 Sarah R. Jacobs Blossom Damania*
Sarah R. Jacobs Blossom Damania*- Department of Microbiology and Immunology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
Kaposi’s sarcoma-associated herpesvirus (KSHV) is a large double-stranded DNA gammaherpesvirus, and the etiological agent for three human malignancies: Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. To establish and maintain infection, KSHV has evolved unique mechanisms to evade the host immune response. Cellular interferon regulatory factors (IRFs) are a critical part of the host anti-viral immune response. KSHV encodes four homologs of IRFs, vIRF1–4, which inhibit the activity of their cellular counterparts. vIRF1, 2, and 3 have been shown to interact directly with cellular IRFs. Additionally, the vIRFs have other functions such as modulation of Myc, p53, Notch, transforming growth factor-β, and NF-κB signaling. These activities of vIRFs may contribute to KSHV tumorigenesis. KSHV vIRF1 and vIRF3 have been implicated as oncogenes, making the understanding of KSHV vIRF function vital to understanding KSHV pathogenesis.
Introduction
Kaposi’s sarcoma-associated herpesvirus (KSHV), also known as human herpes virus 8, is a double-stranded, DNA tumor virus belonging to the gammaherpesvirus subfamily. KSHV has been implicated as the causative agent for multiple human cancers including Kaposi’s sarcoma (KS), multicentric Castleman’s disease (MCD), and primary effusion lymphoma (PEL; Wen and Damania, 2010). KSHV-associated cancers are prevalent in immunocompromised individuals, such as transplant recipients and HIV-infected individuals (Angeletti et al., 2008; Wen and Damania, 2010). Like all herpesviruses, KSHV establishes life-long latency in a variety of cell types including B cells, macrophages, endothelial cells, and monocytes in vivo (Boshoff et al., 1995; Blasig et al., 1997; Sirianni et al., 1998; Monini et al., 1999; Wu et al., 2006).
Similar to other herpesviruses, KSHV establishes infection and exists primarily in the latent state. In a latent infection, KSHV is dormant and persists for the lifetime of the host through viral genome tethering to the host chromosome via the latency-associated nuclear antigen (LANA; Ballestas et al., 1999; Cotter and Robertson, 1999; Barbera et al., 2006; Verma et al., 2007). During latency, only a subset of viral genes is actively transcribed. In contrast, de novo infection results in viral lytic replication for approximately 72–96 h, prior to the establishment of latency (Krishnan et al., 2004). During the lytic cycle, the viral genome is replicated and progeny virions are released from the cell and can subsequently infect neighboring cells. KSHV can also be induced to undergo lytic replication following reactivation by superinfection with another virus (Gregory et al., 2009) or chemical treatment with 12-O-tetradecanoyl-phorbol-13-acetate (Renne et al., 1996).
In order to establish infection and maintain latency, KSHV must evade the host immune response. The virus encodes a variety of immune evasion genes as previously reviewed (West and Damania, 2010). One mechanism through which KSHV evades immunity is by encoding viral homologs of cellular genes that augment or subvert the function of their cellular counterparts. These genes include viral interleukin-6 (IL-6), a viral form of the inflammatory cytokine IL-6 (Neipel et al., 1997a), vBcl-2, a viral protein similar to Bcl-2, which prevents apoptosis (Cheng et al., 1997; Huang et al., 2002), and four viral homologs of cellular interferon regulatory factors (IRFs; Russo et al., 1996; Neipel et al., 1997b; Cunningham et al., 2003), which will be the focus of this review.
Interferon (IFN) is the primary response of a host cell to viral infection and protects the host by inhibiting viral replication, increasing activation of antigen presenting cells, inhibiting cell proliferation, and promoting apoptosis of the infected cell (Pestka et al., 2004). Cellular IRFs mediate part of the IFN response initiated by the host innate immune system and are transcription factors that bind to and activate interferon-responsive promoters. Nine mammalian IRFs operate in coordination to elicit distinct immune responses (Tamura et al., 2008). Activation of the IFN response by viral infection or pathogen detection leads to phosphorylation, dimerization, and nuclear translocation of IRFs. IRF3 and IRF7 are critical for transcription of type I interferons, IFN α and β (Tamura et al., 2008). IRF-mediated transcription creates an anti-viral state characterized by increased antigen presentation, degradation of RNA, cessation of protein processing, and induction of apoptosis (Alsharifi et al., 2008).
Additionally, IRF family members also play a role in cell growth and proliferation. This regulation, combined with cell survival signaling, has implicated IRFs in tumor formation (Tamura et al., 2008). For example, IRF1 deficiency exacerbated tumor predispositions (Nozawa et al., 1999), and constitutively active IRF3 leads to apoptosis (Heylbroeck et al., 2000). Furthermore, IRF3 mediates cell death in response to Sendai virus infection and apoptosis can be blocked by expression of a dominate negative form of IRF3 (Heylbroeck et al., 2000).
Homology and Expression of vIRFs
Examination of the KSHV genome identified the presence of genes that showed similarity to cellular IRFs (Russo et al., 1996). It was subsequently determined that three of these viral genes, vIRF1, 2, and 3, blocked IFN signaling (Lee et al., 2009a; Figure 1). The less studied vIRF4 has not yet been shown to affect IFN signaling, although it shares other functions with vIRF1, 2, and 3 such as inhibition of p53 transcription (Lee et al., 2009b; Table 1). KSHV vIRF1, 2, 3, and 4 are encoded by ORFs K9, K11 and K11.1, K10.5 and K10.6, and K10, respectively (Figure 2 and Table 1). vIRF1 is a lytic gene (Zimring et al., 1998; Burysek et al., 1999a). However, it was found to be transcribed in latently infected KS cells as well (Dittmer, 2003). vIRF1 is also expressed in PEL cells including BCBL-1, BCP-1, and BC-1 cells where expression increased following viral reactivation by chemical induction (Moore et al., 1996; Sarid et al., 1998; Inagi et al., 1999). Lytic expression of vIRF1 in PEL cells required new protein synthesis (Wang et al., 2001). vIRF1 levels peak 48 h after TPA treatment, and the protein has a 3-h half life (Pozharskaya et al., 2004). Similarly, vIRF2 has been mapped as a lytic gene by DNA array and cluster analysis of PEL cells (Jenner et al., 2001). vIRF3 was originally discovered as a latent gene and is also known as LANA2 (Rivas et al., 2001). Unlike vIRF1, vIRF3 expression is not induced during lytic reactivation of PEL cells (Wies et al., 2008). vIRF3 is expressed in nearly all PEL cell lines and in the majority of MCD tumors, but does not appear to be highly expressed in KS (Rivas et al., 2001). vIRF4 expression can be induced by TPA and is expressed mostly in the nucleus (Katano et al., 2000; Kanno et al., 2006).
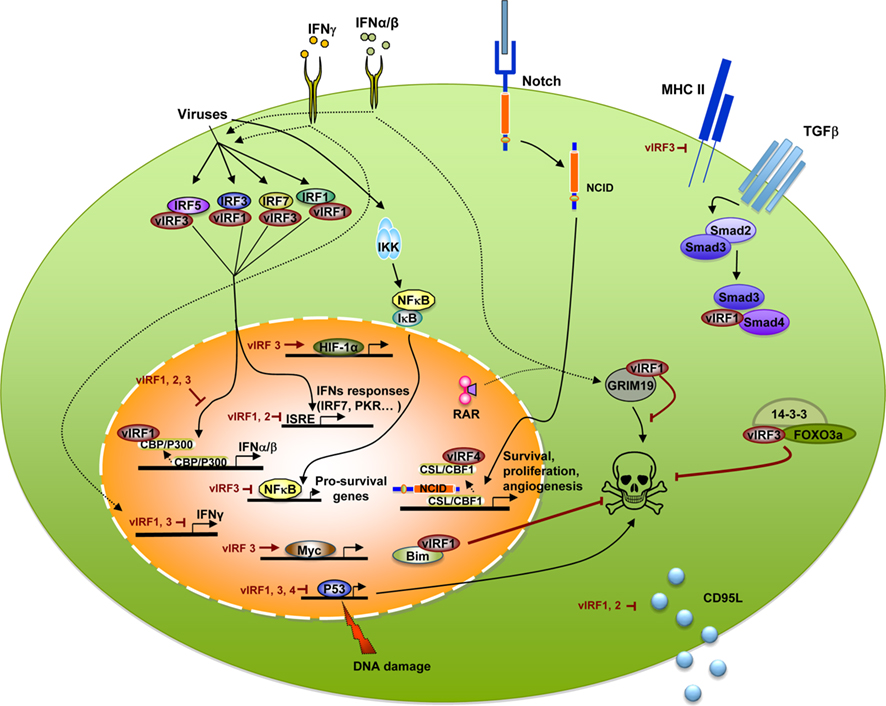
Figure 1. Kaposi’s sarcoma-associated herpesvirus vIRFs bind to cellular proteins, block transcription, and interfere with interferon and cellular signaling pathways. Viral infection activates cellular IRFs, including IRF1, 3, 5, and 7 to initiate IFN transcription. Binding of cellular IRFs by vIRF1 or vIRF3 results in a blockade of IFNα, β, and γ transcription. vIRF1 inhibits type I IFN transcription by binding the CBP/p300 transcriptional coactivator and inhibiting its activity. Viral infection also induces NF-κB transcription that is decreased by vIRF3. vIRF1, 3, and 4 inhibit p53 transcription and impede p53-mediated cell death. Other cell death processes affected by vIRFs include vIRF1 binding to GRIM19, vIRF3 interaction with 14-3-3 and FOXO3a, vIRF1 and 2 blockade of CD95L production, and vIRF1-mediated nuclear localization of Bim. In addition to deregulating immune responses and cell death, vIRFs have other roles such as vIRF3 increasing Myc and HIF-1α transcription and blocking expression of MHC II. vIRF1 blocks TGF-β signaling through binding to Smad3 and Smad4 and vIRF4 binding to the Notch transcription factor, CSL/CBF1 to inhibit activation of Notch target genes.
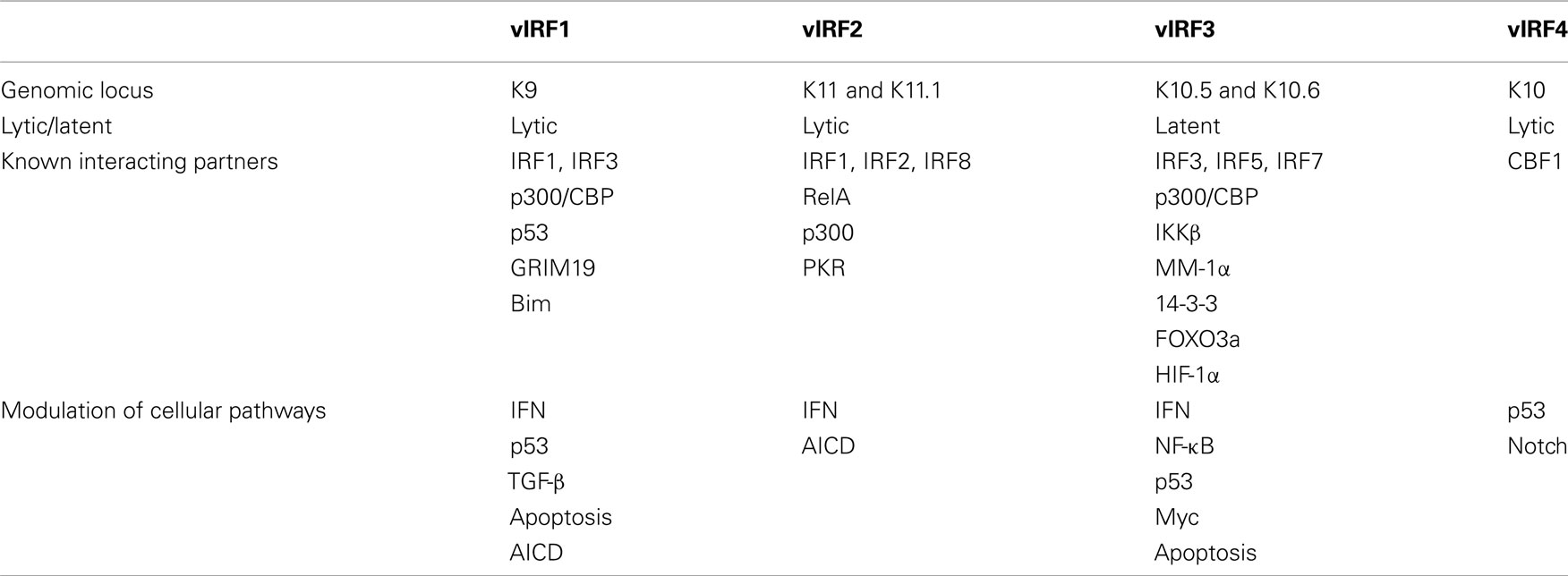
Table 1. Summary of the location, expression, cellular binding partners, and cellular pathways modulated by the KSHV vIRFs.
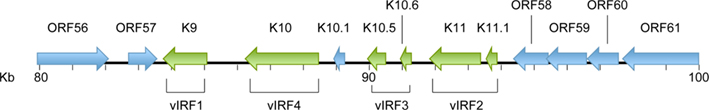
Figure 2. Map of vIRF gene locus in the KSHV genome. The KSHV vIRF genes are encoded by open reading frames K9 (vIRF1), K11 and K11.1 (vIRF2), K10.5 and 10.6 (vIRF3), and K10 (vIRF4).
During latency vIRF1 is localized to pro-myelocytic leukemia (PML) bodies (Pozharskaya et al., 2004), nuclear granules identified by the presence of the PML protein and associated with the regulation of oncogenesis, cell senescence, and anti-viral defenses (Lallemand-Breitenbach and de The, 2010). The vIRF2 protein is present in both the cytoplasm and nucleus in a diffused pattern prior to stimulation of the IFN response (Areste et al., 2009). Activation of the anti-viral response results in enhanced punctate expression in the cytoplasm (Areste et al., 2009). Similarly, vIRF3 is localized to the nucleus in a fine granular pattern (Rivas et al., 2001).
Each cellular IRF contains a well-conserved N-terminal DNA-binding domain (DBD) of approximately 120 amino acids including five conserved tryptophan repeats (Takaoka et al., 2008). KSHV vIRFs share limited homology with the N-terminal DBD of cellular IRFs, but lack several of these crucial tryptophan residues (Takaoka et al., 2008; Figure 3). This renders the vIRFs incapable of binding cellular DNA (Flowers et al., 1998; Zimring et al., 1998; Tamura et al., 2008). Close examination of vIRF1 reveals that it has 13% homology to human IRF family members, much of which is localized to the DBD region, and shows similarity to the IRF binding motif found in IRF8/ICSBP (Russo et al., 1996; Zimring et al., 1998). The cellular proteins that share the most similarity to the KSHV vIRFs by NCBI BLASTP analysis are IRF4 and IRF8/ICSBP (National Center for Biotechnological Information, http://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&BLAST_PROGRAMS=blastp&PAGE_TYPE=BlastSearch&SHOW_DEFAULTS=on&LINK_LOC=blasthome).
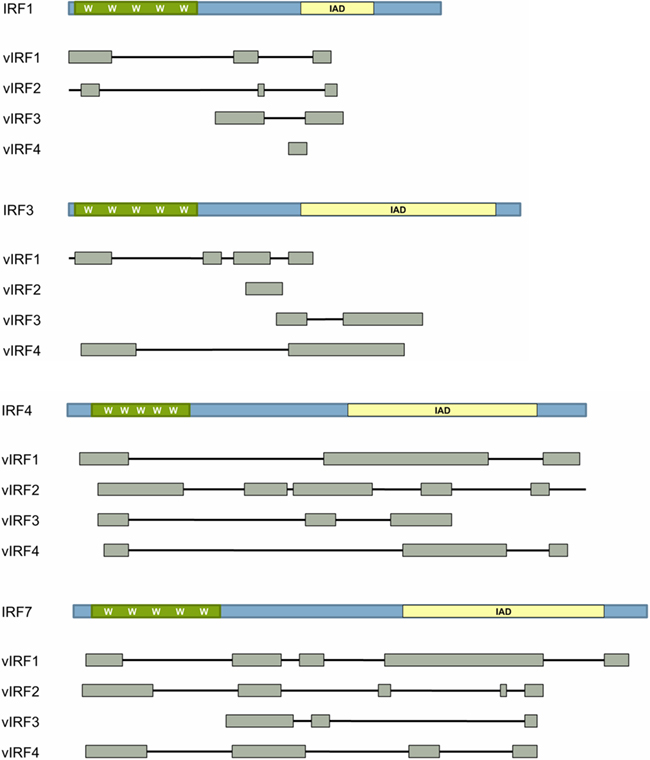
Figure 3. Homology of vIRFs with cellular IRFs. Cellular interferon regulatory factors (IRFs) are composed of a DNA-binding domain (DBD; green) and a regulatory domain (blue). The DBD of cellular IRFs is defined by 5 tryptophan (W) residues. Cellular IRFs may also contain an IRF-association domain (IAD) shown in yellow. Gray boxes indicate domains in the cellular IRF proteins that are homologous to each KSHV vIRF as determined by NCBI BLAST protein analysis (National Center for Biotechnological Information).
vIRF Inhibition of Immune Responses
Due to the homology of the KSHV vIRFs with cellular IRFs, and the requirement of herpesviruses to inhibit immunity to establish life-long infection, it was hypothesized that vIRFs would inhibit the cellular pathway involved in IFN production and the anti-viral state. vIRF expression was found to inhibit transcriptional activation of IRF target genes such IFNα or -β, type II interferon (IFNγ), and inflammatory signals such as RANTES (Taniguchi et al., 2001). IFN-stimulated genes (ISG) often contain an IFN-stimulated response element (ISRE) in their promoter. Several promoter–reporter based studies have shown that vIRF1, 2, and 3 inhibit IFN-induced gene transcription. vIRF1 inhibits ISG54 promoter activity mediated by IFNβ treatment (Gao et al., 1997), (PRD I)4-CAT activity induced by IFNα, -γ or IRF1 (Zimring et al., 1998), IFNA4 expression induced by Newcastle disease virus (Burysek et al., 1999a,b), ISG15-ISRE expression induced by IFNα or β (Li et al., 1998), and IFNγ-stimulated ISRE transactivation (Li et al., 1998). Conversely, knockdown of vIRF1 in BCBL-1 PEL cells stimulated with IFNγ resulted in a four to fivefold increase of gamma-interferon activated sequence (GAS) and ISG15-ISRE reporter activity, demonstrating that vIRF1 reduced IFN responses in latently infected PEL cells (Li et al., 1998).
In addition to reporter assays, vIRF1 stable cell lines infected with Sendai virus had reduced expression of cytokines and chemokine RNA compared to control (Lin et al., 2001). vIRF1-mediated blockade of IFN production has positive effects for viral infection such as the prevention of IFNβ induction of p21WAF1/CIP1, a regulator of cell cycle, which allows KSHV to escape anti-viral cell cycle arrest (Gao et al., 1997).
Reporter assays also demonstrate the ability of vIRF2 to reduce IFN-dependent transcription. vIRF2 inhibited an ISG56K-ISRE reporter in response to recombinant IFN-α2b (Fuld et al., 2006), IRF1- and IRF3-mediated transcriptional activation of an IFNα or ISRE promoter in virus-infected cells (Burysek et al., 1999b; Fuld et al., 2006), IFNβ promoter activity mediated by transfection of a synthetic double-stranded RNA and IRF3 (Areste et al., 2009), and ISRE transactivation mediated by the IFNλ family members IL28A or IL29 (Fuld et al., 2006). However, vIRF2 failed to inhibit IRF7-mediated promoter–reporter activity or the IFNγ promoter (pGAS) activity in response to IFNγ treatment (Fuld et al., 2006).
vIRF3 has been shown to inhibit transactivation of the IFN-α4 and IFN-α6 promoter–reporter upon Sendai virus infection as well as in the presence of overexpressed IRF3 and IRF7 (Lubyova and Pitha, 2000; Joo et al., 2007). vIRF3 could also inhibit IFNγ mediated activation of the GAS promoter (Lubyova et al., 2004). Furthermore, vIRF3 expression led to the reduction of endogenous IFNα1, -α4, and -α6 mRNA levels upon Sendai infection (Joo et al., 2007), and the production of IFN following infection with Newcastle disease virus (Lubyova and Pitha, 2000). These data indicate that vIRF1, 2, and 3 inhibit cellular IRF processes, including transcription and type I IFN production. In contrast to the other three vIRFs, vIRF4 has not been shown to inhibit the IFN signaling pathway (Lee et al., 2009b).
Studies of KSHV vIRF inhibition of the cellular anti-viral IFN response have shown that this occurs through interaction with cellular proteins. It was first hypothesized that vIRF1 would associate with the activating IRF, IRF1, as it was not yet known that IRF3 and IRF7 are the main players in IFN signaling (Tamura et al., 2008). vIRF1 was found to bind strongly to IRF1 in vitro, and in cellular lysates a weak association of vIRF1 with IRF8 was observed. Additionally, binding between vIRF1 and IRF3 was not detected (Burysek et al., 1999a). However, others reported that vIRF1 did not interact with soluble IRF1 in vitro (Zimring et al., 1998). vIRF1 was also unable to interact with IRF1 bound to DNA, although vIRF1 inhibited IRF1 transcriptional activity (Zimring et al., 1998; Burysek et al., 1999a). Overexpression of vIRF1 blocks binding of cellular IRF1 to DNA (Burysek et al., 1999a), and the vIRF1 domain that shows homology to cellular IRFs was not required to inhibit IRF1-mediated transcription (Zimring et al., 1998). vIRF1 did not bind to IRF elements on DNA or alter the ability of IRF1 or IRF2 to bind DNA (Zimring et al., 1998). However, it is unclear how relevant the vIRF-cellular IRF1 interaction is, since IRF1−/− cells do not exhibit altered virus-mediated activation of IFNα and IFNβ gene expression (Matsuyama et al., 1993; Reis et al., 1994). The more relevant interaction appears to be the association of vIRF1 with cellular IRF3 (Lin et al., 2001). This interaction did not inhibit dimerization or nuclear localization of IRF3, but IRF3-mediated transcription was blocked (Burysek et al., 1999a; Lin et al., 2001). Although vIRF1 co-precipitates with IRF7, vIRF1 did not block IRF7-mediated transcriptional activation (Lin et al., 2001). Additionally, the vIRFs appear to interact with each other. In vitro, vIRF1 was found to associate with vIRF1 and vIRF2 (Burysek et al., 1999a,b). It remains unclear if vIRFs require dimerization for functionality similar to their cellular counterparts.
In addition to directly binding cellular IRFs, vIRF1 has been shown to inhibit cellular IRF3 transcription through interaction with transcriptional coactivators. Upon viral infection, cellular IRFs recruit the p300/CBP transcriptional coactivator for efficient production of IFNβ (Wathelet et al., 1998). The C-terminal portion of vIRF1 strongly binds to p300 (Burysek et al., 1999a; Lin et al., 2001). This interaction occurs in the same region as IRF1 binding, suggesting that vIRF1 may function through competition for the binding site (Burysek et al., 1999a). It was later determined that vIRF1’s direct interaction with p300 inhibited p300/CBP-associated-factor (PCAF) binding to p300, and displaces PCAF from p300 complexes (Li et al., 2000). Furthermore, vIRF1 competes with the active form of IRF3 for binding to CBP on its C-terminal end (Lin et al., 2001). Expression of vIRF1 inhibited IRF3 interaction with CBP in a dose dependent manner and reduced IRF3 association with CBP and p300 following viral infection (Lin et al., 2001). However, vIRF1 blockade of IRF3-CBP/p300 complex formation did not affect the DNA-binding activity of IRF3 (Lin et al., 2001).
The mechanism of vIRF2 inhibition of IFN responses is not as well characterized as that of vIRF1. The first report on vIRF2, in which only the first exon of vIRF2 is expressed, demonstrated interactions between vIRF2 and cellular IRF1, IRF2, IRF8, RelA (p65), and p300 (Burysek et al., 1999b). No interaction with IRF3 was observed (Burysek et al., 1999b). Expression of full length vIRF2 was found to associate with IRF3 and inhibit IRF3 transcription of an IFNβ promoter (Areste et al., 2009). In this study vIRF2 was suggested to form a multiprotein complex with IRF3 and caspase-3 and vIRF2 expression enhanced caspase-3 mediated degradation of IRF3, ultimately resulting in decreased anti-viral responses (Areste et al., 2009). However, vIRF2 was also able to inhibit IRF3 activity in a caspase-3 independent manner, suggesting multiple mechanisms of action for vIRF2 (Areste et al., 2009).
Similar to vIRF1 and vIRF2, vIRF3 can associate with multiple cellular IRFs. Like vIRF1, vIRF3 is able to associate with IRF3, but this requires IRF3 to be present in the nucleus, which occurs after Sendai virus infection (Lubyova et al., 2004). vIRF3 also co-immunoprecipitated with IRF7, independent of IRF7 activation (Lubyova et al., 2004; Joo et al., 2007). vIRF3 expression did not prevent IKKε phosphorylation of IRF7, or dimerization of IRF7 (Joo et al., 2007). However, the interaction of vIRF3 with IRF7 inhibited IRF7 DNA-binding, and inhibition was abolished when the vIRF3/IRF7 interaction was disrupted (Joo et al., 2007). In addition to vIRF3’s interaction with IRF3 and IRF7, vIRF3 has also been shown to interact with IRF5 (Wies et al., 2009). IRF5 induces IFN transcription (Tamura et al., 2008). Expression of vIRF3 inhibited IRF5-mediated ISRE and IFNβ reporter activation, and RNA interference (RNAi) knockdown of vIRF3 abolished this effect (Wies et al., 2009). RNAi-mediated knockdown of vIRF3 in PEL cells produced a detectable increase of IRF5 binding to ISRE oligonucleotides (Wies et al., 2009). These studies demonstrate that vIRF3 inhibits IFN transcription and production through direct association with its cellular homologs.
Viral infection activates the IRF pathway as well as the NF-κB pathway. Activation of NF-κB following pathogen infection results in the expression of inflammatory cytokines or apoptosis. NF-κB signaling occurs through activation of the IκB kinases (IKKs), which phosphorylate IκB, thereby inducing its ubiquitination and degradation (Sun and Andersson, 2002). The degradation of IκB allows the NF-κB p65 and p50 subunits to translocate to the nucleus and initiate transcription. The NF-κB pathway is often activated by stimuli that induce IFN signaling. Therefore, it was expected that vIRFs could also inhibit NF-κB activity. Expression of vIRF3 inhibited NF-κB transactivation and DNA-binding in response to TNFα treatment through vIRF3-mediated reduction of IKKβ kinase activity and reduced phosphorylation of IκB (Seo et al., 2004). This was supported by vIRF3 expression resulting in inhibited nuclear localization of p65 NF-κB subunit (Seo et al., 2004). Furthermore, IKKβ and vIRF3 were found to coprecipitate in 293T cells when both proteins were overexpressed, but no interaction was observed in BCBL-1 PEL cells (Seo et al., 2004). These data suggest that vIRF3 may have multiple interaction partners to effectively inhibit various immune signaling pathways. In contrast, a NF-κB driven promoter–reporter construct, (PRD II)4-CAT, was not inhibited by vIRF1 expression, indicating that NF-κB inhibition may be a specific activity of vIRF3 (Zimring et al., 1998). Together, these data suggest that vIRFs vary in their ability to inhibit the NF-κB pathway and support the notion that vIRFs are not redundant.
In addition to inhibition of the IFN response and NF-κB signaling, vIRFs have been shown to inhibit the double-stranded RNA-activated protein kinase (PKR). PKR is activated by double-stranded RNA and affects cell growth, cell differentiation, viral clearance, and induction of apoptosis (Pindel and Sadler, 2011). PKR targets include the phosphorylation of eukaryotic translation initiation factor-2 (eIF-2α) and NF-κB which control protein synthesis and cell survival, respectively. Expression of vIRF3 partially prevented PKR inhibition of protein synthesis by decreasing phosphorylation of eIF-2α, demonstrating vIRF3’s ability to overcome PKR translational control (Esteban et al., 2003). Inhibition of the PKR response by vIRF3 also blocked PKR-mediated apoptosis, but did not affect PKR activation of NF-κB (Esteban et al., 2003). No interaction between PKR and vIRF3 could be detected, so the mechanism of this control remains unclear (Esteban et al., 2003). However, an interaction between vIRF2 and PKR has been reported (Burysek and Pitha, 2001). Expression of vIRF2 inhibited the phosphorylation of PKR, and the phosphorylation of PKR substrates histone 2A and eIF-2α (Burysek and Pitha, 2001).
vIRF Effects on Gene Expression
In addition to evidence that KSHV vIRFs inhibit the host immune response and allow for infection of the host, vIRFs have also been implicated as transcriptional activators. As described above, vIRF1 has been shown to interfere with the formation of the CBP/p300 enhanceosome complex, which resulted in a reduction in cellular IRF transcriptional activity (Li et al., 2000; Lin et al., 2001). vIRF1 interaction with p300 also reduces the histone acetyltransferase (HAT) activity of p300, although vIRF1 itself was not found to have HDAC activity (Li et al., 2000). vIRF1 dependent modulation of HAT activity alters cellular cytokine expression such as macrophage migration inhibitory factor (MIF), normally regulated by acetylation of histones (Li et al., 2000). Hypoacetylation of histones caused by vIRF1 was also observed by a reduction in DNA staining, due to alteration of the DNA structure and its availability to DNA dyes (Li et al., 2000). vIRF3 also interacts with CBP/p300 in both uninfected and infected cells, through the individual C-terminal domains of vIRF3 and p300 (Lubyova et al., 2004). Although vIRF1 can block the association of IRF3 and p300, vIRF3 is unable to do so. Therefore vIRF1 and vIRF3 bind to p300 in a unique manner (Lubyova et al., 2004). Furthermore, vIRF3 binding to p300 does not affect acetylation of histones (Lubyova et al., 2004).
vIRFs have been shown to act as transcriptional activators. When vIRF1 was fused to a Gal4 DBD to direct vIRF1 to DNA, vIRF1 demonstrated an ability to drive transcription (Roan et al., 1999). Indeed, vIRF1 was shown to drive its own expression through two cis elements (Wang and Gao, 2003). Neither cis element contains an ISRE and do not respond to induction with IFNβ or IFNγ, suggesting that vIRF1-mediated transactivation occurs on promoters lacking ISRE-like sequences (Wang and Gao, 2003). Similar to vIRF1, vIRF3 can also drive transcription but through a DNA-independent mechanism. vIRF3 is recruited to IFN-responsive promoters through its association with IRF3 and IRF7 (Lubyova et al., 2004) and contradictory to other reports, appears to stimulate rather than inhibit IFN-responsive genes (Lubyova et al., 2004).
vIRFs and Disruption of Pathways Associated with Oncogenesis
An indication that vIRFs may be involved in carcinogenesis is their inhibitory effects on the tumor suppressor, p53. p53 is a key regulator of many cellular activities such as cell cycle, apoptosis, DNA damage response, differentiation, and angiogenesis (Brady and Attardi, 2010). vIRF1 co-precipitates with p53 and inhibits p53-driven transcription in a dosage dependent manner (Nakamura et al., 2001; Seo et al., 2001). vIRF1 interaction with p53 did not inhibit p53 DNA-binding, but resulted in a decrease in p53 target gene expression and transcription, such as p21 and Bax (Nakamura et al., 2001; Seo et al., 2001). vIRF1 expression also resulted in increased levels of p53 in the cytoplasm compared to normal localization in the nucleus (Shin et al., 2006). Initially, p53 protein levels were reportedly not decreased by vIRF1 expression (Nakamura et al., 2001; Seo et al., 2001), although the phosphorylation and acetylation of p53 was reduced in the presence of vIRF1 (Nakamura et al., 2001). However, later reports indicated that expression of vIRF1 led to a decrease in total p53 levels, due to vIRF1 directed ubiquitination and degradation of p53 (Shin et al., 2006). This activity required the p53 E3 ligase, MDM2, and vIRF1 expression resulted in a reduction of phosphorylation of serine residue 15 of p53. This is a key regulatory site that hinders MDM2 interaction with p53. Loss of serine 15 phosphorylation allowed for increased interaction between p53 and MDM2 leading to increased ubiquitination and degradation of p53 protein (Shin et al., 2006). Etoposide-mediated DNA damage response induction of p53 was reduced by vIRF1 expression, and resulted in increased ubiquitination of p53 (Shin et al., 2006). Importantly, vIRF1 perturbation of the p53 pathway also inhibited p53-mediated apoptosis (Seo et al., 2001). Expression of vIRF1 reduced p53-mediated apoptosis from 75 to 32%, and cells that were protected from apoptosis accumulated at the G2/M transition (Nakamura et al., 2001). Inhibition of p53-mediated apoptosis may be a mechanism through which KSHV is able to establish latency or malignancy. While p53 is rarely mutated in KSHV-associated malignancies, treatment with p53 activators has proved an effective treatment for PEL (Dittmer and Krown, 2007).
In addition to vIRF1, vIRF3 and vIRF4 also interfere with p53 signaling. Like vIRF1, vIRF3 was shown to interact with p53, in vitro (Rivas et al., 2001). vIRF3 expression reduced p53 reporter activity in response to p53 expression (Rivas et al., 2001). Apoptosis and activation of caspase 8 mediated by p53 was also decreased by expression of vIRF3 (Rivas et al., 2001). However, it is not yet clear how vIRF3 targets the p53 pathway. vIRF4 interferes with the p53 pathway by interacting with MDM2 to increase MDM2 protein levels (Lee et al., 2009b). This was accomplished by vIRF4 inhibition of MDM2 autoubiquitination and prevention of MDM2 proteosomal degradation (Lee et al., 2009b). Subsequently, higher levels of MDM2 increased ubiquitination of p53 and degradation of p53 in BCBL-1 cells (Lee et al., 2009b). Thus, both vIRF1 and vIRF4 induce MDM2-mediated p53 degradation but through different mechanisms. Similar to vIRF1, vIRF4 expression reduced apoptosis in response to 5FU or etoposide treatment as a result of inhibition of the p53 pathway (Lee et al., 2009b).
Myc signaling is another pathway that is often deregulated in human cancers and in which KSHV vIRFs have been shown to play a role. vIRF3 increases Myc signaling by interacting with MM-1α (Myc modulator-1; Lubyova et al., 2007), a c-Myc transcriptional repressor (Satou et al., 2001). vIRF3’s interaction with MM-1α reverses its normal suppression of Myc transcription as measured by cdk4 promoter activity (Lubyova et al., 2007). This is accomplished through competition between vIRF3 and c-Myc for binding to MM-1α, which increases the ability of c-Myc to bind to the cdk4 promoter (Lubyova et al., 2007).
The transforming growth factor-β (TGF-β) signaling pathway is also perturbed by vIRFs. The TGF-β family regulates a variety of biological processes including cell growth, differentiation, matrix production, and apoptosis (Meulmeester and Ten Dijke, 2011). TGF-β is activated by a serine/threonine kinase receptor on the cell surface that signals to members of the Smad family including Smad2 and Smad3, which then form a complex with a common mediator, Smad4, and translocate into the nucleus to regulate transcription of target genes. Viral regulation of TGF-β signaling would be advantageous to prevent anti-viral immunity and cell death. vIRF1 has been shown to interfere with TGF-β/Smad signaling. vIRF1 inhibited TGF-β mediated transcription in reporter assays, and vIRF1 stable cell lines were resistant to TGF-β1-induced growth inhibition (Seo et al., 2005). vIRF1 interacts with Smad3 and Smad4 and inhibits TGF-β mediated Smad3/Smad4 DNA-binding, as well as formation of the Smad3/Smad4 complex (Seo et al., 2005). This results in vIRF1 inhibition of the transcriptional activity of Smad proteins (Seo et al., 2005).
Another pathway in which KSHV vIRFs have been implicated is the 14-3-3 family of dimeric regulatory proteins. This family of enzymes, like many associated with vIRFs, is involved in the regulation of cell cycle, apoptosis, and oncogenesis (Fu et al., 2000). 14-3-3 proteins inhibit the forkhead family of winged helix transcription factors, FOXO1, FOXO3a, and FOXO4 by sequestering them in the cytoplasm (Biggs et al., 1999). vIRF3 interacts with 14-3-3s in a manner dependent on the phosphorylation of vIRF3 (Munoz-Fontela et al., 2007). Phosphorylation of vIRF3, the mechanism of which has not been determined, was also required to observe an interaction between vIRF3 and FOXO3a (Munoz-Fontela et al., 2007). It was reported that interaction with vIRF3 facilitated the binding between 14-3-3 and unphosphorylated FOXO3a, which would result in decreased FOXO3a gene transcription (Munoz-Fontela et al., 2007). For example, vIRF3 expression inhibited a G2/M arrest mediated by overexpression of 14-3-3 through blockade of transcriptional activity. Additionally, Bim promoter activation by the FOXO3a transcription factor was inhibited by co-expression of vIRF3 (Munoz-Fontela et al., 2007).
The hypoxia inducible factor 1 transcription factor is another vIRF target that links vIRFs to oncogenesis. HIF-1α is a transcription factor activated in response to low oxygen and other cellular stressors. Upon activation, HIF-1α mediates the transcription of target genes involved in growth, metabolism, invasion, and cell death (Majmundar et al., 2010). These genes have been implicated in the processes of malignant transformation and cancer progression. The most studied HIF-1α target gene is that of vascular endothelial cell growth factor (VEGF), a secreted factor that promotes angiogenesis (Majmundar et al., 2010). Through a screen of viral genes on activity of HIF-1α inducible promoters, it was found that vIRF3 was able to induce transcription (Shin et al., 2008). vIRF3 increased HIF-1α transcription through direct binding and stabilization of the transcription factor (Shin et al., 2008). This interaction leads to increased production of VEGF and increased tube formation in human umbilical vein endothelial cells (Shin et al., 2008). These data suggest that the vIRF3-mediated increase in HIF-1α transcription may contribute to oncogenesis.
The Notch CSL/CBF1 pathway has also been implicated as a KSHV vIRF target. Notch receptors are transmembrane proteins that when bound to their ligand are proteolytically cleaved to generate the intracellular Notch fragment (NCID). The NCID is then translocated to the nucleus and binds to the CSL/CBF1 protein. This DNA-binding factor recruits corepressor and coactivator complexes to its target genes (Kopan and Ilagan, 2009). Activated Notch signaling has been identified in KSHV-infected PEL cells and KS cells and may contribute to cellular survival (Curry et al., 2005, 2007; Lan et al., 2006, 2009). vIRF4 was identified as interacting with CBF1 by yeast two hybrid assays, and confirmed by in vitro pull down assays (Heinzelmann et al., 2010). This interaction resulted in the inhibition of NICD transcription, and may do so by competing for the hydrophobic pocket binding site of CBF1 with NCID (Heinzelmann et al., 2010). No other vIRFs were able to bind to CBF1 (Heinzelmann et al., 2010), but this does not rule out the possibility that other vIRFs inhibit Notch signaling by a different mechanism not yet reported.
Recently, vIRF3 has been implicated in the regulation of type II interferon and type II interferon target genes. Reduction of vIRF3 levels by RNAi in KSHV latently infected PEL cells results in decreased transcription of IFNγ, a type II IFN to which PEL cells are able to respond (Schmidt et al., 2011). Reduction of IFNγ in turn resulted in reduced transcription of the IFNγ-inducible promoter of the major histocompatibility complex class II (MHC II; Schmidt et al., 2011). RNAi silencing of vIRF3 in BC-3 PEL cells increased MHC II transcripts more than twofold and resulted in increased levels of MHC II protein (Schmidt et al., 2011). MHC II is expressed on specialized cell types of the immune system and serves to present antigens for detection by T cells. Transcription of MHC II is directed by the master regulator CIITA (Reith et al., 2005). vIRF3 inhibited MHC II production through inhibition of CIITA (Schmidt et al., 2011). Blockade of IFNγ secretion would prohibit downstream signaling and MHC II antigen presentation, both of which would be beneficial for viral survival. It remains unclear how vIRF3 reduced transcription of IFNγ and what effect reduction of MHC II expression has on maintenance of latency and survival of infected cells.
Inhibition of Cell Death
Although KSHV vIRF’s modulation of the p53 pathway contributes to cell survival, there have also been reports of the interaction of the vIRFs with cell death proteins. Genes associated with retinoid-IFN-induced mortality (GRIM) proteins were originally identified due to their role in IFN/retinoic acid (RA) cell death, a process by which administration of both factors induces apoptosis in tumor cells (Hofmann et al., 1998; Angell et al., 2000). Yeast two hybrid assays identified the GRIM family member GRIM19 as a novel vIRF1 interaction partner (Seo et al., 2002). This interaction was confirmed by mammalian two hybrid, and in latently infected BCBL-1 cells by coimmunoprecipitation and colocalization assays, where vIRF1 and GRIM19 were seen in the nucleus (Seo et al., 2002). vIRF1 interaction with GRIM19 reduced GRIM19 mediated IFN/RA-induced apoptosis (Seo et al., 2002). Additionally, co-expression of GRIM19 and vIRF1 in NIH-3T3 cells resulted in a significant suppression of transformed colonies, and suggested that vIRF1 inhibition of GRIM19 contributes to oncogenesis (Hu et al., 2002).
vIRFs have also been implicated in interfering with activation induced cell death (AICD) mediated by expression of the death receptor CD95. Ligation of CD95 with its cognate ligand, CD95L, initiates extrinsic cell death, caspase cleavage and apoptosis. Expression of CD95L is tightly regulated by multiple transcription factors including NF-κB (Kasibhatla et al., 1999; Li-Weber et al., 2000), TGF-β regulation of c-Myc (Genestier et al., 1999), forkhead transcription factors (Brunet et al., 1999), and IRF1 (Chow et al., 2000; Kirchhoff et al., 2002). As these cellular processes are inhibited by vIRFs, it was hypothesized that vIRFs may reduce the expression of CD95L and effectively reduce AICD. As predicted, transfection of vIRF1 or vIRF2 reduced IRF1 mediated induction of CD95L (Kirchhoff et al., 2002). This action required the presence of two positive regulatory IRF1-dependent domains in the promoter of CD95L (Kirchhoff et al., 2002). Inhibition of CD95L expression resulted in decreased AICD mediated by the T cell receptor in Jurkat T cells (Kirchhoff et al., 2002).
In addition to interfering with IFN/RA- and CD95-mediated cell death, pro-apoptotic Bcl-2 family members, responsible for the activation of caspases, are also affected by vIRFs. vIRF1 associates with the pro-apoptotic BH3-only Bcl2 family member, Bim (Choi and Nicholas, 2010). This interaction leads to the localization of Bim to the nucleus, a location from which Bim cannot induce apoptosis (Choi and Nicholas, 2010). This is of particular interest as Bim is localized to the nucleus during the lytic cycle of KSHV, and vIRF1 expression inhibited Bim-mediated apoptosis (Choi and Nicholas, 2010). Knockdown of vIRF1 in KSHV latently infected endothelial cells resulted in increased cell death compared to control and also reduced viral output upon TPA induction (Choi and Nicholas, 2010). This suggests that vIRF1-mediated nuclear localization of Bim protects cells from apoptosis and thereby increases lytic viral production. Thus, disruption of the Bim/vIRF1 interaction may have a therapeutic effect.
Expression of vIRF3 also affects apoptosis. vIRF3’s interaction with PKR resulted in an inhibition of PKR-mediated cell apoptosis, although PKR activation of NF-κB was not blocked (Esteban et al., 2003). vIRF3 expression blocked activation of caspase-3 downstream of PKR, but did not inhibit activation of caspase 9 (Esteban et al., 2003). However, vIRF3 expression did not inhibit 2-5A synthetase/RNase L system-mediated apoptosis (Esteban et al., 2003). vIRF3 expressing cells died more readily in the presence of TNFα than empty vector, which was demonstrated by increased TUNEL staining (Seo et al., 2004). These data make it unclear as to the conditions under which vIRF3 is pro- or anti-apoptotic. TNFα treatment of vIRF1-expressing NIH-3T3 cells seemed to have the opposite effect, with reduced apoptosis occurring in vIRF1-expressing cells compared to control (Burysek et al., 1999a). Similarly, IFNα treatment of TPA-induced cells accelerated the decline of vIRF1 protein levels and increased apoptosis (Pozharskaya et al., 2004). Likewise, IFNα3 treatment of PEL cells following knockdown of vIRF3 resulted in increased activity of caspase-3/7 (Wies et al., 2009). These data suggest that loss of vIRF or increased IFN production could lead to apoptosis in KSHV-infected cells.
Role of vIRFs in Cancer
As discussed above, KSHV vIRFs manipulate cellular pathways associated with the control of cell cycle, proliferation, and cell death. Viral manipulation of these essential cellular pathways may result in carcinogenesis. It is not surprising, therefore, that vIRF1 and vIRF3 have been implicated as possible KSHV oncogenes. It was first observed that vIRF1 could transform cells in vitro. Expression of vIRF1 in NIH-3T3 cells resulted in loss of contact inhibition, formation of foci prior to confluence, shorter doubling times, and higher growth rates in low serum conditions (Gao et al., 1997; Li et al., 1998). The potential for vIRF1 to contribute to in vivo tumor formation was also investigated by injection of NIH-3T3 cells stably expressing vIRF1 into mice. These cells caused tumors in over 95% of animals with a short latency (Gao et al., 1997). Similar experiments were performed with vIRF2, however, vIRF2 was not able to transform NIH-3T3 cells (Fuld et al., 2006) and BJAB vIRF3 stable cells lines did not exhibit altered morphology, growth rate, or growth in soft agar (Lubyova et al., 2004).
The contribution of vIRF1 to continued cell growth and survival of KSHV latently infected cells was deduced in BCBL-1 cells subjected to vIRF1 knockdown. While vIRF1 knockdown was not particularly efficient in this case, no change in growth rate was observed compared to control (Li et al., 1998). However, siRNAs targeting vIRF3 for destruction in PEL cells, reduced proliferation by 40% (Wies et al., 2008). This decrease in cell growth was attributed to an induction of apoptosis upon silencing of vIRF3 and increased caspase-3 and caspase-7 activities (Wies et al., 2008). These data suggest that both vIRF1 and vIRF3 contribute to the continued survival and deregulated growth of KSHV-infected cells. To this end, Zhang et al. (2001) considered vIRF1 an ideal target for KSHV related cancer treatment and developed a ribozyme capable of cleaving vIRF1. This ribozyme will require further testing before it can be used as a therapeutic.
Conclusion
While the KSHV vIRFs share an ability to block IFN or p53 signaling, each vIRF demonstrates a unique ability to block specific cellular functions. All four vIRFs have been shown to interact with at least one unique cellular protein (Table 1), demonstrating that each vIRF has an individual function during KSHV infection and latency. Why KSHV encodes for multiple vIRFs remains unclear, although the phenomenon seems very specific for this virus. Aside from KSHV, only one other herpesvirus is known to encode multiple vIRFs. It is the closely related simian homolog of KSHV named rhesus monkey rhadinovirus (RRV; Searles et al., 1999; Alexander et al., 2000). Further work is necessary to determine why these viruses encode multiple copies of genes with similar functions and to address the requirement for vIRFs during the viral lifecycle and their contribution to carcinogenesis. Collectively, it is clear the vIRFs evade innate immune responses and also target pathways related to cell survival and cell proliferation. Cellular immune activation and apoptotic pathways are intimately intertwined, and by preventing immune activation and cell death, the KSHV vIRFs activate pro-survival and proliferative pathways that may ultimately contribute to oncogenic transformation and the development of KSHV-associated malignancies. In addition, by inhibiting innate immunity and apoptosis, the KSHV vIRFs play a critical role in the survival of KSHV-infected cells, establishment and maintenance of latent infection, and persistence of the virus in the human population.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Sean Gregory for critical reading of the manuscript. Due to space restrictions, we regret that we had to omit many important references. Blossom Damania is supported by NIH grants DE018281, CA096500, and CA019014 (MPI) and a Burroughs Wellcome Fund grant. Sarah R. Jacobs is supported by NIH training grant T32CA009156. Blossom Damania is a Leukemia and Lymphoma Society Scholar and Burroughs Wellcome Fund Investigator in Infectious Disease.
References
Alexander, L., Denekamp, L., Knapp, A., Auerbach, M. R., Damania, B., and Desrosiers, R. C. (2000). The primary sequence of rhesus monkey rhadinovirus isolate 26–95: sequence similarities to Kaposi’s sarcoma-associated herpesvirus and rhesus monkey rhadinovirus isolate 17577. J. Virol. 74, 3388–3398.
Alsharifi, M., Mullbacher, A., and Regner, M. (2008). Interferon type I responses in primary and secondary infections. Immunol. Cell Biol. 86, 239–245.
Angeletti, P. C., Zhang, L., and Wood, C. (2008). The viral etiology of AIDS-associated malignancies. Adv. Pharmacol. 56, 509–557.
Angell, J. E., Lindner, D. J., Shapiro, P. S., Hofmann, E. R., and Kalvakolanu, D. V. (2000). Identification of GRIM-19, a novel cell death-regulatory gene induced by the interferon-beta and retinoic acid combination, using a genetic approach. J. Biol. Chem. 275, 33416–33426.
Areste, C., Mutocheluh, M., and Blackbourn, D. J. (2009). Identification of caspase-mediated decay of interferon regulatory factor-3, exploited by a Kaposi sarcoma-associated herpesvirus immunoregulatory protein. J. Biol. Chem. 284, 23272–23285.
Ballestas, M. E., Chatis, P. A., and Kaye, K. M. (1999). Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284, 641–644.
Barbera, A. J., Chodaparambil, J. V., Kelley-Clarke, B., Joukov, V., Walter, J. C., Luger, K., and Kaye, K. M. (2006). The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 311, 856–861.
Biggs, W. H. III, Meisenhelder, J., Hunter, T., Cavenee, W. K., and Arden, K. C. (1999). Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. U.S.A. 96, 7421–7426.
Blasig, C., Zietz, C., Haar, B., Neipel, F., Esser, S., Brockmeyer, N. H., Tschachler, E., Colombini, S., Ensoli, B., and Sturzl, M. (1997). Monocytes in Kaposi’s sarcoma lesions are productively infected by human herpesvirus 8. J. Virol. 71, 7963–7968.
Boshoff, C., Schulz, T. F., Kennedy, M. M., Graham, A. K., Fisher, C., Thomas, A., Mcgee, J. O., Weiss, R. A., and O’Leary, J. J. (1995). Kaposi’s sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1, 1274–1278.
Brunet, A., Bonni, A., Zigmond, M. J., Lin, M. Z., Juo, P., Hu, L. S., Anderson, M. J., Arden, K. C., Blenis, J., and Greenberg, M. E. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868.
Burysek, L., and Pitha, P. M. (2001). Latently expressed human herpesvirus 8-encoded interferon regulatory factor 2 inhibits double-stranded RNA-activated protein kinase. J. Virol. 75, 2345–2352.
Burysek, L., Yeow, W. S., Lubyova, B., Kellum, M., Schafer, S. L., Huang, Y. Q., and Pitha, P. M. (1999a). Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J. Virol. 73, 7334–7342.
Burysek, L., Yeow, W. S., and Pitha, P. M. (1999b). Unique properties of a second human herpesvirus 8-encoded interferon regulatory factor (vIRF-2). J. Hum. Virol. 2, 19–32.
Cheng, E. H., Nicholas, J., Bellows, D. S., Hayward, G. S., Guo, H. G., Reitz, M. S., and Hardwick, J. M. (1997). A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. U.S.A. 94, 690–694.
Choi, Y. B., and Nicholas, J. (2010). Bim nuclear translocation and inactivation by viral interferon regulatory factor. PLoS Pathog. 6, e1001031. doi: 10.1371/journal.ppat.1001031
Chow, W. A., Fang, J. J., and Yee, J. K. (2000). The IFN regulatory factor family participates in regulation of Fas ligand gene expression in T cells. J. Immunol. 164, 3512–3518.
Cotter, M. A. II, and Robertson, E. S. (1999). The latency-associated nuclear antigen tethers the Kaposi’s sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264, 254–264.
Cunningham, C., Barnard, S., Blackbourn, D. J., and Davison, A. J. (2003). Transcription mapping of human herpesvirus 8 genes encoding viral interferon regulatory factors. J. Gen. Virol. 84, 1471–1483.
Curry, C. L., Reed, L. L., Broude, E., Golde, T. E., Miele, L., and Foreman, K. E. (2007). Notch inhibition in Kaposi’s sarcoma tumor cells leads to mitotic catastrophe through nuclear factor-kappaB signaling. Mol. Cancer Ther. 6, 1983–1992.
Curry, C. L., Reed, L. L., Golde, T. E., Miele, L., Nickoloff, B. J., and Foreman, K. E. (2005). Gamma secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi’s sarcoma tumor cells. Oncogene 24, 6333–6344.
Dittmer, D. P. (2003). Transcription profile of Kaposi’s sarcoma-associated herpesvirus in primary Kaposi’s sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 63, 2010–2015.
Dittmer, D. P., and Krown, S. E. (2007). Targeted therapy for Kaposi’s sarcoma and Kaposi’s sarcoma-associated herpesvirus. Curr. Opin. Oncol. 19, 452–457.
Esteban, M., Garcia, M. A., Domingo-Gil, E., Arroyo, J., Nombela, C., and Rivas, C. (2003). The latency protein LANA2 from Kaposi’s sarcoma-associated herpesvirus inhibits apoptosis induced by dsRNA-activated protein kinase but not RNase L activation. J. Gen. Virol. 84, 1463–1470.
Flowers, C. C., Flowers, S. P., and Nabel, G. J. (1998). Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor confers resistance to the antiproliferative effect of interferon-alpha. Mol. Med. 4, 402–412.
Fu, H., Subramanian, R. R., and Masters, S. C. (2000). 14-3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40, 617–647.
Fuld, S., Cunningham, C., Klucher, K., Davison, A. J., and Blackbourn, D. J. (2006). Inhibition of interferon signaling by the Kaposi’s sarcoma-associated herpesvirus full-length viral interferon regulatory factor 2 protein. J. Virol. 80, 3092–3097.
Gao, S. J., Boshoff, C., Jayachandra, S., Weiss, R. A., Chang, Y., and Moore, P. S. (1997). KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene 15, 1979–1985.
Genestier, L., Kasibhatla, S., Brunner, T., and Green, D. R. (1999). Transforming growth factor beta1 inhibits Fas ligand expression and subsequent activation-induced cell death in T cells via downregulation of c-Myc. J. Exp. Med. 189, 231–239.
Gregory, S. M., West, J. A., Dillon, P. J., Hilscher, C., Dittmer, D. P., and Damania, B. (2009). Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. U.S.A. 106, 11725–11730.
Heinzelmann, K., Scholz, B. A., Nowak, A., Fossum, E., Kremmer, E., Haas, J., Frank, R., and Kempkes, B. (2010). Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 4 (vIRF4/K10) is a novel interaction partner of CSL/CBF1, the major downstream effector of Notch signaling. J. Virol. 84, 12255–12264.
Heylbroeck, C., Balachandran, S., Servant, M. J., Deluca, C., Barber, G. N., Lin, R., and Hiscott, J. (2000). The IRF-3 transcription factor mediates Sendai virus-induced apoptosis. J. Virol. 74, 3781–3792.
Hofmann, E. R., Boyanapalli, M., Lindner, D. J., Weihua, X., Hassel, B. A., Jagus, R., Gutierrez, P. L., and Kalvakolanu, D. V. (1998). Thioredoxin reductase mediates cell death effects of the combination of beta interferon and retinoic acid. Mol. Cell. Biol. 18, 6493–6504.
Hu, J., Angell, J. E., Zhang, J., Ma, X., Seo, T., Raha, A., Hayashi, J., Choe, J., and Kalvakolanu, D. V. (2002). Characterization of monoclonal antibodies against GRIM-19, a novel IFN-beta and retinoic acid-activated regulator of cell death. J. Interferon Cytokine Res. 22, 1017–1026.
Huang, Q., Petros, A. M., Virgin, H. W., Fesik, S. W., and Olejniczak, E. T. (2002). Solution structure of a Bcl-2 homolog from Kaposi sarcoma virus. Proc. Natl. Acad. Sci. U.S.A. 99, 3428–3433.
Inagi, R., Okuno, T., Ito, M., Chen, J., Mori, Y., Haque, M., Zou, P., Yagi, H., Kiniwa, S., Saida, T., Ueyama, Y., Hayashi, K., and Yamanishi, K. (1999). Identification and characterization of human herpesvirus 8 open reading frame K9 viral interferon regulatory factor by a monoclonal antibody. J. Hum. Virol. 2, 63–71.
Jenner, R. G., Alba, M. M., Boshoff, C., and Kellam, P. (2001). Kaposi’s sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 75, 891–902.
Joo, C. H., Shin, Y. C., Gack, M., Wu, L., Levy, D., and Jung, J. U. (2007). Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol. 81, 8282–8292.
Kanno, T., Sato, Y., Sata, T., and Katano, H. (2006). Expression of Kaposi’s sarcoma-associated herpesvirus-encoded K10/10.1 protein in tissues and its interaction with poly(A)-binding protein. Virology 352, 100–109.
Kasibhatla, S., Genestier, L., and Green, D. R. (1999). Regulation of fas-ligand expression during activation-induced cell death in T lymphocytes via nuclear factor kappaB. J. Biol. Chem. 274, 987–992.
Katano, H., Sato, Y., Kurata, T., Mori, S., and Sata, T. (2000). Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi’s sarcoma, and multicentric Castleman’s disease. Virology 269, 335–344.
Kirchhoff, S., Sebens, T., Baumann, S., Krueger, A., Zawatzky, R., Li-Weber, M., Meinl, E., Neipel, F., Fleckenstein, B., and Krammer, P. H. (2002). Viral IFN-regulatory factors inhibit activation-induced cell death via two positive regulatory IFN-regulatory factor 1-dependent domains in the CD95 ligand promoter. J. Immunol. 168, 1226–1234.
Kopan, R., and Ilagan, M. X. (2009). The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233.
Krishnan, H. H., Naranatt, P. P., Smith, M. S., Zeng, L., Bloomer, C., and Chandran, B. (2004). Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi’s sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78, 3601–3620.
Lallemand-Breitenbach, V., and de The, H. (2010). PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2, a000661.
Lan, K., Choudhuri, T., Murakami, M., Kuppers, D. A., and Robertson, E. S. (2006). Intracellular activated Notch1 is critical for proliferation of Kaposi’s sarcoma-associated herpesvirus-associated B-lymphoma cell lines in vitro. J. Virol. 80, 6411–6419.
Lan, K., Murakami, M., Bajaj, B., Kaul, R., He, Z., Gan, R., Feldman, M., and Robertson, E. S. (2009). Inhibition of KSHV-infected primary effusion lymphomas in NOD/SCID mice by gamma-secretase inhibitor. Cancer Biol. Ther. 8, 2136–2143.
Lee, H. R., Kim, M. H., Lee, J. S., Liang, C., and Jung, J. U. (2009a). Viral interferon regulatory factors. J. Interferon Cytokine Res. 29, 621–627.
Lee, H. R., Toth, Z., Shin, Y. C., Lee, J. S., Chang, H., Gu, W., Oh, T. K., Kim, M. H., and Jung, J. U. (2009b). Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 4 targets MDM2 to deregulate the p53 tumor suppressor pathway. J. Virol. 83, 6739–6747.
Li, M., Damania, B., Alvarez, X., Ogryzko, V., Ozato, K., and Jung, J. U. (2000). Inhibition of p300 histone acetyltransferase by viral interferon regulatory factor. Mol. Cell. Biol. 20, 8254–8263.
Li, M., Lee, H., Guo, J., Neipel, F., Fleckenstein, B., Ozato, K., and Jung, J. U. (1998). Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor. J. Virol. 72, 5433–5440.
Lin, R., Genin, P., Mamane, Y., Sgarbanti, M., Battistini, A., Harrington, W. J. Jr., Barber, G. N., and Hiscott, J. (2001). HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene 20, 800–811.
Li-Weber, M., Laur, O., Dern, K., and Krammer, P. H. (2000). T cell activation-induced and HIV tat-enhanced CD95(APO-1/Fas) ligand transcription involves NF-kappaB. Eur. J. Immunol. 30, 661–670.
Lubyova, B., Kellum, M. J., Frisancho, A. J., and Pitha, P. M. (2004). Kaposi’s sarcoma-associated herpesvirus-encoded vIRF-3 stimulates the transcriptional activity of cellular IRF-3 and IRF-7. J. Biol. Chem. 279, 7643–7654.
Lubyova, B., Kellum, M. J., Frisancho, J. A., and Pitha, P. M. (2007). Stimulation of c-Myc transcriptional activity by vIRF-3 of Kaposi sarcoma-associated herpesvirus. J. Biol. Chem. 282, 31944–31953.
Lubyova, B., and Pitha, P. M. (2000). Characterization of a novel human herpesvirus 8-encoded protein, vIRF-3, that shows homology to viral and cellular interferon regulatory factors. J. Virol. 74, 8194–8201.
Majmundar, A. J., Wong, W. J., and Simon, M. C. (2010). Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 40, 294–309.
Matsuyama, T., Kimura, T., Kitagawa, M., Pfeffer, K., Kawakami, T., Watanabe, N., Kundig, T. M., Amakawa, R., Kishihara, K., Wakeham, A., Potter, J., Furlonger, C. L., Narendran, A., Suzuki, H., Ohashi, P. S., Paige, C. J., Taniguchi, T., and Mak, T. W. (1993). Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell 75, 83–97.
Meulmeester, E., and Ten Dijke, P. (2011). The dynamic roles of TGF-beta in cancer. J. Pathol. 223, 205–218.
Monini, P., Colombini, S., Sturzl, M., Goletti, D., Cafaro, A., Sgadari, C., Butto, S., Franco, M., Leone, P., Fais, S., Melucci-Vigo, G., Chiozzini, C., Carlini, F., Ascherl, G., Cornali, E., Zietz, C., Ramazzotti, E., Ensoli, F., Andreoni, M., Pezzotti, P., Rezza, G., Yarchoan, R., Gallo, R. C., and Ensoli, B. (1999). Reactivation and persistence of human herpesvirus-8 infection in B cells and monocytes by Th-1 cytokines increased in Kaposi’s sarcoma. Blood 93, 4044–4058.
Moore, P. S., Boshoff, C., Weiss, R. A., and Chang, Y. (1996). Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 274, 1739–1744.
Munoz-Fontela, C., Marcos-Villar, L., Gallego, P., Arroyo, J., Da Costa, M., Pomeranz, K. M., Lam, E. W., and Rivas, C. (2007). Latent protein LANA2 from Kaposi’s sarcoma-associated herpesvirus interacts with 14-3-3 proteins and inhibits FOXO3a transcription factor. J. Virol. 81, 1511–1516.
Nakamura, H., Li, M., Zarycki, J., and Jung, J. U. (2001). Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J. Virol. 75, 7572–7582.
Neipel, F., Albrecht, J. C., Ensser, A., Huang, Y. Q., Li, J. J., Friedman-Kien, A. E., and Fleckenstein, B. (1997a). Human herpesvirus 8 encodes a homolog of interleukin-6. J. Virol. 71, 839–842.
Neipel, F., Albrecht, J. C., and Fleckenstein, B. (1997b). Cell-homologous genes in the Kaposi’s sarcoma-associated rhadinovirus human herpesvirus 8: determinants of its pathogenicity? J. Virol. 71, 4187–4192.
Nozawa, H., Oda, E., Nakao, K., Ishihara, M., Ueda, S., Yokochi, T., Ogasawara, K., Nakatsuru, Y., Shimizu, S., Ohira, Y., Hioki, K., Aizawa, S., Ishikawa, T., Katsuki, M., Muto, T., Taniguchi, T., and Tanaka, N. (1999). Loss of transcription factor IRF-1 affects tumor susceptibility in mice carrying the Ha-ras transgene or nullizygosity for p53. Genes Dev. 13, 1240–1245.
Pestka, S., Krause, C. D., and Walter, M. R. (2004). Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202, 8–32.
Pindel, A., and Sadler, A. (2011). The role of protein kinase R in the interferon response. J. Interferon Cytokine Res. 31, 59–70.
Pozharskaya, V. P., Weakland, L. L., Zimring, J. C., Krug, L. T., Unger, E. R., Neisch, A., Joshi, H., Inoue, N., and Offermann, M. K. (2004). Short duration of elevated vIRF-1 expression during lytic replication of human herpesvirus 8 limits its ability to block antiviral responses induced by alpha interferon in BCBL-1 cells. J. Virol. 78, 6621–6635.
Reis, L. F., Ruffner, H., Stark, G., Aguet, M., and Weissmann, C. (1994). Mice devoid of interferon regulatory factor 1 (IRF-1) show normal expression of type I interferon genes. EMBO J. 13, 4798–4806.
Reith, W., Leibundgut-Landmann, S., and Waldburger, J. M. (2005). Regulation of MHC class II gene expression by the class II transactivator. Nat. Rev. Immunol. 5, 793–806.
Renne, R., Zhong, W., Herndier, B., Mcgrath, M., Abbey, N., Kedes, D., and Ganem, D. (1996). Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2, 342–346.
Rivas, C., Thlick, A. E., Parravicini, C., Moore, P. S., and Chang, Y. (2001). Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 75, 429–438.
Roan, F., Zimring, J. C., Goodbourn, S., and Offermann, M. K. (1999). Transcriptional activation by the human herpesvirus-8-encoded interferon regulatory factor. J. Gen. Virol. 80(Pt 8), 2205–2209.
Russo, J. J., Bohenzky, R. A., Chien, M. C., Chen, J., Yan, M., Maddalena, D., Parry, J. P., Peruzzi, D., Edelman, I. S., Chang, Y., and Moore, P. S. (1996). Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U.S.A. 93, 14862–14867.
Sarid, R., Flore, O., Bohenzky, R. A., Chang, Y., and Moore, P. S. (1998). Transcription mapping of the Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 72, 1005–1012.
Satou, A., Taira, T., Iguchi-Ariga, S. M., and Ariga, H. (2001). A novel transrepression pathway of c-Myc. Recruitment of a transcriptional corepressor complex to c-Myc by MM-1, a c-Myc-binding protein. J. Biol. Chem. 276, 46562–46567.
Schmidt, K., Wies, E., and Neipel, F. (2011). Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 3 inhibits IFNΓ and MHC II expression. J. Virol. 85, 4530–4537.
Searles, R. P., Bergquam, E. P., Axthelm, M. K., and Wong, S. W. (1999). Sequence and genomic analysis of a Rhesus macaque rhadinovirus with similarity to Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 73, 3040–3053.
Seo, T., Lee, D., Shim, Y. S., Angell, J. E., Chidambaram, N. V., Kalvakolanu, D. V., and Choe, J. (2002). Viral interferon regulatory factor 1 of Kaposi’s sarcoma-associated herpesvirus interacts with a cell death regulator, GRIM19, and inhibits interferon/retinoic acid-induced cell death. J. Virol. 76, 8797–8807.
Seo, T., Park, J., and Choe, J. (2005). Kaposi’s sarcoma-associated herpesvirus viral IFN regulatory factor 1 inhibits transforming growth factor-beta signaling. Cancer Res. 65, 1738–1747.
Seo, T., Park, J., Lee, D., Hwang, S. G., and Choe, J. (2001). Viral interferon regulatory factor 1 of Kaposi’s sarcoma-associated herpesvirus binds to p53 and represses p53-dependent transcription and apoptosis. J. Virol. 75, 6193–6198.
Seo, T., Park, J., Lim, C., and Choe, J. (2004). Inhibition of nuclear factor kappaB activity by viral interferon regulatory factor 3 of Kaposi’s sarcoma-associated herpesvirus. Oncogene 23, 6146–6155.
Shin, Y. C., Joo, C. H., Gack, M. U., Lee, H. R., and Jung, J. U. (2008). Kaposi’s sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1 alpha to induce vascular endothelial growth factor expression. Cancer Res. 68, 1751–1759.
Shin, Y. C., Nakamura, H., Liang, X., Feng, P., Chang, H., Kowalik, T. F., and Jung, J. U. (2006). Inhibition of the ATM/p53 signal transduction pathway by Kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J. Virol. 80, 2257–2266.
Sirianni, M. C., Vincenzi, L., Fiorelli, V., Topino, S., Scala, E., Uccini, S., Angeloni, A., Faggioni, A., Cerimele, D., Cottoni, F., Aiuti, F., and Ensoli, B. (1998). Gamma-Interferon production in peripheral blood mononuclear cells and tumor infiltrating lymphocytes from Kaposi’s sarcoma patients: correlation with the presence of human herpesvirus-8 in peripheral blood mononuclear cells and lesional macrophages. Blood 91, 968–976.
Takaoka, A., Tamura, T., and Taniguchi, T. (2008). Interferon regulatory factor family of transcription factors and regulation of oncogenesis. Cancer Sci. 99, 467–478.
Tamura, T., Yanai, H., Savitsky, D., and Taniguchi, T. (2008). The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 26, 535–584.
Taniguchi, T., Ogasawara, K., Takaoka, A., and Tanaka, N. (2001). IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 19, 623–655.
Verma, S. C., Lan, K., and Robertson, E. (2007). Structure and function of latency-associated nuclear antigen. Curr. Top. Microbiol. Immunol. 312, 101–136.
Wang, X. P., and Gao, S. J. (2003). Auto-activation of the transforming viral interferon regulatory factor encoded by Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8). J. Gen. Virol. 84, 329–336.
Wang, X. P., Zhang, Y. J., Deng, J. H., Pan, H. Y., Zhou, F. C., Montalvo, E. A., and Gao, S. J. (2001). Characterization of the promoter region of the viral interferon regulatory factor encoded by Kaposi’s sarcoma-associated herpesvirus. Oncogene 20, 523–530.
Wathelet, M. G., Lin, C. H., Parekh, B. S., Ronco, L. V., Howley, P. M., and Maniatis, T. (1998). Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol. Cell 1, 507–518.
Wen, K. W., and Damania, B. (2010). Kaposi sarcoma-associated herpesvirus (KSHV): molecular biology and oncogenesis. Cancer Lett. 289, 140–150.
West, J. A., and Damania, B. (2010). Kaposi’s sarcoma-associated herpesvirus and innate immunity. Future Virol. 5, 185–196.
Wies, E., Hahn, A. S., Schmidt, K., Viebahn, C., Rohland, N., Lux, A., Schellhorn, T., Holzer, A., Jung, J. U., and Neipel, F. (2009). The Kaposi’s Sarcoma-associated Herpesvirus-encoded vIRF-3 Inhibits Cellular IRF-5. J. Biol. Chem. 284, 8525–8538.
Wies, E., Mori, Y., Hahn, A., Kremmer, E., Sturzl, M., Fleckenstein, B., and Neipel, F. (2008). The viral interferon-regulatory factor-3 is required for the survival of KSHV-infected primary effusion lymphoma cells. Blood 111, 320–327.
Wu, W., Vieira, J., Fiore, N., Banerjee, P., Sieburg, M., Rochford, R., Harrington, W. Jr., and Feuer, G. (2006). KSHV/HHV-8 infection of human hematopoietic progenitor (CD34+) cells: persistence of infection during hematopoiesis in vitro and in vivo. Blood 108, 141–151.
Zhang, Y. J., Wang, X. P., Deng, J. H., Salinas, R. A., Oishi, N., and Gao, S. J. (2001). Suppression of oncogenic viral interferon regulatory factor (vIRF) of Kaposi’s sarcoma-associated herpesvirus by ribozyme-mediated cleavage. Cancer Gene Ther. 8, 285–293.
Keywords: KSHV, vIRF, HHV-8
Citation: Jacobs SR and Damania B (2011) The viral interferon regulatory factors of KSHV: immunosuppressors or oncogenes?. Front. Immun. 2:19. doi: 10.3389/fimmu.2011.00019
Received: 13 March 2011;
Paper pending published: 11 April 2011;
Accepted: 24 May 2011;
Published online: 17 June 2011.
Edited by:
Murali Krishna Kaja, University of Washington, USAReviewed by:
Fanxiu Zhu, Florida State University, USAMichael Lagunoff, University of Washington, USA
Copyright: © 2011 Jacobs and Damania. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Blossom Damania, Lineberger Cancer Center, University of North Carolina at Chapel Hill, 450 West Drive, Campus Box 7295, Chapel Hill, NC 27599, USA. e-mail: damania@med.unc.edu