
- 1 School of Biomedical Sciences and Pharmacy, Faculty of Health, University of Newcastle, NSW, Australia
- 2 Department of Immunology, Hunter Area Pathology Service, John Hunter Hospital, Newcastle, NSW, Australia
Chronic obstructive pulmonary disease (COPD) a term based on the demonstration of irreversible airways obstruction, introduced to unify a range of chronic progressive diseases of the airways consequent upon inhalation of toxins. While disease is initiated and progressed by inhaled toxins, an additional pathway of damage has emerged, with particular relevance to acute exacerbations. Exacerbations of disease due to an increase in the level of intrabronchial inflammation have taken on a new significance as their role in determining both acute and chronic outcomes is better understood. This “second pathway” of disease is a consequence of bacterial colonization of damaged airways. Although bacteria have been linked to acute episodes in COPD over 50 years, only recently has quality data on antibiotic usage and the detection of “exacerbation isolates” of non-typeable Haemophilus influenzae (NTHi) provided strong argument in support of a pathogenic role. Yet a poor correlation between detection of colonizing bacteria and clinical status remained a concern in attempts to explain a role for bacteria in a classical infection model. This presentation discusses a hypothesis that acute exacerbations reflect a T cell-dependent hypersensitivity response to colonizing bacteria, with IL-17 dependent accumulation of neutrophils within the bronchus, as the main outcome measure. Critical protection against exacerbations following oral administration of NTHi, an immunotherapy that drives a TH17 T cell response from Peyer’s patches, reduces the load of intrabronchial bacteria while preventing access of inhaled bacteria into small airways. Immunotherapy augments a physiological “loop” based on aspiration of bronchus content into the gut. A second “hypersensitivity” mechanism may cause bronchospasm – in both COPD and treatment-resistant asthma – due to specific IgE antibody directed against colonizing bacteria, as oral NTHi abrogates wheeze in subjects with recurrent “wheezy bronchitis.”
Introduction
Chronic obstructive pulmonary disease (COPD) has a long history of neglect, being seen as a self-induced lung disease of elderly smokers for which little could be done. Recognition of the extent of personal and economic costs of the disease together with detection of inflammation as a process relevant to disease progression, has heightened interest in COPD and encouraged new strategies for disease control (Barnes, 2008). The term “COPD” was introduced to unify divergent opinion about smoking-related airways disease (SRAD), focusing on irreversible airways obstruction defined by spirometry as the common denominator (Briscoe and Nash, 1965). In the 1950s and 1960s opinion was divided across the Atlantic. In the USA interest concentrated on the structural and functional outcomes of toxin inhalation (Petty, 2006), whereas in the UK experience was influenced by epidemiological analysis of smog-related bronchitis (Fletcher and Peto, 1977). These latter studies recognized productive cough (simple bronchitis), frequent chest illnesses (infective bronchitis), and impairment of ventilating capacity (obstructive bronchitis; Fletcher and Peto, 1977). The British view was that inhaled toxin-induced hypersecretion encouraged infection which damaged bronchi and lungs causing “obstructive bronchitis” and emphysema (Fletcher and Peto, 1977). At the same time, argument existed for and against the idea that asthma and COPD reflected different extremes of a common airways disease (known as the “Dutch Hypothesis”; Kraft, 2006). There was only limited support for bacterial infection as a cause of acute episodes came from microbiological, serological, and antibiotic studies (Tager and Speizer, 1975). The candidate pathogens identified most commonly from sputum cultures were non-typeable Haemophilus influenzae (NTHi) and Streptococcus pneumoniae (May, 1953). When Fletcher failed to demonstrate that “infection” episodes influenced change in chronic airflow obstruction (Fletcher and Peto, 1977), support waned for bacterial colonization as a contributory cause of airways damage in COPD. Interest both sides of the Atlantic was in toxin inhalation as the cause of the intrabronchial inflammatory response. A recent British review of airway inflammation in COPD saw this response only in terms of smoking (Barnes, 2008). Twenty-five years ago, the view that bacteria were not a significant “driver” of intrabronchial inflammation in COPD was challenged when subjects with moderate–severe COPD had a significant reduction in both positive sputum cultures and frequency of “acute episodes” following oral immunization with inactivated NTHi (Clancy et al., 1985). No increase in specific IgA was detected in secretions, which at the time was the mechanism expected to prevent the downward spread of pathogens from the upper airways (Clancy et al., 1985). At the time, the reduction in incidence of exacerbations following oral immunization with NTHi had no impact on the view that inhaled toxins were sufficient to account for COPD. Currently there has been a revision of the view that bacteria play little role in the pathogenesis of COPD, based on better quality antibiotic trials and detection of “exacerbation isolates” of NTHi (Sethi and Murphy, 2008; Sethi et al., 2008). Sethi and his colleagues have proposed a “vicious cycle” hypothesis whereby primary damage to airways leads to bacterial colonization, with added damage begetting damage (Sethi and Murphy, 2008). While bacteria may directly contribute to airways disease (Sethi et al., 2008), current theories still inadequately explain the poor “fit” between microbiological and clinical data. For example, isolation of “pathogens” from sputum collected in an exacerbation is similar to the pattern obtained in specimens from stable disease (Butt et al., 1990; Sethi et al., 2007). We review a hypothesis that has acute exacerbations of COPD reflecting a particular balance of the host–parasite relationship involving NTHi and the mucosal immune response. Thus exacerbations occur as an uncontrolled and inappropriate inflammatory response to bacteria colonizing damaged airways due to an ineffective Peyer’s patch-derived T lymphocyte response. A corollary to this hypothesis is that oral immunization with NTHi improves the efficiency of gut-derived bronchus protection, with a consequent reduction in the frequency and severity of exacerbations of COPD.
The Hypothesis
Current thinking as well as changes in management strategies in COPD have largely flowed from the “asthma” model, substituting the neutrophil for the eosinophil as the dominant cell type in the intrabronchial inflammatory exudate (Barnes, 2008). As a consequence, search for new therapies has focused on key regulatory molecules and cells in inflammation. The key questions become: “To what extent is inflammation the cause of damage?” and “What are the drivers of this inflammatory response?” Directing therapy toward removal of the stimulus of inflammation has the advantages of avoiding the complications of both drug-induced immune suppression, and broad based suppression of the inflammatory process. We discuss a hypothesis that focuses on a host–parasite relationship involving colonizing bacteria and the mucosal immune response where the balance of this relationship determines the level of intrabronchial inflammation. An acute shift in the balance leads to an excessive inflammatory response. If this exceeds a threshold it is detected clinically as an “acute exacerbation.” Simply put, an acute exacerbation of COPD is a hypersensitivity response, involving antigens and effector immune responses (particularly Th17 CD4+ T cells) within the mucosal compartment. A corollary of this hypothesis is that a shift favoring host mucosal protection would buffer against acute exacerbations. As with any hypersensitivity reaction, it is the inappropriate and excessive production of components of the innate immune system, beyond that which is directed against pathogens and which is therefore protective, that cause non-specific target damage. The sequence of events in the proposed model is summarized in Figure 1:
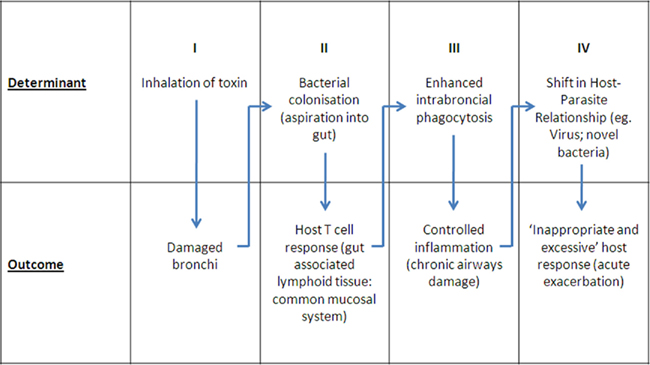
Figure 1. Proposed sequence of events in pathogenesis of COPD.
(I) Inhalation of toxins damage bronchus mucosa: This has been well documented and requires no further discussion.
(II) Bacterial colonization and antigen presentation to gut-associated lymphoid tissue (GALT): It has been long recognized that damaged bronchus mucosa becomes colonized by bacteria, most commonly NTHi (Butt et al., 1990; Sethi et al., 2007) and when present NTHi represent the majority of bacterial isolates (Butt et al., 1990). Argument has centered on the significance of these bacteria with respect to acute exacerbations, the question often framed in terms of “colonization” versus “infection.” Recent reviews conclude that a link exists between colonization and acute exacerbations, claiming that 30–50% of acute exacerbations are due to bacterial infection (Sethi and Murphy, 2008). Uncertainty about these conclusions, however, comes from concern that the infection parameters assessed are more appropriate to invasive monobacterial infection, and there are notorious difficulties in obtaining reproducible and representative sputum samples. Of importance was the observation that detection of pathogens in sputum varied little between acute exacerbations and chronic disease (Butt et al., 1990; Sethi et al., 2007), which is not consistent with invasive infection. Quantitative analysis showed little difference in density of colonization in acute and stable disease (Butt et al., 1990; Lehmann et al., 1991), requiring a review of the mechanism whereby bacteria contribute to inflammation. Sputum cultures underestimate the true incidence of colonization given the results of cultures from bronchial brush samples (Bandi et al., 2001), and recent molecular analysis of lower airways secretions which suggest nearly all with COPD are colonized with NTHi. Aspiration of bronchus content may deliver up to 1011 live bacteria per day into the gut (Lehmann et al., 1991). A prospective study in smokers over a winter period showed a significant seasonal increase in antigen-reactive T cells, consistent with the idea that swallowed sputum leads to antigen presentation to the GALT as a physiological mechanism of antigen handling (Clancy and Dunkley, 2010). This was the first evidence that bronchus protection is a consequence of antigen handling via the gut, emphasizing a critical role for the common mucosal system in man.
(III) Controlled inflammation: Central to the hypothesis is identification of antigen (bacteria) within the bronchus lumen where it interacts with a mucosal immune response. Only if these bacteria penetrate as far as the small airways and the broncho-alveolar apparatus, is a serum IgG antibody detected. The classical “marker” of local immunity, an IgA antibody response, is downregulated in subjects with mucosal damage and inflammation (Clancy et al., 1995). The key effector mechanism responsible for control of mucosal colonization is T cell-dependent recruitment and activation of neutrophils, and secretion of antibacterial factors (Khader et al., 2009). These T cells are generated in the GALT and transit to the bronchus mucosa as a component of the common mucosal system (Wallace et al., 1989, 1991; Dunkley et al., 1994, 1995). Secondary stimulation by colonizing bacteria of T cells relocated to the bronchus mucosa may be required if the immunogenic signal to the GALT is weak (Wallace et al., 1989, 1991, 1991). T cell-activated neutrophils within sputum undergo phenotypic change characterized by secretion of large amounts of IL-8 and TNFα, and increased phagocytic capacity. Viability is maintained by an autocrine loop involving IL-1 (Pang et al., 1997). Recent Studies on the mucosal immune system have focused on regional induction and effector mucosal sites as a “sub-network” with particular “homing” of B- and T-lymphocytes determined by chemokines, integrins, and cytokines that are differentially expressed between the mucosal tissues (Otczyk and Cripps, 2010). This review summarizes the current but limited knowledge with respect to the airways including potential roles for CD62L, α4B1 integrin, and LFA-1 adhesion pathways (Xu et al., 2003), and the importance of antigen at target mucosal sites (Qimron et al., 2004). The importance of dendritic cells and secretion of retinoic acid in the production of mucosa – imprinting homing molecules has been described (Iwata et al., 2004) and specific dendritic cells direction TH17 cells homing to the airways have been identified (Khader et al., 2009).
The concept of “balance” of the host–parasite relationship being relevant to acute exacerbations is supported by study of subjects with an exacerbation-prone phenotype. These subjects have more cell-attached bacteria, and a reduced lysozyme level in secretions (Taylor et al., 1990; Clancy et al., 1995). Thus chronic inflammation within the airways in part involves a direct T cell/neutrophil response to colonizing bacteria and, indirectly, a persistence of activated neutrophils maintained by autocrine mechanisms. The role of gut-driven Th17 cells is critical to mucosal protection through recruitment of neutrophils and the secretion of antibacterial factors from the bronchus epithelium, with detection of IL-17 in sputum, and Th17 cells in the draining nodes (Khader et al., 2009). It has been shown that bacteria trapped in a biofilm can induce Th17 cells that contribute to damage – validating the concept of hypersensitivity reactions to colonizing bacteria (Dubin et al., 2007). As few with moderate–severe COPD continue to smoke (Tandon et al., 2010), hypersensitivity to persistent bacteria within the airways is a credible explanation for continuing intrabronchial inflammation. Recent studies in emphysema identify phenotypic changes in dendritic cells obtained from resected lung from ex-smokers that drive Th1 and Th17 cells (Shan et al., 2009), and a significant increase in IL-17, IL-22, and IL-23 positive immunoreactive cells within the bronchial mucosa (Shan et al., 2009), while intestinal immunization of rats with inactivated NTHi increases specific Th17 cells in both mesenteric lymph nodes and in airways (Figure 2).
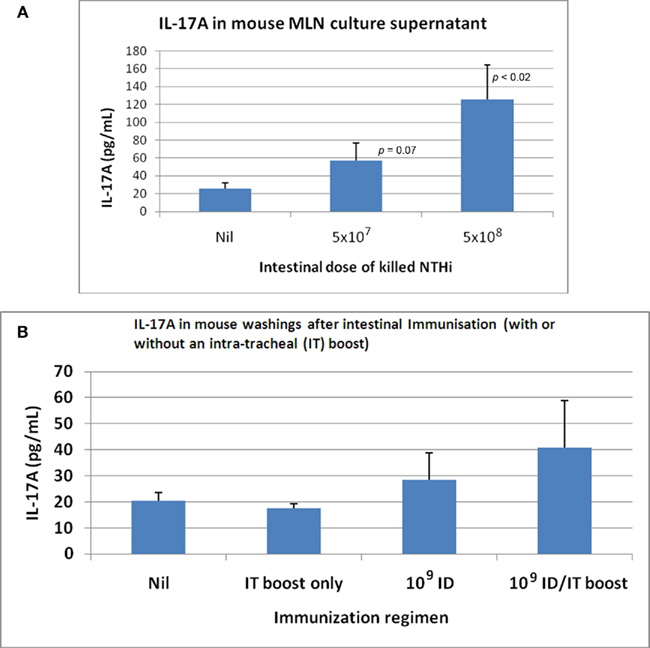
Figure 2. IL-17A produced by in vitro re-stimulated mesenteric lymph node (MLN) lymphocytes and detected in broncho-alveolar lavage fluid following intestinal immunization with killedNTHi. (A) IL-17 produced by re-stimulated MLN (lymphocytes) following oral immunization of C57BL/6 mice with killed whole cells of NTHi. (B) IL-17 in C57BL/6 mouse lung following oral immunization with killed whole cells of NTHi.
(IV) Shifts in the host–parasite relationship: Acute exacerbation: (shift favors parasite). Acute exacerbations of COPD are periods of accentuated intrabronchial inflammation (Wedzicha and Seemungal, 2007) due to a failure of the host to contain colonizing bacteria, causing an excessive and inappropriate inflammatory response. Increase in inflammation may follow acquisition of pathogenic “exacerbation” isolates of NTHi (Sethi et al., 2008), other bacterial pathogens, or an increase in resident bacteria due to an intercurrent virus infection (Sethi and Murphy, 2008). Co-infection of mice with NTHi and influenza virus increases titer of bacteria 100-fold and virus 10-fold (Dunkley and Clancy, 1997). These observations support the idea that colonizing bacteria in COPD are a common denominator of acute exacerbations, by upregulation of antigenic stimulus directly due to acquisition of novel “exacerbation” strains of bacteria, or indirectly as a consequence of a virus infection. Bronchus brush biopsy analysis confirms a difference in colonization level with 33% of stable COPD samples positive for NTHi compared to 87% of biopsies taken during an exacerbation (Bandi et al., 2001). Clinical studies show a subtle but significant increase in colonizing bacteria during exacerbations (Sethi et al., 2007). Damage is due to direct toxicity by pathogenic bacteria (Sethi et al., 2008), and the inappropriate and excessive recruitment of activated neutrophils (Wallace et al., 1990, 1991; Wedzicha and Seemungal, 2007). This conclusion is supported by the presence in sputum of increased levels of neutrophils, IL-8, TNFα, and neutrophil elastase at times of clinical exacerbations Wedzicha and Seemungal, 2007).
The Corollary
If acute exacerbations reflect ineffective containment of colonizing bacteria, any therapy improving the efficiency of host protection should reduce their frequency and severity. Reduction in colonizing pressure reduces the level of intraluminal inflammation below threshold levels for “severe” exacerbations before falling below a lower threshold for “any exacerbation” – hence a dominant effect on reducing severity of episodes (Figure 3).
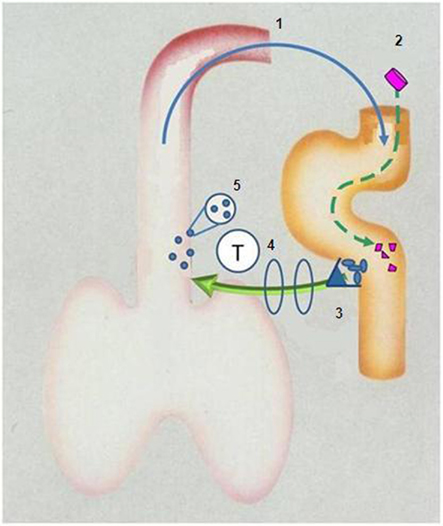
Figure 3. Diagrammatic representation of aspiration of bronchus content (including bacteria) into gut (1) to stimulate Peyer’s patches (3) to release T-lymphocytes which “home” to the bronchus (4) where they – directly or indirectly – secrete cytokines and chemokines which augment recruitment and activation of phagocytes (5) which reduce colonizing load of damaged bronchus mucosa. Ingestion of inactivated non-typeable Haemophilus influenzae (2) augments this protective “loop”.
Immunotherapy with Oral NTHi (Shift Favors Host)
Reduction in frequency and severity of exacerbations in COPD, and in reduction of pathogens in sputum following specific oral immunotherapy with oral NTHi, are strong arguments in support of the postulate that colonizing bacteria play a critical role in the pathogenesis of acute exacerbations. Results from four published trials (Clancy et al., 1985; Clancy et al., 1990; Lehmann et al., 1991; Tandon and Gebski, 1991) showed a reduction in exacerbations (defined as an increase in volume and purulence of sputum), and reduced use of antibiotics. These were the basis of a positive Cochrane Report (Foxwell et al., 2006). A recent study in severe COPD showed a reduction in total exacerbations of 16%, but when exacerbations were defined in terms of a requirement for therapeutic intervention (capturing the more severe episodes), a significant reduction of 63% (corticosteroid-treated patients), and 90% (hospital-treated) followed oral NTHi immunization (Tandon et al., 2010). This “shift” toward more severe exacerbations would be expected if oral NTHi reduced the level of intrabronchial inflammation in the majority of recipients. Further, quantitative bacteriology of sputum following oral NTHi noted a 3 log reduction in bacteria (Lehmann et al., 1991), and reduction involved all pathogens (Tandon and Gebski, 1991; Tandon et al., 2010). Non-specific effector function based on phagocytosis is responsible for reduction of all pathogens. Oral immunization of smokers with NTHi prevents inhaled NTHi from penetrating into terminal airways (Clancy and Dunkley, 2010). In this latter study, protection correlated with an enhanced circulating specific antigen-reactive T cell response and a reduction in level of salivary lysozyme, a marker of luminal inflammation in COPD (Clancy and Dunkley, 2010). These human studies follow the demonstration in rodents that specific T cells obtained from thoracic duct drainage after Peyer’s patch immunization with NTHi, can transfer accelerated clearance of bacteria from the bronchus (Clancy et al., 1995). Oral immunization with NTHi of mice co-infected with influenza virus and NTHi abrogated the increase in titer of both microbes (Dunkley and Clancy, 1997), indicating that colonizing NTHi can amplify the inflammatory response to virus infections. Thus by reducing the level of colonization, oral immunization with NTHi protects against infection initiated by bacteria and viruses. It is unclear as to whether most subjects with COPD require re-stimulation of T cells migrating to the bronchi by colonizing bacteria as recent data suggests most if not all COPD patients have NTHi in their lower airways (Bandi et al., 2001).
Discussion and Conclusion
This hypothesis identifies colonizing bacteria as an “antigen sump” that on one hand drives a T cell response following aspiration of bronchus content into the gut, while on the other is the target of that response. The outcome is determined by the extent of the antigen load – a low antigen load reflects protection and containment of infection by T cell-activated phagocytosis, while a high load reflects ineffective containment and a hypersensitivity reaction (seen clinically as an acute exacerbation). An upward shift in antigen occurs by inhaling novel “exacerbation” isolates of bacteria (in particular NTHi; Sethi et al., 2008) or following a virus infection, in particular influenza (Dunkley and Clancy, 1997). These ideas place bacteria – especially NTHi – on a final common pathway that determines the level of intrabronchial inflammatory exudate. Progression of COPD and exacerbations occur whether or not there is continued inhalation of toxic material. This hypothesis can be tested using quantitative cellular and molecular assays of bronchial biopsies and broncho-alveolar washings, to analyze the host–parasite relationship as it pertains to the Th17 response to colonizing bacteria (and associated inflammation) in subjects with stable COPD, with an acute exacerbation, and following immunotherapy with oral NTHi. Oral immunotherapy with NTHi augments antigen delivery to the Peyer’s patches, with a consequent increase in T cells delivered to the mucosa, increasing the efficiency of phagocytosis. Protection is refected in a reduction in the load of bacteria colonizing damaged bronchi, reflecting a “shift” in outcome away from the hypersensitivity pro-inflammatory response characteristic of a less efficient T cell response.
Clinical studies demonstrate a new approach to management of one of the major unmet medical needs, while providing strong evidence that abnormal colonization of airways plays a significant role in the pathogenesis of exacerbations of COPD. Important and effective strategies for controlling mucosal infection through oral immunization have likely been neglected due to an inappropriate focus on IgA antibody as a monitor of vaccine efficacy.
Conflict of Interest Statement
Robert Clancy owns shares and is on the Board of Hunter Immunology Ltd. Margaret Dunkley is an indirect employee of Hunter Immunology Ltd and owns share options.
References
Bandi, V., Apicella, M. A., Mason, E., Murphy, T. F., Siddiqi, A., Atmar, R. L., and Greenberg, S. V. (2001). Nontypeable Haemophilus influenzae in the lower respiratory tract of patients with chronic bronchitis. Am. J. Respir. Crit. Care Med. 164, 2114–2119.
Barnes, P. J. (2008). Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 8, 183–192.
Briscoe, W. A., and Nash, E. S. (1965). The slow space in chronic obstructive pulmonary disease. Ann. N. Y. Acad. Sci. 121, 706–722.
Butt, H. L., Clancy, R. L., Cripps, A. W., Murree-Allen, K., Saunders, N. A., Sutherland, D. C., and Hensley, M. J. (1990). Bacterial colonisation of the respiratory tract in chronic bronchitis. Aust. N. Z. J. Med. 20, 35–38.
Clancy, R. L., Cripps, A. W., Murree-Allen, K., Yeung, S., and Engel, M. (1985). Oral immunisation with killed Haemophilus influenzae for protection against acute bronchitis in chronic obstructive lung disease. Lancet 2, 1395–1397.
Clancy, R. L., Pang, G., Dunkley, M. L., Taylor, D., and Cripps, A. W. (1995). Acute on chronic bronchitis: a model of mucosal immunology. Immunol. Cell Biol. 73, 414–417.
Clancy, R. L., Cripps, A. W., and Gebski, V. (1990). Protection against recurrent acute bronchitis following oral immunization with killed Haemophilus influenzae. Med. J. Aust. 152, 413–416.
Clancy, R. L., and Dunkley, M. L. (2010). Oral non-typeable Haemophilus influenzae enhances physiological mechanism of airways protection. Clin. Exp. Immunol. 161, 127–133.
Dubin, P. J., McAllister, F., and Kolls, J. K. (2007). Is cystic fibrosis a TH17 disease? Inflamm. Res. 56, 221–227.
Dunkley, M., and Clancy, R. L. (1997). “A rodent model of concurrent respiratory infection with influenza virus and Gram-negative bacteria: synergistic infection and protection by oral immunization.” In Mucosal Solutions: Advances in Mucosal Immunology, Vol. 1, eds A. J. Husband, K. W. Beagley, R. L. Clancy, and A. W. Cripps (Sydney: University of Sydney Press), 279–288.
Dunkley, M., Pabst, R., and Cripps, A. (1995). An important role for intestinally-derived T cells in respiratory defence. Immunol. Today 16, 231–236.
Dunkley, M. L., Clancy, R. L., and Cripps, A. W. (1994). A role for CD4+ T cells from orally immunized rats in enhanced clearance of Pseudomonas aeruginosa from the lung. Immunology 83, 362–369.
Fletcher, C., and Peto, R. (1977). The natural history of chronic airflow obstruction. Br. Med. J. 1, 1645–1648.
Foxwell, A. R., Cripps, A. W., and Dear, K. B. G. (2006). Haemophilus influenzae oral whole cell vaccination for preventing acute exacerbations of chronic bronchitis. Cochrane Database Syst. Rev. 4, CD001958.
Iwata, M., Hirakiyama, A., Eshima, Y., Kagechika, H., Kato, C., and Song, S. Y. (2004). Retinoic acid imprints gut-homing specificity on T cells. Immunity 21, 257–238.
Khader, S. A., Gaffen, S. L., and Kolls, J. K. (2009). Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. 5, 403–411.
Kraft, M. (2006). Asthma and chronic obstructive pulmonary disease exhibit common origins in any country! Am. J. Respir. Crit. Care Med. 174, 238–240.
Lehmann, D., Coakley, K. J., Coakley, C. A., Spooner, V., Montgomery, J.M., Michael, A., Riley, I. D., Smith, T., Clancy, R. L., and Cripps, A. W. (1991). Reduction in the incidence of acute bronchitis by an oral Haemophilus influenzae vaccine in patients with chronic bronchitis in the Highlands of Papua New Guinea. Am. Rev. Respir. Dis. 144, 324–330.
Otczyk, D., and Cripps, A. (2010). Mucosal immunisation: a realistic alternative. Hum. Vaccin. 6, 1–29.
Pang, G., Ortega, M., Zighang, R., Reeves, G., and Clancy, R. L. (1997). Autocrine modulation of IL-8 production by sputum neutrophils in chronic bronchial sepsis. Am. J. Respir. Crit. Care Med. 155, 726–731.
Qimron, U., Paul, L., Bar-Haim, E., Bloushtain, N., Eisenbach, L., Staats, H.F., and Porgador, A. (2004). Non-replicating mucosal and systematic vaccines: quantitative and qualitative differences in the Ag-specific CD8(+) T cell population in different tissues. Vaccine 22, 1390–1394.
Sethi, S., and Murphy, T. F. (2008). Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N. Engl. J. Med. 359, 2355–2365.
Sethi, S., Sethi, R., Eschberger, K., Lobbions, P., Cai, X., Grant, B. J. B., and Murphy, T. F. (2007). Airway bacterial concentrations and exacerbations of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 176, 356–361.
Sethi, S., Wrona, C., Eschberger, K., Lobbins, P., Cai, X., Grant, B. J. B., and Murphy, T. F. (2008). Inflammatory profile of new bacterial strain exacerbations of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 177, 491–497.
Shan, M., Cheng, H. F., Song, L. Z., Roberts, L., Green, L., Hacken-Bitar, J., Huh, J., Bakaeen, S., Coxson, H. O., Storness-Bliss, C., Ramchandani, M., Lee, S. H., Corry, D., and Kheradmand, F. (2009). Lung myeloid dendritic cells coordinately induce Th1 and Th17 responses in human emphysema. Sci. Transl. Med. 1.4, 1–9.
Tager, I., and Speizer, F. E. (1975). Role of infection in chronic bronchitis. N. Engl. J. Med. 292, 563–571.
Tandon, M. K., and Gebski, V. (1991). A controlled trial of a killed Haemophilus influenzae vaccine for prevention of acute exacerbations of chronic bronchitis. Aust. N. Z. J. Med. 21, 427–432.
Tandon, M. K., Phillips, M., Waterer, G., Dunkley, M. L., Coman, P., and Clancy, R. L. (2010). Oral immunotherapy with inactivated nontypeable Haemophilus influenzae reduces severity of acute exacerbations in severe COPD. Chest 137, 805–811.
Taylor, D. C., Cripps, A. W., and Clancy, R. L. (1990). Inhibition of adhesion of Haemophilus influenzae to buccal cells by respiratory secretions. Immunol. Cell Biol. 68, 335–342.
Wallace, F. J., Clancy, R. L., and Cripps, A. W. (1989). An animal model demonstration of enhanced clearance of nontypeable Haemophilus influenzae from the respiratory tract after antigen stimulation of gut associated lymphoid tissue. Am. Rev. Respir. Dis. 140, 311–316.
Wallace, F. J., Clancy, R. L., and Cripps, A. W. (1990). “Haemophilus influenzae immunity in the respiratory tract.” In Advances in Mucosal Immunology, eds T. T. MacDonald, S. J. Challacombe, and P. W. Bland (London: Kluwer Academic Publishers), 859–860.
Wallace, F. J., Cripps, A. W., Clancy, R. L., Husband, A. J., and Witt, C. S. (1991). A role for intestinal T lymphocytes in bronchus mucosal immunity. Immunology 74, 68–73.
Wedzicha, J. A., and Seemungal, T. A. R. (2007). COPD exacerbations: defining their cause and prevention. Lancet 370, 786–796.
Keywords: COPD, chronic obstructive pulmonary disease, emphysema, respiratory, airways
Citation: Clancy RL, and Dunkley M (2011) Acute exacerbations in COPD and their control with oral immunization with non-typeable Haemophilus influenzae. Front. Immun. 2:7. doi:10.3389/fimmu.2011.00007
Received: 23 December 2010; Accepted: 21 February 2011;
Published online: 15 March 2011.
Edited by:
Allan Cripps, Griffith University, AustraliaReviewed by:
Ruth Foxwell, Unversity of Canberra, AustraliaCarlos Alberto Guzman, Helmholtz Centre for Infection Research, Germany
Copyright: © 2011 Clancy and Dunkley. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Robert L. Clancy, Department of Immunology and Microbiology, Level 4, David Maddison Clinical Sciences Building, University of Newcastle, Newcastle, NSW 2308, Australia e-mail: robert.clancy@ newcastle.edu.au