 Rodrigo C. de Almeida
Rodrigo C. de Almeida Vinícius S. Chagas
Vinícius S. Chagas Mauro A. A. Castro
Mauro A. A. Castro Maria L. Petzl-Erler
Maria L. Petzl-Erler- 1Human Molecular Genetics Laboratory, Department of Genetics, Federal University of Paraná, Curitiba, Brazil
- 2Molecular Epidemiology, Department of Biomedical Data Sciences, Leiden University Medical Center, Leiden, Netherlands
- 3Bioinformatics and Systems Biology Laboratory, Federal University of Paraná, Curitiba, Brazil
Genome-wide and fine mapping studies have shown that more than 90% of genetic variants associated with autoimmune diseases (AID) are located in non-coding regions of the human genome and especially in regulatory sequences, including microRNAs (miRNA) target sites. MiRNAs are small endogenous noncoding RNAs that modulate gene expression at the post-transcriptional level. Single nucleotide polymorphisms (SNPs) located within the 3′ untranslated region of their target mRNAs (miRSNP) can alter miRNA binding sites. Yet, little is known about their effect on regulation by miRNA and the consequences for AID. Conversely, it is well known that two or more AID may share part of their genetic background. Here, we hypothesized that miRSNPs could be associated with more than one AID. To identify miRSNPs associated with AID, we integrated results from three different prediction tools (Polymirts, miRSNP, and miRSNPscore) using a naïve Bayes classifier approach to identify miRSNPs predicted to affect binding sites of miRNAs. Further, to detect miRSNPs associated with two or more AID, we integrated predictions with summary statistics from 12 AID studies. In addition, to prioritize miRSNPs, miRNAs and AID-associated target genes, we used public expression quantitative trait locus (eQTL) data and mRNA-seq and small RNA-seq data. We identified 34 miRNSPs associated with at least two AID. Furthermore, we found 86 miRNAs predicted to target 18 of the associated gene's mRNAs. Our integrative approach revealed new insights into miRNAs and AID associated target genes. Thus, it helped to prioritize AID noncoding risk SNPs that might be involved in the causal mechanisms, providing valuable information for further functional studies.
Introduction
Genome-wide association studies (GWAS) and fine mapping studies have identified approximately 250 loci associated with autoimmune disease (AID), and some of these loci are shared between two or more diseases (Ricaño-Ponce and Wijmenga, 2013). The recent results of fine mapping studies performed by the Immunochip platform, showed that the majority of the associated variants are located in non-coding regions, especially in regulatory sequences such as microRNAs (miRNAs) target sites (Ricaño-Ponce and Wijmenga, 2013).
MiRNAs are small molecules about 22 nt long that act by imperfect base-paring to 3′ untranslated regions (3′ UTR) of target messenger RNAs (mRNAs), leading to translation repression, degradation of the mRNA, or both (Bartel, 2009). MiRNAs negatively regulate their target mRNAs primarily through Watson and Crick base-pairing interactions, and the seed sequence located at positions 2–8 within the miRNA sequence is key for this action. Single nucleotide polymorphisms (SNPs) within the seed site, or in the target mRNA at sites complementary to miRNA seed sites, referred to as miRSNPs, may reduce effectiveness or abolish miRNA-mediated repression (Saunders et al., 2007), which could have functional consequences for complex diseases (Sethupathy and Collins, 2008), including AID (Hrdlickova et al., 2014).
In addition, it has been recently shown that numerous miRSNPs have expression quantitative trait loci (eQTL) effect on GWAS reported genes (Võsa et al., 2015). Moreover, the number of miRSNPs reported to be associated with human diseases increased in the past few years (Wei et al., 2012; Ghanbari et al., 2015; Stegeman et al., 2015; Cipolla et al., 2016), therefore, several in silico approaches has been developed to identify the potential impact of these polymorphisms in miRNA target genes (reviewed in Moszynska et al., 2017). Nevertheless, a systematically approach that integrates predictions from different miRSNPs algorithms has not yet been developed.
Here, we hypothesized that miRSNPs could be associated with more than one AID and that a comprehensive analysis of miRSNPs could identify additional still unknown risk variants. To investigate genetic associations of miRSNPs with AID, we applied an approach that integrates information provided by different algorithms, which uses several different data types. First, we used a naïve Bayes classifier to integrate results from three major prediction tools. Next, we intersected these results with summary statistics from GWAS of 12 common autoimmune diseases (AID), where 10 of these studies used the high-density platform Immunochip (Cortes and Brown, 2011). Moreover, by integrating small and mRNA-seq with eQTL public data, we were able to prioritize miRSNPs, miRNAs, and AID associated target genes. The results of our analyses provide valuable information for further functional studies.
Materials and Methods
Data Integration
In order to find SNPs predicted to have an effect on miRNA binding sites (miRSNPs) results from three different prediction tools were integrated:
(1) PolymiRts v.3.0 (http://compbio.uthsc.edu/miRSNP/; Bhattacharya et al., 2014); (2) miRNASNP2 (http://www.bioguo.org/miRNASNP2; Liu C. et al., 2012); and (3) miRSNPscore (http://www.bigr.medisin.ntnu.no/mirsnpscore; Thomas et al., 2011).
PolymiRTS v3.0 (accessed 15 Feb 2016) uses dbSNP v137, miRBase v20, and TargetScan (Friedman et al., 2009) algorithms to predict miRNA binding sites. To predict efficacy of targeting, this algorithm considers two major features, the probability of targeting conservation and the perfect match between the target and the seed sequence on the miRNA, that is, the at least 7 nt long canonical seed region (7mer-A1, 7mer-m8, or 8mer).
miRNASNP2 (accessed 15 Feb 2016) integrates data from miRBase v19, dbSNP v137 and uses two prediction tools: TargetScan and the miRanda (John et al., 2004) algorithm with stringent 2–8 nt pairing for miRNA binding site prediction. MiRanda assumes strict complementarity between nucleotides 2 and 8 and uses a score of alignment quality and free energy for miRNA binding site identification.
MirSNPscore uses miRBase v16, SNP data from HapMap 3 Project, and its own score algorithm to miRNA-biding site predictions. This tool uses haplotype information to calculate a score that corresponds to the probability of the SNP interfering with miRNA-binding.
To integrate results from these tools, a naïve Bayesian approach, adapted from previously successful data integration strategies (von Mering et al., 2005; Szklarczyk et al., 2017), was used. A naïve Bayes classifier was implemented in R (R Development Core Team, 2017) and a combined score was calculated for each miRSNP under the assumption of independence for the various data sources (data harmonization was obtained by z-score transformation of the results from each prediction algorithm):
MiRSNPs with naïve Bayes combined (NBC) score >0.7 were considered candidates to affect the miRNA-binding site.
Summary Statistics From Autoimmune Diseases (Immunochip Studies)
To investigate whether miRSNPs are associated with AID, we downloaded summary statistics for 12 diseases available at the Immunobase database (https://www.immunobase.org/, accessed 16 May 2016) and integrated them with the naïve Bayes classifier results. Cohort participants and studies are shown in Table 1. Of these studies, 10 used high-density genotype array Immunochip and two used the first generation of chip arrays (Illumina HumanHapMap 5, Illumina HumanHapMap3 and Affymetrix Gene Chip 500). Quality control procedures are described in detail in the original papers of each study (references are cited in Table 1).
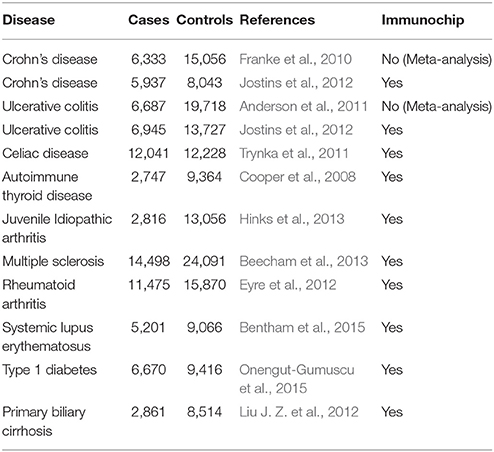
Table 1. Cohorts description.
To capture also SNPs in linkage disequilibrium (LD) that were not reported as associated by the study, we used LDlink tool (Machiela and Chanock, 2014) using data from the 1000 Genomes Project phase 3 (Abecasis et al., 2012) with standard parameters (r2 ≥ 0.8 and D' = 1) and the CEU population as reference panel.
For our purpose, only miRSNPs that either had genome-wide threshold P-value (P < 5 × 10−8) or were in strong LD (r2 > 0.8) with a significantly associated SNP but not evaluated in the study, were considered as candidates. In addition, only miRSNPs associated with at least two AID were considered for further analysis.
eQTL Data Integration
eQTL data was acquired from three different databases, BloodeQTLBrowser (http://genenetwork.nl/bloodeqtlbrowser/; Westra et al., 2013), the Geuvadis project (Lappalainen et al., 2013), and the GTEx v6 portal (http://www.gtexportal.org/home; Lonsdale et al., 2013).
BloodeQTLBrowser contains data from peripheral blood of 5,311 individuals obtained with microarray chips. The Geuvadis project (Lappalainen et al., 2013) used RNA-seq data from 465 lymphoblastoid cell lines (LCL) from 5 populations of the 1000 Genomes Project: the CEPH (CEU), Finns (FIN), British (GBR), Toscani (TSI), and Yoruba (YRI) to perform its eQTL analysis. Data from GTEx v6 portal (Lonsdale et al., 2013) derived from 44 different tissues in 7,051 RNA-seq samples.
BloodeQTLBrowser and Geuvadis data were filtered using false discovery rate (FDR) correction at the 0.05 level, which resulted in 514,000 cis-eQTLs from BloodeQTL and 7,714 cis-eQTLs from the Geuvadis project. The GTEx v6 data used a FDR ≤ 0.05, containing 27,159 unique cis-eQTL genes.
Small RNA-Seq, mRNA-Seq and Genotype Data From the GEUVADIS and the 1000 Genomes Projects
mRNA-seq and small RNA-seq data from 465 LCL were downloaded from the Geuvadis Project (Lappalainen et al., 2013). After matching samples that have both the miRNA and the mRNA profiles, 321 RNA-seq samples remained, which were further log2 normalized. Further, genotype data were downloaded from the 1000 Genomes Project (Abecasis et al., 2012). After matching with individuals from the Geuvadis project, 309 samples with genotype and RNA-seq (small RNA and mRNA) data were used for Pearson correlation analysis between the expression levels of the miRNAs and their mRNAs targets, where P < 0.05 was considered the limit for statistical significance.
Experimentally Validated miRNA-Target Interactions Databases
Experimentally validated miRNA target sites were downloaded from miRTarBase v7.0 (Chou et al., 2016; http://mirtarbase.mbc.nctu.edu.tw/php/index.php) and Tarbase v7 (Vlachos et al., 2015; under authorization on http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=tarbase/index). These databases contain hundreds of thousands of published experimentally manually curated validated miRNA-target interactions. These databases include data generated by the main types of functional experiments currently used to validate miRNA-mRNA interaction, such as, western blot, microarray, luciferase report assays and high-throughput sequencing of immunoprecipitated RNAs after cross-linking (CLIP-seq), submitted by many other researchers.
Results
Data Integration Showed That Autoimmune Diseases Share Associations With miRSNPs
To identify SNPs that affect the binding site of miRNAs (miRSNPs) in genes associated with AID, we implemented a naïve Bayes classifier approach to integrate different miRSNPs prediction tools and further intersection with summary statistics from GWAS high density genotyped data (Immunochip).
We integrated three miRSNPs prediction tools with summary statistics from 12 GWAS of AID available in the Immunobase database (Table 1).
However, even studies using a dense genotyping custom-made array designed to fine-map immune-related diseases, such as the Immunochip, may still miss associations due to differences in linkage disequilibrium (LD) pattern between populations, or because of differences of quality control steps. To minimize this bias, we also included miRSNPs that were in high LD (r2 ≥ 0.8 and D' = 1 in the CEU population Abecasis et al., 2012) with the most associated SNP of the corresponding target gene reported by each study (Supplementary Table 1).
Additionally, to identify miRSNPs that might affect expression levels of the reported disease-associated genes, we intersected our results with publicly available eQTL data from the GTEx project (Lonsdale et al., 2013), Blood eQTL Browser (Westra et al., 2013), and the Geuvadis project (Lappalainen et al., 2013) (Supplementary Table 2).
We found 34 miRSNPs that may alter the binding sites of 86 miRNAs predicted to target mRNAs of 18 genes (Table 2). Of these, 28 miRSNPs displayed an eQTL effect on 13 target genes in several tissues (Table 2). Moreover, from the 34 miRNSPs, four (rs1054037, rs2070197, rs727088, rs7444) had been reported as the most associated SNPs by the original studies (P < 5 × 10−8) (Table 2). The remaining 30 miRSNPs were in strong LD with the most associated SNP reported, but were not evaluated by the study. For instance, two miRSNPs (rs60474474 and rs45450798), both located in the 3′ UTR of PTPN2, were not reported in any of the GWAS. However, these two miRSNPs display maximum possible LD (r2 = 1 and D' = 1) (Supplementary Table 1) with the most associated SNP reported for five AIDs inflammatory bowel disease (IBD), Crohn's disease (CD), type 1 diabetes (T1D), celiac disease (CeD), juvenile idiopathic arthritis (JIA) (Table 2). Both miRSNPs were predicted to alter miRNA binding sites in the 3′ UTR of the PTPN2 mRNA: miRSNP rs60474474 disrupts the binding site of miR-4290 (NBC Score = 0.99) and miRSNP rs45450798 creates a new binding site for miR-4531 (NBC Score = 0.73) (Table 2).
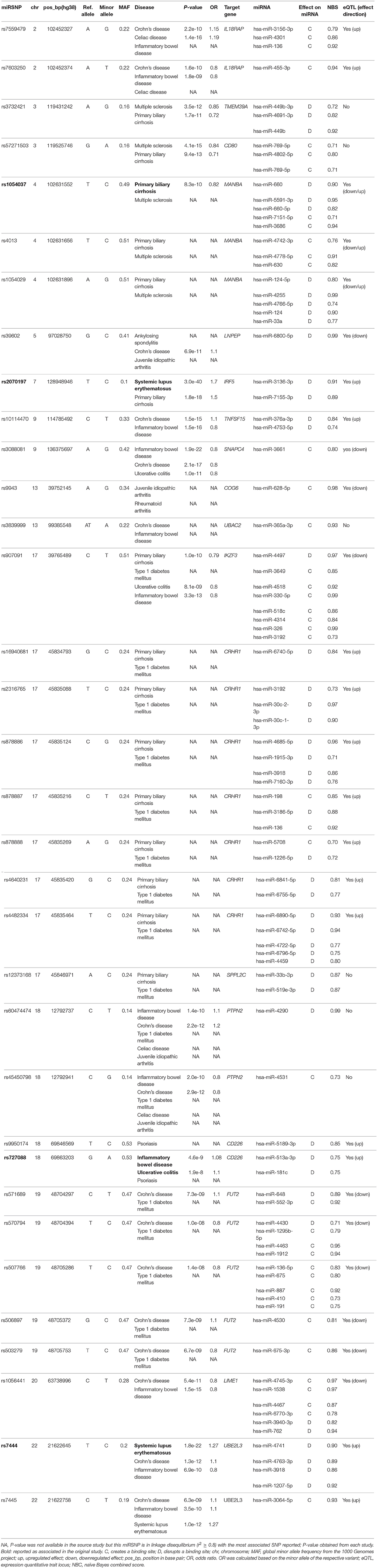
Table 2. MiRSNPs associated with autoimmune disease.
Among the investigated 12 diseases, Crohn's disease was the one that displayed more associated miRSNPs. In total, we found 16 miRSNPs associated with CD, affecting the binding sites of 31 miRNAs in the mRNAs of the CD-associated genes (Table 2).
The most significantly associated miRSNP (rs2070197) was associated with two diseases, systemic lupus erythematosus (SLE) (P = 3.04 × 10−40) and primary biliary cirrhosis (PBC) (P = 1.8 × 10−18) (Table 2). This miRSNP is located in the 3′ UTR of the IRF5 gene and may disrupt the binding sites of two miRNAs (miR-3136-3p NBC Score = 0.91 and miR-7155-3p NBC Score = 0.89). Thus, it is an eQTL predicted to downregulate the expression of IRF5 at least in LCL.
The miRSNP rs7444, which is in the 3′ UTR of the UBE2L3 gene, was associated with three AIDs (SLE, CD, and IBD). This miRSNP was predicted to affect binding of four miRNAs (miR-4741 NBC Score = 0.9, miR-4763-3p NBC Score = 0.89, miR-3918 NBC Score = 0.86, miR-1207-5p NBC Score = 0.92) and is an eQTL that upregulates the target gene in whole blood (Figure 1).
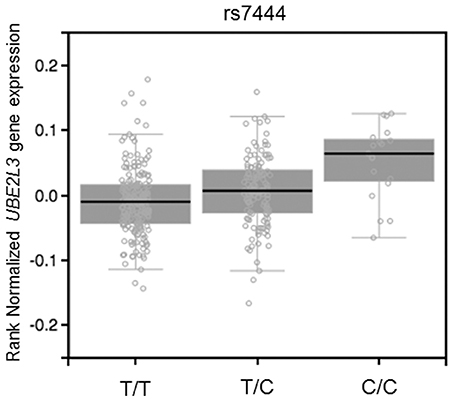
Figure 1. eQTL effect on whole blood of SNP rs7444 genotypes on expression of UBE2L3 mRNA (P = 1.7 × 10−7, effect size = 0.16). Figure adapted from the Gtex database (https://www.gtexportal.org).
Moreover, the gene FUT2, which was associated with three AID (CD, IBD, and T1D), had five miRSNPs affecting 13 miRNAs. These miRSNPs are eQTL with a downregulation effect on the target mRNAs in several tissues, including small intestine and whole blood (Table 2).
To verify if the expression levels of miRNAs could be correlated with the mRNA levels of the associated target genes, we used public available RNA-seq data from the Geuvadis Project (Lappalainen et al., 2013). First, we matched all samples that had both, small RNA-seq and mRNA-seq data (N = 321 samples). Further, we performed Pearson correlation analysis of the expression of all miRNAs and the mRNA of their targets. Of the 86 miRNAs in our dataset (Table 2) we found 19 whose expression levels were correlated with those of 15 predicted targets (Figure 2). Nevertheless, only 9 of these 19 miRNAs were correlated with expression levels of their target genes in the same direction (up- or downregulated) corresponding to that predicted by the algorithms (Figure 3).
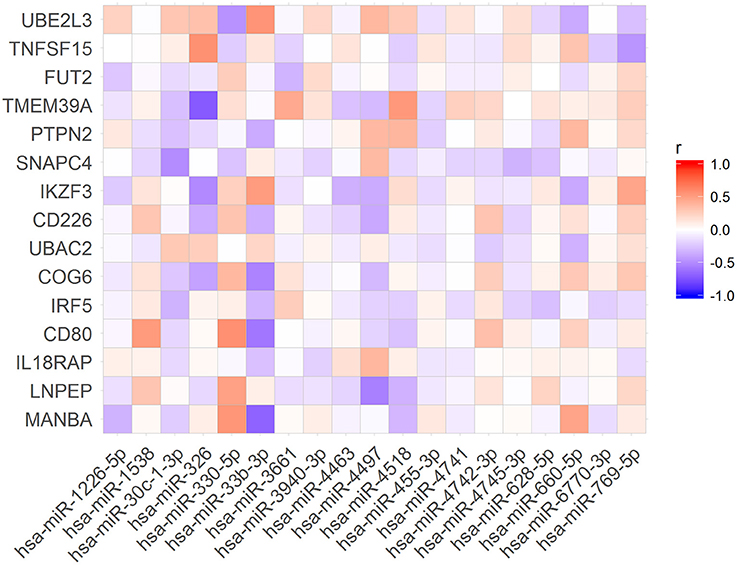
Figure 2. Pearson correlations between the expression levels of the miRNAs and their target mRNAs.
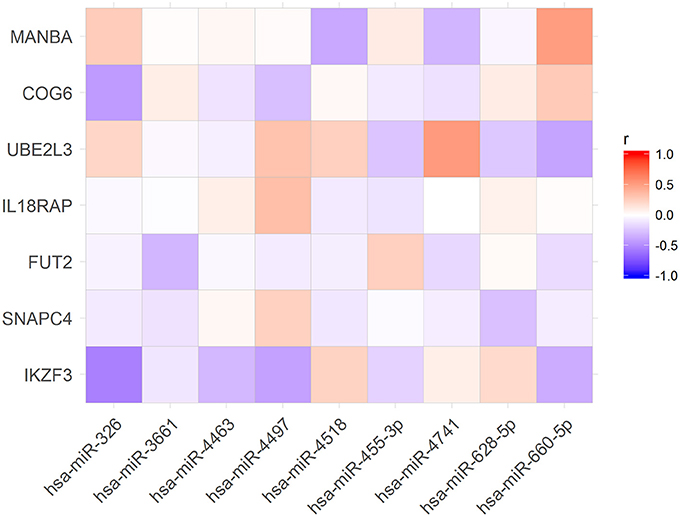
Figure 3. Expression levels of nine microRNA were correlated with expression levels of their target mRNAs as expected according with TargetScan and miRanda predictions.
To investigate whether correlation of expression levels of these 9 miRNAs and their targets were dependent on the genotype, we extracted genotype data from the 1000 Genomes Project, and RNA-seq data (small RNA and mRNA) from the Geuvadis project. We then performed Pearson correlation analysis with only homozygous individuals for the minor and the reference alleles of each miRSNP. We found that expression levels of three miRNAs correlated with those of two target genes (P < 0.05) depending on the genotype (Figures 4–6). Nevertheless, when comparing the correlation between the mRNA and microRNA expression data between genotypes, the predictions of the miRSNP's effect on the expression of the target gene could not be clearly demonstrated. In the sample of individuals homozygous for the minor allele (T) of miRSNP rs907091 located in the 3′ UTR of IKZF3, expression of miR-326, and IKZF3 were negatively correlated (P = 0.006479, r2 = −0.35) (Figure 4). This miRSNP is predicted to create a binding site for miR-326 with a high NBC Score (NBC Score = 0.99), which fits the negative correlation observed between expression of miR-326 and the IKZF3 mRNA. Yet, although not significant, the correlation follows the same trend in the sample of individuals homozygous for allele C (Figure 4). Furthermore, the expression levels of miR-4518 and the IKZF3 mRNA were positively correlated (P = 0.02, r2 = 0.2) in the presence of homozygosity for allele rs907091 T, which is predicted to create a binding site for miR-4518 in the 3′ UTR of that mRNA.

Figure 4. (A) Individuals homozygous for the protective allele T (T/T). (B) Individuals homozygous for the reference allele C (C/C).
In addition, miRSNP rs9943 allele G, which is an eQTL associated with downregulation of the target gene COG6 (Figure 6), was predicted to create a binding site for miR-628-5p. Notwithstanding, expression of this miRNA and of the mRNA of its target COG6 are positively correlated (P = 0.01, r2 = 0.43) for genotype G/G but are not significantly correlated for genotype A/A (Figure 5). It should be noticed that data from the Geuvadis project derive from LCL only and may not reflect the expression pattern in cells relevant for pathogenesis of the AID. Altogether, these results reveal the complexity of inferences based on predictions and the importance of functional validation of the hypotheses.
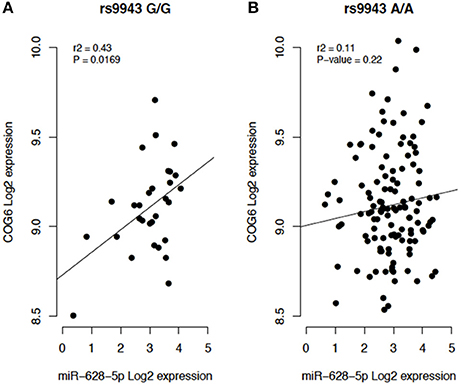
Figure 5. (A) Individuals homozygous for the risk allele rs9943 G (G/G). (B) Individuals homozygous for the reference allele A (A/A).
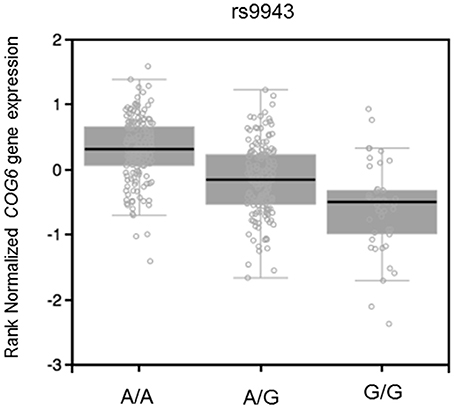
Figure 6. eQTL effect on muscle skeletal of rs9943 genotypes on expression of COG6 mRNA (P = 2.0e-25, effect size = −0.53). Figure adapted from the Gtex database (https://www.gtexportal.org).
Furthermore, in order to verify if the predictions of miRNA-target interactions had been experimentally validated, we integrated our results with data available in the DIANA-Tarbase v7 (Vlachos et al., 2015) and the MiRTarbase (Chou et al., 2016). We found two miRNA—target interactions (UBAC2—miR-365a-3p and SNAPC4—miR-3661) which were experimentally validated by the CLIP-seq method (Supplementary Table 3). Variants of both validated target genes are associated with IBD and CD (Table 2), and SNAPC4 is also associated with UC. In addition, the rs3088081 miRSNP is an eQTL with a downregulation effect of gene SNAPC4 in thyroid and skeletal muscle tissues (Supplementary Table 2).
Discussion
We used an approach to integrate miRNAs targets predictions, GWAS summary statistics and RNA-seq publicly available data, to investigate whether SNPs associated with two or more AID may affect miRNAs binding sites. Furthermore, we integrated eQTL results with RNA sequencing data (miRNA-seq and mRNA-seq) to verify if the genotypes of the selected miRSNPs influence the expression levels of the miRNAs and the mRNAs of their predicted target genes.
We identified 34 miRNSPs that may affect the binding sites of 86 miRNAs in 18 target genes, of which 30 were not previously reported by any of the original GWAS.
The gene PTPN2 displayed highest number of associated diseases, five in total. This gene had two miRSNPs in its 3′ UTR. The risk allele (T) of miRSNP rs60474474 is predicted to disrupt the binding of miR-4290. On the other hand, the miRSNP rs45450798 also located in the 3′ UTR of PTPN2, has its protective allele (G) predicted to create a binding site for miR-4531. Tyrosine-protein phosphatase non-receptor type 2 (PTPN2) attenuates JAK/STAT signaling, among other effects. In mice, PTPN2 deficiency results in perturbations of T cell tolerance and enhanced T cell and B cell responses, resulting in severe inflammatory disease and autoimmunity (Wiede et al., 2017). Although we could not find evidence of an eQTL effect for these miRSNPs in any of the public databases, it has been shown that non-coding SNPs repressing PTPN2 are associate with several immune related diseases, including T1D (Bottini et al., 2004), RA (Begovich et al., 2004), and CD (Festen et al., 2011). This agrees with our findings, once deregulation of PTPN2 expression by the creation of a miRNA binding site by the risk allele could eventually reduce expression PTPN2, favoring autoimmune disease. Additionally, miR-4290 is present in exosomes (Leidinger et al., 2014) but not in any blood cell type or whole blood, suggesting that ectopic or increased expression of this miRNA could be a candidate biomarker for immune-related diseases.
eQTL mapping studies have shown that SNPs associated with complex diseases, detected in GWAS, are more likely to be an eQTL compared to non-associated SNPs (Nicolae et al., 2010). In autoimmune disease it has been shown that approximately 12% of causal non-coding SNPs are eQTL (Farh et al., 2015). In addition, eQTL data integration with prediction of miRSNPs can help to link causal non-coding disease variants to specific genes (Võsa et al., 2015). We found that 28 miRSNPs associated with AID were eQTLs for their target genes. Interestingly, these results agree with results obtained with our naïve Bayes classifier predictions. One interesting example of this scenario is the miRSNP rs7444. This miRSNP is an eQTL that may upregulate UBE2L3 (the gene that encodes the ubiquitin-conjugating enzyme E2 L3) and is predicted to disrupt the binding of four miRNAs to the mRNA of this target gene. The risk allele (C) disrupts binding of miRNAs with the UBE2L3 mRNA, resulting in overexpression of UBE2L3, which is in perfect agreement with the eQTL results. Although UBE2L3 polymorphisms have been associated with seven AID (Ricaño-Ponce and Wijmenga, 2013), after applying our approach and considering the stringent genome-wide P-value threshold (P < 5 × 10−8), we found three AID (SLE, CD, and IBD) sharing association with UBE2L3, indicating deregulation of miRNAs pathways at least in these three diseases. UBE2L3 participates of ubiquitination of the NF-κB precursor (Whiteside, 1995), a major transcription factor for genes involved in inflammation and immune responses. Loss of normal regulation of NF-κB is a major contributor to a variety of diseases, including AID. In addition, a SNP (rs140490) in absolute LD (r2 = 1 and D' = 1) with miRSNP rs7444, was correlated with basal NF-kB activation in unstimulated B cells and monocytes (Lewis et al., 2015), suggesting an effect of miRSNP rs7444 in the regulation of this gene through miRNAs.
Moreover, the UBE2L3 protein has been described as involved in the cytotoxic function of NK cells, which is a key cell type in the innate immune response (Fortier and Kornbluth, 2006). Hence, it is conceivable that if any of the miRNAs predicted to target this gene are expressed in NK cells, they could regulate UBE2L3 expression in this cell type. Although it is not known if these four miRNAs are expressed in NK cells, according to miRmine database (Panwar et al., 2017), apart from miR-4741 that presents low levels in blood, the other miRNAs are highly expressed in blood and in plasma, and display normal levels of expression in T-cells (miRmine database Panwar et al., 2017; accessed July 2017). Functional NK cell-specific assays might help to confirm these predictions.
Another interesting example is the miRSNP rs907091 which is an eQTL for IKZF3 and was previously reported as one of the possible causal SNPs of this autoimmune associated region (Farh et al., 2015). We found that the allele T of this SNP was predicted to affect the binding site of eight miRNAs to IKZF3 mRNA. The same allele T had a protective effect in four AID (T1D, PBC, UC, and IBD). By integrating genotype and RNA-seq (mRNA and microRNA) data, we found miR-326 levels negatively correlated with IKZF3 mRNA levels, in homozygous (T/T) individuals. Additionally, miR-326 displayed a high NBC Score (0.99) and is highly expressed in whole blood and plasma (miRmine database; Panwar et al., 2017, accessed July 2017), in agreement with the possible effect of this miRNA on IKZF3 expression levels, which is also expressed in these tissues. The IKZF3 gene (also known as AIOLOS) encodes an IKAROS family transcription factor involved in regulation of lymphocyte development (Cortes et al., 1999). Loss of IKZF3 in mice can prevent autoreactive B cells and decrease peritoneal, marginal and recirculating B cells (Wang et al., 1998; Cariappa et al., 2001), suggesting that low expression of IKZF3 could limit autoimmunity. This again matched our predictions, and eQTL results available in public databases. Furthermore, our results suggest that the downregulation of IKZF3 showed by the eQTL results for blood (P = 3.9 × 10−5, Z-score = −4.11, Blood eQTL database Westra et al., 2013; accessed July 2017) could be caused through the interaction between miRNA and mRNA of this target gene. Functional experiments are necessary to confirm these results. We hypothesize that knockdown of miR-326 in blood cells would counteract the deregulation of IKZF3. Anyhow, this miRNA could be a candidate for future therapy of AID.
Although our correlation analyses did not show that levels of all 86 miRNAs correlated with expression levels of their respective predicted target genes, we still have found correlations with 9 target genes. Since the miRNA and mRNA expression analyses were performed on LCL, the lack of correlation could be explained by tissue-specific expression, either of the target genes, the miRNAs, or other interacting proteins and non-coding RNAs. Moreover, predicted tissue-specific target genes are typically expressed in the same tissue as the miRNA but at significantly lower levels than in tissues where the miRNA is not present (Sood et al., 2006). Therefore, levels of miRNAs and the cognate target mRNA would correlate only in certain tissues. In addition, under certain circumstances, the effect of an miRNA on its target can only be observed when the protein level is measured (Li et al., 2015; Seo et al., 2017).
Furthermore, after integrating our results with data of experimentally validated assays available in two databases, we found two genes and two miRNAs (Supplementary Table 3) reported as functionally validated in these databases. Interestingly, the rs3088081 miRSNP of the SNAPC4 gene is an eQTL that affects interaction with only one miRNA (miR-3661). Although little is known about this gene, this observation indicates the need of further functional validation of our results in the specific cell types, which could confirm whether these miRNAs should be considered candidates to regulate AID associated genes.
GWAS has identified more than 250 susceptibility loci for AID (Ricaño-Ponce and Wijmenga, 2013). Many risk loci are shared between these diseases, which is consistent with them having an overlapping genetic background. The majority of genetic variants identified by GWAS were located in non-coding regions of the genome. A few AID studies reported association of differential susceptibility with SNPs at miRNA binding sites, such as rs3190930 in the PTPRK locus in CeD that alters the binding site for miR-1910 (Trynka et al., 2011). However, our study is the first that showed, in a systematic integrative manner, that immune associated non-coding SNPs could alter miRNAs binding sites. Overall, our integrative approach allowed us to find possible functional SNPs that were not described by the original GWAS. In addition, this approach could be extended to other complex diseases where GWAS summary statistics data are available. Thus, we highlighted miRNAs and genetic variarion at their binding sites as new candidates to be involved in the development of the AID.
Author Contributions
RdA study design, data analysis and manuscript writing. VC and MC study design and data analysis. MP-E study design, data interpretation and critical revision of the manuscript. All authors approved the version to be published.
Funding
This work was supported by the National Research Council (CNPq) of Brazil. Young Talent fellowship CNPq to RdA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Daniel Weingaertner and the Department of Informatics from the Federal University of Paraná in Brazil, for computational infrastructure and technical support in this project.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00139/full#supplementary-material
References
Abecasis, G. R., Auton, A., Brooks, L. D., DePristo, M. A., Durbin, R. M., Handsaker, R. E., et al. (2012). An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65. doi: 10.1038/nature11632
Anderson, C. A., Boucher, G., Lees, C. W., Franke, A., D'Amato, M., Taylor, K. D., et al. (2011). Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 43, 246–252. doi: 10.1038/ng.764
Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. doi: 10.1016/j.cell.2009.01.002
Beecham, A. H., Patsopoulos, N. A., Xifara, D. K., Davis, M. F., Kemppinen, A., Cotsapas, C., et al. (2013). Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 45, 1353–1360. doi: 10.1038/ng.2770
Begovich, A. B., Carlton, V. E. H., Honigberg, L. A., Schrodi, S. J., Chokkalingam, A. P., Alexander, H. C., et al. (2004). A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 75, 330–337. doi: 10.1086/422827
Bentham, J., Morris, D. L., Cunninghame Graham, D. S., Pinder, C. L., Tombleson, P., Behrens, T. W., et al. (2015). Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 47, 1457–1464. doi: 10.1038/ng.3434
Bhattacharya, A., Ziebarth, J. D., and Cui, Y. (2014). PolymiRTS Database 3.0: linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 42, 86–91. doi: 10.1093/nar/gkt1028
Bottini, N., Musumeci, L., Alonso, A., Rahmouni, S., Nika, K., Rostamkhani, M., et al. (2004). A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat. Genet. 36, 337–338. doi: 10.1038/ng1323
Cariappa, A., Tang, M., Parng, C., Nebelitskiy, E., Carroll, M., Georgopoulos, K., et al. (2001). The follicular versus marginal zone B lymphocyte cell fate decision is regulated by Aiolos, Btk, and CD21. 14, 603–615. doi: 10.1016/S1074-7613(01)00135-2
Chou, C. H., Chang, N. W., Shrestha, S., Hsu, S. D., Lin, Y. L., Lee, W. H., et al. (2016). miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 44, D239–D247. doi: 10.1093/nar/gkv1258
Cipolla, G. A., Park, J. K., de Oliveira, L. A., Lobo-Alves, S. C., de Almeida, R. C., Farias, T. D. J., et al. (2016). A 3′UTR polymorphism marks differential KLRG1 mRNA levels through disruption of a miR-584-5p binding site and associates with pemphigus foliaceus susceptibility. Biochim. Biophys. Acta Gene Regul. Mech. 1859, 1306–1313. doi: 10.1016/j.bbagrm.2016.07.006
Cooper, J. D., Smyth, D. J., Smiles, A. M., Plagnol, V., Walker, N. M., Allen, J. E., et al. (2008). Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat. Genet. 40, 1399–1401. doi: 10.1038/ng.249
Cortes, A., and Brown, M. A (2011). Promise and pitfalls of the immunochip. Arthritis Res. Ther. 13:101. doi: 10.1186/ar3204
Cortes, M., Wong, E., Koipally, J., and Georgopoulos, K. (1999). Control of lymphocyte by the lkaros gene family. Curr. Opin. Immunol. 11, 167–171.
Eyre, S., Bowes, J., Diogo, D., Lee, A., Barton, A., Martin, P., et al. (2012). High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet. 44, 1336–1340. doi: 10.1038/ng.2462
Farh, K. K., Marson, A., Zhu, J., Kleinewietfeld, M., Housley, W. J., Beik, S., et al. (2015). Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343. doi: 10.1038/nature13835
Festen, E. A., Goyette, P., Green, T., Boucher, G., Beauchamp, C., Trynka, G., et al. (2011). A meta-analysis of genome-wide association scans identifies IL18RAP, PTPN2, TAGAP, and PUS10 as shared risk loci for crohn's disease and celiac disease. PLoS Genet. 7:e1001283. doi: 10.1371/journal.pgen.1001283
Fortier, J. M., and Kornbluth, J. (2006). NK lytic-associated molecule, involved in NK cytotoxic function, is an E3 ligase. J. Immunol. 176, 6454–6463. doi: 10.4049/jimmunol.176.11.6454
Franke, A., McGovern, D. P. B., Barrett, J. C., Wang, K., Radford-Smith, G. L., Ahmad, T., et al. (2010). Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet. 42, 1118–1125. doi: 10.1038/ng.717
Friedman, R. C., Farh, K. K., Burge, C. B., and Bartel, D. P. (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105. doi: 10.1101/gr.082701.108
Ghanbari, M., Franco, O. H., De Looper, H. W. J., Hofman, A., Erkeland, S. J., and Dehghan, A. (2015). Genetic variations in MicroRNA-binding sites affect microrna-mediated regulation of several genes associated with cardio-metabolic phenotypes. Circ. Cardiovasc. Genet. 8, 473–486. doi: 10.1161/CIRCGENETICS.114.000968
Hinks, A., Cobb, J., Marion, M. C., Prahalad, S., Sudman, M., Bowes, J., et al. (2013). Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat. Genet. 45, 664–669. doi: 10.1038/ng.2614
Hrdlickova, B., de Almeida, R. C., Borek, Z., and Withoff, S. (2014). Genetic variation in the non-coding genome: involvement of micro-RNAs and long non-coding RNAs in disease. Biochim. Biophys. Acta 1842, 1910–1922. doi: 10.1016/j.bbadis.2014.03.011
John, B., Enright, A. J., Aravin, A., Tuschl, T., Sander, C., and Marks, D. S. (2004). Human microRNA targets. PLoS Biol. 2:e363. doi: 10.1371/journal.pbio.0020363
Jostins, L., Ripke, S., Weersma, R. K., Duerr, R. H., McGovern, D. P., Hui, K. Y., et al. (2012). Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124. doi: 10.1038/nature11582
Lappalainen, T., Sammeth, M., Friedländer, M. R., ‘t Hoen, P. A., Monlong, J. C., Rivas, M. A., et al. (2013). Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511. doi: 10.1038/nature12531
Leidinger, P., Backes, C., Meder, B., Meese, E., and Keller, A. (2014). The human miRNA repertoire of different blood compounds. BMC Genomics 15:474. doi: 10.1186/1471-2164-15-474
Lewis, M. J., Vyse, S., Shields, A. M., Boeltz, S., Gordon, P. A., Spector, T. D., et al. (2015). UBE2L3 polymorphism amplifies NF-kB activation and promotes plasma cell development, linking linear ubiquitination to multiple Autoimmune diseases. Am. J. Hum. Genet. 96, 221–234. doi: 10.1016/j.ajhg.2014.12.024
Li, Z., Qin, T., Wang, K., Hackenberg, M., Yan, J., Gao, Y., et al. (2015). Integrated microRNA, mRNA, and protein expression profiling reveals microRNA regulatory networks in rat kidney treated with a carcinogenic dose of aristolochic acid. BMC Genomics 16:365. doi: 10.1186/s12864-015-1516-2
Liu, C., Zhang, F., Li, T., Lu, M., Wang, L., Yue, W., et al. (2012). MirSNP, a database of polymorphisms altering miRNA target sites, identifies miRNA-related SNPs in GWAS SNPs and eQTLs. BMC Genomics 13:661. doi: 10.1186/1471-2164-13-661
Liu, J. Z., Almarri, M. A., Gaffney, D. J., Mells, G. F., Jostins, L., Cordell, H. J., et al. (2012). Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat. Genet. 44, 1137–1141. doi: 10.1038/ng.2395
Lonsdale, J., Thomas, J., Salvatore, M., Phillips, R., Lo, E., Shad, S., et al. (2013). The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585. doi: 10.1038/ng.2653
Machiela, M. J., and Chanock, S. J. (2014). LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557. doi: 10.1093/bioinformatics/btv402
Moszynska, A., Gebert, M., Collawn, J. F., and Bartoszewski, R. (2017). SNPs in microRNA target sites and their potential role in human disease. Open Biol. 7:170019. doi: 10.1098/rsob.170019
Nicolae, D. L., Gamazon, E., Zhang, W., Duan, S., Dolan, M. E., and Cox, N. J. (2010). Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 6:e1000888. doi: 10.1371/journal.pgen.1000888
Onengut-Gumuscu, S., Chen, W.-M., Burren, O., Cooper, N. J., Quinlan, A. R., Mychaleckyj, J. C., et al. (2015). Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet. 47, 381–386. doi: 10.1038/ng.3245
Panwar, B., Omenn, G. S., and Guan, Y. (2017). miRmine: a database of human miRNA expression profiles. Bioinformatics 33, 1554–1560. doi: 10.1093/bioinformatics/btx019
R Development Core Team (2017). R: A Language and Environment for Statistical Computing. Vienna: The R Foundation for Statistical Computing. Available online at: http://www.r-project.org
Ricaño-Ponce, I., and Wijmenga, C. (2013). Mapping of immune-mediated disease genes. Annu. Rev. Genomics Hum. Genet. 14, 325–353. doi: 10.1146/annurev-genom-091212-153450
Saunders, M. A., Liang, H., and Li, W.-H. (2007). Human polymorphism at microRNAs and microRNA target sites. Proc. Natl. Acad. Sci. U.S.A. 104, 3300–3305. doi: 10.1073/pnas.0611347104
Seo, J., Jin, D., Choi, C., and Lee, H. (2017). Integration of MicroRNA, mRNA, and protein expression data for the identification of cancer-related MicroRNAs. PLoS ONE 12:e0168412. doi: 10.1371/journal.pone.0168412
Sethupathy, P., and Collins, F. S. (2008). MicroRNA target site polymorphisms and human disease. Trends Genet. 24, 489–497. doi: 10.1016/j.tig.2008.07.004
Sood, P., Krek, A., Zavolan, M., Macino, G., and Rajewsky, N. (2006). Cell-type-specific signatures of microRNAs on target mRNA expression. Proc. Natl. Acad. Sci. U.S.A. 103, 2746–2751. doi: 10.1073/pnas.0511045103
Stegeman, S., Amankwah, E., Klein, K., O'Mara, T. A., Kim, D., Lin, H. Y., et al. (2015). A large scale analysis of genetic variants within putative miRNA binding sites in prostate cancer. Cancer Discov. 5, 368–379. doi: 10.1158/2159-8290.CD-14-1057
Szklarczyk, D., Morris, J. H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., et al. (2017). The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368. doi: 10.1093/nar/gkw937
Thomas, L. F., Saito, T., and Sætrom, P. (2011). Inferring causative variants in microRNA target sites. Nucleic Acids Res. 39:e109. doi: 10.1093/nar/gkr414
Trynka, G., Hunt, K. A., Bockett, N. A., Romanos, J., Mistry, V., Szperl, A., et al. (2011). Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 43, 1193–1201. doi: 10.1038/ng.998
Vlachos, I. S., Paraskevopoulou, M. D., Karagkouni, D., Georgakilas, G., Vergoulis, T., Kanellos, I., et al. (2015). DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 43, D153–D159. doi: 10.1093/nar/gku1215
von Mering, C., Jensen, L. J., Snel, B., Hooper, S. D., Krupp, M., Foglierini, M., et al. (2005). STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 33, 433–437. doi: 10.1093/nar/gki005
Võsa, U., Esko, T., Kasela, S., and Annilo, T. (2015). Altered gene expression associated with microRNA binding site polymorphisms. PLoS ONE 10:e0141351. doi: 10.1371/journal.pone.0141351
Wang, J., Avitahl, N., Cariappa, A., Friedrich, C., Ikeda, T., Renold, A., et al. (1998). Aiolos regulates B cell activation and maturation to effector state. Immunity 9, 543–553. doi: 10.1016/S1074-7613(00)80637-8
Wei, R., Yang, F., Urban, T. J., Li, L., Chalasani, N., Flockhart, D. A., et al. (2012). Impact of the interaction between 3′-UTR SNPs and microRNA on the expression of human xenobiotic metabolism enzyme and transporter genes. Front. Genet. 3:248. doi: 10.3389/fgene.2012.00248
Westra, H.-J., Peters, M. J., Esko, T., Yaghootkar, H., Schurmann, C., Kettunen, J., et al. (2013). Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243. doi: 10.1038/ng.2756
Whiteside, S. (1995). Ubiquitin-mediated processing of NF-kappa B transcriptional activator precursor p105. J. Biol. Chem. 270, 21707–21714.
Keywords: microRNA, SNP, autoimmunity, eQTL, GWAS, data integration
Citation: de Almeida RC, Chagas VS, Castro MAA and Petzl-Erler ML (2018) Integrative Analysis Identifies Genetic Variants Associated With Autoimmune Diseases Affecting Putative MicroRNA Binding Sites. Front. Genet. 9:139. doi: 10.3389/fgene.2018.00139
Received: 10 January 2018; Accepted: 04 April 2018;
Published: 26 April 2018.
Edited by:
Stefano Volinia, University of Ferrara, ItalyReviewed by:
Venugopal Thayanithy, University of Minnesota, United StatesIgor Jurak, University of Rijeka, Croatia
Copyright © 2018 de Almeida, Chagas, Castro and Petzl-Erler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria L. Petzl-Erler, perler@ufpr.br