 Jonathan S. Wall1,2* Stephen J. Kennel1,2 Tina Richey1 Amy Allen1 Alan Stuckey2 Deborah T. Weiss1 Sallie D. Macy1 Robin Barbour3 Peter Seubert3 Alan Solomon1 Dale Schenk3
Jonathan S. Wall1,2* Stephen J. Kennel1,2 Tina Richey1 Amy Allen1 Alan Stuckey2 Deborah T. Weiss1 Sallie D. Macy1 Robin Barbour3 Peter Seubert3 Alan Solomon1 Dale Schenk3
- 1 Human Immunology and Cancer Program, Department of Medicine, University of Tennessee Graduate School of Medicine, Knoxville, TN, USA
- 2 Department of Radiology, University of Tennessee Graduate School of Medicine, Knoxville, TN, USA
- 3 Neotope Biosciences, Elan Pharmaceuticals, South San Francisco, CA, USA
AA amyloidosis results from the pathologic deposition in the kidneys and other organs of fibrils composed of N-terminal fragments of serum amyloid A protein (SAA). Given that there are only limited means to visualize these deposits, we have developed a series of mAbs, 2A4, 7D8, and 8G9, that bind specifically with nanomolar affinity to a carboxy-terminal epitope generated following proteolysis of SAA that yields the predominant component of AA amyloid deposits. Notably, these antibodies do not recognize native SAA, they retain their immunoreactivity when radiolabeled with I-125 and, after injection into AA amyloidotic mice, localize, as evidenced by autoradiography and micro-single photon emission computed tomography imaging, to histologically confirmed areas of amyloid deposition; namely, spleen, liver, and pancreas. The results of our in vitro and in vivo studies demonstrate the AA fibril-selectivity of mAbs 2A4, 7D8, and 8G9 and warrant further investigation into their role as novel diagnostic agents for patients with AA amyloidosis.
Introduction
AA amyloidosis is characterized by the pathological deposition in various body tissues, particularly the kidneys, spleen, and liver (Gillmore et al., 2001), of fibrils formed from N-terminal fragments of serum amyloid A (SAA) protein. This disorder occurs in man and various mammalian and avian species, typically as a consequence of chronic inflammation, e.g., rheumatoid arthritis, tuberculosis, Familial Mediterranean fever, etc., that results in a sustained elevation of the amyloidogenic SAA precursor. The relentless accumulation of fibrillar proteins in affected tissues leads to progressive organ failure and eventually death. Experimentally, this disease can be induced in susceptible strains of mice that are given silver nitrate, casein, or lipopolysaccharide, and also occurs spontaneously in animals carrying the human IL-6 transgene (Solomon et al., 1999; Wall et al., 2005). Amyloid deposition in these in vivo models is markedly accelerated by concomitant intravenous (i.v.) injection or oral administration of miniscule amounts of AA fibrils that serve as a “seed,” i.e., an amyloid enhancing factor (AEF), to promote fibril formation (Kisilevsky et al., 1984; Varga et al., 1986; Senthilkumar et al., 2008; Westermark and Westermark, 2009).
From a clinical standpoint, there is a need for an objective means to document the extent of AA amyloid deposition or its resolution in order to ascertain a patient’s response to treatment and/or if relapse has occurred. In this regard, routine radiographic techniques (CT, MRI, and ultrasound) are not particularly informative or “amyloid specific”; furthermore, the deposits are rarely visualized using available nuclear medicine agents. Although European investigators have imaged AA amyloid by planar gamma scintigraphy and single photon emission computed tomography (SPECT) using 123I-labeled serum amyloid P-component (Hawkins et al., 1988, 1998; Hazenberg et al., 2006), this compound is not clinically available in the United States and this technique does not provide quantitative data.
Another strategy involves use of specific radiolabeled fibril-reactive antibodies as imaging agents. A precedent for this approach has been established utilizing mAb 11-1F4, which recognizes an amyloid fibril-dependent, conformational epitope on immunoglobulin light chains, but is non-reactive with the natively folded molecules. This mAb, when radiolabeled with the positron-emitting isotope I-124, has been shown by PET/CT to image AL amyloid, first in an animal model (Wall et al., 2006a), and then in patients with AL amyloidosis enrolled in a Phase I clinical trial (Wall et al., 2010).
Given the necessity to monitor the presence and biodistribution of AA amyloid in the major target organs of patients with AA amyloidosis, we have developed a series of mAbs, designated 2A4, 7D8, and 8G9, that bind specifically to AA fibrils, but not the normal circulating precursor protein, and have defined the structural basis for their specificity. Notably, these reagents, when radiolabeled with I-125, visualized fibrillar deposits in a transgenic murine model of AA amyloidosis (Solomon et al., 1999), as shown by micro-SPECT imaging. Based on the results of our studies, we posit that these antibodies could be utilized clinically as imaging agents for individuals with AA amyloidosis.
Materials and Methods
Antibodies
Preparation of the immunogen
A peptide containing amino acids 71–75 of murine SAA, with two artificial N-terminal amino acids added for ease of coupling (CGGHEDT), was synthesized by AnaSpec Inc. The peptide was conjugated to sheep anti-mouse IgG using N-[e-Maleimidocaproyloxy]succinimide ester (EMCS) to crosslink the cysteine to the tertiary amines on lysyl residues in the antibody.
Immunization and fusion
AJ mice (Jackson Labs) were immunized with 50 μg of the peptide conjugate in complete Freund’s adjuvant, followed by boosts 14 and 28 days later with another 50 μg in incomplete Freund’s adjuvant. Animals were bled on day 35, where the anti-peptide titer, as determined by ELISA, was >1/10000. Splenocytes from one such mouse were fused to SP2/0 cells and the resulting hybridomas screened by ELISA for reactivity to the CGGHEDT component, as well as for lack of reactivity to a peptide that included an additional five downstream residues of the murine SAA sequence (GHEDTMADQE). Three hybridomas, 2A4, 7D8, and 8G9, were identified, stabilized, and cloned by three rounds of limiting dilution. Antibodies were generated in ascites and following delipidation were isolated from a 50% (w/v) ammonium sulfate precipitate by centrifugation. The antibody precipitate was dialyzed against PBS and then purified on a Protein G column.
Biotinylation of purified antibodies
Purified antibodies were diluted to 1 mg/ml in PBS and biotinylated using EZ-Link NHS-PEG4-Biotin, No Weigh Format (ThermoFisher Scientific), according to the manufacturer’s specifications, except that the ratio of biotin to antibody was reduced to a 10-fold molar excess.
Murine and Human AA Amyloid
Murine and human AA amyloid extracts were isolated from amyloidotic tissues by a serial flotation procedure, as described previously (Pras et al., 1968). Human AA was kindly provided by Dr. Per Westermark (University of Uppsala, Sweden). All procedures using human-derived materials were approved by the University of Tennessee Institutional Review Board. AA amyloidosis was induced in 8-week-old transgenic H2-Ld-huIL-6 Tg Balb/c (Wall et al., 2008) mice by injection into the lateral tail vein of 100 μl PBS containing 100 μg of a spleen-derived AA extract.
All animal experiments were performed in accordance with Institutional Animal Care and Use Committee-approved protocols.
Immunohistochemistry
Six micrometer-thick sections, cut from formalin-fixed, paraffin-embedded tissues, were subjected to antigen retrieval using the High pH Target Retrieval™ system (Dako). After a 30-min incubation at 90°C, followed by 20 min at room temperature, the tissues were immunostained overnight at 4°C with a 3-μg/ml solution of biotinylated anti-AA amyloid-specific mAb in 0.05% Tween/PBS. The reaction was developed using the Vectastain ABC kit (Vector Labs). The presence of amyloid was confirmed by staining with a 0.1% alkaline Congo red solution, where the characteristic green birefringence was seen upon examination using a Leica DM500 light microscope fitted with cross-polarizing filters. Digital microscopic images were acquired with a cooled CCD camera (SPOT, Diagnostic Instruments).
ELISA
Wells of NUNC Maxisorb plates (eBioscience) were coated with extracts of murine AA amyloid fibrils (8 μg/ml) or the two synthetic peptides (3 μg/ml) and incubated overnight at 37°C. The dried wells were washed ×1 with assay buffer (PBS containing 0.05% Tween 20) and blocked with 200 μl of 1% BSA in PBS at 37°C for 1 h, followed by addition of the anti-AA amyloid-specific and control mAbs titrated in triplicate over the range of 1 M × 10−11 M–1 M × 10−6 M (to determine background values, wells were filled only with antibody). After a 1-h incubation at 37°C, the wells were washed ×2, filled with 100 μl of a 1:5000 dilution of goat anti-mouse HRP conjugate and, following another 1-h incubation at 37°C and washes, 100 μl of ABTS in 0.06% H2O2 were added. The absorbance at 405 nm was measured 30 min later with a Wallac Victor3 plate reader (PerkinElmer) and the data analyzed using SigmaPlot (SPSS Inc.).
Competition ELISA
Quadruplicate rows of NUNC Maxisorb wells were coated with 50 μl of murine or human renal or splenic AA fibrils suspended in PBS (8 μg/ml), the plates dried overnight at 37°C, and the wells blocked for 1 h at 37°C with 1% BSA/PBS (200 μl/well). The AA samples used for competition were suspended in assay buffer containing 1% BSA to make a 2× stock solution (800 μg/ml) and then serially diluted to a final concentration of 1 × 10−11 M. Anti-AA amyloid-specific and control mAbs were diluted with assay buffer to form a 2× stock solution and added to the human and murine AA fibril preparations to yield a final concentration of 50 nM and a starting 400 μg titer of competitor. Next, the competitor-mAb solutions were transferred to the AA fibril-coated plates and left for 1 h at 37°C [to establish maximum binding, one row of wells contained mAb alone (no competitor) and non-specific (background) binding was measured in those with antibody, but no fibrils]. After two washes, 100 μl of a 1:5000 dilution of goat anti-mouse HRP-conjugated secondary antibody was added and the plates incubated for 1 h at 37°C, washed, and the reaction developed by addition of 100 μl of ABTS in 0.06% H2O2. After 30 min, the absorbance was measured at 405 nm with the microplate reader. Data were analyzed using Excel and SigmaPlot, with each point representing the mean of triplicate values corrected for non-specific binding.
Radiolabeling
Forty nanomolar of anti-AA and control mAbs were labeled with 2 mCi of reductant-free 125I (Perkin Elmer), using limiting amounts of chloramine T (Wall et al., 2006b). The reaction was quenched with a two-fold molar excess of sodium metabisulfite and the labeled reagents were then suspended in PBS containing 5 mg/ml BSA; unbound isotope and protein aggregates were removed by size-exclusion liquid chromatography via an Ultrogel AcA34 column (Amersham Pharmacia). Fractions containing the IgG monomer were pooled for imaging experiments. Radiolabeling efficiency was qualitatively assessed by Cyclone phosphor imaging (Packard Instruments) of 125I-labeled mAb in 10% SDS-PAGE gels, under native and reducing conditions.
Micro-SPECT/CT
The biodistribution of the radiolabeled mAbs was determined in groups composed of three control or amyloidotic mice. Each animal was injected with 6 μg of 125I-labeled mAb (∼150 μCi) in the lateral tail vein and, after 48 or 72 h, given a 200-μl i.v. dose of Fenestra VC™ (Advanced Research Technologies). Thirty minutes later, the mice were euthanized by isoflurane overdose and SPECT/CT images acquired.
Single photon emission computed tomography data were collected with a microCAT II + SPECT dual modality imaging apparatus equipped with a 1-mm-pore diameter pinhole collimator (Siemens Preclinical Solutions). For imaging, the two detectors (composed of a 50-mm-diameter Hamamatsu R2486-02 multi-anode photo-multiplier tube coupled to a 1-mm × 1-mm × 8-mm CsI (Tl) crystal array arranged on a 1.2-mm2 grid) were positioned ∼45 mm from the center of rotation. Each SPECT dataset comprised 45 projections collected over 360° during the course of ∼50 min. Images were reconstructed using an implementation of the expectation maximization–maximum likelihood (EM–ML) algorithm.
After acquisition of SPECT data, high-resolution CT images were obtained with the microCAT II scanner that had a circular orbit cone beam geometry and was equipped with a 20- to 80-kVp microfocus X-ray source that captured a 90-mm × 60-mm field of view using a 2048 × 3072 CCD array detector. Each CT dataset, composed of 360 projections at 1° azimuths, was procured over 8 min. Images were reconstructed in real-time on isotropic 77-μm voxels using an implementation of the Feldkamp backprojection algorithm. To facilitate co-registration of the reconstructed SPECT and CT images, Co-57-sealed sources were placed on the imaging bed and datasets were visualized and co-registered manually with a 3-D image analysis software package (Amira, Version 3.1: Mercury Computer Systems).
Biodistribution
Tissue samples harvested from AA amyloid-bearing and control mice injected with the 125I-labeled mAbs were placed into tared vials, weighed, and the radioactivity measured. The primary index values were expressed as % injected dose/g tissue (% ID/g).
Autoradiography
Six micrometer-thick sections cut from formalin-fixed, paraffin-embedded blocks of tissue obtained from mice sacrificed 48 h post-injection of 125I-labeled mAb were placed on Probond microscopic slides (ThermoFisher), dipped in NTB-2 emulsion (Eastman Kodak), stored in the dark, and developed after a 96-h exposure. The sections were counterstained with hematoxylin and eosin, cover-slipped with Permount (ThermoFisher), and examined by light microscopy. In addition, consecutively cut sections were stained with Congo red and viewed under cross-polarized illumination or immunostained using the AA-reactive mAbs as primary reagents. Digital photographs were taken and evaluated with the Image Pro Plus software package (MediaCybernetics).
Results
The primary structures of mouse, human, and three other mammalian AA proteins are provided in Figure 1. To generate mAbs specific for the AA amyloid fibril, a 7-amino acid peptide containing the 5 carboxy-terminal residues (GHEDT) of the cleaved murine SAA that forms AA amyloid fibrils was used as the immunogen and the resultant mAbs were screened for reactivity against this component. Three monoclonal antibodies, designated 2A4, 7D8, and 8G9 (all of the IgG2 isotype), exhibited saturable binding over a molarity of 10−7–10−11 M, with EC50 values of 3.4, 1.84, and 4.0 nM, respectively (Figure 2A). In contrast, there was no affinity observed, even at 10−7 M, when the mAbs were titrated over the same concentration range using, as substrate, the longer peptide, GHEDTMADQE, that contained an additional five residues not present in the AA fibrils. To determine if the interaction of the mAbs with AA amyloid was similar to that of the immunizing peptide, mouse and human AA amyloid extracts were adsorbed onto microplate wells and antibody reactivity measured by ELISA (Figures 2B,C). In both cases, the three reagents bound with saturation values of <10−7 M; however, there was greater affinity for mouse AA, with EC50s comparable to that for the GHEDT peptide (Table 1). Similar binding signal amplitudes were observed for mouse AA and the GHEDTMADQE molecule, indicating an equivalent epitope density. For human AA amyloid, the EC50s were ∼10–30 nM. Notably, irrespective of the substrate (GHEDT peptide, mouse or human AA), the binding of the three mAbs varied, i.e., 7D8 > 2A4 > 8G9, with 7D8 having a three-fold lower EC50 than 8G9.

Figure 1. Amino acid sequences of AA amyloid from five mammalian species. The five-residue murine C-terminal AA peptide, GHEDT, spanning residues 71–75 and used as immunogen to generate mAbs 2A4, 7D8, and 8G9 is underlined (the cheetah, cat, and dog proteins contain an 8-amino acid insertion between positions 65 and 66).
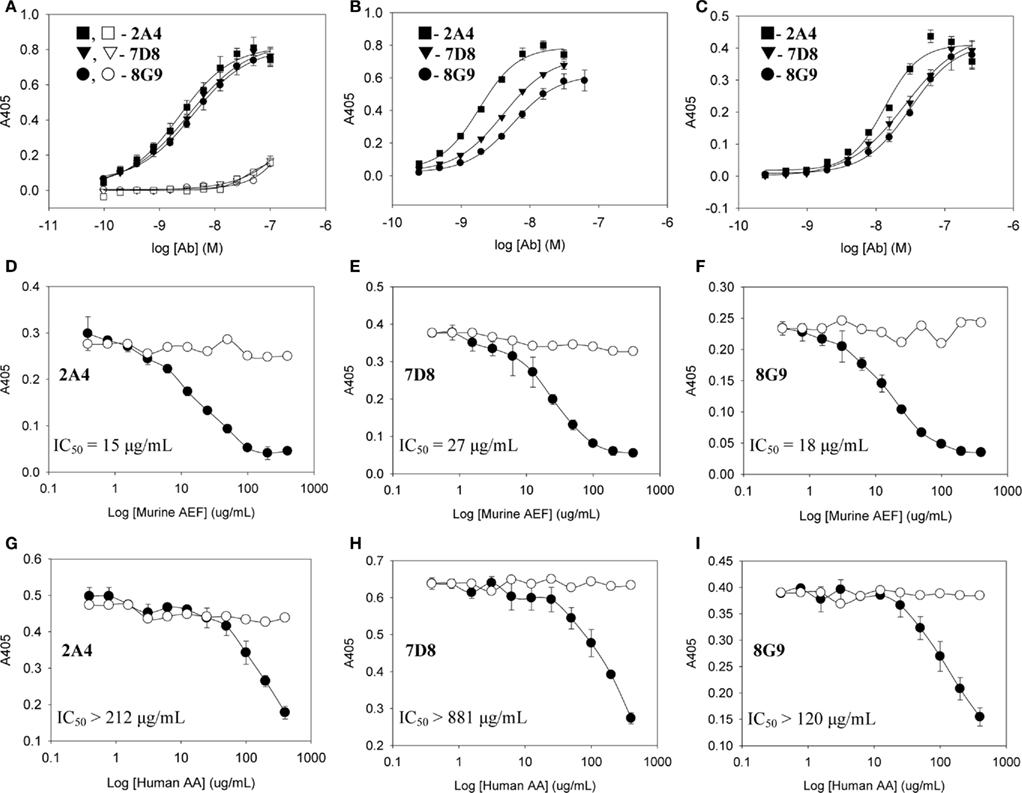
Figure 2. Interactions of mAbs 2A4, 7D8, and 8G9 with murine and human AA amyloid and synthetic AA-related peptides. (A) Binding of 2A4, 7D8, and 8G9 using GHEDT (filled) or, as a control for full length SAA, the GHEDTMADQE peptide (open), as substrate in the ELISA. (B,C) Reactivity of the three mAbs with murine or human AA amyloid extracts, respectively. (D–F) Competitive inhibition of binding of mAbs 2A4, 7D8, and 8G9 to human and (G–I) murine AA amyloid in the absence (○) or presence (●) of increasing concentrations of the heterologous AA extract.
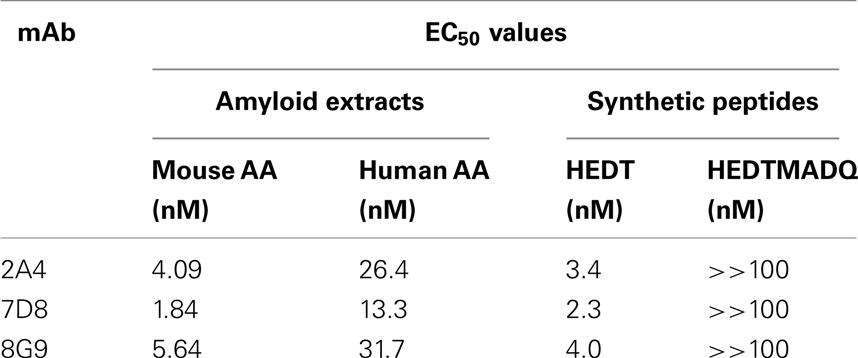
Table 1. Binding of mAbs to AA amyloid extracts and synthetic AA peptides.
To ensure that the interaction of the mAbs with AA fibrils did not result from an “artificial” neo-epitope formed as a consequence of adsorbing the material onto the microplate well, competitive binding studies were performed using soluble mouse and human AA extracts (Figures 2D–I). In these studies, although both were capable of inhibiting mAb binding to the heterologous AA amyloid on the microplate, the murine AA was more effective, as evidenced by the finding that the IC50 for 7D8 binding to human AA, when mouse AA was used as a competitor, was 27 versus 15 and 18 μg/ml for mAbs 2A4 and 8G9, respectively. Similarly, as compared to 2A4 and 8G9, 7D8 required four- and seven-fold more soluble human AA extract to achieve 50% inhibition of binding. These data were consistent with the stronger affinity of mAb 7D8 to the GHEDT peptide, as well as to murine and human AA amyloid.
Although the mAbs were generated using the murine SAA GHEDT peptide as the immunogen, all showed high avidity for mouse and human AA amyloid with slightly higher affinity for the murine (HEDT) versus human (AEDS) sequences. To better understand the nature of the antibody binding sites, we performed studies using alanine (Ala)-substituted peptides as substrates in the ELISA (Figure 3). mAb 2A4 bound AEDT and HEDA peptides equally well, with an EC50 equivalent to that of the unmodified HEDT molecule. In contrast, substitution of Ala for Glu in the HADT sequence resulted in a shift in the EC50 from ∼3 to 100 nM (Figure 3). Furthermore, reactivity was completely abrogated by replacement of Ala for the Asp residue (HEAT). Disruption of the binding to both HADT and HEAT peptides also occurred using mAbs 7D8 and 8G9. Although the presence of glutamyl and aspartyl residues (E73 and D74) was required for optimal antibody binding, the presence of D74 was absolutely necessary to maintain a high-affinity interaction.
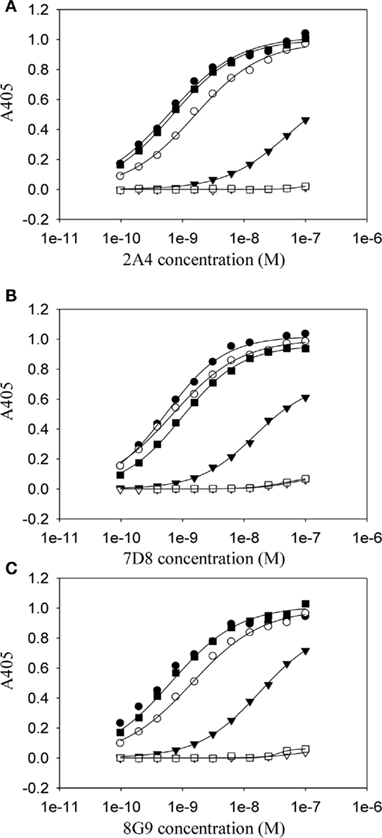
Figure 3. Dependency on residues E73 and D74 for the high-affinity binding of anti-AA fibril-specific mAbs. Measurement by ELISA of the reactivity of mAbs 2A4 (A) 7D8 (B) and 8G9 (C) at different concentrations against microplate well adsorbed peptides (●, HEDT; ○, AEDT; ▼, HADT; ▽, HEAT; ■, HEDA; □, HEDTMADQE).
The ex vivo reactivity of mAbs 2A4, 7D8, and 8G9 with amyloid was demonstrated immunohistochemically, as seen when the congophilic birefringent deposits present in the liver, spleen, kidney, and pancreas of AA mice, as well as human AA amyloid-containing tissues, were immunostained with these reagents. In all instances, the 2A4 antibody exhibited the most intense binding that, notably, was abolished by absorption with the GHEDT-containing peptide used for immunization (Figure 4).
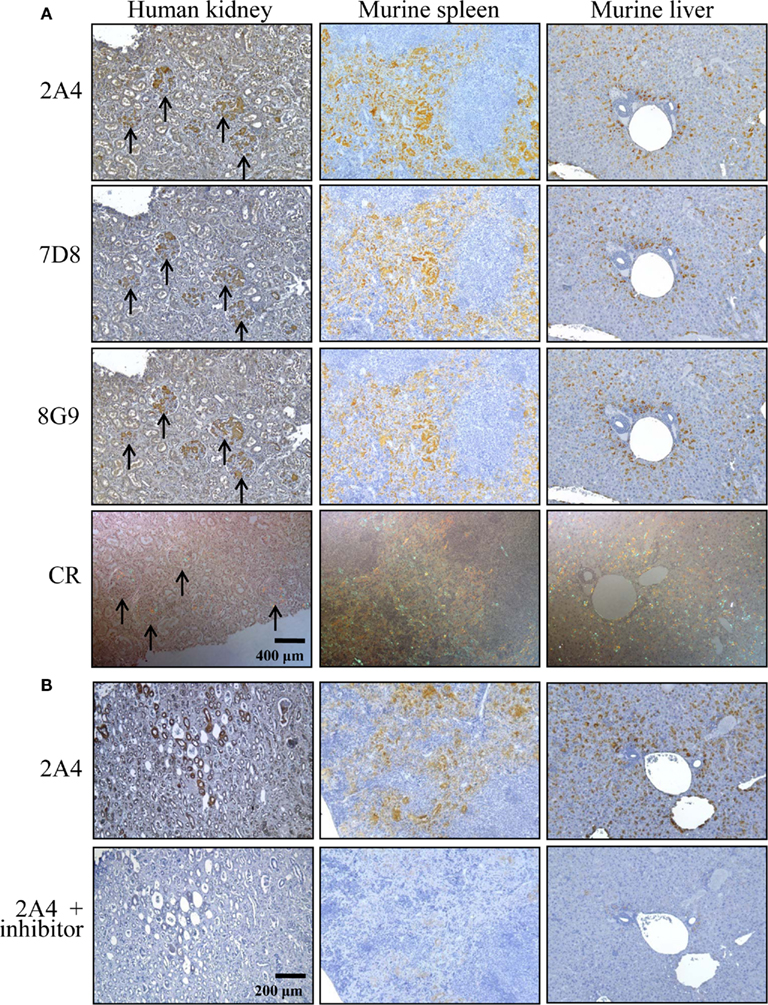
Figure 4. Immunostaining of human and murine AA deposits by mAbs 2A4, 7D8, and 8G9. (A) Interaction of the antibodies with AA amyloid contained in sections of H2-Ld-huIL-6 mouse-derived spleen and liver, as well as human kidney, compared favorably with the distribution of amyloid evidenced by Congo red staining. (B) Binding of mAb 2A4 absorbed with (upper) the GHEDTMADQE peptide or (lower) the GHEDT immunogen. Original magnifications, ×40.
The capability of mAbs 2A4, 7D8, and 8G9 to bind AA amyloid in vivo also was assessed using radioiodinated derivatives of the three reagents which, when labeled with I-125, had a radiochemical yield of ∼50% and a specific activity of ∼25 μCi/μg. H2-Ld-huIL-6 mice with extensive systemic AA amyloidosis were given 200 μl tail vein injections of a solution containing ∼10 μg of radiolabeled antibody (∼250 μCi) in PBS and imaged 48 h later. As seen by high-resolution SPECT, there was marked uptake of the radiotracers in the liver, spleen (Figure 5) and, to a lesser extent, other amyloid-containing areas, e.g., kidneys and intestine. In contrast, there was no visualization of the amyloid in transgenic mice that received the control radioiodinated antibody (since the animals were not pre-treated with Lugol’s solution, there was thyroidal uptake of free radioiodide liberated during antibody catabolism). Further, the radiolabeled mAbs did not bind to amyloid-free organs or tissues from healthy mice (Figure 6).
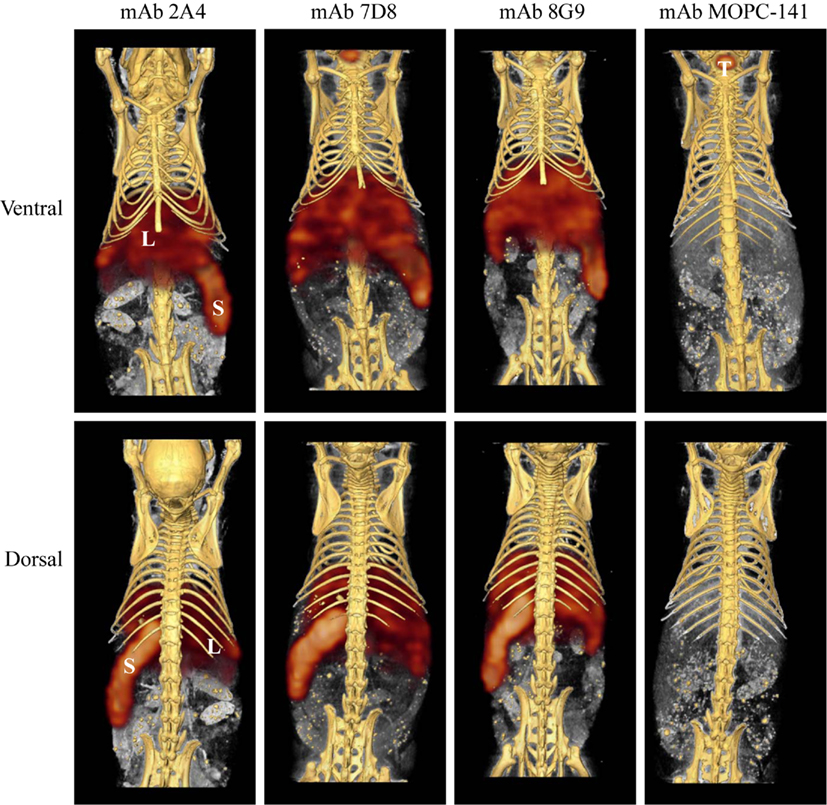
Figure 5. Radioimmunoimaging of AA amyloid. SPECT/CT images obtained 48 h after AA amyloidotic mice were injected with ∼250 μCi of 125I-labeled mAbs 2A4, 7D8, 8G9, or the isotype-matched control antibody, MOPC 141. Accumulation of the radioiodinated antibodies in amyloid-laden liver (L) and spleen (S) is indicated, as is uptake of free iodide in the thyroid (T) of a mouse given the control mAb.
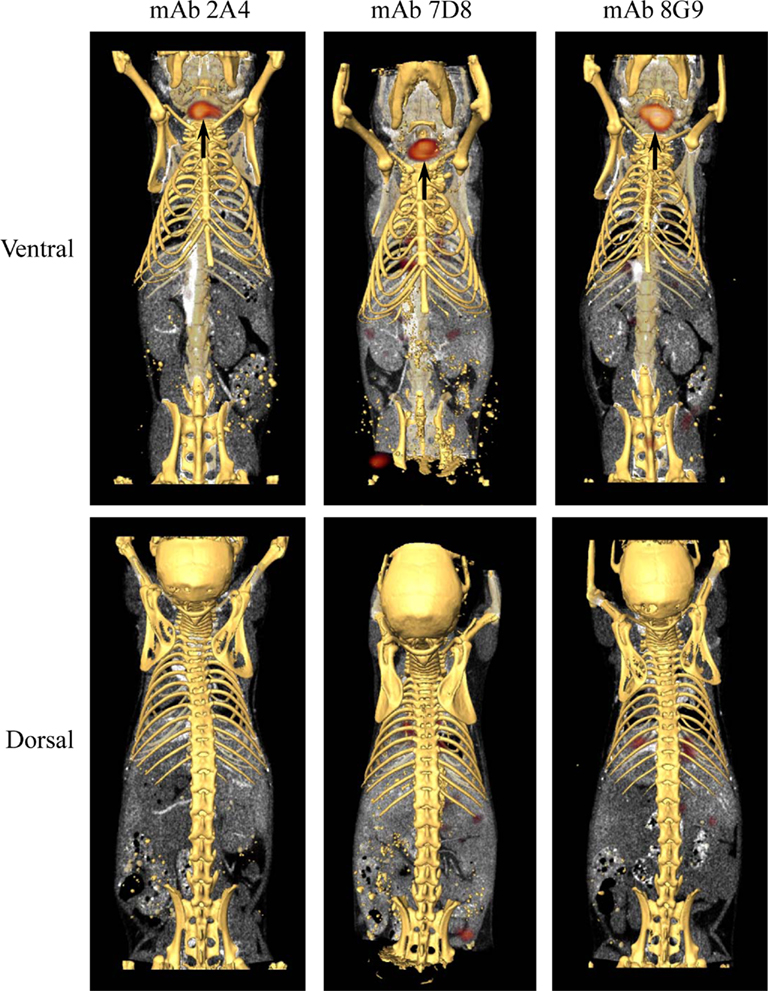
Figure 6. Biodistribution of mAbs in healthy, amyloid-free mice. SPECT/CT images obtained 48 h after animals were injected with ∼250 μCi of 125I-labeled mAbs 2A4, 7D8, and 8G9. No accumulation of the radiotracers was detected in healthy organs. Arrows indicate the presence in the thyroid of free iodide liberated during catabolism.
The specific localization of the 125I-labeled mAbs within amyloid was evidenced autoradiographically, where dense black deposits were seen around hepatic vasculature and sinusoids, splenic follicles, and renal papilla (Figure 7), as well as other anatomic sites deemed to contain this pathologic material based on Congo red staining. The control radioiodinated mAb did not label the amyloid deposits and was evident only within renal tubules. These results were confirmed by biodistribution measurements that revealed the liver and spleen of animals injected with 125I-labeled mAbs 2A4, 7D8, and 8G9 to contain ∼25 and ∼45% ID/g of mAb, respectively, as compared to <1% ID/g with the control (Table 2). The ratio of tissue to muscle uptake in these two organs also was significantly greater in mice that received the three mAbs (∼100:1 and 200:1, respectively; Table 3). In the case of the pancreas and small intestine, the ratios were two- to three-fold higher, as well.
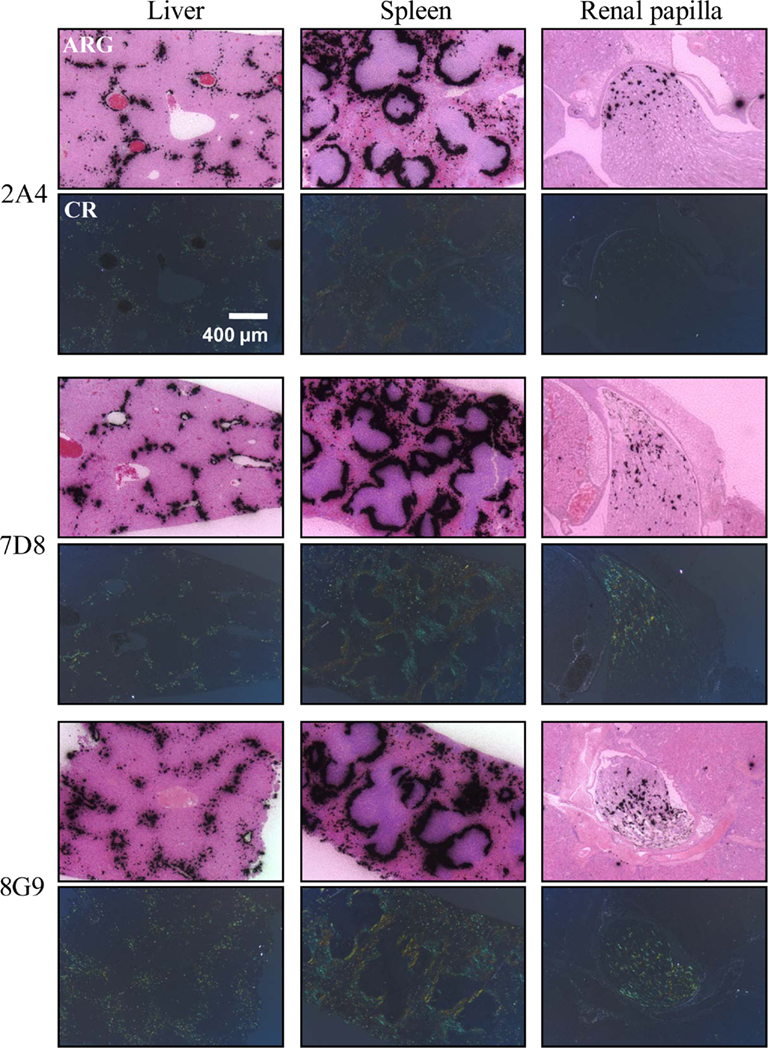
Figure 7. Specific in vivo binding of radiolabeled AA fibril-specific mAbs to AA amyloid. Comparison of Congo red (CR)-stained tissue sections with autoradiographs (ARG) of murine AA deposits in liver, spleen, and renal papilla from H2-Ld-huIL-6 mice given i.v. injections of 125I-labeled mAbs 2A4, 7D8, or 8G9.
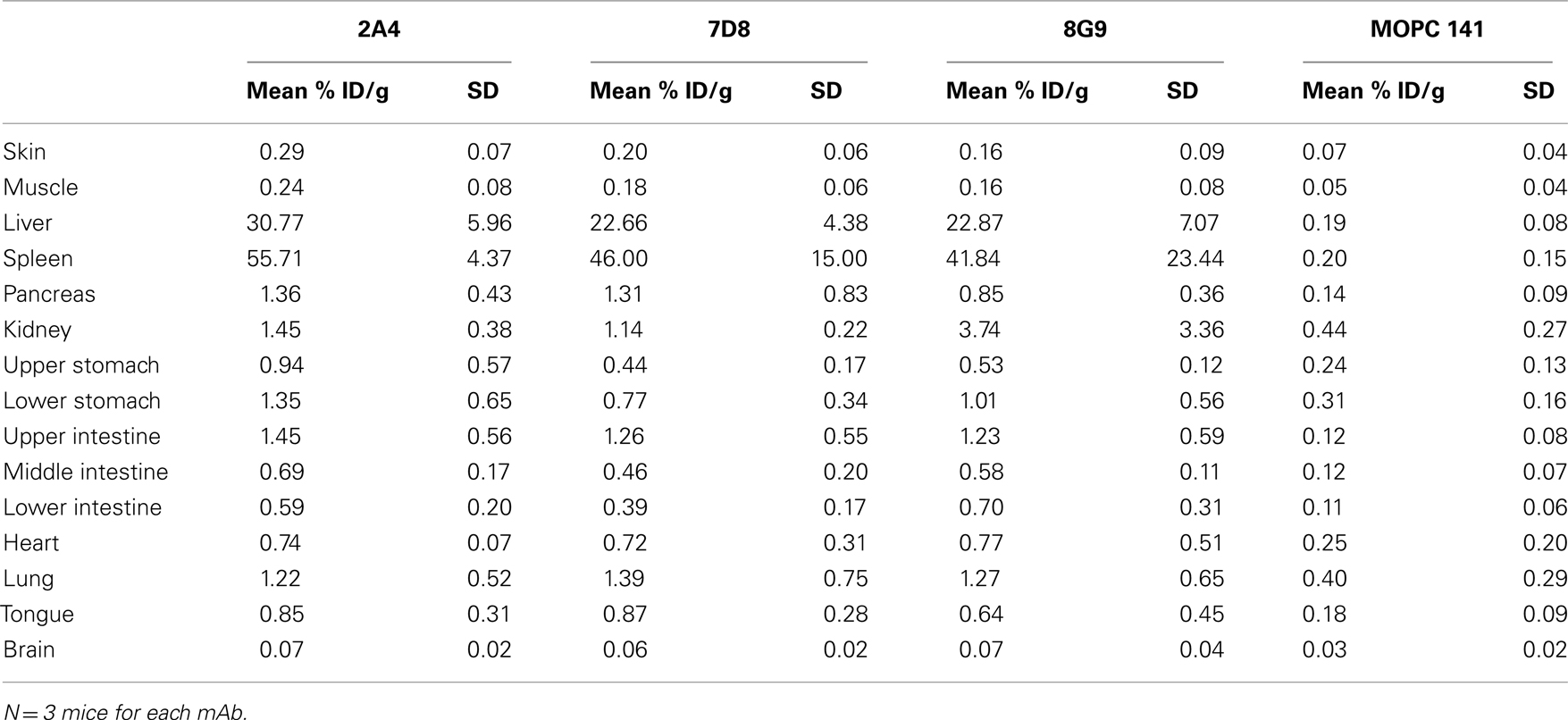
Table 2. Biodistribution of radiolabeled mAbs in AA amyloidotic or control mice.
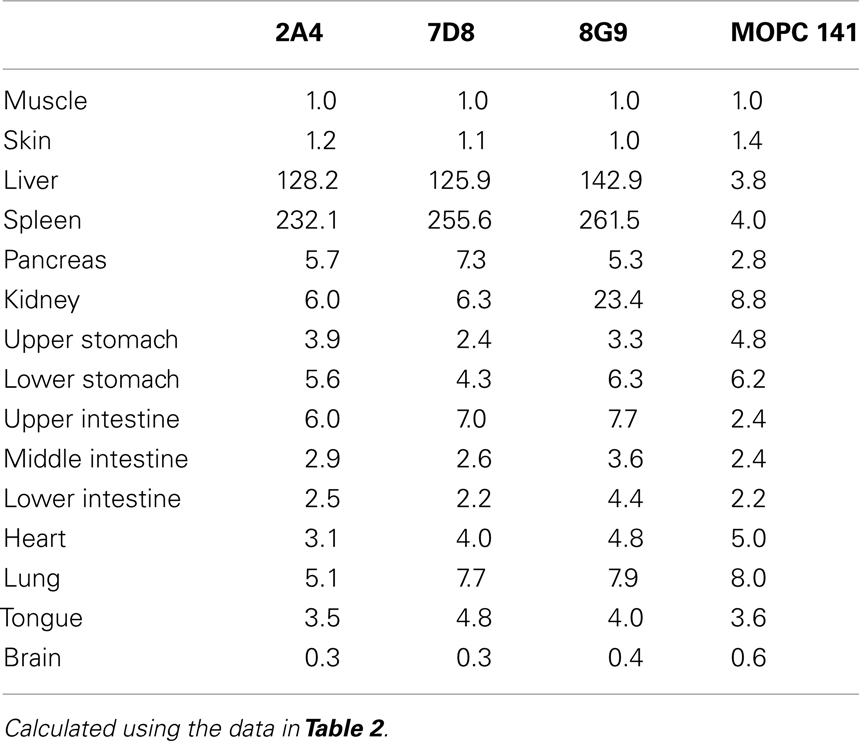
Table 3. Tissue:muscle ratios for mice with AA amyloidosis injected with 125I-labeled anti-AA or control mAbs.
Discussion
The amyloidogenic SAA protein precursor is a 103 amino acid, acute phase reactant lipoprotein synthesized in the liver; when isolated from amyloid deposits, it has been cleaved proteolytically by cathepsin-like enzymes into N-terminal fragments consisting predominantly of ∼75 residues (Yamada et al., 1995; Röcken et al., 2005, 2006). The mAbs 2A4, 7D8, and 8G9 were generated, using as immunogen the GHEDT sequence representing the 5 C-terminal residues of murine AA (71–75), and their specificity for this site was evidenced by the nanomolar EC50 binding values achieved with murine, as well as human AA, and similar to that of the peptide used for immunization. Notably, these two proteins, as well as SAA of other mammals (e.g., domestic cat, cheetah, and Shar-Pei dog), have identical amino acids at positions 73 and 74 (Figure 1), i.e., glutamic acid and aspartic acid. Given the comparable affinity of the mAbs for murine and human AA amyloid, we posit that their binding is based upon an interaction with this common ED sequence, as supported by results with Ala-substituted peptides where E73A and, in particular, D74A dramatically reduced the reactivity of each of the mAbs with the immunogen.
The fibril-related specificity of the three mAbs also was evidenced by the fact that they bound only to the truncated AA peptide found in amyloid fibrils, but not the circulating native protein; furthermore, the longer peptide, GHEDTMADQE spanning the cleavage site, did not block the interaction between the mAbs and AA amyloid adsorbed onto microplate wells or that present in tissue sections. Notably, the AA epitope recognized by mAbs 2A4, 7D8 and 8G9 located between residues 71–75 is structurally accessible, and not blocked by molecules typically associated with amyloid deposits, e.g., proteoglycans, apolipoproteins, and serum amyloid P-component (Alexandrescu, 2005), that could potentially hinder or prevent antibody-fibril binding. Although proteolysis of the precursor protein is common in amyloid fibrils (Bohne et al., 2004; Monti et al., 2005; Röcken et al., 2006; Enqvist et al., 2009), this is the first example of fibril-specific mAbs that target a neo-epitope formed as a result of proteolytic cleavage. In contrast, other reported anti-fibril-mAbs bind conformational epitopes present on the non-native, fibrillar form of the amyloid precursor protein, but not the native, soluble, circulating molecule (O’Nuallain et al., 2005, 2007).
Small animal imaging, together with micro-autoradiography, provide an exquisitely sensitive method to determine the in vivo biodistribution and specificity of amyloid-binding reagents. Using the transgenic H2-Ld-huIL-6 Tg Balb/C murine model of AA amyloidosis, in which animals develop spontaneous, extensive, systemic deposits and have circulating SAA levels of >1 mg/ml (Wall et al., 2008), we demonstrated that each of our mAbs localized specifically to the amyloid occurring in the liver, spleen, pancreas, kidneys, heart, and intestines. The concentration of the antibodies in the major sites of deposition, i.e., liver and spleen, was sufficient to be readily visualized by SPECT imaging and was further demonstrated by increased tissue:muscle ratios.
Conclusion
The highly specific targeting of amyloid by our three mAbs, along with the lack of binding to circulating SAA or healthy (amyloid-free) tissues, suggest that these radiolabeled antibodies have diagnostic utility as imaging agents in patients with AA amyloidosis. Additionally, given that other fibril-specific mAbs have been shown to opsonize and effect removal of pathologic light chain or Aβ amyloid (Bard et al., 2000, 2003; Hrncic et al., 2000; Schroeter et al., 2008), our anti-AA reagents also may have therapeutic potential.
Conflict of Interest Statement
Robin Barbour, Peter Seubert, and Dale Schenk are employes of Elan Pharmaceuticals.
Acknowledgments
This work was supported by a collaborative research grant from Elan Pharmaceuticals, South San Francisco, CA, USA. Alan Solomon is an American Cancer Society Clinical Research Professor.
References
Bard, F., Barbour, R., Cannon, C., Carretto, R., Fox, M., Games, D., Guido, T., Hoenow, K., Hu, K., Johnson-Wood, K. Khan, K. Kholodenko, D., Lee, C., Lee, M., Motter, R. Nguyen, M., Reed, A., Schenk, D., Tang, P., Vasquez, N., Seubert, P., and Yednock, T. (2003). Epitope and isotype specificities of antibodies to beta-amyloid peptide for protection against Alzheimer’s disease-like neuropathology. Proc. Natl. Acad. Sci. U.S.A. 100, 2023–2028.
Bard, F., Cannon, C., Barbour, R., Burke, R. L., Games, D., Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., Khan, K., Kholodenko, D., Lee, M., Lieberburg, I., Motter, R., Nguyen, M., Soriano, F., Vasquez, N., Weiss, K., Welch, B., Seubert, P., Schenk, D., and Yednock, T. (2000). Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 6, 916–919.
Bohne, S., Sletten, K., Menard, R., Bühling, F., Vöckler, S., Wrenger, E., Roessner, A., and Röcken, C. (2004). Cleavage of AL amyloid proteins and AL amyloid deposits by cathepsins B, K, and L. J. Pathol. 203, 528–537.
Enqvist, S., Sletten, K., and Westermark, P. (2009). Fibril protein fragmentation pattern in systemic AL-amyloidosis. J. Pathol. 219, 473–480.
Gillmore, J. D., Lovat, L. B., Persey, M. R., Pepys, M. B., and Hawkins, P. N. (2001). Amyloid load and clinical outcome in AA amyloidosis in relation to circulating concentration of serum amyloid A protein. Lancet 358, 24–29.
Hawkins, P. N., Aprile, C., Capri, G., Viganò, L., Munzone, E., Gianni, L., Pepys, M. B., and Merlini, G. (1998). Scintigraphic imaging and turnover studies with iodine-131 labelled serum amyloid P component in systemic amyloidosis. Eur. J. Nucl. Med. 25, 701–708.
Hawkins, P. N., Myers, M. J., Lavender, J. P., and Pepys, M. B. (1988). Diagnostic radionuclide imaging of amyloid: biological targeting by circulating human serum amyloid P component. Lancet 1, 1413–1418.
Hazenberg, B. P., van Rijswijk, M. H., Piers, D. A., Lub-de Hooge, M. N., Vellenga, E., Haagsma, E. B., Hawkins, P. N., and Jager, P. L. (2006). Diagnostic performance of 123I-labeled serum amyloid P component scintigraphy in patients with amyloidosis. Am. J. Med. 119, 355.e315–e324.
Hrncic, R., Wall, J., Wolfenbarger, D. A., Murphy, C. L., Schell, M., Weiss, D. T., and Solomon, A. (2000). Antibody-mediated resolution of light chain-associated amyloid deposits. Am. J. Pathol. 157, 1239–1246.
Kisilevsky, R., Tan, R., Subrahmanyan, L., and Snow, A. (1984). Are elevated serum amyloid A levels and amyloid-enhancing factor sufficient to induce inflammation-associated amyloid deposition? Appl. Pathol. 2, 308–315.
Monti, M., Amoresano, A., Giorgetti, S., Bellotti, V., and Pucci, P. (2005). Limited proteolysis in the investigation of beta2-microglobulin amyloidogenic and fibrillar states. Biochim. Biophys. Acta 1753, 44–50.
O’Nuallain, B., Allen, A., Kennel, S. J., Weiss, D. T., Solomon, A., and Wall, J. S. (2007). Localization of a conformational epitope common to non-native and fibrillar immunoglobulin light chains. Biochemistry 46, 1240–1247.
O’Nuallain, B., Murphy, C. L., Wolfenbarger, D. A., Kennel, S. J., Solomon, A., and Wall, J. S. (2005). “The amyloid-reactive monoclonal antibody 11-1F4 binds a cryptic epitope on fibrils and partially denatured immunoglobulin light chains and inhibits fibrillogenesis,” in Amyloid and Amyloidosis: Proceedings of the Xth International Symposium on Amyloidosis, eds G. Grateau, R. A. Kyle, and M. Skinner (Tours: CRC Press), 482–484.
Pras, M., Schubert, M., Zucker-Franklin, D., Rimon, A., and Franklin, E. C. (1968). The characterization of soluble amyloid prepared in water. J. Clin. Invest. 47, 924–933.
Röcken, C., Fändrich, M., Stix, B., Tannert, A., Hortschansky, P., Reinheckel, T., Saftig, P., Kähne, T., Menard, R., Ancsin, J. B., and Bühling, F. (2006). Cathepsin protease activity modulates amyloid load in extracerebral amyloidosis. J. Pathol. 210, 478–487.
Röcken, C., Menard, R., Bühling, F., Vöckler, S., Raynes, J., Stix, B., Krüger, S., Roessner, A., and Kähne, T. (2005). Proteolysis of serum amyloid A and AA amyloid proteins by cysteine proteases: cathepsin B generates AA amyloid proteins and cathepsin L may prevent their formation. Ann. Rheum. Dis. 64, 808–815.
Schroeter, S., Khan, K., Barbour, R., Doan, M., Chen, M., Guido, T., Gill, D., Basi, G., Schenk, D., Seubert, P., and Games, D. (2008). Immunotherapy reduces vascular amyloid-beta in PDAPP mice. J. Neurosci. 28, 6787–6793.
Senthilkumar, S., Chang, E., and Jayakumar, R. (2008). Diffusible amyloid oligomers trigger systemic amyloidosis in mice. Biochem. J. 415, 207–215.
Solomon, A., Weiss, D. T., Schell, M., Hrncic, R., Murphy, C. L., Wall, J., McGavin, M. D., Pan, H. J., Kabalka, G. W., and Paulus, M. J. (1999). Transgenic mouse model of AA amyloidosis. Am. J. Pathol. 154, 1267–1272.
Varga, J., Flinn, M. S., Shirahama, T., Rodgers, O. G., and Cohen, A. S. (1986). The induction of accelerated murine amyloid with human splenic extract. Probable role of amyloid enhancing factor. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 51, 177–185.
Wall, J. S., Kennel, S. J., Paulus, M. J., Gleason, S., Gregor, J., Baba, J., Schell, M., Richey, T., O’Nuallain, B., Donnell, R., Hawkins, P. N., Weiss, D. T., and Solomon, A. (2005). Quantitative high-resolution microradiographic imaging of amyloid deposits in a novel murine model of AA-amyloidosis. Amyloid 12, 149–156.
Wall, J. S., Kennel, S. J., Stuckey, A. C., Long, M. J., Townsend, D. W., Smith, G. T., Wells, K. J., Fu, Y., Stabin, M. G., Weiss, D. T., and Solomon, A. (2010). Radioimmunodetection of amyloid deposits in patients with AL amyloidosis. Blood 116, 2241–2244.
Wall, J. S., Paulus, M. J., Gleason, S., Gregor, J., Solomon, A., and Kennel, S. J. (2006a). Micro-imaging of amyloid in mice. Meth. Enzymol. 412, 161–182.
Wall, J. S., Kennel, S. J., Paulus, M., Gregor, J., Richey, T., Avenell, J., Yap, J., Townsend, D., Weiss, D. T., and Solomon, A. (2006b). Radioimaging of light chain amyloid with a fibril-reactive monoclonal antibody. J. Nucl. Med. 47, 2016–2024.
Wall, J. S., Richey, T., Allen, A., Donnell, R., Kennel, S. J., and Solomon, A. (2008). Quantitative tomography of early-onset spontaneous AA amyloidosis in interleukin 6 transgenic mice. Comp. Med. 58, 542–550.
Westermark, G. T., and Westermark, P. (2009). Serum amyloid A and protein AA: molecular mechanisms of a transmissible amyloidosis. FEBS Lett. 583, 2685–2690.
Keywords: monoclonal antibodies, AA amyloidosis, radioimmunoimaging, immunotherapy
Citation: Wall JS, Kennel SJ, Richey T, Allen A, Stuckey A, Weiss DT, Macy SD, Barbour R, Seubert P, Solomon A and Schenk D (2011) Generation and characterization of anti-AA amyloid-specific monoclonal antibodies. Front. Immun. 2:32. doi: 10.3389/fimmu.2011.00032
Received: 25 April 2011;
Paper pending published: 18 May 2011;
Accepted: 24 July 2011;
Published online: 08 August 2011.
Edited by:
Harry W. Schroeder, University of Alabama at Birmingham, USAReviewed by:
Jayanta Chaudhuri, Memorial Sloan Kettering Cancer Center, USAAntonio La Cava, University of California Los Angeles, USA
Copyright: © 2011 Wall, Kennel, Richey, Allen, Stuckey, Weiss, Macy, Barbour, Seubert, Solomon and Schenk. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jonathan S. Wall, University of Tennessee Graduate School of Medicine, 1924 Alcoa Highway, Knoxville, TN 37920, USA. e-mail: jwall@utmck.edu