuous synthesis of cDNA on non-continuous RNA templates. J. Mol. Biol. 316, 459–473. doi: 10.1006/jmbi.2001.5369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blask, D. E., Brainard, G. C., Dauchy, R. T., Hanifin, J. P., Davidson, L. K., Krause, J. A.,et al. (2005). Melatonin-depleted blood from premenopausal women exposed to light at night stimulates growth of human breast cancer xenografts in nude rats. Cancer Res. 65, 11174–11184. doi: 10.1158/0008-5472.CAN-05-1945
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blask, D. E., Dauchy, R. T., Dauchy, E. M., Mao, L., Hill, S. M., Greene, M. W.,et al. (2014). Light exposure at night disrupts host/cancer circadian regulatory dynamics: impact on the Warburg effect, lipid signaling and tumor growth prevention. PLoS ONE 9:e102776. doi: 10.1371/journal.pone.0102776
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blask, D. E., Hill, S. M., Dauchy, R. T., Xiang, S., Yuan, L., Duplessis, T.,et al. (2011). Circadian regulation of molecular, dietary, and metabolic signaling mechanisms of human breast cancer growth by the nocturnal melatonin signal and the consequences of its disruption by light at night. J. Pineal. Res. 51, 259–269. doi: 10.1111/j.1600-079X.2011.00888.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bonde, J. P., Hansen, J., Kolstad, H. A., Mikkelsen, S., Olsen, J. H., Blask, D. E.,et al. (2012). Work at night and breast cancer - report on evidence-based options for preventive actions. Scand. J. Work Environ. Health 38, 380–390. doi: 10.5271/sjweh.3282
Bratthauer, G. L., Cardiff, R. D., and Fanning, T. G. (1994). Expression of LINE-1 retrotransposons in human breast cancer. Cancer 73, 2333–2336. doi: 10.1002/1097-0142(19940501)73:9<2333::AID-CNCR2820730915>3.0.CO;2-4
Bratthauer, G. L., and Fanning, T. G. (1992). Active line-1 retrotransposons in human testicular cancer. Oncogene 7, 507–510.
Campisi, J. (2005). Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120, 513–522. doi: 10.1016/j.cell.2005.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Case, M. E., Griffith, J., Dong, W., Tigner, I. L., Gaines, K., Jiang, J. C.,et al. (2014). The aging biological clock in Neurospora crassa. Ecol. Evol. 4, 3494–3507. doi: 10.1002/ece3.1202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chang, H. C., and Guarente, L. (2013). SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 153, 1448–1460. doi: 10.1016/j.cell.2013.05.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chang, H. M., Wu, U. I., and Lan, C. T. (2009). Melatonin preserves longevity protein (sirtuin 1) expression in the hippocampus of total sleep-deprived rats. J. Pineal Res. 47, 211–220. doi: 10.1111/j.1600-079X.2009.00704.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Coppede, F., and Migliore, L. (2010). DNA repair in premature aging disorders and neurodegeneration. Curr. Aging Sci. 3, 3–19. doi: 10.2174/1874609811003010003
Dauchy, R. T., Dauchy, E. M., Tirrell, R. P., Hill, C. R., Davidson, L. K., Greene, M. W.,et al. (2010). Dark-phase light contamination disrupts circadian rhythms in plasma measures of endocrine physiology and metabolism in rats. Comp. Med. 60, 348–356.
Davis, S., Mirick, D. K., and Stevens, R. G. (2001). Night shift work, light at night, and risk of breast cancer. J. Natl. Cancer Inst. 93, 1557–1562. doi: 10.1093/jnci/93.20.1557
De Cecco, M., Criscione, S. W., Peterson, A. L., Neretti, N., Sedivy, J. M., and Kreiling, J. A. (2013). Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging (Albany. NY) 5, 867–883.
Deharo, D., Kines, K. J., Sokolowski, M., Dauchy, R. T., Streva, V. A., and Hill, S. M. (2014). Regulation of L1 expression and retrotransposition by melatonin and its receptor: implications for cancer risk associated with light exposure at night. Nucleic Acids Res. 42, 7694–7707. doi: 10.1093/nar/gku503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dewannieux, M., Esnault, C., and Heidmann, T. (2003). LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 35, 41–48. doi: 10.1038/ng1223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dibner, C., Schibler, U., and Albrecht, U. (2010). The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu. Rev. Physiol. 72, 517–549. doi: 10.1146/annurev-physiol-021909-135821
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eckel-Mahan, K. L., Patel, V. R., de Mateo, S., Orozco-Solis, R., Ceglia, N. J., Sahar, S.,et al. (2013). Reprogramming of the circadian clock by nutritional challenge. Cell 155, 1464–1478. doi: 10.1016/j.cell.2013.11.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Erol, A. (2010). Systemic DNA damage response and metabolic syndrome as a premalignant state. Curr. Mol. Med. 10, 321–334. doi: 10.2174/156652410791065282
Evrony, G. D., Cai, X., Lee, E., Hills, L. B., Elhosary, P. C., Lehmann, H. S.,et al. (2012). Single-neuron sequencing analysis of l1 retrotransposition and somatic mutation in the human brain. Cell 151, 483–496. doi: 10.1016/j.cell.2012.09.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feng, Q., Moran, J. V., Kazazian, H. H. Jr., and Boeke, J. D. (1996). Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 87, 905–916. doi: 10.1016/S0092-8674(00)81997-2
Feng, Z., Liu, L., Zhang, C., Zheng, T., Wang, J., Lin, M.,et al. (2012). Chronic restraint stress attenuates p53 function and promotes tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 109, 7013–7018. doi: 10.1073/pnas.1203930109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Froy, O., Chapnik, N., and Miskin, R. (2008). Relationship between calorie restriction and the biological clock: lessons from long-lived transgenic mice. Rejuvenation Res. 11, 467–471. doi: 10.1089/rej.2008.0669
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fu, L., Pelicano, H., Liu, J., Huang, P., and Lee, C. (2002). The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 111, 41–50. doi: 10.1016/S0092-8674(02)00961-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gamsby, J. J., Loros, J. J., and Dunlap, J. C. (2009). A phylogenetically conserved DNA damage response resets the circadian clock. J. Biol. Rhythms 24, 193–202. doi: 10.1177/0748730409334748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gasior, S. L., Roy-Engel, A. M., and Deininger, P. L. (2008). ERCC1/XPF limits L1 retrotransposition. DNA Repair 7, 983–989. doi: 10.1016/j.dnarep.2008.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gasior, S. L., Wakeman, T. P., Xu, B., and Deininger, P. L. (2006). The human LINE-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol. 357, 1383–1393. doi: 10.1016/j.jmb.2006.01.089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gorbunova, V., Hine, C., Tian, X., Ablaeva, J., Gudkov, A. V., Nevo, E.,et al. (2012). Cancer resistance in the blind mole rat is mediated by concerted necrotic cell death mechanism. Proc. Natl. Acad. Sci. U.S.A. 109, 19392–19396. doi: 10.1073/pnas.1217211109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gorbunova, V., and Seluanov, A. (2005). Making ends meet in old age: DSB repair and aging. Mech. Ageing Dev. 126, 621–628. doi: 10.1016/j.mad.2005.02.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gorbunova, V., Seluanov, A., Mao, Z., and Hine, C. (2007). Changes in DNA repair during aging. Nucleic Acids Res. 35, 7466–7474. doi: 10.1093/nar/gkm756
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gotoh, T., Vila-Caballer, M., Santos, C. S., Liu, J., Yang, J., and Finkielstein, C. V. (2014). The circadian factor Period 2 modulates p53 stability and transcriptional activity in unstressed cells. Mol. Biol. Cell 25, 3081–3093. doi: 10.1091/mbc.E14-05-0993
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grimaldi, B., Bellet, M. M., Katada, S., Astarita, G., Hirayama, J., Amin, R. H.,et al. (2010). PER2 controls lipid metabolism by direct regulation of PPARgamma. Cell Metab. 12, 509–520. doi: 10.1016/j.cmet.2010.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hancks, D. C., Goodier, J. L., Mandal, P. K., Cheung, L. E., and Kazazian, H. H. Jr. (2011). Retrotransposition of marked SVA elements by human L1s in cultured cells. Hum. Mol. Genet. 20, 3386–3400. doi: 10.1093/hmg/ddr245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hill, S. M., Cheng, C., Yuan, L., Mao, L., Jockers, R., Dauchy, B.,et al. (2010). Declining melatonin levels and MT1 receptor expression in aging rats is associated with enhanced mammary tumor growth and decreased sensitivity to melatonin. Breast Cancer Res. Treat. 27, 91–98. doi: 10.1007/s10549-010-0958-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hill, S. M., Frasch, T., Xiang, S., Yuan, L., Duplessis, T., and Mao, L. (2009). Molecular mechanisms of melatonin anticancer effects. Integr. Cancer Ther. 8, 337–346. doi: 10.1177/1534735409353332
Hsieh, J. C., Van Den, B. D., Kang, H., Hsieh, C. L., and Lieber, M. R. (2013). Large chromosome deletions, duplications, and gene conversion events accumulate with age in normal human colon crypts. Aging Cell 12, 269–279. doi: 10.1111/acel.12053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jazwinski, S. M., Kim, S., Dai, J., Li, L., Bi, X., Jiang, J. C.,et al. (2010). HRAS1 and LASS1 with APOE are associated with human longevity and healthy aging. Aging Cell 9, 698–708. doi: 10.1111/j.1474-9726.2010.00600.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jeyapalan, J. C., Ferreira, M., Sedivy, J. M., and Herbig, U. (2007). Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 128, 36–44. doi: 10.1016/j.mad.2006.11.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jung-Hynes, B., Reiter, R. J., and Ahmad, N. (2010). Sirtuins, melatonin and circadian rhythms: building a bridge between aging and cancer. J. Pineal Res. 48, 9–19. doi: 10.1111/j.1600-079X.2009.00729.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kang, T. H., Reardon, J. T., Kemp, M., and Sancar, A. (2009). Circadian oscillation of nucleotide excision repair in mammalian brain. Proc. Natl. Acad. Sci. U.S.A. 106, 2864–2867. doi: 10.1073/pnas.0812638106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kang, T. H., Reardon, J. T., and Sancar, A. (2011). Regulation of nucleotide excision repair activity by transcriptional and post-transcriptional control of the XPA protein. Nucleic Acids Res. 39, 3176–3187. doi: 10.1093/nar/gkq1318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keane, M., Craig, T., Alfoldi, J., Berlin, A. M., Johnson, J., Seluanov, A.,et al. (2014). The naked mole rat genome resource: facilitating analyses of cancer and longevity-related adaptations. Bioinformatics 30, 3558–3560. doi: 10.1093/bioinformatics/btu579
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, S., Bi, X., Czarny-Ratajczak, M., Dai, J., Welsh, D. A., Myers, L.,et al. (2012). Telomere maintenance genes SIRT1 and XRCC6 impact age-related decline in telomere length but only SIRT1 is associated with human longevity. Biogerontology 13, 119–131. doi: 10.1007/s10522-011-9360-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kines, K. J., Sokolowski, M., deHaro, D. L., Christian, C. M., and Belancio, V. P. (2014). Potential for genomic instability associated with retrotranspositionally-incompetent L1 loci. Nucleic Acids Res. 42, 10488–10502. doi: 10.1093/nar/gku687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lai, L., Yuan, L., Cheng, Q., Dong, C., Mao, L., and Hill, S. M. (2009). Alteration of the MT1 melatonin receptor gene and its expression in primary human breast tumors and breast cancer cell lines. Breast Cancer Res. Treat. 118, 293–305. doi: 10.1007/s10549-008-0220-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J.,et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. doi: 10.1038/35057062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, E., Iskow, R., Yang, L., Gokcumen, O., Haseley, P., Luquette, L. J. III,et al. (2012). Landscape of somatic retrotransposition in human cancers. Science 337, 967–971. doi: 10.1126/science.1222077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lewy, A. J., Wehr, T. A., Goodwin, F. K., Newsome, D. A., and Markey, S. P. (1980). Light suppresses melatonin secretion in humans. Science 210, 1267–1269. doi: 10.1126/science.7434030
Masana, M. I., and Dubocovich, M. L. (2001). Melatonin receptor signaling: finding the path through the dark. Sci. STKE 2001, e39. doi: 10.1126/stke.2001.107.pe39
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Masri, S., Rigor, P., Cervantes, M., Ceglia, N., Sebastian, C., Xiao, C.,et al. (2014). Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cell 158, 659–672. doi: 10.1016/j.cell.2014.06.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Masri, S., and Sassone-Corsi, P. (2014). Sirtuins and the circadian clock: bridging chromatin and metabolism. Sci. Signal. 7, re6. doi: 10.1126/scisignal.2005685
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mathias, S. L., Scott, A. F., Kazazian, H. H. Jr., Boeke, J. D., and Gabriel, A. (1991). Reverse transcriptase encoded by a human transposable element. Science 254, 1808–1810. doi: 10.1126/science.1722352
Megdal, S. P., Kroenke, C. H., Laden, F., Pukkala, E., and Schernhammer, E. S. (2005). Night work and breast cancer risk: a systematic review and meta-analysis. Eur. J. Cancer 41, 2023–2032. doi: 10.1016/j.ejca.2005.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moran, J. V., Holmes, S. E., Naas, T. P., DeBerardinis, R. J., Boeke, J. D., and Kazazian, H. H. Jr. (1996). High frequency retrotransposition in cultured mammalian cells. Cell 87, 917–927. doi: 10.1016/S0092-8674(00)81998-4
Pfeffer, M., Rauch, A., Korf, H. W., and von Gall, C. (2012). The endogenous melatonin (MT) signal facilitates reentrainment of the circadian system to light-induced phase advances by acting upon MT2 receptors. Chronobiol. Int. 29, 415–429. doi: 10.3109/07420528.2012.667859
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Poirel, V. J., Cailotto, C., Streicher, D., Pevet, P., Masson-Pevet, M., and Gauer, F. (2003). MT1 melatonin receptor mRNA tissular localization by PCR amplification. Neuro Endocrinol. Lett. 24, 33–38.
Raiz, J., Damert, A., Chira, S., Held, U., Klawitter, S., Hamdorf, M.,et al. (2012). The non-autonomous retrotransposon SVA is trans-mobilized by the human LINE-1 protein machinery. Nucleic Acids Res. 40, 1666–1683. doi: 10.1093/nar/gkr863
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reiter, R. J., Tan, D. X., and Fuentes-Broto, L. (2010). Melatonin: a multitasking molecule. Prog. Brain Res. 181, 127–151. doi: 10.1016/S0079-6123(08)81008-4
Reiter, R. J., Tan, D. X., Mayo, J. C., Sainz, R. M., and Lopez-Burillo, S. (2002). Melatonin, longevity and health in the aged: an assessment. Free Radic. Res. 36, 1323–1329. doi: 10.1080/1071576021000038504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roth, G. S., Lesnikov, V., Lesnikov, M., Ingram, D. K., and Lane, M. A. (2001). Dietary caloric restriction prevents the age-related decline in plasma melatonin levels of rhesus monkeys. J. Clin. Endocrinol. Metab. 86, 3292–3295. doi: 10.1210/jcem.86.7.7655
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schernhammer, E. S., Laden, F., Speizer, F. E., Willett, W. C., Hunter, D. J., Kawachi, I.,et al. (2001). Rotating night shifts and risk of breast cancer in women participating in the nurses’ health study. J. Natl. Cancer Inst. 93, 1563–1568. doi: 10.1093/jnci/93.20.1563
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sedelnikova, O. A., Horikawa, I., Zimonjic, D. B., Popescu, N. C., Bonner, W. M., and Barrett, J. C. (2004). Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 6, 168–170. doi: 10.1038/ncb1095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Solyom, S., Ewing, A. D., Rahrmann, E. P., Doucet, T. T., Nelson, H. H., Burns, M. B.,et al. (2012). Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res. 2, 2328–2338. doi: 10.1101/gr.145235.112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stevens, R. G., Brainard, G. C., Blask, D. E., Lockley, S. W., and Motta, M. E. (2013). Adverse health effects of nighttime lighting: comments on American Medical Association policy statement. Am. J. Prev. Med. 45, 343–346. doi: 10.1016/j.amepre.2013.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stevens, R. G., Brainard, G. C., Blask, D. E., Lockley, S. W., and Motta, M. E. (2014). Breast cancer and circadian disruption from electric lighting in the modern world. CA Cancer J. Clin. 64, 207–218. doi: 10.3322/caac.21218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tian, X., Azpurua, J., Hine, C., Vaidya, A., Myakishev-Rempel, M., Ablaeva, J.,et al. (2013). High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 499, 346–349. doi: 10.1038/nature12234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tubio, J. M., Li, Y., Ju, Y. S., Martincorena, I., Cooke, S. L., Tojo, M.,et al. (2014). Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science 345, 1251343. doi: 10.1126/science.1251343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vijg, J., and Dolle, M. E. (2002). Large genome rearrangements as a primary cause of aging. Mech. Ageing Dev. 123, 907–915. doi: 10.1016/S0047-6374(02)00028-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vijg, J., and Suh, Y. (2013). Genome instability and aging. Annu. Rev. Physiol. 75, 645–668. doi: 10.1146/annurev-physiol-030212-183715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vinogradova, I. A., Anisimov, V. N., Bukalev, A. V., Semenchenko, A. V., and Zabezhinski, M. A. (2009). Circadian disruption induced by light-at-night accelerates aging and promotes tumorigenesis in rats. Aging (Albany NY) 1, 855–865.
Wood, P. A., Yang, X., and Hrushesky, W. J. (2009). Clock genes and cancer. Integr. Cancer Ther. 8, 303–308. doi: 10.1177/1534735409355292
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, J., Dauchy, R. T., Tirrell, P. C., Wu, S. S., Lynch, D. T., Jitawatanarat, P.,et al. (2011). Light at night activates IGF-1R/PDK1 signaling and accelerates tumor growth in human breast cancer xenografts. Cancer Res. 71, 2622–2631. doi: 10.1158/0008-5472.CAN-10-3837
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xiang, S., Coffelt, S. B., Mao, L., Yuan, L., Cheng, Q., and Hill, S. M. (2008). Period-2: a tumor suppressor gene in breast cancer. J. Circadian Rhythms 6, 4. doi: 10.1186/1740-3391-6-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, L., Sun, Y., Cheng, L., Jin, Z., Yang, Y., Zhai, M.,et al. (2014). Melatonin receptor-mediated protection against myocardial ischemia/reperfusion injury: role of SIRT1. J. Pineal Res. 57, 228–238. doi: 10.1111/jpi.12161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhdanova, I. V., Masuda, K., Quasarano-Kourkoulis, C., Rosene, D. L., Killiany, R. J., and Wang, S. (2011). Aging of intrinsic circadian rhythms and sleep in a diurnal nonhuman primate, Macaca mulatta. J. Biol. Rhythms 26, 149–159. doi: 10.1177/0748730410395849
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhu, Y., Stevens, R. G., Hoffman, A. E., Fitzgerald, L. M., Kwon, E. M., Ostrander, E. A.,et al. (2009). Testing the circadian gene hypothesis in prostate cancer: a population-based case-control study. Cancer Res. 69, 9315–9322. doi: 10.1158/0008-5472.CAN-09-0648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: aging, light exposure at night, retroelements, LINE-1, metabolism
Citation: Belancio VP, Blask DE, Deininger P, Hill SM and Jazwinski SM (2015) The aging clock and circadian control of metabolism and genome stability. Front. Genet. 5:455. doi: 10.3389/fgene.2014.00455
Received: 26 September 2014; Accepted: 11 December 2014;
Published online: 14 January 2015.
Edited by:
Nektarios Tavernarakis, University of Crete – Foundation for Research and Technology-Hellas, GreeceReviewed by:
Hugo Aguilaniu, Centre National de la Recherche Scientifique – Ecole Normale Supérieure de Lyon, FranceMarta Artal Sanz, Andalusian Centre for Developmental Biology – University Pablo de Olavide, Spain
Pidder Jansen-Duerr, Innsbruck University, Austria
Copyright © 2015 Belancio, Blask, Deininger, Hill and Jazwinski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victoria P. Belancio, Department of Structural and Cellular Biology, Tulane School of Medicine, Tulane University, SL-49, New Orleans, LA 70112, USA e-mail: vperepe@tulane.edu