 Kejian Guo1†
Kejian Guo1† Xuan Zhou
Xuan Zhou Yili Wu
Yili Wu- 1Jining Maternal and Child Health Care Hospital, Jining, China
- 2Department of Psychiatry, Jining Medical University, Jining, China
- 3Shandong Key Laboratory of Behavioral Medicine, Jining Medical University, Jining, China
- 4Collaborative Innovation Center for Birth Defect Research and Transformation of Shandong Province, Jining Medical University, Jining, China
- 5Department of Biochemistry, Jining Medical University, Jining, China
The incidence of inborn errors of metabolisms (IEMs) varies dramatically in different countries and regions. Expanded newborn screening for IEMs by tandem mass spectrometry (MS/MS) is an efficient approach for early diagnosis and presymptomatic treatment to prevent severe permanent sequelae and death. To determine the characteristics of IEMs and IEMs-associated mutations in newborns in Jining area, China, 48,297 healthy neonates were recruited for expanded newborn screening by MS/MS. The incidence of IEMs was 1/1178 in Jining, while methylmalonic acidemia, phenylketonuria, and primary carnitine deficiency ranked the top 3 of all detected IEMs. Thirty mutations in nine IEMs-associated genes were identified in 28 confirmed cases. As 19 cases with the mutations in phenylalanine hydroxylase (PAH), solute carrier family 22 member 5 (SLC22A5), and methylmalonic aciduria (cobalamin deficiency) cblC type with homocystinuria (MMACHC) genes, respectively, it suggested that mutations in the PAH, SLC22A5, and MMACHC genes are the predominant causes of IEMs, leading to the high incidence of phenylketonuria, primary carnitine deficiency, and methylmalonic acidemia, respectively. Our work indicated that the overall incidence of IEMs is high and the mutations in PAH, SLC22A5, and MMACHC genes are the leading causes of IEMs in Jining area. Therefore, it is critical to increase the coverage of expanded newborn screening by MS/MS and prenatal genetic consulting in Jining area.
Introduction
Inborn errors of metabolisms (IEMs) are a group of genetic diseases leading to severe complications in newborns, infants, children, adolescents, or adults. IEMs are caused by the defect of an enzyme, its cofactor or a transporter resulting in the accumulation of a substrate and/or the deficiency of its downstream products. Up to now, more than 500 IEMs have been detected. Although individual IEM is rare, the incidence of overall IEMs is high and varies dramatically in different countries and regions (Campeau et al., 2008). For example, the incidence of IEMs was reported to be 1/667 in Saudi Arabia (Moammar et al., 2010), 1/784 in United Kingdom (Sanderson et al., 2006), 1/2500 in Canada (Applegarth et al., 2000), 1/2900 in Germany (Lindner et al., 2011), 1/1944 in Egypt (Hassan et al., 2016), 1/2916 in Malaysia (Yunus et al., 2016), 1/2800 in South Korea (Yoon et al., 2005), and 1/3165 in Singapore (Lim et al., 2014). The incidence of IEMs is much lower in Japan, approximately 1/9000 (Yamaguchi, 2008; Yamaguchi et al., 2013).
Newborn screening for IEMs is an efficient approach for early diagnosis and presymptomatic treatment, which could improve the survival and well-being of affected infants. In addition to the classical newborn screening, expanded newborn screening by tandem mass spectrometry (MS/MS) has been applied widely since last decade. By analyzing amino acids and acylcarnitines in blood, three groups of IEMs could be simultaneously detected, including amino acid disorders, organic acid disorders, and fatty acid oxidation disorders. In China, expanded newborn screening started in 2004 and the incidence of IEMs was 1/3795 in a pilot study (Shi et al., 2012). However, later studies showed that the incidence of IEMs varied dramatically in different cities and regions of China, from 1/1683 to 1/8304 (Huang et al., 2011; Fan et al., 2013; Zhu et al., 2015; Lu et al., 2016). Expanded newborn screening for 25 IEMs is widely used in Jining since late 2014. To determine the incidence and characteristics of IEMs in Jining, 48,297 healthy neonates were recruited for expanded newborn screening of 25 IEMs by MS/MS. In the present study, we showed that the incidence of IEMs was 1/1178 in Jining, which is higher the incidence in China. In addition, 30 mutations in nine IEMs-associated genes were identified in the 28 confirmed cases and 19 cases with the mutations in phenylalanine hydroxylase (PAH), solute carrier family 22 member 5 (SLC22A5), and methylmalonic aciduria (cobalamin deficiency) cblC type with homocystinuria (MMACHC) genes, respectively, leading to the high incidence of phenylketonuria, primary carnitine deficiency, and methylmalonic acidemia, respectively. Our work indicated that the overall incidence of IEMs is high and the mutations in PAH, SLC22A5, and MMACHC genes are the leading causes of IEMs in Jining area. Therefore, it is critical to increase the coverage of expanded newborn screening by MS/MS and prenatal genetic consulting in Jining area.
Materials and Methods
Subjects
A total of 48,297 infants born in Jining, China, between January 2015 and December 2015, were recruited for expanded newborn screening by MS/MS. In all, 15,731 infants were born in the urban area and 32,566 infants were born in the suburban and rural areas. Informed and written consent was obtained from the parents. This study was approved by the Ethical Committee of Jining Medical University.
Procedures of Screening Test and Follow-Up Testing
All the procedures were carried out in accordance with the Jining Newborn Screening Programme (2015) and Technical Guide of Newborn Screening in China (2010). Briefly, blood taken from heelstick was spotted on Whatman 903 filter paper, which was dried and sent by mail to the screening laboratory. The time of sampling was between the 3rd to 10th day of life. Dried blood spots were pre-processed following the instruction of NeoBaseTM non-derivatized MS/MS kit (PerkinElmer, MA, United States), and then they were analyzed by using TQD tandem mass spectrometry system (Waters, MA, United States) and NeoBase non-derivatized MS/MS kit (PerkinElmer, United States). The analytes include alanine (ALA), arginine (ARG), citrulline (CIT), glycine (GLY), leucine (LEU), methionine (MET), ornithine (ORN), phenylalanine (PHE), tyrosine (TYR), valine (VAL), free carnitine (C0), acylcarnitines (C2, C3, C3DC, C4, C4DC, C4OH, C5, C5:1, C5DC, C5OH, C6, C8, C8:1, C10, C10:1, C10:2, C12, C12:1, C14, C14:1, C14:2, C14OH, C16, C16:1, C16OH, C16:1 OH, C18, C18:1, C18:2, C18OH, C18:1OH) and succinylacetone. The normal ranges and cut-off values of analytes in the manual of NeoBaseTM non-derivatized MS/MS kit (Cat. 3040-001Z, PerkinElmer, MA, United States) and from the worldwide collaborative project were applied (McHugh et al., 2011). Suspected positive cases were recalled for the repeated test by MS/MS. The follow-up testing commences for the second time positive cases, including biochemical tests or genetic analysis. The recall and follow-up protocol in the guidelines “Follow-Up Testing for Metabolic Disease Identified by Expanded Newborn Screening Using Tandem Mass Spectrometry” was applied in our study (Bennett, 2009; Dietzen et al., 2009). Definitive diagnosis is made by specialists based on the clinical symptoms, screening test, and biochemical and genetic analysis. The parents of all cases with definitive diagnosis were informed and referred to specialists for the treatment.
Screened Panel of IEMs
Twenty-five IEMs were screened in this study. Nomenclature and abbreviations are adapted from the guidelines “Follow-Up Testing for Metabolic Disease Identified by Expanded Newborn Screening Using Tandem Mass Spectrometry” (Bennett, 2009). The screened IEMs included hypermethioninemia, phenylketonuria, homocystinuria, maple syrup urine disease, tyrosinemia, citrullinemia, argininemia, very long-chain acyl-CoA dehydrogenase deficiency, long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency, medium-chain acyl-CoA dehydrogenase deficiency, short-chain acyl-CoA dehydrogenase deficiency, glutaric acidemia type II, primary carnitine deficiency, carnitine palmitoyltransferase I (CPT I) deficiency, carnitine palmitoyltransferase II (CPT II) deficiency, trifunctional protein deficiency, multiple carboxylase deficiency, methylmalonic acidemia, glutaric acidemia type I, isovaleric acidemia, 3-hydroxy-3-methylglutaric aciduria, propionic acidemia, malonic aciduria, 3-methylcrotonyl-CoA carboxylase deficiency, and beta-ketothiolase deficiency.
Genetic Analysis
Genetic analysis was performed by BioSan Biochemical Technologies (Hangzhou, Zhejiang, China) and Be Creative Lab (Beijing, China) using the Sequenom MassARRAY iPLEX platform (Sequenom, San Diego, CA, United States), respectively. Briefly, genomic DNA was extracted from whole blood using QIAamp DNA Micro Kit (Qiagen, Hilden, Germany). Primers were designed by using Typer 4.0 software (Sequenom, San Diego, CA, United States). A total of 942 sites in IEMs-associated genes, ACADL, ACADM, acyl-CoA dehydrogenase, C-2 to C-3 short chain (ACADS), ACADVL, ACAT1, BTD, CPT2, ETFA, electron transfer flavoprotein dehydrogenase (ETFDH), glutaryl-CoA dehydrogenase (GCDH), HLCS, HMGCL, IVD, MCCC1, MCCC2, MMACHC, methylmalonyl-CoA mutase (MUT), PCCA, PCCB, SLC22A5, SLC25A20, ARG1, ASS1, BCKDHA, BCKDHB, CBS, DBT, FAH, MAT1A, MTHFR, OTC, PAH, 6-pyruvoyltetrahydropterin synthase (PTS), SLC25A13, are included based on the mutations in PubMed, OMIM, and HGMD databases. The protocol of Sequenom iPLEX genotyping was strictly followed. One picomole PCR primers, 5 ng genomic DNA, and reaction mix (Sequenom, San Diego, CA, United States) were applied in each PCR. After SAP enzyme treatment and single-base extension, the products were spotted onto a SpectroCHIP with the Sequenom MassARRAY RS1000 Nanodispenser (Sequenom, San Diego, CA, United States). Mass determination was done with the Sequenom MassARRAY mass spectrometer (Sequenom, San Diego, CA, United States).
Statistical Analysis
The data was presented descriptively and Chi-square tests were performed to compare the incidences or rates in the urban and suburban/rural areas. Statistical significance is accepted when P < 0.05.
Results
Incidence and Characteristics of IEMs
To investigate the incidence of IEMs in Jining, 48,297 infants were recruited for expanded newborn screening by MS/MS in Jining. Of 769, 725 positive cases were successfully recalled for the second examination by MS/MS. The follow-up testing immediately commences for the positive cases in the second MS/MS test, including plasma amino acid analysis, urine amino acid analysis, urine pterin metabolite analysis, enzyme activity analysis, and/or genetic analysis (Bennett, 2009; Dietzen et al., 2009). The parents of all cases with definitive diagnosis were informed and referred to specialists for the treatment. Forty-one cases with IEMs were confirmed by follow-up testing and clinical investigation. The incidence of IEMs was 1/1178 and the corrected incidence was 1/1110 (Table 1).
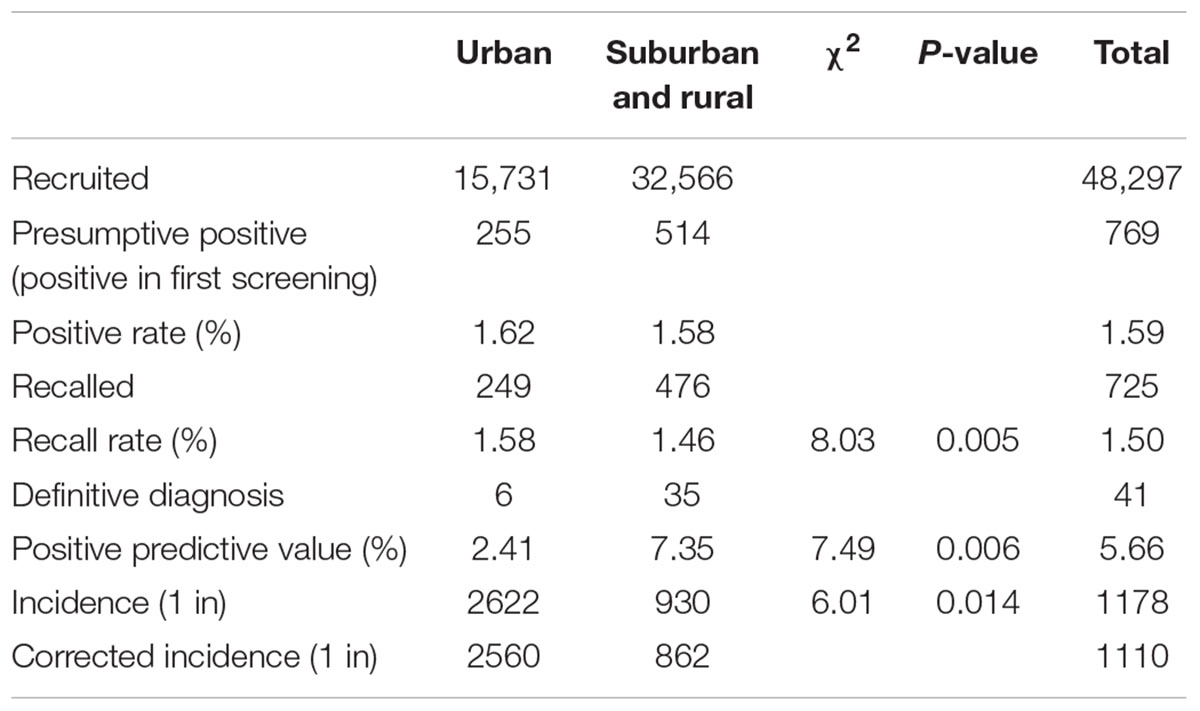
TABLE 1. Incidence and characteristics of IEMs.
A total of 15,731 neonates in the urban area and 32,566 neonates in the suburban or rural area were screened. Six and 35 confirmed cases were detected in the urban area and the suburban or rural area, respectively. The incidence of IEMs in the suburban or rural area was significantly higher than that in the urban area, 1/930 vs. 1/2622 (P < 0.05; Table 1). Based on the positive predictive values, 2.41 and 7.35% (P < 0.01), the corrected incidence was 1/862 and 1/2560 (P < 0.05) in the suburban or rural area and urban area, respectively (Table 1).
Nine types of IEMs were diagnosed in 41 cases. The incidences of amino acid disorders, fatty acid disorders, and organic acid disorders were 1/2841, 1/5366, and 1/3019, respectively. Methylmalonic acidemia, phenylketonuria, and primary carnitine deficiency ranked the top 3 of all detected IEMs with the incidence of 1/3220, 1/4391, and 1/9659, respectively. The incidences of maple syrup urine disease, argininemia, ornithine transcarbamylase deficiency, short-chain acyl-CoA dehydrogenase deficiency, glutaric acidemia type II, and glutaric acidemia type I were 1/24,149, 1/16,099, 1/48,297, 1/12,074, 1/48,297, and 1/48,297, respectively.
Genetic Analysis in the Confirmed Cases and Carriers
As IEMs are mainly caused by IEMs-associated mutations, genetic analysis is a golden approach to figure out the etiology of IEMs. Among the 41 confirmed cases, the parents of 28 cases were consent to perform genetic analysis. Thirty mutations in nine genes were detected, which resulted in seven types of IEMs (Table 2). Pathogenic mutations in the PAH and PTS genes were detected in four and one phenylketonuria cases, respectively. Pathogenic mutations in the MMACHC and MUT genes were detected in 11 and 2 cases of methylmalonic acidemia, respectively. Moreover, four cases of primary carnitine deficiency had mutations in the SLC22A5 gene while no genetic mutation was detected in one primary carnitine deficiency case. In addition, mutations in ACADS gene were detected in four cases of short-chain acyl-CoA dehydrogenase deficiency, and two of them were accompanied with glutaric acidemia (type I or type II ) carried mutations in the GCDH and ETFDH genes, respectively. Interestingly, most cases were heterozygous except that two cases were homozygous, case 12 with short-chain acyl-CoA dehydrogenase deficiency and case 25 with methylmalonic acidemia (Table 2).
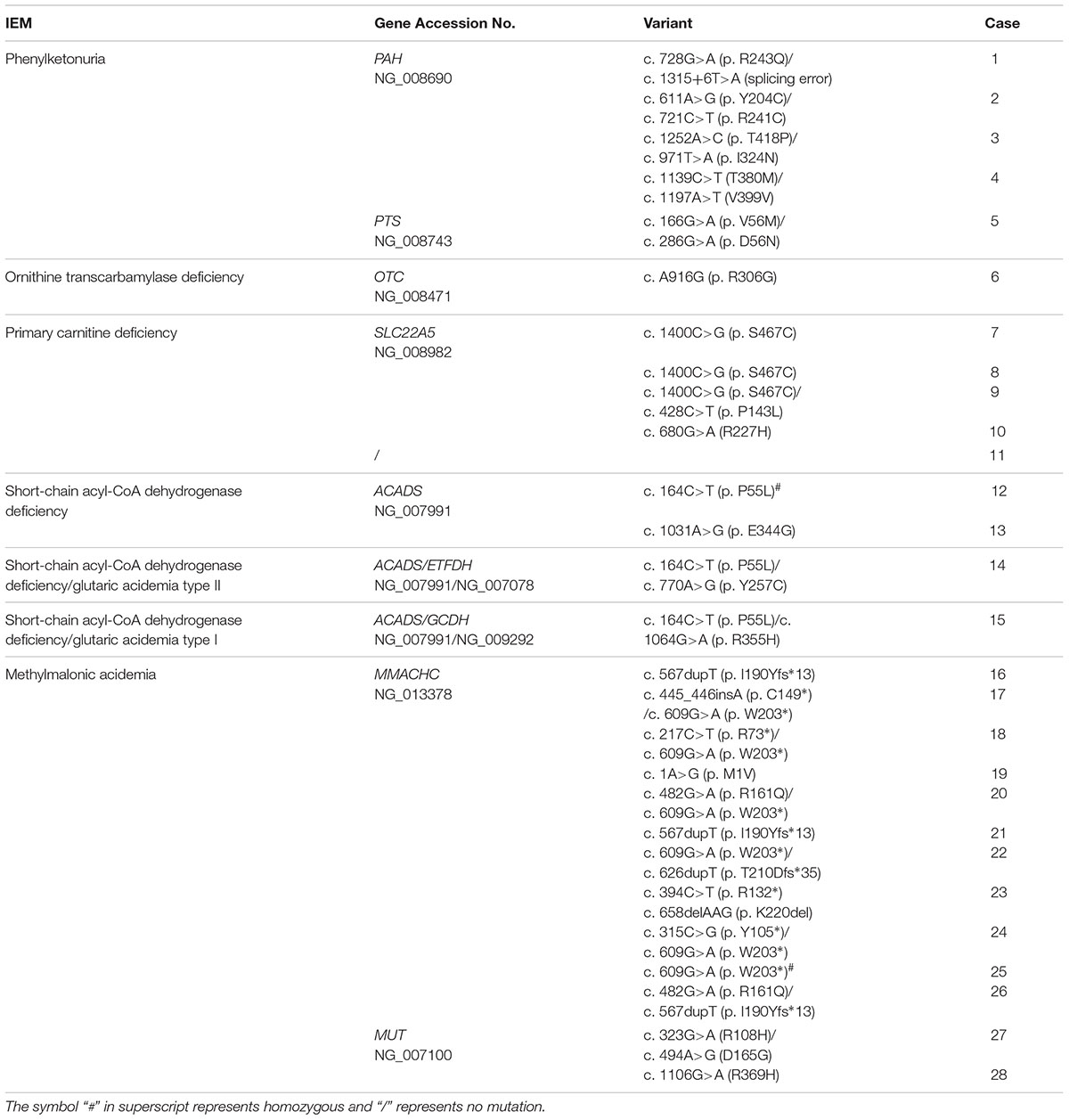
TABLE 2. Spectrum of variants in confirmed cases.
Discussion
A total of 48,297 neonates were recruited for expanded newborn screening by MS/MS and the corrected incidence of IEMs is 1/1110, which was higher than the average incidences of IEMs in China (1/3795) (Shi et al., 2012) and in many other countries, such as 1/2500 in Canada (Applegarth et al., 2000), 1/2900 in Germany (Lindner et al., 2011), 1/1944 in Egypt (Hassan et al., 2016), 1/2916 in Malaysia (Yunus et al., 2016), 1/2800 in South Korea (Yoon et al., 2005), and 1/3165 in Singapore (Lim et al., 2014). Moreover, the incidence of IEMs in Jining was higher than that in many areas or cities of China, e.g., 1/1683 in Zibo, 1/4384 in Yancheng, 1/5626 in Zhejiang province, and 1/8304 in Nanning (Huang et al., 2011; Fan et al., 2013; Zhu et al., 2015; Lu et al., 2016). It indicated that the incidence of IEMs was greater in Jining area than in many regions.
It is known that the genetic variants related to IEMs are mainly detected by Sanger sequencing and MassARRAY (Wright et al., 2008; Zhu et al., 2010; Shibbani et al., 2014; Mutlu-Albayrak et al., 2015). Recently, next generation sequencing (NGS) started to be applied for the clinical genetic analysis of IEMs (Qian et al., 2017; Smon et al., 2018). Sanger sequencing is the golden standard to determine the DNA sequence, which is applied for the known gene locus in genetic analysis. Both MassARRAY and NGS are high throughput sequencing methods, which can be used for genetic analysis of a spectrum of genes. Compared with NGS, MassARRAY has been used more widely and longer to detect known mutations. In addition, it is reliable and the cost is less. Therefore, 942 sites in IEMs-associated genes were detected by MassARRAY in this study. However, NGS has the advantage to detect the novel mutations. Thus, it may be a good option for the case 11 to discover unknown mutations.
Although the corrected incidence was significantly higher in the suburban and rural area compared with that in the urban area, 1/862 vs. 1/2560, it is unlikely that the high incidence of IEMs in the suburban and rural area is caused by consanguineous marriage and close genetic relationship. First, consanguineous marriage including cousin marriage is prohibited by Chinese Marital Law, which is widely accepted and strictly abided by the residence. Moreover, genetic analysis showed that only one case was homozygous in 22 cases in the suburban area, while one in six cases was homozygous in the urban area. The aforementioned evidence indicated that the close genetic relationship is not the cause of high incidence of IEMs in the suburban/rural area. However, the underlying reasons of the difference need to be further investigated, such as environmental factors and life style.
The incidence of phenylketonuria varies in countries and regions (Williams et al., 2008). For example, the incidence of phenylketonuria varies from 1/7325 to 1/39,338 in countries of southeastern Europe, while the incidences are 1/11,400 in the United States and 1/4172 in Turkey (Hertzberg et al., 2011; Zerjav Tansek et al., 2015). The incidence of phenylketonuria is 1/11,614 in China based on the newborn screening data of past 30 years (Shi et al., 2012). However, we found that the incidence of phenylketonuria was 1/4391 in Jining, which was much higher than the incidence of phenylketonuria in China. Genetic analysis showed that six cases carried PAH mutants and one case carried PTS mutant in patients with phenylketonuria, suggesting that mutations in PAH gene are the predominant cause of phenylketonuria in Jining.
The incidence of methylmalonic acidemia was 1/3220 in Jining, which was close to 1/3920 in Jinan, a nearby city (Han et al., 2016). However, it was significantly higher than that in other cities of China, e.g., 1/26,000 in Shanghai/Beijing, and that in other countries, e.g., approximately 1/15,155 in an Italy population and 1/67,376 in Japan (Yamaguchi, 2008; Tu, 2011; Scolamiero et al., 2015). Genetic analysis was performed in 13 confirmed cases, among which 11 cases carried mutations in the MMACHC gene, suggesting that mutations in the MMACHC gene are the predominant cause of methylmalonic acidemia in Jining.
The incidence of primary carnitine deficiency was 1/9659, which was higher than that in other regions of China, e.g., 1/32,354 in Zhejiang province and 1/31,773 in Guangxi province (Huang et al., 2011). It was also higher than that in other countries, e.g., approximately 1/40,000 in Japan and 1/50,000 in the United States (Koizumi et al., 1999; Magoulas and El-Hattab, 2012). Among the five primary carnitine deficiency cases, four cases carried SLC22A5 mutants. It suggested that SLC22A5 mutants are the predominant cause of primary carnitine deficiency in Jining. However, no known mutation was detected in SLC22A5 gene in case 11. It has to be noted that only the known mutations in the exons of SLC22A5 gene can be detected by MassARRAY method in this study. Thus, novel mutations in the exons of SLC22A5 gene may not be detected by this method. In addition, it is possible that mutations in exons of other genes may cause primary carnitine deficiency. Therefore, whole exon sequencing may need to be performed to discover the novel mutations in the exons of SLC22A5 gene and/or other genes. In addition to mutations in the exons, mutations existing in the 5′-UTR, introns and 3′-UTR may contribute to the dysfunction of SLC22A by altering the expression level or splicing process. For example, an SNP in the intron 4 of TMP21 gene significantly affects the splicing efficiency leading to the dysregulation of TMP21 expression, contributing to the pathogenesis of Alzheimer’s disease (Zhang et al., 2018). Therefore, the 5′-UTR, introns and 3′-UTR may also need to be sequenced in the later study.
In the present study, we showed that the incidence of IEMs was 1/1178 in Jining, while the incidence was significantly higher in the suburban and rural area compared with that in the urban area, 1/862 vs. 1/2560. The incidence of methylmalonic acidemia, phenylketonuria, and primary carnitine deficiency ranked the top 3 of all detected IEMs. In addition, 30 mutations in nine IEMs-associated genes were identified in the 28 confirmed cases and 19 cases with the mutations in PAH, SLC22A5, and MMACHC genes, respectively. Our work indicated that the overall incidence of IEMs is higher in Jining than that in China. Methylmalonic acidemia, phenylketonuria, and primary carnitine deficiency were the major IEMs and the mutations in PAH, SLC22A5, and MMACHC genes are the leading causes of IEMs resulting in phenylketonuria, primary carnitine deficiency, and methylmalonic acidemia, respectively, in Jining area. Therefore, it is critical to increase the coverage of expanded newborn screening by MS/MS and prenatal genetic consulting in Jining area. Furthermore, the underlying reasons of higher incidence of IEMs in Jining and particularly higher incidence of IEMs in the suburban area need to be investigated to achieve the decrease of the incidence of IEMs in the future.
Author Contributions
KG and XC performed screening by MS/MS and collected the data. YW and XZ formulated the study, analyzed the data, wrote the manuscript, and designed the tables. CL and QK provided intellectual thoughts, revised the manuscript, and led the project.
Funding
This work was supported by grants from the Natural Science Foundation of Shandong Province (ZR2016HM30, ZR2017PH011, and ZR2013CM031), National Natural Science Foundation of China (81771147), the Jining Science and Technology Program for Public Wellbeing (2017SMNS006, 2014kjhm-10), the Development of Medical Science and Technology Project of Shandong Province (2013WSB33004), Youth Fund Project of Jining Medical University (JY2017JS002), and Science and Technology Project of Higher Education of Shandong Province (J12LL12).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer RTA and handling Editor declared their shared affiliation.
Acknowledgments
We thank Xin Li and Shuai Wang for the critical comments and helpful suggestions.
References
Applegarth, D. A., Toone, J. R., and Lowry, R. B. (2000). Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics 105:e10. doi: 10.1542/peds.105.1.e10
Bennett, M. J. (2009). Follow-up testing for metabolic disease identified by expanded newborn screening using tandem mass spectrometry. Clin. Biochem. 55, 1615–1626. doi: 10.1373/clinchem.2009.131300
Campeau, P. M., Scriver, C. R., and Mitchell, J. J. (2008). A 25-year longitudinal analysis of treatment efficacy in inborn errors of metabolism. Mol. Genet. Metab. 95, 11–16. doi: 10.1016/j.ymgme.2008.07.001
Dietzen, D. J., Rinaldo, P., Whitley, R. J., Rhead, W. J., Hannon, W. H., Garg, U. C., et al. (2009). National academy of clinical biochemistry laboratory medicine practice guidelines: follow-up testing for metabolic disease identified by expanded newborn screening using tandem mass spectrometry; executive summary. Clin. Chem. 55, 1615–1626. doi: 10.1373/clinchem.2009.131300
Fan, X., Chen, S. B., Luo, J., Li, W., Lin, F., Luo, C., et al. (2013). Application of tandem mass spectrometry in genetic metabolism screening in Nanning, Guangxi. J. Guangxi Med. Univ. 30, 756–758. doi: 10.16190/j.cnki.45-1211/r.2013.05.055
Han, B., Cao, Z., Tian, L., Zou, H., Yang, L., Zhu, W., et al. (2016). Clinical presentation, gene analysis and outcomes in young patients with early-treated combined methylmalonic acidemia and homocysteinemia (cblC type) in Shandong province. China. Brain Dev. 38, 491–497. doi: 10.1016/j.braindev.2015.10.016
Hassan, F. A., El-Mougy, F., Sharaf, S. A., Mandour, I., Morgan, M. F., Selim, L. A., et al. (2016). Inborn errors of metabolism detectable by tandem mass spectrometry in Egypt: the first newborn screening pilot study. J. Med. Screen. 23, 124–129. doi: 10.1177/0969141315618229
Hertzberg, V. S., Hinton, C. F., Therrell, B. L., and Shapira, S. K. (2011). Birth prevalence rates of newborn screening disorders in relation to screening practices in the United States. J. Pediatr. 159, 555–560. doi: 10.1016/j.jpeds.2011.04.011
Huang, X. W., Yang, J. B., Tong, F., Yang, R. L., Mao, H. Q., Zhou, X. L., et al. (2011). [Screening for neonatal inborn errors of metabolism by electrospray ionization-tandem mass spectrometry and follow-up]. Zhonghua Er Ke Za Zhi 49, 765–770.
Koizumi, A., Nozaki, J., Ohura, T., Kayo, T., Wada, Y., Nezu, J., et al. (1999). Genetic epidemiology of the carnitine transporter OCTN2 gene in a Japanese population and phenotypic characterization in Japanese pedigrees with primary systemic carnitine deficiency. Hum. Mol. Genet. 8, 2247–2254. doi: 10.1093/hmg/8.12.2247
Lim, J. S., Tan, E. S., John, C. M., Poh, S., Yeo, S. J., Ang, J. S., et al. (2014). Inborn Error of Metabolism (IEM) screening in Singapore by electrospray ionization-tandem mass spectrometry (ESI/MS/MS): an 8 year journey from pilot to current program. Mol. Genet. Metab. 113, 53–61. doi: 10.1016/j.ymgme.2014.07.018
Lindner, M., Gramer, G., Haege, G., Fang-Hoffmann, J., Schwab, K. O., Tacke, U., et al. (2011). Efficacy and outcome of expanded newborn screening for metabolic diseases–report of 10 years from South-West Germany. Orphanet J. Rare Dis. 6:44. doi: 10.1186/1750-1172-6-44
Lu, H., Zheng, J., Yao, Y., Yang, M., Yao, S., Shi, N., et al. (2016). A retrospective analysis of tandem mass spectrometry screening for neonatal genetic metabolic diseases in Yancheng area. J. Birth Health Hered. 24, 83–85. doi: 10.13404/j.cnki.cjbhh.2016.03.036
Magoulas, P. L., and El-Hattab, A. W. (2012). Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet J. Rare Dis. 7:68. doi: 10.1186/1750-1172-7-68
McHugh, D., Cameron, C. A., Abdenur, J. E., Abdulrahman, M., Adair, O., Al Nuaimi, S. A., et al. (2011). Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: a worldwide collaborative project. Genet. Med. 13, 230–254. doi: 10.1097/GIM.0b013e31820d5e67
Moammar, H., Cheriyan, G., Mathew, R., and Al-Sannaa, N. (2010). Incidence and patterns of inborn errors of metabolism in the Eastern Province of Saudi Arabia, 1983-2008. Ann. Saudi Med. 30, 271–277. doi: 10.4103/0256-4947.65254
Mutlu-Albayrak, H., Bene, J., Oflaz, M. B., Tanyalcin, T., Caksen, H., and Melegh, B. (2015). Identification of SLC22A5 gene mutation in a family with carnitine uptake defect. Case Rep. Genet. 2015:259627. doi: 10.1155/2015/259627
Qian, J., Wang, X., Liu, J., Zhong, J., Le, Y., Melchior Tellier, L. C. A., et al. (2017). Applying targeted next generation sequencing to dried blood spot specimens from suspicious cases identified by tandem mass spectrometry-based newborn screening. J. Pediatr. Endocrinol. Metab. 30, 979–988. doi: 10.1515/jpem-2017-0003
Sanderson, S., Green, A., Preece, M. A., and Burton, H. (2006). The incidence of inherited metabolic disorders in the West Midlands, UK. Arch. Dis. Child. 91, 896–899. doi: 10.1136/adc.2005.091637
Scolamiero, E., Cozzolino, C., Albano, L., Ansalone, A., Caterino, M., Corbo, G., et al. (2015). Targeted metabolomics in the expanded newborn screening for inborn errors of metabolism. Mol. Biosyst. 11, 1525–1535. doi: 10.1039/c4mb00729h
Shi, X. T., Cai, J., Wang, Y. Y., Tu, W. J., Wang, W. P., Gong, L. M., et al. (2012). Newborn screening for inborn errors of metabolism in mainland china: 30 years of experience. JIMD Rep. 6, 79–83. doi: 10.1007/8904_2011_119
Shibbani, K., Fahed, A. C., Al-Shaar, L., Arabi, M., Nemer, G., Bitar, F., et al. (2014). Primary carnitine deficiency: novel mutations and insights into the cardiac phenotype. Clin. Genet. 85, 127–137. doi: 10.1111/cge.12112
Smon, A., Repic Lampret, B., Groselj, U., Zerjav Tansek, M., Kovac, J., Perko, D., et al. (2018). Next generation sequencing as a follow-up test in an expanded newborn screening programme. Clin. Biochem. 52, 48–55. doi: 10.1016/j.clinbiochem.2017.10.016
Tu, W. J. (2011). Methylmalonic acidemia in mainland China. Ann. Nutr. Metab. 58:281. doi: 10.1159/000331469
Williams, R. A., Mamotte, C. D., and Burnett, J. R. (2008). Phenylketonuria: an inborn error of phenylalanine metabolism. Clin. Biochem. Rev. 29, 31–41.
Wright, W. T., Heggarty, S. V., Young, I. S., Nicholls, D. P., Whittall, R., Humphries, S. E., et al. (2008). Multiplex MassARRAY spectrometry (iPLEX) produces a fast and economical test for 56 familial hypercholesterolaemia-causing mutations. Clin. Genet. 74, 463–468. doi: 10.1111/j.1399-0004.2008.01071.x
Yamaguchi, S. (2008). Newborn screening in Japan: restructuring for the new era. Ann. Acad. Med. Singapore 37, 13–15.
Yamaguchi, S., Purevsuren, J., Hasegawa, Y., Kobayashi, H., Mushimoto, Y., Yamada, K., et al. (2013). “Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency and expanded newborn screening in Japan,” in Proceedings of the Genetic Testing Symposium and the International Society for Neonatal Screening and Featured presentation at Newborn Screening, Atlanta.
Yoon, H. R., Lee, K. R., Kang, S., Lee, D. H., Yoo, H. W., Min, W. K., et al. (2005). Screening of newborns and high-risk group of children for inborn metabolic disorders using tandem mass spectrometry in South Korea: a three-year report. Clin. Chim. Acta 354, 167–180. doi: 10.1016/j.cccn.2004.11.032
Yunus, Z. M., Rahman, S. A., Choy, Y. S., Keng, W. T., and Ngu, L. H. (2016). Pilot study of newborn screening of inborn error of metabolism using tandem mass spectrometry in Malaysia: outcome and challenges. J. Pediatr. Endocrinol. Metab. 29, 1031–1039. doi: 10.1515/jpem-2016-0028
Zerjav Tansek, M., Groselj, U., Angelkova, N., Anton, D., Baric, I., Djordjevic, M., et al. (2015). Phenylketonuria screening and management in southeastern Europe - survey results from 11 countries. Orphanet J. Rare Dis. 10:68. doi: 10.1186/s13023-015-0283-0
Zhang, X., Wu, Y., Cai, F., Liu, S., Bromley-Brits, K., Xia, K., et al. (2018). A novel Alzheimer-associated SNP in Tmp21 increases amyloidogenesis. Mol. Neurobiol. 55, 1862–1870. doi: 10.1007/s12035-017-0459-9
Zhu, F., Xu, J., Dong, L., and Mou, K. (2015). Analysis of 25243 cases of neonatal genetic metabolic disease screening by tandem mass spectrometry. J. Binzhou Med. Univ. 38, 300–302.
Keywords: newborn screening, inborn errors of metabolism, incidence of IEMs, IEMs-associated gene mutation
Citation: Guo K, Zhou X, Chen X, Wu Y, Liu C and Kong Q (2018) Expanded Newborn Screening for Inborn Errors of Metabolism and Genetic Characteristics in a Chinese Population. Front. Genet. 9:122. doi: 10.3389/fgene.2018.00122
Received: 26 January 2018; Accepted: 26 March 2018;
Published: 20 April 2018.
Edited by:
Haranatha R. Potteti, University of Illinois at Chicago, United StatesReviewed by:
Rand Talal Akasheh, University of Illinois at Chicago, United StatesMuhammad Jawad Hassan, National University of Sciences and Technology, Pakistan
Copyright © 2018 Guo, Zhou, Chen, Wu, Liu and Kong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yili Wu, yili_wu2004@yahoo.ca Chuanxin Liu, liuchuanxin2003b@163.com Qingsheng Kong, jnyxykqs@163.com
†These authors have contributed equally to this work.