 Walid Beghdadi1,2 Lydia Célia Madjene1,2 Marc Benhamou1,2 Nicolas Charles1,2 Gregory Gautier1,2 Pierre Launay1,2 Ulrich Blank1,2*
Walid Beghdadi1,2 Lydia Célia Madjene1,2 Marc Benhamou1,2 Nicolas Charles1,2 Gregory Gautier1,2 Pierre Launay1,2 Ulrich Blank1,2*- 1 INSERM U699, Paris, France
- 2 Université Paris-Diderot UMR-S699, Paris, France
Mast cells are localized in tissues. Intense research on these cells over the years has demonstrated their role as effector cells in the maintenance of tissue integrity following injury produced by infectious agents, toxins, metabolic states, etc. After stimulation they release a sophisticated array of inflammatory mediators, cytokines, and growth factors to orchestrate an inflammatory response. These mediators can directly initiate tissue responses on resident cells, but they have also been shown to regulate other infiltrating immune cell functions. Research in recent years has revealed that the outcome of mast cell actions is not always detrimental for the host but can also limit disease development. In addition, mast cell functions highly depend on the physiological context in the organism. Depending on the genetic background, strength of the injurious event, the particular microenvironment, mast cells direct responses ranging from pro- to anti-inflammatory. It appears that they have evolved as cellular sensors to discern their environment in order to initiate an appropriate physiological response either aimed to favor inflammation for repair or at the contrary limit the inflammatory process to prevent further damage. Like every sophisticated machinery, its dysregulation leads to pathology. Given the broad distribution of mast cells in tissues this also explains their implication in many inflammatory diseases.
Introduction
Mast cells are tissue-localized cells of hematopoietic origin. They have originally been named by Paul Ehrlich as “Mastzellen,” which in German stands for well-fed cells. He thought that their numerous cytoplasmic granules visualized after metachromatic staining with aniline dyes, were a feature of intense ingestion of nutrients by phagocytes (Ehrlich, 1878). Riley and West (1953) published that mast cell numbers in tissues are correlated with the tissue histamine content and that exceptional high amounts are found in dog mastocytomas. Histamine had been described somewhat earlier by Dale and Laidlaw (1910) to mediate the anaphylactic shock defined by Portier and Richet (1902) as an unusual reaction of dogs to the injection of toxins isolated from the sea anemone Physalia. These landmark studies established mast cells as important inflammatory effectors, a concept that has largely evolved ever since, with mast cells being now well-recognized effector cells in immunity and inflammatory responses.
Since then, mast cells have been found to be largely distributed throughout virtually every tissue of the body where upon stimulation they produce a whole set of biologically active products. This enables them to mediate tissue responses, fulfill immunoregulatory functions, participate in the attraction of other immune effector cells or organize tissue remodeling (Marshall, 2004; Galli et al., 2005, 2008a; Bischoff, 2007; Abraham and St John, 2010). As mast cells express high affinity IgE receptors (FcεRI) that interact with IgE antibodies, research on these cells has initially been driven by their implication in IgE-mediated type I immediate hypersensitivity reactions and allergic diseases (Metcalfe et al., 1997; Beaven, 2009; Hakim-Rad et al., 2009), the focus has now evolved extending to the study of their role in different physiological and pathophysiological settings (Maurer et al., 2003; Marshall, 2004; Galli et al., 2005, 2008a; Bischoff, 2007; Abraham and St John, 2010). This was at least in part made possible by the availability of mast cell-deficient animal models (Galli et al., 2005; Grimbaldeston et al., 2005). The accumulated evidence obtained so far show that mast cells are an integral part of the immunological surveillance system that act together with other immune cells to mount specific reactions in response to an inflammatory insult. Most importantly, while they have been considered initially as inflammatory effector cells whose main role was to contribute to pathologies such as allergies, many mast cell physiologic functions have now been unveiled (Marshall, 2004; Galli et al., 2005, 2008a; Bischoff, 2007; Abraham and St John, 2010). These studies have made clear that physiology and pathology are often closely delimited and that small differences in mast cell environment yields variant and even opposite functions. In the following review we present some of the recent advances made in our understanding of mast cell functions with a particular emphasis on the multiple faces and spectrum of responses these cells can deliver following inflammatory signals.
Fundamental Notions about Mast Cells
Mast cells originate from a common precursor in the bone marrow. They are poorly characterized in the blood as they rapidly enter tissues under the influence of various growth factors to complete their final differentiation (Metcalfe et al., 1997; Hallgren and Gurish, 2007). The major growth factor is c-kit-binding stem cell factor (SCF). However, other factors including IL-3, IL-4, IL-9, NGF, and TGFβ1 also support to various degrees mast cell growth and differentiation (Kitamura, 1989; Galli et al., 2005; Ryan et al., 2007). Depending on the particular tissue, mast cells further differentiate into subpopulations, which led to the concept of mast cell heterogeneity. Tentatively, two types of mast cell populations have been distinguished including connective tissue-type mast cells and mucosal mast cells in rodents or tryptase/chymase and tryptase positive mast cells in humans (Enerback, 1966; Metcalfe et al., 1997; Beaven, 2009; Tsuboi et al., 2011). Additional specific features can be acquired depending on the microenvironment in tissues. The phenotypic heterogeneity also translates into a functional heterogeneity governed for example by the expression of various sets of receptors (Metcalfe et al., 1997; Beaven, 2009). Mast cells are preponderant in all tissues lining outer surfaces such as the skin, the airways, or the gut. Often, they can be found closely associated to blood vessels and nerve endings. However, mast cells exist in noticeable quantities in virtually every tissue. Even in tissues sparsely populated with mast cells, their number can increase markedly under inflammatory conditions i.e., in kidneys during the development of chronic kidney diseases (CKD; Blank et al., 2007). Mast cells express a large set of receptors on the cell surface or intracellularly allowing them to respond to a large variety of stimuli in tissues. The best studied mechanism of activation is the aggregation of the high affinity IgE receptor (FcεRI) implicated in allergic reactions. However, numerous other activation mechanisms have been characterized including, for instance, stimulation through innate immune receptors such as TLRs or complement receptors, or receptors for inflammatory peptides, hormones, and other compounds. Interestingly, while IgE receptor stimulation usually launches a complete program of stimulation with release of inflammatory products from cytoplasmic granules followed by the release of newly synthesized products, some stimuli, like TLR-ligands, CD30 ligand or vitamin D, selectively activate mast cells to release particular sets of cytokines (Marshall, 2004; Fischer et al., 2006; Galli et al., 2008a; Beaven, 2009; Biggs et al., 2010). Some of these stimuli also have a priming function to increase the secretory response or alter the response profile to a subsequent stimulus (Fifadara et al., 2009; Andrade et al., 2011). Mast cells store many of their inflammatory products, like histamine, proteoglycans or specific proteases, in cytoplasmic granules, where they are ready for immediate release within a few minutes. Others, such as arachidonic acid metabolites or chemokines/cytokines are newly synthesized and in consequence are released somewhat later in the range of 10 min up to several hours or even days. Some cytokines such as TNF can also be found in cytoplasmic granules where they can be released immediately. In addition new types of responses have been discovered recently such as the formation of extracellular nets, previously described in neutrophils (Brinkmann et al., 2004). These are composed of DNA, histones, tryptase, and the antimicrobial peptide LL-37, with growth inhibitory effects for Streptococcus pyogenes (Von Kockritz-Blickwede et al., 2008), Furthermore, synergistic cross-talk between FcεRI and chemokine receptors was shown to promote F-actin-rich cytoneme extensions between mast cells that may play a role in intercellular communication (Fifadara et al., 2010).
Research on the role of mast cells in health and disease has largely benefited from the availability of several mast cell-deficient animal models, the most utilized being the W/Wv mouse and more recently the Wsh/Wsh mouse (Galli et al., 2005; Grimbaldeston et al., 2005). Both carry natural, albeit different, mutations in the c-kit gene, a major growth factor receptor involved in mast cell differentiation. These strains of mice have been used in various physiological settings and experimental disease models to probe for the role of mast cells. Although these mice are profoundly deficient in mast cells as compared to wild-type mice, they carry other deficiencies that differ somewhat between the two strains (Nocka et al., 1990; Grimbaldeston et al., 2005; Zhou et al., 2007; Nigrovic et al., 2008; Mancardi et al., 2011; see Table 1). In addition, described deficiencies do not always coincide between studies carried out by various investigators, which may at least in part be due to colony conditions or the age of mice at time of investigations. Therefore, to effectively evaluate whether these cells are essential or contribute to an observed biological effect, it is always necessary to selectively reconstitute these mice with mast cells. Without such engraftment studies, sometimes important differences in experimental results obtained with these strains that could be related to the accompanying abnormalities might be misinterpreted as mast cell-dependent. For example, due to the relative neutrophilia observed in Wsh mice, mast cell dependent effects could be masked in case of an important contribution of neutrophils in pathological conditions, whereas this is less the case in W/Wv mice, which have neutropenia. This indicates that mast cells do not act alone but that they highly depend on their interaction with other components of their physiopathological environment. It should be noted that engraftment with mast cells does not exactly reproduce the distribution found in wild-type mice and reconstitution times largely differ between various tissues (Tanzola et al., 2003). Therefore, in addition to these classically used mouse models new transgenic mast cell-specific Cre mice have been under development (Musch et al., 2008; Scholten et al., 2008) where Cre is under control of the Mcpt5 and baboon a-chymase promoter, respectively. However, their complete characterization is still in progress. In addition, several mouse models that lack mediators or receptors that are relatively mast cell specific have been used to test the impact of deficiency in various experimental set-ups and disease models. They include for example mice lacking high affinity IgE receptor alpha or beta chains as well as mice deficient in various proteases such as mast cell protease 1 (MCP-1), MCP-4, MCPT6 (β2-tryptase), or carboxypeptidase A3 (CPA3; Knight et al., 2000; Tchougounova et al., 2004; Abonia et al., 2005; McNeil et al., 2008; Hamilton et al., 2011).
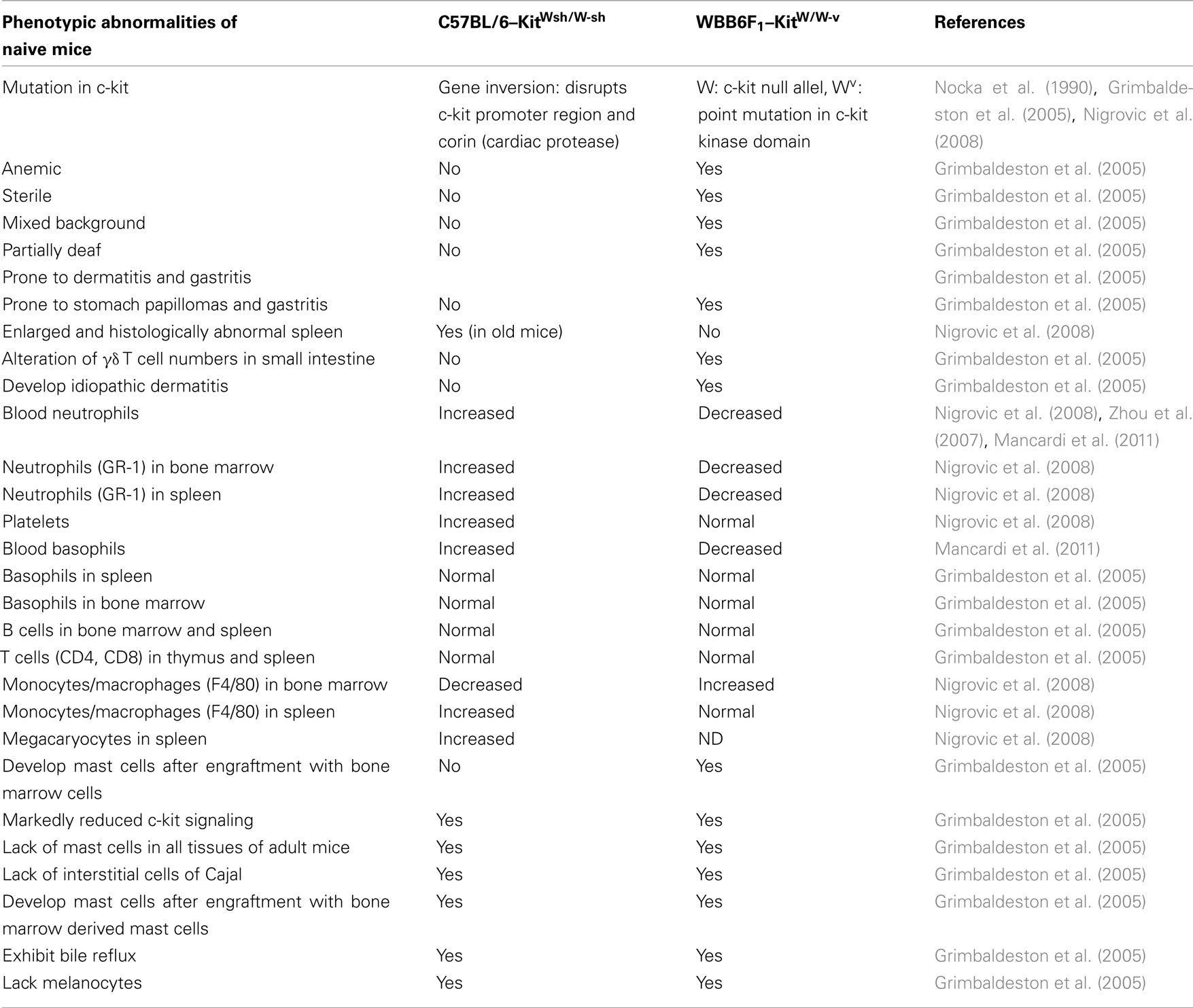
Table 1. Comparison of phenotypic abnormalities in mast cell-deficient C57BL/6–KitWsh/W-sh and WBB6F1–KitW/W-v mice as compared to wild-type animals.
Mast Cells as Actors of Immediate Hypersensitivity Reactions in Tissues
For a long time, mast cells together with basophils have been recognized as effectors of the classically defined IgE-mediated immediate type I hypersensitivity response characterized by the massive release of inflammatory mediators by degranulation (Metcalfe et al., 1997; Bradding, 2003; Blank and Rivera, 2004; Galli et al., 2008b). Similar types of responses can be triggered by various other agents and tissue factors including complement fragments, inflammatory products, and neuropeptides (Table 2; Maurer et al., 2003; Theoharides et al., 2010). This reactivity contributes importantly to the immunosurveillance function of these cells ready to respond instantly to tissue injury of traumatic, toxic, auto-immune or infectious origin. It appears that immunological alteration through the production of immunoglobulin E (IgE) antibodies to normally harmless substances has somehow hijacked this type of inflammatory response of mast cells to cause allergic disease. Indeed, together with blood basophils, mast cells express high affinity IgE receptors (FcεRI; Mendoza and Metzger, 1976), IgE being identified a few years earlier as the serum factor responsible for anaphylaxis, allergies, and asthma (Frick and Ishizaka, 1970; Lichtenstein et al., 1970). The study of their implications and the mechanisms of pathophysiology in IgE-mediated type I immediate type hypersensitivity has for a long time predominated in the mast cell research field. Besides leading to the characterization of the molecular structure of this receptor (Blank et al., 1989; Miller et al., 1989) and its signaling pathways (Kinet, 1999; Metcalfe et al., 2009; Benhamou and Blank, 2010), they also revealed that besides histamine and other granule-stored mediators, mast cells can produce a large set of inflammatory mediators including prostaglandins, leukotrienes, and many different chemokines, cytokines, and growth factors (Gurish and Austen, 2001; Galli et al., 2008a; Beaven, 2009). This attributed a role to mast cells also in the late phase allergic reactions including chronic inflammatory and remodeling responses during development of asthma. However, these studies did not resolve the enigmatic question raised at the turn of the twentieth century by Paul Portier and Charles Richet on the role of anaphylaxis in immunity as this contradicted immunity’s protective function (Portier and Richet, 1902). Indeed, it was difficult to conceive that the unique goal of the IgE/mast cells axis is to cause hay fever, food allergies, urticaria, asthma, etc. At least under some circumstances and particularly during parasite infections, evidence has now accumulated that IgE-mediated effector mechanisms of mast cells participate in host protection (see below).
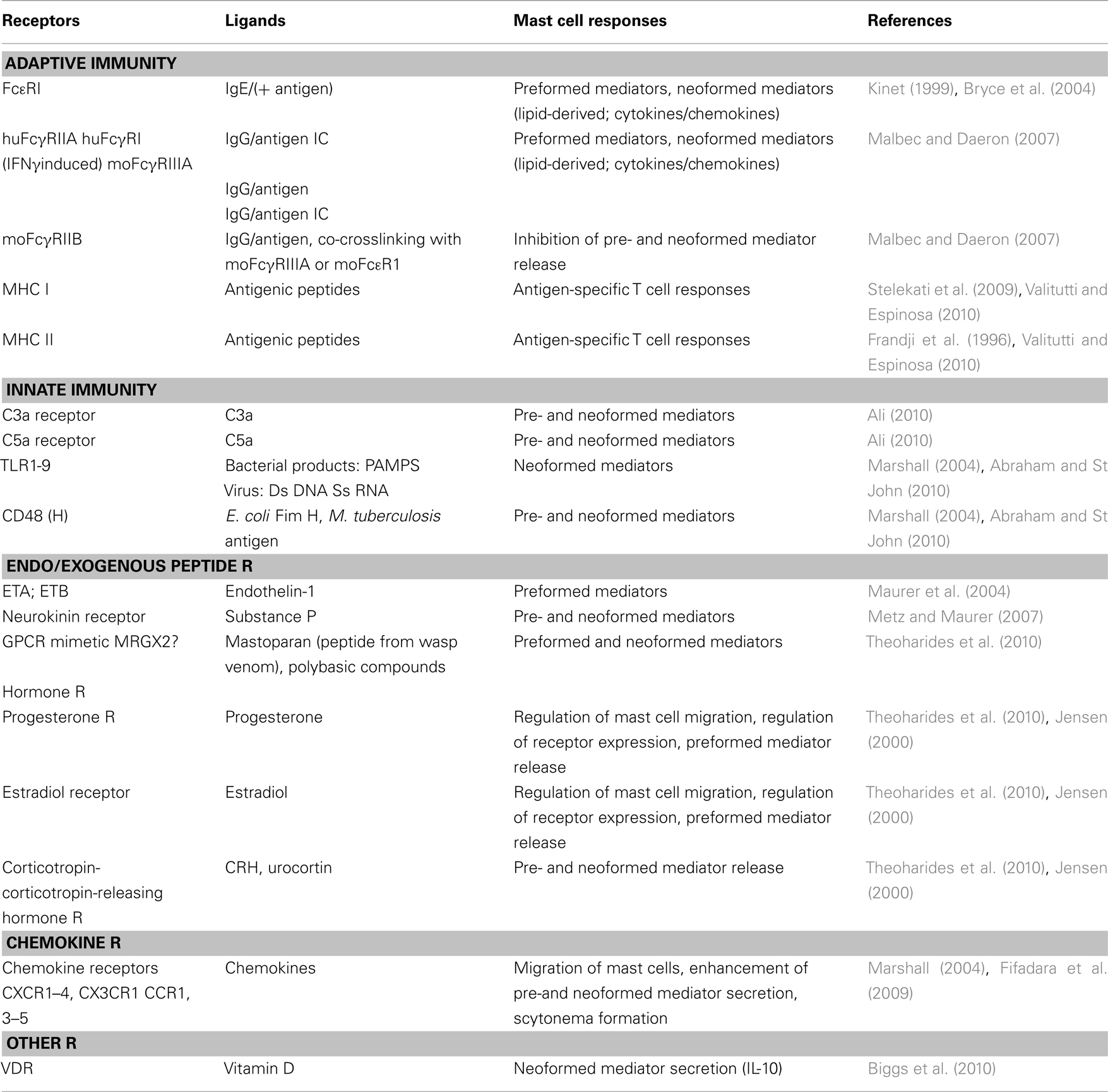
Table 2. Some of the receptors (R) expressed by mast cells.
Furthermore, recent works nicely offer some new clues about how hypersensitivity reactions participate in host protection. Endothelin-1 (ET-1), a 21 aa inflammatory and potent vasoconstrictor peptide released by endothelial cells, is implicated in a wide spectrum of pathological processes including sepsis, asthma, peripheral, acute, and chronic renal failure, cutaneous disorders, etc. (Maurer et al., 2004). Work by Marcus Maurer in the laboratory of Stephen Galli elegantly showed that ET-1 can be rapidly inactivated by mast cell-released carboxypeptidase A (CPA), a protease stored in cytoplasmic granules. ET-1 itself can activate mast cells to release CPA suggesting a negative feed-back loop of regulation. The high homology of this peptide with sarafotoxins, a potent venom component of the Israeli mole viper led to another intriguing set of data revealing the protective action of mast cell granule components. Metz et al. (2006) in the same laboratory showed that released CPA was responsible for diminished circulating levels of sarafotoxin. Following injection, the latter remained highly elevated in mast cell-deficient mice, mice treated with CPA inhibitors or mast cell-deficient mice reconstituted with CPA-deficient mast cells. This protective function was also confirmed against a variety of other snake venoms. Most strikingly, while honeybee venom can lead to severe IgE-mediated anaphylactic shock responses in susceptible IgE-sensitized patients, the authors provided evidence that injection of honeybee venom, which contains many mast cell degranulating agents, causes mortality with extensive hematuria in mast cell-deficient mice. More recently, mast cells were also shown to reduce neurotensin peptide levels, which contributes to protect against sepsis-related mortality in mice in the cecum ligation and puncture (CLP) model. Again this involved mast cell released proteases in particular neurolysin and to a minor extent CPA (Piliponsky et al., 2008). Taken together, these studies clearly sustain that mast cells have evolved as specialized effectors that via immediate type hypersensitivity-like reactions and ensuing release of inflammatory products such as proteases stored in mast cell granules play an important role in venom detoxification (Figure 1). The other side of the coin is that this type of reaction can also endanger certain individuals notably those exhibiting exaggerated IgE-mediated hypersensitivity reactions to honey bee venoms. They also endanger individuals developing immune responses to “harmless” environmental antigens (allergens; Galli et al., 2008b), which could be the result of the increasing inadequacy of the immunological adaption to a rapidly changing modern hygienic society (Bach, 2002).
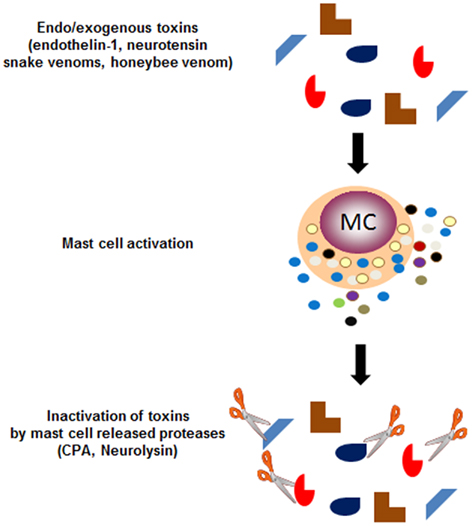
Figure 1. Immediate hypersensitivity responses to inactivate endo- or exogenous venoms. While IgE-dependent hypersensitivity reactions have been known for long to initiate allergic responses, recent research has unveiled that this type of response can also be activated by certain endogenous or exogenous toxins, which cause the mast cells to release granule-stored proteases, which in a feed-back mechanism can inactivate these toxins through proteolytic cleavage. Thus endogenously produced highly active pharmacological peptides such as endothelin-1 and neurotensin can be cleaved with mast cell-released carboxypeptidase A and neurolysin, respectively. Similarly certain snake venoms such as sarafotoxins, which bear homology to endothelin-1 can also be cleaved by CPA. Interestingly, while honeybee venom has been shown in some cases to cause IgE-dependent deadly anaphylactic reactions, mast cell released products also serve to inactivate this venom and limit its toxicity.
Mast Cells and Adaptive Immunity
Mast cells have the capacity to directly participate in adaptive immunity via the expression of Fc receptors that function as signaling modules that relay the antigen binding specificity of immunoglobulin ligands. This type of activation may contribute to pathology, in particular autoimmunity. Besides the high affinity IgE receptor (FcεRI), mast cells also express receptors for IgG subclasses conferring mast cells responsiveness to stimulation with IgG immune complexes. Mouse mast cells express the activating FcΓRIIIA, human MCs express both the activating FcΓRIIA and under certain conditions FcΓRI, the latter being induced by low concentrations of IFNΓ. Mouse mast cells also express FcΓRIIB, while its expression in human MCs is not firmly established (Malbec and Daeron, 2007). FcΓRIIB functions as a general inhibitor of cell activation when co-aggregated with another activating immune receptor. Mast cells promote adaptive immunity also by their ability to present antigen through MHC class I and II molecules (Frandji et al., 1996; Stelekati et al., 2009; Valitutti and Espinosa, 2010). They also may participate indirectly by releasing immunoregulatory cytokines or by their ability to recruit and regulate other cells of the adaptive immune system as shown for example for T lymphocytes, regulatory T lymphocytes or B lymphocytes (Ott et al., 2003; Lu et al., 2006; Merluzzi et al., 2010; see also below) and by their ability to function as adjuvants for the elaboration of immune responses (McLachlan et al., 2008).
Early studies published in the 1960s (Ogilvie, 1964) showed that “reagin-like” IgE antibodies were elevated during helminth infections, giving some hints of the participation of IgE-mediated effector mechanisms in the defense against parasites. Many other studies confirmed the marked elevation of IgE levels in an environment with endemic helminth parasite infections and the sites of infection have often been associated with an important recruitment and/or the proliferation of IgE-receptor-bearing mast cells (Lynch et al., 1984; Bell, 1996). Although, this could be secondary to the parasite-induced promotion of TH2 responses characterized by several other features (IL-4, IL-5, IL-13, eosinophilia, basophilia, etc.), at least in some cases both IgE and mast cells contribute to antiparasitic defense mechanisms (Finkelman and Urban, 2001; Anthony et al., 2007). Thus, transfer of IgE from Trichinella spiralis-infected animals conferred resistance to naïve animals (Ahmad et al., 1991). Studies conducted with IgE- and mast cell-deficient mice showed that both mast cells and IgE can actively participate to the resistance to various nematodes (Abe and Nawa, 1987; Lantz et al., 1998), ticks (Haemaphysalis longicornis; Matsuda et al., 1990), and protozoans (Li et al., 2004). Some of their actions rely on proteases such as the mast cell tryptase mMCP-6 (Shin et al., 2008) and mucosal mast cell chymase mMCP-1 (Knight et al., 2000). However, results on this host protective function were variable and largely depended on the parasite species under study leading in some cases to conclude to an absence of a mast cell role or even to a disease-promoting function, such as the case for the tick Dermacentor variabilis (Denhollander and Allen, 1985; Anthony et al., 2007). Furthermore, while in some cases mast cells showed efficiency during primary IgE-independent infection, other studies showed that they were efficient only during secondary infections, i.e., adaptive immunity (Finkelman and Urban, 2001). A striking example of the complexity of the response profile was provided recently in the model of tick infection with H. longicornis using both mast cell and basophil-deficient mice (Wada et al., 2010). The authors confirmed the role of mast cells in secondary infection, but showed at the same time that basophils were also necessary to confer resistance to secondary infection and likely are the effectors of the IgE and/or FcR-mediated response. While resistance was ablated in FcRΓ-deficient mice devoid of any activating FcR (FcεRI, FcΓRIII), mice reconstituted with FcRΓ-deficient mast cells were still able to restore protection after engraftment supporting a role of basophils in FcΓR-mediated effects and concomitant cooperation with mast cells in the mechanisms leading to host resistance. They also show that adaptive IgE-mediated effector mechanisms do not necessarily need to involve mast cells, but could rely also on basophils. These studies make clear that it is the combined action of various TH2-dependent effector mechanisms that contribute to host protection during parasite infection. Such effector mechanisms may involve direct killing by tissue-localized and/or recruited effector cells through released mediators (proteases, enzymes, ROS, etc.), enhanced mucus production with entrapment of parasites (weep and sweep mechanism) promoted by various inflammatory mediators including TH2 cytokines, the activation of smooth muscle contraction promoting expulsion as well as the coordination of an efficient inflammatory cell cross-talk (Anthony et al., 2007). In addition to antigen-triggered protective functions the interaction of mast cells with IgE may also support antigen-independent functions since IgE alone can enhance mast cell survival and prime mast cells to respond more efficiently during certain types of cutaneous hypersensitivity responses (Asai et al., 2001; Kalesnikoff et al., 2001; Bryce et al., 2004).
Although mast cells can clearly be triggered through IgG receptors in vitro, few studies have examined IgG-mediated mast cell responses. In the 1950s Zoltan Ovary established that IgG and antigen trigger passive cutaneous anaphylaxis reactions (Ovary, 1958), that were reconsidered as mast cell-mediated Arthus reactions mediated through FcΓR many years later (Hazenbos et al., 1996; Sylvestre and Ravetch, 1996). Results obtained in FcεRI- or mast cell-deficient mice established IgE-dependent stimulation of mast cells as major effector mechanism in a passive systemic anaphylaxis model with the effect being largely mediated by histamine release (Dombrowicz et al., 1993; Miyajima et al., 1997; Makabe-Kobayashi et al., 2002), although IgG immune complexes were also shown to induce such reactions. However, in the latter case, basophils, and more recently neutrophils activated through either FcΓRIII or FcΓRIV were reported to be the relevant effector cells and the effect was mediated by Platelet-Activating Factor (Tsujimura et al., 2008; Jonsson et al., 2011). It is possible that, after systemic injection, the high concentration of IgG immune complexes required to stimulate mast cells are unable to reach tissues (such as the skin) where mast cells are localized and, therefore, activate circulating cells instead. This may be less the case with the IgE system, as minute amounts of specific IgE are sufficient to sensitize mast cells through their steady binding to high affinity IgE receptors. Thereafter, minute amounts of antigen alone, even intravenously injected, will be able to reach mast cells and activate them preferentially in this context.
In other cases, even after injection into the skin of newborn mice of large amounts of antibodies directed against desmosomal proteins to induce experimental bullous pemphigoid auto-immune disease, mast cell degranulation did not involve FcR stimulation but rather was triggered by complement fragments (Chen et al., 2001). Nevertheless, recently the possible implication of FcΓRIII in the protective effects of mast cells in certain types of cutaneous hypersensitivity responses was discussed (Grimbaldeston et al., 2007). Furthermore, several other studies support a role of mast cell FcΓR in pathology, notably in IgG-mediated autoimmunity. Therefore, it is possible that at least under some circumstances, IgG immune complexes may be involved in triggering mast cell responses. Examples of experimentally induced auto-immune diseases with implication of FcΓR include EAE (Secor et al., 2000; Sayed et al., 2011) and rheumatoid arthritis (RA; Lee et al., 2002). In these models, which depend either on the injection of pathologic IgG (RA) or on the development of pathologic IgG (EAE), W/Wv mice are less susceptible to disease and their reconstitution with WT, but not with FcRΓ-deficient mast cells, restores disease susceptibility. However, again the picture is complex as, for example, the increased susceptibility to RA was apparent in W/Wv, but not Wsh/Wsh mast cell-deficient mice. This holds true also for both the passive K/BxN (Tsuboi et al., 2011) or active collagen-induced arthritis model (Zhou et al., 2007) pointing out that the physiological context in each animal model is important. In agreement, as W/Wv mice show neutropenia (see Table 1), reconstitution with neutrophils restored susceptibility to RA in these mice (Tsuboi et al., 2011). In addition, the K/BxN model also involved FcΓRIV, a receptor not expressed by mouse mast cells (Mancardi et al., 2011). Similarly in the EAE model, disease susceptibility could not be shown in Wsh/Wsh mice and discordances exist even in W/Wv mice regarding the implication of mast cells (Bennett et al., 2009; Sayed et al., 2011) indicating additional factors in colonies that may influence disease initiation.
Taken together, these data reveal a complex picture of the role of mast cells in adaptive immune defense mechanisms but also autoimmunity. There is no doubt that IgE-dependent mechanisms play a major role in allergies but some studies also agree on a prime role of mast cells in IgE-dependent protection to parasitic infections. Furthermore, new data concerning the contribution of FcΓR are emerging. The simultaneous engagement of FcεRI and inhibitory FcΓRIIB may lead to inhibition of responses although a convincing in vivo demonstration remains to be established as well as its significance in human mast cells (Daeron et al., 1995). It also has become clear that such mast cell mediated actions are highly dependent on the endogenous environment and that mast cells are rather an integral part of the effector response attributing an important role to cross-talk with other cells to organize efficient adaptive defense mechanisms.
Mast Cells and Innate Immunity
Although, there is no doubt today that the adaptive and innate immune systems are highly intertwined, it has become clear that mast cells are highly efficient effectors of primary innate immunity against infectious agents (Marshall, 2004; Abraham and St John, 2010). While, as shown above, adaptive immunity clearly plays a role in the defense mechanisms against parasites, the rapid action of mast cells response during primary infection speaks for their important role as actors of innate immunity. Emerging data show that parasites express a variety of surface molecules or secrete products that may be able to activate mast cells, although these have not been studied in detail so far (Robinson et al., 2010).
Following the studies by Echtenacher et al. (1996) and Malaviya et al. (1996) showing a protective action of mast cells in an acute model of sepsis induced by CLP, many other studies have followed demonstrating a protective function of mast cells in bacterial infection (Abraham and St John, 2010). These include bacterial infections in various compartments of the human body including peritoneum, skin bladder, airways, and lung, clearly implicating mast cells as highly relevant cells in the body internal surveillance system. Several types of innate immune receptors have been implicated in these anti-bacterial defense mechanisms. They include TLRs (TLR 2, TLR4, and TLR9) recognizing pathogen associated molecular patterns, CD48 recognizing E. coli FimH and complement fragments C3a and C5a or complement receptors CD21/CD35 and CD19 responding to byproducts of the complement cascade (Marshall, 2004; Abraham and St John, 2010; Ali, 2010).
Although multiple studies support mast cell participation in bacterial host defense, recent data reveal again a more complex picture indicating that mast cell-mediated actions depend on the pathophysiological context (Piliponsky et al., 2010). Thus, in the experimental model of sepsis induced by CLP, the mast cell mediated protective function highly depends on the severity of bacterial infection. In moderate sepsis induced by CLP (1/2 ligation, 22 gage needle) mast cells clearly mediated protection both in reconstituted W/Wv and Wsh/Wsh mice. In the latter, the effect was TNFα-independent, as reconstitution of Wsh/Wsh mice with either WT or TNF-deficient mast cells was equally effective in restoring protection. The mast cell mediated protection was not apparent in severe CLP (2/3 ligation, 22 gage needle). Although, mast cell-deficient W/Wv were more susceptible, protection could not be restored by engraftment of mast cells suggesting the participation of additional c-kit-dependent effects. Surprisingly, it seems that in the severe sepsis model, mast cell-deficient Wsh mice were less susceptible than WT mice. The enhanced pathology could be attributed to mast cell derived TNF, as in this background engraftment with WT mast cells but not TNF-deficient mast cells restored disease susceptibility. Thus, while under certain circumstances TNF will protect the host, by contributing to the recruitment of other immune effector cells with protective function such as neutrophils, under circumstances of excessive production it may also contribute to sustain a disease-aggravating inflammatory response. A similar deleterious effect of mast cell-derived TNF was also seen after intraperitoneal inoculation of Salmonella typhimurium. Thus, like in adaptive immunity, innate immune functions of mast cells contribute to protection or aggravation of disease depending on many additional factors that include strain background, the nature of the mutation resulting in mast cell deficiency as well as the type and severity of infection.
Mast cells may also participate in anti-viral defense mechanisms. For example, they express TLR3, a receptor recognizing double-stranded RNA (dsRNA), a molecular pattern associated with viral infections (Kulka et al., 2004). Such interaction induces release of various cytokines including IFN-α and IFN-β, which are implicated in the induction of anti-viral defense mechanisms. Upon interaction with dsRNA or reovirus infection, human mast cells produce a variety of chemokines (CCL4, CXCL8, and CXCL10; Burke et al., 2008). CXCL8 released by low levels of reovirus infection induces substantial anti-viral human NK cell chemotaxis (Burke et al., 2008). In mouse mast cells, TLR3 ligands or Newcastle disease virus induced anti-viral IFN-β and chemokine release and induced the recruitment of cytotoxic T cells (Orinska et al., 2005). Thus, mast cells could represent an important effector arm in the defense against viruses. However further in vivo studies are necessary to confirm such an implication. Mast cells, may be together with basophils, respond also to some HIV proteins including gp120 and Tat (Marone et al., 2001b). Gp120 interacted with the VH3 domain of IgE and induced the specific release of the TH2 promoting cytokines IL-4 and IL-13, while the HIV Tat protein induced chemotaxis of mast cells and enhanced the expression of CCR3 (Marone et al., 2001a). The latter serves as a receptor for viral entry. Based on this it was proposed that mast cells can also promote viral infection by serving as a reservoir for HIV. However, recent systematic colocalization studies in HIV-infected patients did not confirm active viral presence in various tissue mast cells (Nelson et al., 2009).
Physiological Roles of Mast Cells in Inflammation
Mast Cells as Crucial Immunoregulators of Tissue Repair
Besides functioning in host defense mechanisms in the context of infection, mast cells also participate more generally in the orchestration of inflammatory responses initiated after tissue injury (Laffargue et al., 1999; Maurer et al., 2003; Blank et al., 2007), in response to certain metabolic conditions (Liu et al., 2009), through hormonal signals (Matsumoto et al., 2001; Rudolph et al., 2004; Jensen et al., 2010), during cancer (Theoharides and Conti, 2004; Metz and Maurer, 2007; Galinsky and Nechushtan, 2008), or by signals emanating from the nervous system (Mori et al., 2002; Van Diest et al., 2011). Participation is made possible by the expression of numerous types of receptors enabling appropriate responses of mast cells (Table 2). Indeed, after stimulation mast cells directly evoke a large variety of tissue responses via the release of specific inflammatory mediators, the best known example of which being probably the vasodilatory action of released histamine. They also secrete many products involved in the orchestration of tissue remodeling including some proteases and cytokines. Many new studies also highlight their ability to migrate to the inflammatory site where they interact with other tissue resident or infiltrating cells. They also show their ability to mediate immunoregulatory functions through the release of cytokines and/or by fostering the cross-talk with other immune cells through the release of chemokines or small molecular weight chemotactic compounds (Metz and Maurer, 2007; Galli et al., 2008b).
The presence of immune cells during inflammatory responses is often considered as being causally linked to the disease process. It has become clear, however, that infiltrating cells including mast cells are also necessary to the resolution of inflammation and maintenance of tissue homeostasis. Although mast cell-deficient mice do not show grossly disturbed tissue integrity, an impact can readily be measured upon specific challenge. It has been shown in various experimental settings that mast cells can play pivotal roles in tissue protection and repair (Maurer et al., 2003). Some striking examples have already been mentioned above showing that their capacity to rapidly release granule-contained mediators contributes to detoxification against potentially dangerous exogenous or endogenous substances such as venoms or inflammatory peptides. They also participate in the prevention of acute and chronic damage induced by ultraviolet radiation. This involves their ability to suppress certain immune effector cells via the secretion of immunoregulatory cytokines such as IL-10 (see below). A physiological role of mast cells was also demonstrated both in in vitro and in in vivo models of wound healing (Weller et al., 2006). Wound healing is a complex event that involves interactions of various cell types, such as lymphocytes, monocytes, epithelial cells, and fibroblasts. Mast cells influence the wound healing process by increasing fibroblast migration and proliferation (Levi-Schaffer and Kupietzky, 1990), an effect partially mediated by histamine, IL-4 and tryptase (Pombo et al., 2001; Levi-Schaffer and Piliponsky, 2003). In addition, mast cells also recruit other inflammatory cells engaged in the wound healing process via the secretion of various chemokines.
Protective Functions of Mast Cells in Kidney Disease
Recently, a protective function of mast cells was reported in kidney disease. It was known for quite some time that mast cell numbers increase during various types of inflammatory CKD. Although this was usually interpreted as a disease contributing factor several recent studies support a more subtle view. Two studies conducted in W/Wv mice reported that mast cells are protective in an experimental model of glomerulonephritis induced by the injection of antibodies against the glomerular basement membrane (GBM; Hochegger et al., 2005; Kanamaru et al., 2006). The first study attributed the protective action of mast cells to their ability to initiate remodeling responses limiting the deposition of pathologic fibrin and collagen within glomeruli. In agreement, mast cell-deficient mice were unable to maintain high levels of tPA and uPA activity found in urine during the healing process. The second study rather supported an immunoregulatory role of mast cells, which dampens inflammatory cell infiltration and the inflammatory process. Interestingly, a third study, despite being conducted in the same background of W/Wv mice, reached the opposite conclusion supporting a disease-aggravating role of mast cells by increasing inflammatory TH1 responses through the upregulation of adhesion molecules, which promotes inflammatory cell infiltration (Timoshanko et al., 2006). Although the three studies lead to different conclusions, these differences may again reveal the importance of the pathophysiological context leading to contrasting functions of the MC as an effector of inflammation. Thus, we postulated that the mast cell-dependent action in glomerulonephritis relies on the speed of disease development (Blank et al., 2007; Tiwari et al., 2008). While the first two studies employed an accelerated model of anti-GBM nephritis promoting the surveillance function of mast cells, the third study used a non-accelerated model allowing mast cells to support a TH1-directed immune response that promoted injury. The complexity is even further illustrated by follow-up studies showing that in the accelerated model, while the overall action of mast cells was found to be protective, examination of the role of a particular mast cell mediator, the mMCP-4 chymase, was found to be disease-promoting, notably by supporting an inflammatory response, likely sparked by the ability of mMCP-4 to produce increased amounts of nephrotoxic angiotensin II (Scandiuzzi et al., 2010).
Role of Mast Cells in Skin Homeostasis
Mast cells are highly predominant in the skin, where they play an important roles in maintaining skin homeostasis. Although mast cells can participate as effector cells in contact hypersensitivity reaction to irritants (Mackenzie et al., 1981; Nakae et al., 2005), rather surprisingly, new results show that mast cells in the long-term instead of promoting inflammatory skin reactions can also limit their magnitude and duration in some experimental set-ups and thus contribute to the maintenance of homeostasis (Grimbaldeston et al., 2007). This was shown in two examples of adaptive immune responses of contact hypersensitivity to urushiol or dinitrofluorobenzene (DNFB) and in innate response to chronic low-dose UVB irradiation. Indeed, while after primary immunization no differences were observed after rechallenge (up to 48 hrs) between WT and mast cell-deficient mice (Wsh and W/Wv), the latter became much more susceptible after longer time periods. In all disease models protection could be restored after reconstitution with cultured WT mast cells, but not IL-10-deficient mast cells suggesting that local production of immunosuppressive IL-10 by mast cells limits the extent of pro-inflammatory cell infiltrate into the skin. Additional studies indicated a possible implication of Fc receptors, likely FcΓRIII, in the adaptive model, as reconstitution with FcRΓ-deficient mast cells was less efficient to restore protection. By contrast, in the innate UVB-induced model they were equally effective as WT cells. Given that FcR-mediated stimulation also induces release of disease-promoting mediators such as TNF-α (Nakae et al., 2005), the exact mechanism still needs to be further defined, but could depend on the timing of stimulation as well as additional cofactors. In the UVB-induced model, evidence was presented that UV-generated Vitamin D3 activates mast cells via its nuclear receptor VDR to induce IL-10 production in the absence of degranulation, which in consequence curtails pro-inflammatory mediator production (particularly TNF-α) and immune cell infiltration (Biggs et al., 2010). While these data show the ability of mast cells to limit inflammation, other products have been shown to rather enhance contact hypersensitivity responses. This includes for example the hapten oxazalone which, via an IgE/FcεRI mediated axis, promotes CHS responses (Bryce et al., 2004). However, new studies again indicate that this pro-inflammatory action can be shifted to an anti-inflammatory action under specific circumstances. Thus, while CHS elicited with low-dose oxazalone led to a mast cell mediated CHS, elicitation with high dose oxazolone promoted an immunosuppressive action of mast cells that was able to limit the ensuing T cell response (Kobayashi et al., 2010). Similar immunosuppressive actions have also been reported in a model of experimental malaria induced by bites of Anopheles stephensi, where mast cells where shown to suppress the development of an adaptive T cell response. This was associated with increased IL-10 levels in draining lymph nodes (Depinay et al., 2006). Similarly, mast cells by their ability to interact with regulatory T cells can promote allograft tolerance, an effect that was shown to require IL-9 produced by regulatory T cells (Lu et al., 2006). Interestingly, regulatory T cells have also been shown to limit mast cell degranulation and allergic-type reaction, an effect that involved direct cell–cell interaction mediated via OX40–OX40L (Gri et al., 2008) further indicating that an intense cross-talk between inflammatory effector cells is operant in tissues. Taken together, these data and those described above on kidney disease favor a scenario whereby under strong stimulatory conditions (accelerated glomerulonephritis model, strong irritants) or chronic stimulation (chronic UVB), mast cells act as sensors that initiate an appropriate repair response and/or initiate immunoregulatory functions in order to prevent further damage. Under low or moderate stimulatory conditions (non-accelerated glomerulonephritis model, low-dose irritant), they rather shift the balance to a pro-inflammatory action in order to fight the mechanisms leading to injury (Figure 2). Thus, besides their role in bona fide immune defense mechanisms mast cells may have evolved as regulatory cellular sensors that, depending on the particular circumstances, can limit further damage caused by an exaggerated inflammatory response or boost inflammatory responses when they are insufficiently robust. This opens the possibility that these opposite functions could also be timely activated in the course of an inflammatory reaction with amplification as the main early function and dampening as the main late function of mast cells, thus favoring a smooth resolution of the insulting damage.
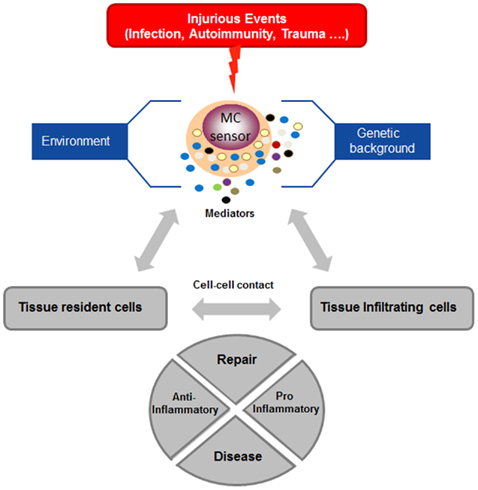
Figure 2. Mast cells function as cellular sensors. Mast cells are components of the innate and adaptive immune surveillance system in tissues. Following an injurious event, mast cells become activated and release a whole array of pro-inflammatory mediators. Their activation will promote a tissue inflammatory response characterized by the cellular cross-talk with tissue resident cells and other tissue infiltrating cells through released products or direct cell–cell contact. Mast cell activation is controlled by multiple additional factors that highly depend on a given genetic background and the local environment. Studies in recent years have revealed that mast cells are able to differentially control this response by initiating either a pro- or anti-inflammatory program destined for tissue repair and conditioned by the nature of the initial injurious event (strength, type, etc., for explanation see text). Mast cells can therefore be viewed as cellular sensors that direct tissue responses in inflammation and immunity as a function of the stimulus and physiological/pathological context. Dysregulation of this sensor function by chronic stimulation or defective control mechanisms can promote pathology and lead to inflammatory and auto-immune diseases.
Pathological Role of Mast Cells in Inflammatory Diseases
Although the idea of a sensor function for the strength of an inflammatory response likely serves to reduce pathological consequences, the danger remains that its dysregulation promotes inflammatory diseases. Such dysregulation may be the result of chronic stimulation or the consequence of an inadequate regulatory process (Nathan, 2002). Thus, in addition to their well-recognized role in IgE-mediated allergic diseases, evidence has emerged pointing to the involvement of mast cells in a variety of human inflammatory diseases such as RA, multiple sclerosis, fibrosis, and even cancer (Theoharides et al., 2010). Based on their wide tissue distribution, their ability to increase in number after inflammation or to release mediators that act locally and systemically, mast cells could play a role in virtually every tissue and inflammatory disease process. Therefore, over the last 15 years our view has rapidly expanded to suggest that mast cells are integral players of the inflammatory process and can contribute to a wide range of pathologies.
Mast Cells as Essential Players in the Long-Term Consequences of Asthma
Besides their role in the initiation of allergic disease and allergic asthma recent research has clearly unveiled that mast cells also contribute to the progression of such diseases. Their participation may involve multiple features, which again may be largely driven by the pathophysiological context. Allergic asthma, a chronic inflammatory disease is characterized by airway inflammation, bronchial hyperreactivity, and airway obstruction. In humans, strong evidences exist indicating that mast cells participate in all of these responses (Bradding et al., 2006). Thus, via IgE-dependent degranulation, synthesis of lipid compound and cytokine/chemokine production mast cells can promote the onset of an inflammatory response favoring the influx of other cell types such as T cells and eosinophils (Galli et al., 2008b). Mast cell derived mediators such as histamine also favor mucus production by goblets cells (Shimizu et al., 2003). While mucus production is initially intended to protect lung epithelium from harmful substances, it will promote airway obstruction in case of chronicity. Mast cells also contribute to smooth muscle contractions either directly via prostaglandins and leukotrienes or via the transformation of smooth muscle cells into a more contractile phenotype via tryptase-dependent upregulation of TGFβ (Tatler et al., 2008). While these features have been evaluated in humans and are already part of asthma therapies, mast cell participation in experimental animal model has not become obvious in all models. Thus, models which employ immunization protocols with alum which strongly activate the NALP3 inflammasome were found to be mast cell independent, while immunization with antigen in the absence of adjuvant clearly reveals the implication of mast cells (Williams and Galli, 2000). In addition, asthma has been described as a chronic disease involving an amplification loop based on excessive tissue remodeling which finally leads to tissue destruction (Holgate, 2010) that cannot easily be transferred to experimental murine models. However, establishment of a long-term protocol enabled to show, at least in part, that mast cells participate in key features of chronic asthma such as the initiation and maintenance of an inflammatory cell infiltrate, goblet cell hyperplasia and collagen deposition (Yu et al., 2006). Interestingly, reconstitution experiments using WT and FcRΓ-deficient mast cells showed that many aspects only partially involved an IgE or IgG-dependent process and show that mast cells also participated via FcR-independent activating mechanisms. Again, the situation may be complex as some of the mediators produced by mast cells were shown to have opposite protective effects. Thus, in a mouse model of allergic asthma in mice deficient for the mast cell chymase mMCP-4 airway hyperresponsiveness and lung airway inflammation was significantly increased. This was correlated with an increased thickening of the smooth muscle cell layer indicating that this mMCP-4 chymase could regulate smooth muscle cell activity (Waern et al., 2009).
Role of Mast Cells in Inflammatory Skin Diseases
Mast cells are also implicated in skin diseases such as chronic urticaria, atopic dermatitis, contact hypersensitivity and psoriasis (Navi et al., 2007; Harvima et al., 2008; Kaplan and Greaves, 2009; Kawakami et al., 2009; Liu et al., 2011). Although these diseases are complex and involve multiple etiologic factors, there is evidence that mast cells participate in multiple features of disease development. Generally, these diseases have in common an increase of either mast cells or mast cell-derived mediators in the skin (Navi et al., 2007). Mast cells may therefore contribute to disease development either directly or indirectly via the production of specific mediators. For example, mast cell-released histamine plays a major role in the generation of skin rash, wheals, and hives that become apparent in urticaria (Mlynek et al., 2008). In contact hypersensitivity, mast cell-produced TNF may play a role in the initiation and translation of the T cell mediated delayed type hypersensitivity responses (Nakae et al., 2005). In atopic dermatitis, mast cells contribute to disease development by the generation of pruritus symptoms through release of histamine and tryptase (Ikoma et al., 2003; Steinhoff et al., 2003). Mast cells were also shown to become activated by thymic stromal lymphopoietin (TSLP) that is produced in high amounts by keratinocytes in AD skin lesions (Yoo et al., 2005; Allakhverdi et al., 2007). TSLP stimulation promotes the release of TH2 cytokines involved in the development of atopic dermatitis (He and Geha, 2010). In psoriasis, in early lesions infiltrated mast cells show a degranulated phenotype and contain at high frequencies IFN-Γ (Harvima et al., 2008), while in AD the majority stains positive for IL-4 suggesting that mast cells via differential cytokine production influence immune response parameters (Horsmanheimo et al., 1994). A common feature of these diseases is also the involvement of emotional stress. It has been known that mast cells can often be found in close functional and anatomical association with sensory nerves in the skin (Arck et al., 2006; Harvima et al., 2010). During emotional stress, mast cells can therefore function as neuroinflammatory sensors that become activated through peptides and hormones released by the neuroendocrine system and peripheral sensory nerves, which release neuropeptides, neurotrophins, corticotropin-releasing hormone and α-melanocyte-stimulating hormone, which are capable of activating mast cells (Arck et al., 2006; Theoharides et al., 2010). Inversely, mast cell-derived mediators (histamine, tryptase) can in turn excite and stimulate surrounding neuropeptide-containing C-fibers possibly resulting in an amplification loop of neurogenic inflammation (Harvima et al., 2010). However, as stated above, mast cells, when present in chronic lesions, may also have immunosuppressive functions via the production of the immunosuppressive cytokine IL-10 in order to limit disease. In this context it may be interesting to note that psoriasis lesions are treated with Vitamin D analogs and that Vitamin D has been shown to selectively induce the production of IL-10 by mast cells.
Mast Cells and Gastrointestinal Disease
Mast cells are also found in large numbers in the gastrointestinal (GI) tract, where they may play an important function in the regulation of GI barrier functions (Bischoff, 2009). Given its large contact surfaces with the external environment and colonization with billions of microbes the GI barrier comprises multiple defense mechanisms which maintain integrity. This includes epithelial cell functions (mucus production, secretory functions, defensin release, etc.), regulation by innate and adaptive immune cells and the enteric nervous system (Bischoff, 2009). Dysregulation or breakdown of a single system results in disease. For example, impairment of defensin production has been associated with inflammatory bowel disease and ulcerative colitis (Wehkamp et al., 2009). Mast cells can interact in multiple ways with systems of the GI barrier. Release of mast cell mediators, such as histamine, can directly increase mucosal permeability and, as seen in the lung, increase mucus production (Bischoff, 2009). We have already seen above that mucus production plays an important role in the expulsion of intestinal parasites. Mast cell mediators also directly influence epithelial cell secretion. Evidence comes from studies in systemic mastocytosis patients where 70–80% of the patients report GI symptoms including diarrhea (Jensen, 2000). In pathophysiology, diarrhea is part of the mucosal defense system against microbes, parasites, toxins, or other harmful substances. Mast cells via release of multiple cytokines and chemokines are also known to regulate in multiple ways innate or adaptive immune effector cells, for example by promoting their activities, proliferation or by initiating their recruitment. Part of the GI tract-mediating actions may also be related to the capacity of mast cells to respond to signals of the enteric nervous system. Based on these multiple functions it is clear that chronic stimulation or dysregulation of mast cells can be associated with GI disease such as seen in the case of food allergies (Brandtzaeg, 2002). Apart from their role in allergies mast cells have been suggested to be involved in other pathologies of the GI tract. In celiac disease patients, local challenge with gliadin led to a decrease in mast cell numbers and histamine with a concomitant increase in CD4 T lymphocytes number (Lahteenoja et al., 2000). The decrease in mast cell numbers is likely the result of IgE-independent degranulation diminishing the number of countable mast cells with granule-staining methods. Similarly, in inflammatory bowel syndrome, although no differences in mast cell numbers were found, specific analysis of locally inflamed tissue showed an apparent decrease in their numbers, again likely due to degranulation (Bischoff et al., 1996), which suggests that in affected areas mast cells are activated. By contrast, patients with irritable inflammatory bowel syndrome, an affection with still unknown origins characterized by low grade inflammation, show mast cell hyperplasia (De Giorgio and Barbara, 2008). They may become activated under certain conditions and this could be directly related to the generation of symptoms by their bioactive mediators. At present, the precise triggers of mast cell activation (except for allergies) in these manifestations remain unknown. Animal models for these complex GI diseases are also missing, which may at least in part depend on altered microbial colonization of the human gut and thus could depend on certain microbial gene products that yet need to be identified (Qin et al., 2010).
Therapeutic Applications
Development of therapies relating to mast cell functions was initially largely driven by their role they play in allergies. They were aimed to limit the effects of mast cell mediators, in particular histamine, leading to the generation of new classes of anti-histamines with less sedating side-effects (Banerji et al., 2007), anti-leukotrienes (Calhoun, 2001), but included also the development of mast cell stabilizers (Gebbie et al., 1972). More recently, this has also led to the development of humanized anti-IgE antibodies (Omalizumab), although such treatment stays reserved for severe cases of allergies and asthma that are uncontrollable with corticosteroids due to its costs (Busse, 2001). However, the recent data of the broad influence of MCs and their many products in exacerbating inflammatory disorders, call for further research to limit mast cell activity and suggest that certain drugs may also be applicable for other types of applications. In this respect, tyrosine kinase inhibitors targeting the mast cell growth factor receptor c-kit such as masitinib and imatinib have shown promising results in several inflammatory diseases as well as in certain cancers (Eklund and Joensuu, 2003; Dubreuil et al., 2009; Humbert et al., 2009). On the other hand, mast cells have also been shown to exhibit many beneficial actions suggesting that development of drugs enhancing mast cell functions may also be useful in certain disease set-ups, although care has to be taken with regard to the potential side-effects of anaphylactic type of reactions, as for example noted in the treatment with omalizumab (Busse, 2001).
Concluding Remarks
Mast cell or mast cell like cells have emerged early in evolution appearing already in primitive organisms like tunicates (Cavalcante et al., 2000) before other classical immune effector cells like neutrophils, monocytes, and lymphocytes (Rowley, 1996). They may be part of an ancient defense system that protected tissues against foreign invaders or harmful substances and that have been maintained during evolution acquiring additional properties, enzymes, and mediators to fulfill these functions, eventually in concert with other immune effector cells. As evidenced by the multiple examples described in this review they continue to play an important role in a number of defense and repair mechanisms due to their strategic location in tissues at the interface with the environment. Within the vertebrate immune system mast cells are, together with basophils, the prime mediators of IgE-mediated hypersensitivity responses that probably have co-evolved together with a need for defense against parasitic infections. We have also seen that their inappropriate activation can result in inflammatory and auto-immune disease where mast cells can be either prime effectors, such as for example in allergic diseases, or participating actors. New data also show that their degree of participation in physiology and pathology very much depends on a given pathophysiological context. Thus, genetic factors, the local microenvironment, the presence, and interaction with other immune cells or tissue resident cells are important and can interfere with or overtake certain functions. This explains some differences which have been observed in various experimental mouse models of disease. Mast cells also release a whole array of inflammatory products some of which may act in an opposite manner when compared to the general action of mast cells in a particular pathological disease model. This illustrates the complexity of an inflammatory response, which is composed of pro-inflammatory but also regulatory anti-inflammatory components. The action of mediators also strongly depends on the disease context. Some mediators like TNF-α may have a prime protective function in host defense mechanisms against bacteria but can also significantly contribute to pathogenesis of skin diseases. Most intriguingly, these new studies have made clear that mast cells can initiate a whole spectrum of responses and thus that they act as sensors of their tissue environment to undertake appropriate actions. Thus, while one would think that mast cells engage an inflammatory response to restore homeostasis, several studies now also show that they can also limit inflammation by the promotion of immunosuppressive functions, likely to limit the negative side-effects of an inflammatory overshot. These two-face functions unveil mast cells as important microenvironment sensors involved in the maintenance of tissue homeostasis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work in our laboratory was supported by the French National Research Agency (R08171HS), the Association pour l’Utilisation du Rein Artificiel. Walid Beghdadi was supported by a post-doctoral fellowship from Region Ile-de-France (CODDIM).
References
Abe, T., and Nawa, Y. (1987). Reconstitution of mucosal mast cells in W/WV mice by adoptive transfer of bone marrow-derived cultured mast cells and its effects on the protective capacity to Strongyloides ratti-infection. Parasite Immunol. 9, 31–38.
Abonia, J. P., Friend, D. S., Austen, W. G. Jr., Moore, F. D. Jr., Carroll, M. C., Chan, R., Afnan, J., Humbles, A., Gerard, C., Knight, P., Kanaoka, Y., Yasuda, S., Morokawa, N., Austen, K. F., Stevens, R. L., and Gurish, M. F. (2005). Mast cell protease 5 mediates ischemia-reperfusion injury of mouse skeletal muscle. J. Immunol. 174, 7285–7291.
Abraham, S. N., and St John, A. L. (2010). Mast cell-orchestrated immunity to pathogens. Nat. Rev. Immunol. 10, 440–452.
Ahmad, A., Wang, C. H., and Bell, R. G. (1991). A role for IgE in intestinal immunity. Expression of rapid expulsion of Trichinella spiralis in rats transfused with IgE and thoracic duct lymphocytes. J. Immunol. 146, 3563–3570.
Ali, H. (2010). Regulation of human mast cell and basophil function by anaphylatoxins C3a and C5a. Immunol. Lett. 128, 36–45.
Allakhverdi, Z., Comeau, M. R., Jessup, H. K., Yoon, B. R., Brewer, A., Chartier, S., Paquette, N., Ziegler, S. F., Sarfati, M., and Delespesse, G. (2007). Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med. 204, 253–258.
Andrade, M. V., Iwaki, S., Ropert, C., Gazzinelli, R. T., Cunha-Melo, J. R., and Beaven, M. A. (2011). Amplification of cytokine production through synergistic activation of NFAT and AP-1 following stimulation of mast cells with antigen and IL-33. Eur. J. Immunol. 41, 760–772.
Anthony, R. M., Rutitzky, L. I., Urban, J. F. Jr., Stadecker, M. J., and Gause, W. C. (2007). Protective immune mechanisms in helminth infection. Nat. Rev. Immunol. 7, 975–987.
Arck, P. C., Slominski, A., Theoharides, T. C., Peters, E. M., and Paus, R. (2006). Neuroimmunology of stress: skin takes center stage. J. Invest. Dermatol. 126, 1697–1704.
Asai, K., Kitaura, J., Kawakami, Y., Yamagata, N., Tsai, M., Carbone, D. P., Liu, F. T., Galli, S. J., and Kawakami, T. (2001). Regulation of mast cell survival by IgE. Immunity 14, 791–800.
Bach, J. F. (2002). The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 347, 911–920.
Banerji, A., Long, A. A., and Camargo, C. A. Jr. (2007). Diphenhydramine versus nonsedating antihistamines for acute allergic reactions: a literature review. Allergy Asthma Proc. 28, 418–426.
Beaven, M. A. (2009). Our perception of the mast cell from Paul Ehrlich to now. Eur. J. Immunol. 39, 11–25.
Bell, R. G. (1996). IgE, allergies and helminth parasites: a new perspective on an old conundrum. Immunol. Cell Biol. 74, 337–345.
Benhamou, M., and Blank, U. (2010). Stimulus-secretion coupling by high-affinity IgE receptor: new developments. FEBS Lett. 584, 4941–4948.
Bennett, J. L., Blanchet, M. R., Zhao, L., Zbytnuik, L., Antignano, F., Gold, M., Kubes, P., and Mcnagny, K. M. (2009). Bone marrow-derived mast cells accumulate in the central nervous system during inflammation but are dispensable for experimental autoimmune encephalomyelitis pathogenesis. J. Immunol. 182, 5507–5514.
Biggs, L., Yu, C., Fedoric, B., Lopez, A. F., Galli, S. J., and Grimbaldeston, M. A. (2010). Evidence that vitamin D(3) promotes mast cell-dependent reduction of chronic UVB-induced skin pathology in mice. J. Exp. Med. 207, 455–463.
Bischoff, S. C. (2007). Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat. Rev. Immunol. 7, 93–104.
Bischoff, S. C. (2009). Physiological and pathophysiological functions of intestinal mast cells. Semin. Immunopathol. 31, 185–205.
Bischoff, S. C., Wedemeyer, J., Herrmann, A., Meier, P. N., Trautwein, C., Cetin, Y., Maschek, H., Stolte, M., Gebel, M., and Manns, M. P. (1996). Quantitative assessment of intestinal eosinophils and mast cells in inflammatory bowel disease. Histopathology 28, 1–13.
Blank, U., Essig, M., Scandiuzzi, L., Benhamou, M., and Kanamaru, Y. (2007). Mast cells and inflammatory kidney disease. Immunol. Rev. 217, 79–95.
Blank, U., Ra, C., Miller, L., White, K., Metzger, H., and Kinet, J. P. (1989). Complete structure and expression in transfected cells of high affinity IgE receptor. Nature 337, 187–189.
Blank, U., and Rivera, J. (2004). The ins and outs of IgE-dependent mast-cell exocytosis. Trends Immunol. 25, 266–273.
Bradding, P. (2003). The role of the mast cell in asthma: a reassessment. Curr. Opin. Allergy Clin. Immunol. 3, 45–50.
Bradding, P., Walls, A. F., and Holgate, S. T. (2006). The role of the mast cell in the pathophysiology of asthma. J. Allergy Clin. Immunol. 117, 1277–1284.
Brandtzaeg, P. E. (2002). Current understanding of gastrointestinal immunoregulation and its relation to food allergy. Ann. N. Y. Acad. Sci. 964, 13–45.
Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D. S., Weinrauch, Y., and Zychlinsky, A. (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535.
Bryce, P. J., Miller, M. L., Miyajima, I., Tsai, M., Galli, S. J., and Oettgen, H. C. (2004). Immune sensitization in the skin is enhanced by antigen-independent effects of IgE. Immunity 20, 381–392.
Burke, S. M., Issekutz, T. B., Mohan, K., Lee, P. W., Shmulevitz, M., and Marshall, J. S. (2008). Human mast cell activation with virus-associated stimuli leads to the selective chemotaxis of natural killer cells by a CXCL8-dependent mechanism. Blood 111, 5467–5476.
Busse, W. W. (2001). Anti-immunoglobulin E (omalizumab) therapy in allergic asthma. Am. J. Respir. Crit. Care Med. 164, S12–S17.
Cavalcante, M. C., Allodi, S., Valente, A. P., Straus, A. H., Takahashi, H. K., Mourao, P. A., and Pavao, M. S. (2000). Occurrence of heparin in the invertebrate Styela plicata (Tunicata) is restricted to cell layers facing the outside environment. An ancient role in defense? J. Biol. Chem. 275, 36189–36186.
Chen, R., Ning, G., Zhao, M. L., Fleming, M. G., Diaz, L. A., Werb, Z., and Liu, Z. (2001). Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J. Clin. Invest. 108, 1151–1158.
Daeron, M., Malbec, O., Latour, S., Arock, M., and Fridman, W. H. (1995). Regulation of high-affinity IgE receptor-mediated mast cell activation by murine low-affinity IgG receptors. J. Clin. Invest. 95, 577–585.
Dale, H. H., and Laidlaw, P. P. (1910). The physiological action of beta-iminazolylethylamine. J. Physiol. (Lond.) 41, 318–344.
De Giorgio, R., and Barbara, G. (2008). Is irritable bowel syndrome an inflammatory disorder? Curr. Gastroenterol. Rep. 10, 385–390.
Denhollander, N., and Allen, J. R. (1985). Dermacentor variabilis: resistance to ticks acquired by mast cell-deficient and other strains of mice. Exp. Parasitol. 59, 169–179.
Depinay, N., Hacini, F., Beghdadi, W., Peronet, R., and Mecheri, S. (2006). Mast cell-dependent down-regulation of antigen-specific immune responses by mosquito bites. J. Immunol. 176, 4141–4146.
Dombrowicz, D., Flamand, V., Brigman, K. K., Koller, B. H., and Kinet, J. P. (1993). Abolition of anaphylaxis by targeted disruption of the high affinity immunoglobulin E receptor alpha chain gene. Cell 75, 969–976.
Dubreuil, P., Letard, S., Ciufolini, M., Gros, L., Humbert, M., Casteran, N., Borge, L., Hajem, B., Lermet, A., Sippl, W., Voisset, E., Arock, M., Auclair, C., Leventhal, P. S., Mansfield, C. D., Moussy, A., and Hermine, O. (2009). Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE 4, e7258.
Echtenacher, B., Mannel, D. N., and Hultner, L. (1996). Critical protective role of mast cells in a model of acute septic peritonitis. Nature 381, 75–77.
Ehrlich, P. (1878). Beiträge zur Theorie und Praxis histologischer Färbung. Deutschland: Universität Leipzig.
Eklund, K. K., and Joensuu, H. (2003). Treatment of rheumatoid arthritis with imatinib mesylate: clinical improvement in three refractory cases. Ann. Med. 35, 362–367.
Enerback, L. (1966). Mast cells in rat gastrointestinal mucosa. 2. Dye-binding and metachromatic properties. Acta Pathol. Microbiol. Scand. 66, 303–312.
Fifadara, N. H., Aye, C. C., Raghuwanshi, S. K., Richardson, R. M., and Ono, S. J. (2009). CCR1 expression and signal transduction by murine BMMC results in secretion of TNF-alpha, TGFbeta-1 and IL-6. Int. Immunol. 21, 991–1001.
Fifadara, N. H., Beer, F., Ono, S., and Ono, S. J. (2010). Interaction between activated chemokine receptor 1 and FcepsilonRI at membrane rafts promotes communication and F-actin-rich cytoneme extensions between mast cells. Int. Immunol. 22, 113–128.
Finkelman, F. D., and Urban, J. F. Jr. (2001). The other side of the coin: the protective role of the TH2 cytokines. J. Allergy Clin. Immunol. 107, 772–780.
Fischer, M., Harvima, I. T., Carvalho, R. F., Möller, C., Naukkarinen, A., Enblad, G., and Nilsson, G. (2006). Mast cell CD30 ligand is upregulated in cutaneous inflammation and mediates degranulation-independent chemokine secretion. J. Clin. Invest. 116, 2748–2756.
Frandji, P., Tkaczyk, C., Oskeritzian, C., David, B., Desaymard, C., and Mecheri, S. (1996). Exogenous and endogenous antigens are differentially presented by mast cells to CD4+ T lymphocytes. Eur. J. Immunol. 26, 2517–2528.
Frick, O. L., and Ishizaka, K. (1970). Association of IgE with reaginic activity in sera from grass pollen- and horse dander-sensitive individuals. J. Allergy 45, 220–233.
Galinsky, D. S., and Nechushtan, H. (2008). Mast cells and cancer – no longer just basic science. Crit. Rev. Oncol. Hematol. 68, 115–130.
Galli, S. J., Grimbaldeston, M., and Tsai, M. (2008a). Immunomodulatory mast cells: negative, as well as positive, regulators of immunity. Nat. Rev. Immunol. 8, 478–486.
Galli, S. J., Tsai, M., and Piliponsky, A. M. (2008b). The development of allergic inflammation. Nature 454, 445–454.
Galli, S. J., Kalesnikoff, J., Grimbaldeston, M. A., Piliponsky, A. M., Williams, C. M., and Tsai, M. (2005). Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu. Rev. Immunol. 23, 749–786.
Gebbie, T., Harris, E. A., O’Donnell, T. V., and Spears, G. F. (1972). Multicentre, short-term therapeutic trial of disodium cromoglycate, with and without prednisone, in adults with asthma. Br. Med. J. 4, 576–580.
Gri, G., Piconese, S., Frossi, B., Manfroi, V., Merluzzi, S., Tripodo, C., Viola, A., Odom, S., Rivera, J., Colombo, M. P., and Pucillo, C. E. (2008). CD4 ( CD25 ( regulatory T cells suppress mast cell degranulation and allergic responses through OX40-OX40L interaction. Immunity 29, 771–781.
Grimbaldeston, M. A., Chen, C. C., Piliponsky, A. M., Tsai, M., Tam, S. Y., and Galli, S. J. (2005). Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 167, 835–848.
Grimbaldeston, M. A., Nakae, S., Kalesnikoff, J., Tsai, M., and Galli, S. J. (2007). Mast cell-derived interleukin 10 limits skin pathology in contact dermatitis and chronic irradiation with ultraviolet B. Nat. Immunol. 8, 1095–1104.
Hakim-Rad, K., Metz, M., and Maurer, M. (2009). Mast cells: makers and breakers of allergic inflammation. Curr. Opin. Allergy Clin. Immunol. 9, 427–430.
Hallgren, J., and Gurish, M. F. (2007). Pathways of murine mast cell development and trafficking: tracking the roots and routes of the mast cell. Immunol. Rev. 217, 8–18.
Hamilton, M. J., Sinnamon, M. J., Lyng, G. D., Glickman, J. N., Wang, X., Xing, W., Krilis, S. A., Blumberg, R. S., Adachi, R., Lee, D. M., and Stevens, R. L. (2011). Essential role for mast cell tryptase in acute experimental colitis. Proc. Natl. Acad. Sci. U.S.A. 108, 290–295.
Harvima, I. T., Nilsson, G., and Naukkarinen, A. (2010). Role of mast cells and sensory nerves in skin inflammation. G. Ital. Dermatol. Venereol. 145, 195–204.
Harvima, I. T., Nilsson, G., Suttle, M. M., and Naukkarinen, A. (2008). Is there a role for mast cells in psoriasis? Arch. Dermatol. Res. 300, 461–478.
Hazenbos, W. L., Gessner, J. E., Hofhuis, F. M., Kuipers, H., Meyer, D., Heijnen, I. A., Schmidt, R. E., Sandor, M., Capel, P. J., Daeron, M., Van De Winkel, J. G., and Verbeek, J. S. (1996). Impaired IgG-dependent anaphylaxis and Arthus reaction in Fc gamma RIII (CD16) deficient mice. Immunity 5, 181–188.
Hochegger, K., Siebenhaar, F., Vielhauer, V., Heininger, D., Mayadas, T. N., Mayer, G., Maurer, M., and Rosenkranz, A. R. (2005). Role of mast cells in experimental anti-glomerular basement membrane glomerulonephritis. Eur. J. Immunol. 35, 3074–3082.
Holgate, S. T. (2010). Has the time come to rethink the pathogenesis of asthma? Curr. Opin. Allergy Clin. Immunol. 10, 48–53.
Horsmanheimo, L., Harvima, I. T., Jarvikallio, A., Harvima, R. J., Naukkarinen, A., and Horsmanheimo, M. (1994). Mast cells are one major source of interleukin-4 in atopic dermatitis. Br. J. Dermatol. 131, 348–353.
Humbert, M., De Blay, F., Garcia, G., Prud’Homme, A., Leroyer, C., Magnan, A., Tunon-De-Lara, J. M., Pison, C., Aubier, M., Charpin, D., Vachier, I., Purohit, A., Gineste, P., Bader, T., Moussy, A., Hermine, O., and Chanez, P. (2009). Masitinib, a c-kit/PDGF receptor tyrosine kinase inhibitor, improves disease control in severe corticosteroid-dependent asthmatics. Allergy 64, 1194–1201.
Ikoma, A., Rukwied, R., Stander, S., Steinhoff, M., Miyachi, Y., and Schmelz, M. (2003). Neuronal sensitization for histamine-induced itch in lesional skin of patients with atopic dermatitis. Arch Dermatol 139, 1455–1458.
Jensen, F., Woudwyk, M., Teles, A., Woidacki, K., Taran, F., Costa, S., Malfertheiner, S. F., and Zenclussen, A. C. (2010). Estradiol and progesterone regulate the migration of mast cells from the periphery to the uterus and induce their maturation and degranulation. PLoS ONE 5, e14409.
Jensen, R. T. (2000). Gastrointestinal abnormalities and involvement in systemic mastocytosis. Hematol. Oncol. Clin. North Am. 14, 579–623.
Jonsson, F., Mancardi, D. A., Kita, Y., Karasuyama, H., Iannascoli, B., Van Rooijen, N., Shimizu, T., Daeron, M., and Bruhns, P. (2011). Mouse and human neutrophils induce anaphylaxis. J. Clin. Invest. 121, 1484–1496.
Kalesnikoff, J., Huber, M., Lam, V., Damen, J. E., Zhang, J., Siraganian, R. P., and Krystal, G. (2001). Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity 14, 801–811.
Kanamaru, Y., Scandiuzzi, L., Essig, M., Brochetta, C., Guerin-Marchand, C., Tomino, Y., Monteiro, R. C., Peuchmaur, M., and Blank, U. (2006). Mast cell-mediated remodeling and fibrinolytic activity protect against fatal glomerulonephritis. J. Immunol. 176, 5607–5615.
Kaplan, A. P., and Greaves, M. (2009). Pathogenesis of chronic urticaria. Clin. Exp. Allergy 39, 777–787.
Kawakami, T., Ando, T., Kimura, M., Wilson, B. S., and Kawakami, Y. (2009). Mast cells in atopic dermatitis. Curr. Opin. Immunol. 21, 666–678.
Kinet, J. P. (1999). The high-affinity IgE receptor (Fc epsilon RI): from physiology to pathology. Annu. Rev. Immunol. 17, 931–972.
Kitamura, Y. (1989). Heterogeneity of mast cells and phenotypic change between subpopulations. Annu. Rev. Immunol. 7, 59–76.
Knight, P. A., Wright, S. H., Lawrence, C. E., Paterson, Y. Y., and Miller, H. R. (2000). Delayed expulsion of the nematode Trichinella spiralis in mice lacking the mucosal mast cell-specific granule chymase, mouse mast cell protease-1. J. Exp. Med. 192, 1849–1856.
Kobayashi, M., Nunomura, S., Gon, Y., Endo, D., Kishiro, S., Fukunaga, M., Kitahata, Y., Terui, T., and Ra, C. (2010). Abrogation of high-affinity IgE receptor-mediated mast cell activation at the effector phase prevents contact hypersensitivity to oxazolone. J. Invest. Dermatol. 130, 725–731.
Kulka, M., Alexopoulou, L., Flavell, R. A., and Metcalfe, D. D. (2004). Activation of mast cells by double-stranded RNA: evidence for activation through Toll-like receptor 3. J. Allergy Clin. Immunol. 114, 174–182.
Laffargue, M., Ragab-Thomas, J. M., Ragab, A., Tuech, J., Missy, K., Monnereau, L., Blank, U., Plantavid, M., Payrastre, B., Raynal, P., and Chap, H. (1999). Phosphoinositide 3-kinase and integrin signalling are involved in activation of Bruton tyrosine kinase in thrombin-stimulated platelets. FEBS Lett. 443, 66–70.
Lahteenoja, H., Maki, M., Viander, M., Raiha, I., Vilja, P., Rantala, I., Toivanen, A., and Syrjanen, S. (2000). Local challenge on oral mucosa with an alpha-gliadin related synthetic peptide in patients with celiac disease. Am. J. Gastroenterol. 95, 2880–2887.
Lantz, C. S., Boesiger, J., Song, C. H., Mach, N., Kobayashi, T., Mulligan, R. C., Nawa, Y., Dranoff, G., and Galli, S. J. (1998). Role for interleukin-3 in mast-cell and basophil development and in immunity to parasites. Nature 392, 90–93.
Lee, D. M., Friend, D. S., Gurish, M. F., Benoist, C., Mathis, D., and Brenner, M. B. (2002). Mast cells: a cellular link between autoantibodies and inflammatory arthritis. Science 297, 1689–1692.
Levi-Schaffer, F., and Kupietzky, A. (1990). Mast cells enhance migration and proliferation of fibroblasts into an in vitro wound. Exp. Cell Res. 188, 42–49.
Levi-Schaffer, F., and Piliponsky, A. M. (2003). Tryptase, a novel link between allergic inflammation and fibrosis. Trends Immunol. 24, 158–161.
Li, E., Zhou, P., Petrin, Z., and Singer, S. M. (2004). Mast cell-dependent control of Giardia lamblia infections in mice. Infect. Immun. 72, 6642–6649.
Lichtenstein, L. M., Levy, D. A., and Ishizaka, K. (1970). In vitro reversed anaphylaxis: characteristics of anti-IgE mediated histamine release. Immunology 19, 831–842.
Liu, F. T., Goodarzi, H., and Chen, H. Y. (2011). IgE, mast cells, and eosinophils in atopic dermatitis. Clin. Rev. Allergy Immunol. [Epub ahead of print]..
Liu, J., Divoux, A., Sun, J., Zhang, J., Clement, K., Glickman, J. N., Sukhova, G. K., Wolters, P. J., Du, J., Gorgun, C. Z., Doria, A., Libby, P., Blumberg, R. S., Kahn, B. B., Hotamisligil, G. S., and Shi, G. P. (2009). Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat. Med. 15, 940–945.
Lu, L. F., Lind, E. F., Gondek, D. C., Bennett, K. A., Gleeson, M. W., Pino-Lagos, K., Scott, Z. A., Coyle, A. J., Reed, J. L., Van Snick, J., Strom, T. B., Zheng, X. X., and Noelle, R. J. (2006). Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature 442, 997–1002.
Lynch, N. R., Medouze, L., Di Prisco-Fuenmayor, M. C., Verde, O., Lopez, R. I., and Malave, C. (1984). Incidence of atopic disease in a tropical environment: partial independence from intestinal helminthiasis. J. Allergy Clin. Immunol. 73, 229–233.
Mackenzie, A. R., Pattison, J., and Hillier, M. A. (1981). Early and late reactions in contact sensitivity in the mouse. Int. Arch. Allergy Appl. Immunol. 65, 187–197.
Makabe-Kobayashi, Y., Hori, Y., Adachi, T., Ishigaki-Suzuki, S., Kikuchi, Y., Kagaya, Y., Shirato, K., Nagy, A., Ujike, A., Takai, T., Watanabe, T., and Ohtsu, H. (2002). The control effect of histamine on body temperature and respiratory function in IgE-dependent systemic anaphylaxis. J. Allergy Clin. Immunol. 110, 298–303.
Malaviya, R., Ikeda, T., Ross, E., and Abraham, S. N. (1996). Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature 381, 77–80.
Malbec, O., and Daeron, M. (2007). The mast cell IgG receptors and their roles in tissue inflammation. Immunol. Rev. 217, 206–221.
Mancardi, D. A., Jonsson, F., Iannascoli, B., Khun, H., Van Rooijen, N., Huerre, M., Daeron, M., and Bruhns, P. (2011). The murine high-affinity IgG receptor Fc(gamma)RIV is sufficient for autoantibody-induced arthritis. J. Immunol. 186, 1899–1903.
Marone, G., De Paulis, A., Florio, G., Petraroli, A., Rossi, F. W., and Triggiani, M. (2001a). Are mast cells MASTers in HIV-1 infection? Int. Arch. Allergy Immunol. 125, 89–95.
Marone, G., Florio, G., Petraroli, A., Triggiani, M., and De Paulis, A. (2001b). Human mast cells and basophils in HIV-1 infection. Trends Immunol. 22, 229–232.
Matsuda, H., Watanabe, N., Kiso, Y., Hirota, S., Ushio, H., Kannan, Y., Azuma, M., Koyama, H., and Kitamura, Y. (1990). Necessity of IgE antibodies and mast cells for manifestation of resistance against larval Haemaphysalis longicornis ticks in mice. J. Immunol. 144, 259–262.
Matsumoto, I., Inoue, Y., Shimada, T., and Aikawa, T. (2001). Brain mast cells act as an immune gate to the hypothalamic-pituitary-adrenal axis in dogs. J. Exp. Med. 194, 71–78.
Maurer, M., Theoharides, T., Granstein, R. D., Bischoff, S. C., Bienenstock, J., Henz, B., Kovanen, P., Piliponsky, A. M., Kambe, N., Vliagoftis, H., Levi-Schaffer, F., Metz, M., Miyachi, Y., Befus, D., Forsythe, P., Kitamura, Y., and Galli, S. (2003). What is the physiological function of mast cells? Exp. Dermatol. 12, 886–910.
Maurer, M., Wedemeyer, J., Metz, M., Piliponsky, A. M., Weller, K., Chatterjea, D., Clouthier, D. E., Yanagisawa, M. M., Tsai, M., and Galli, S. J. (2004). Mast cells promote homeostasis by limiting endothelin-1-induced toxicity. Nature 432, 512–516.
McLachlan, J. B., Shelburne, C. P., Hart, J. P., Pizzo, S. V., Goyal, R., Brooking-Dixon, R., Staats, H. F., and Abraham, S. N. (2008). Mast cell activators: a new class of highly effective vaccine adjuvants. Nat. Med. 14, 536–541.
McNeil, H. P., Shin, K., Campbell, I. K., Wicks, I. P., Adachi, R., Lee, D. M., and Stevens, R. L. (2008). The mouse mast cell-restricted tetramer-forming tryptases mouse mast cell protease 6 and mouse mast cell protease 7 are critical mediators in inflammatory arthritis. Arthritis Rheum. 58, 2338–2346.
Mendoza, G., and Metzger, H. (1976). Distribution and valency of receptor for IgE on rodent mast cells and related tumour cells. Nature 264, 548–550.
Merluzzi, S., Frossi, B., Gri, G., Parusso, S., Tripodo, C., and Pucillo, C. (2010). Mast cells enhance proliferation of B lymphocytes and drive their differentiation toward IgA-secreting plasma cells. Blood 115, 2810–2817.
Metcalfe, D. D., Peavy, R. D., and Gilfillan, A. M. (2009). Mechanisms of mast cell signaling in anaphylaxis. J. Allergy Clin. Immunol. 124, 639–646.
Metz, M., and Maurer, M. (2007). Mast cells–key effector cells in immune responses. Trends Immunol. 28, 234–241.
Metz, M., Piliponsky, A. M., Chen, C. C., Lammel, V., Abrink, M., Pejler, G., Tsai, M., and Galli, S. J. (2006). Mast cells can enhance resistance to snake and honeybee venoms. Science 313, 526–530.
Miller, L., Blank, U., Metzger, H., and Kinet, J.-P. (1989). Expression of high-affinity binding of human immunoglobulin E by tansfected cells. Science 244, 334–337.
Miyajima, I., Dombrowicz, D., Martin, T. R., Ravetch, J. V., Kinet, J. P., and Galli, S. J. (1997). Systemic anaphylaxis in the mouse can be mediated largely through IgG1 and Fc gammaRIII. Assessment of the cardiopulmonary changes, mast cell degranulation, and death associated with active or IgE- or IgG1- dependent passive anaphylaxis. J. Clin. Invest. 99, 901–914.
Mlynek, A., Maurer, M., and Zalewska, A. (2008). Update on chronic urticaria: focusing on mechanisms. Curr. Opin. Allergy Clin. Immunol. 8, 433–437.
Mori, N., Suzuki, R., Furuno, T., Mckay, D. M., Wada, M., Teshima, R., Bienenstock, J., and Nakanishi, M. (2002). Nerve-mast cell (RBL) interaction: RBL membrane ruffling occurs at the contact site with an activated neurite. Am. J. Physiol. Cell Physiol. 283, C1738–C1744.
Musch, W., Wege, A. K., Mannel, D. N., and Hehlgans, T. (2008). Generation and characterization of alpha-chymase-Cre transgenic mice. Genesis 46, 163–166.
Nakae, S., Suto, H., Kakurai, M., Sedgwick, J. D., Tsai, M., and Galli, S. J. (2005). Mast cells enhance T cell activation: importance of mast cell-derived TNF. Proc. Natl. Acad. Sci. U.S.A. 102, 6467–6472.
Navi, D., Saegusa, J., and Liu, F. T. (2007). Mast cells and immunological skin diseases. Clin. Rev. Allergy Immunol. 33, 144–155.
Nelson, A. M., Auerbach, A., and Man, Y. G. (2009). Failure to detect active virus replication in mast cells at various tissue sites of HIV patients by immunohistochemistry. Int. J. Biol. Sci. 5, 603–610.
Nigrovic, P. A., Gray, D. H., Jones, T., Hallgren, J., Kuo, F. C., Chaletzky, B., Gurish, M., Mathis, D., Benoist, C., and Lee, D. M. (2008). Genetic inversion in mast cell-deficient (Wsh) mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am. J. Pathol. 173, 1693–1701.
Nocka, K., Tan, J. C., Chiu, E., Chu, T. Y., Ray, P., Traktman, P., and Besmer, P. (1990). Molecular bases of dominant negative and loss of function mutations at the murine c-kit/white spotting locus: W37, Wv, W41 and W. EMBO J. 9, 1805–1813.
Ogilvie, B. M. (1964). Reagin-like antibodies in animals immune to helminth parasites. Nature 204, 91–92.
Orinska, Z., Bulanova, E., Budagian, V., Metz, M., Maurer, M., and Bulfone-Paus, S. (2005). TLR3-induced activation of mast cells modulates CD8( T-cell recruitment. Blood 106, 978–987.
Ott, V. L., Cambier, J. C., Kappler, J., Marrack, P., and Swanson, B. J. (2003). Mast cell-dependent migration of effector CD8( T cells through production of leukotriene B4. Nat. Immunol. 4, 974–981.
Piliponsky, A. M., Chen, C. C., Grimbaldeston, M. A., Burns-Guydish, S. M., Hardy, J., Kalesnikoff, J., Contag, C. H., Tsai, M., and Galli, S. J. (2010). Mast cell-derived TNF can exacerbate mortality during severe bacterial infections in C57BL/6-KitW-sh/W-sh mice. Am. J. Pathol. 176, 926–938.
Piliponsky, A. M., Chen, C. C., Nishimura, T., Metz, M., Rios, E. J., Dobner, P. R., Wada, E., Wada, K., Zacharias, S., Mohanasundaram, U. M., Faix, J. D., Abrink, M., Pejler, G., Pearl, R. G., Tsai, M., and Galli, S. J. (2008). Neurotensin increases mortality and mast cells reduce neurotensin levels in a mouse model of sepsis. Nat. Med. 14, 392–398.
Pombo, I., Martin-Verdeaux, S., Iannascoli, B., Le Mao, J., Deriano, L., Rivera, J., and Blank, U. (2001). IgE receptor type I-dependent regulation of a Rab3D-associated kinase: a possible link in the calcium-dependent assembly of SNARE complexes. J. Biol. Chem. 276, 42893–42900.
Portier, P., and Richet, C. (1902). Nouveaux Faits d’Anaphylaxie ou Sensibilisation aux Venins par Doses Réitérées. C. R. Soc. Biol. 54, 548–551.
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., Nielsen, T., Pons, N., Levenez, F., Yamada, T., Mende, D. R., Li, J., Xu, J., Li, S., Li, D., Cao, J., Wang, B., Liang, H., Zheng, H., Xie, Y., Tap, J., Lepage, P., Bertalan, M., Batto, J. M., Hansen, T., Le Paslier, D., Linneberg, A., Nielsen, H. B., Pelletier, E., Renault, P., Sicheritz-Ponten, T., Turner, K., Zhu, H., Yu, C., Jian, M., Zhou, Y., Li, Y., Zhang, X., Qin, N., Yang, H., Wang, J., Brunak, S., Dore, J., Guarner, F., Kristiansen, K., Pedersen, O., Parkhill, J., Weissenbach, J., Bork, P., and Ehrlich, S. D. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65.
Riley, J. F., and West, G. B. (1953). The presence of histamine in tissue mast cells. J. Physiol. (Lond.) 120, 528–537.
Robinson, M. W., Hutchinson, A. T., Donnelly, S., and Dalton, J. P. (2010). Worm secretory molecules are causing alarm. Trends Parasitol. 26, 371–372.
Rudolph, M. I., Rojas, I. G., and Penissi, A. B. (2004). Uterine mast cells: a new hypothesis to understand how we are born. Biocell 28, 1–11.
Ryan, J. J., Kashyap, M., Bailey, D., Kennedy, S., Speiran, K., Brenzovich, J., Barnstein, B., Oskeritzian, C., and Gomez, G. (2007). Mast cell homeostasis: a fundamental aspect of allergic disease. Crit. Rev. Immunol. 27, 15–32.
Sayed, B. A., Walker, M. E., and Brown, M. A. (2011). Cutting edge: mast cells regulate disease severity in a relapsing-remitting model of multiple sclerosis. J. Immunol. 186, 3294–3298.
Scandiuzzi, L., Beghdadi, W., Daugas, E., Abrink, M., Tiwari, N., Brochetta, C., Claver, J., Arouche, N., Zang, X., Pretolani, M., Monteiro, R. C., Pejler, G., and Blank, U. (2010). Mouse mast cell protease-4 deteriorates renal function by contributing to inflammation and fibrosis in immune complex-mediated glomerulonephritis. J. Immunol. 185, 624–633.
Scholten, J., Hartmann, K., Gerbaulet, A., Krieg, T., Muller, W., Testa, G., and Roers, A. (2008). Mast cell-specific Cre/loxP-mediated recombination in vivo. Transgenic Res. 17, 307–315.
Secor, V. H., Secor, W. E., Gutekunst, C. A., and Brown, M. A. (2000). Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J. Exp. Med. 191, 813–822.
Shimizu, T., Shimizu, S., Hattori, R., and Majima, Y. (2003). A mechanism of antigen-induced goblet cell degranulation in the nasal epithelium of sensitized rats. J. Allergy Clin. Immunol. 112, 119–125.
Shin, K., Watts, G. F., Oettgen, H. C., Friend, D. S., Pemberton, A. D., Gurish, M. F., and Lee, D. M. (2008). Mouse mast cell tryptase mMCP-6 is a critical link between adaptive and innate immunity in the chronic phase of Trichinella spiralis infection. J. Immunol. 180, 4885–4891.
Steinhoff, M., Neisius, U., Ikoma, A., Fartasch, M., Heyer, G., Skov, P. S., Luger, T. A., and Schmelz, M. (2003). Proteinase-activated receptor-2 mediates itch: a novel pathway for pruritus in human skin. J. Neurosci. 23, 6176–6180.
Stelekati, E., Bahri, R., D’Orlando, O., Orinska, Z., Mittrucker, H. W., Langenhaun, R., Glatzel, M., Bollinger, A., Paus, R., and Bulfone-Paus, S. (2009). Mast cell-mediated antigen presentation regulates CD8( T cell effector functions. Immunity 31, 665–676.
Sylvestre, D. L., and Ravetch, J. V. (1996). A dominant role for mast cell Fc receptors in the Arthus reaction. Immunity 5, 387–390.
Tanzola, M. B., Robbie-Ryan, M., Gutekunst, C. A., and Brown, M. A. (2003). Mast cells exert effects outside the central nervous system to influence experimental allergic encephalomyelitis disease course. J. Immunol. 171, 4385–4391.
Tatler, A. L., Porte, J., Knox, A., Jenkins, G., and Pang, L. (2008). Tryptase activates TGFbeta in human airway smooth muscle cells via direct proteolysis. Biochem. Biophys. Res. Commun. 370, 239–242.
Tchougounova, E., Lundequist, A., Fajardo, I., Winberg, J. O., Abrink, M., and Pejler, G. (2004). A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and -2. J. Biol. Chem. 279, 32339–32344.
Theoharides, T. C., Alysandratos, K. D., Angelidou, A., Delivanis, D. A., Sismanopoulos, N., Zhang, B., Asadi, S., Vasiadi, M., Weng, Z., Miniati, A., and Kalogeromitros, D. (2010). Mast cells and inflammation. Biochim. Biophys. Acta. [Epub ahead of print].
Theoharides, T. C., and Conti, P. (2004). Mast cells: the Jekyll and Hyde of tumor growth. Trends Immunol. 25, 235–241.
Timoshanko, J. R., Kitching, R., Semple, T. J., Tipping, P. G., and Holdsworth, S. R. (2006). A pathogenetic role for mast cells in experimental crescentic glomerulonephritis. J. Am. Soc. Nephrol. 17, 150–159.
Tiwari, N., Wang, C. C., Brochetta, C., Ke, G., Vita, F., Qi, Z., Rivera, J., Soranzo, M. R., Zabucchi, G., Hong, W., and Blank, U. (2008). VAMP-8 segregates mast cell preformed mediator exocytosis from cytokine trafficking pathways. Blood 111, 3665–3674.
Tsuboi, N., Ernandez, T., Li, X., Nishi, H., Cullere, X., Mekala, D., Hazen, M., Kohl, J., Lee, D. M., and Mayadas, T. N. (2011). Human neutrophil FcgammaRIIA regulation by C5aR promotes inflammatory arthritis in mice. Arthritis Rheum. 63, 467–478.
Tsujimura, Y., Obata, K., Mukai, K., Shindou, H., Yoshida, M., Nishikado, H., Kawano, Y., Minegishi, Y., Shimizu, T., and Karasuyama, H. (2008). Basophils play a pivotal role in immunoglobulin-G-mediated but not immunoglobulin-E-mediated systemic anaphylaxis. Immunity 28, 581–589.
Valitutti, S., and Espinosa, E. (2010). Cognate interactions between mast cells and helper T lymphocytes. Self Nonself 1, 114–122.
Van Diest, S. A., Stanisor, O. I., Boeckxstaens, G. E., De Jonge, W. J., and Van Den Wijngaard, R. M. (2011). Relevance of mast cell-nerve interactions in intestinal nociception. Biochim. Biophys. Acta. [Epub ahead of print].
Von Kockritz-Blickwede, M., Goldmann, O., Thulin, P., Heinemann, K., Norrby-Teglund, A., Rohde, M., and Medina, E. (2008). Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 111, 3070–3080.
Wada, T., Ishiwata, K., Koseki, H., Ishikura, T., Ugajin, T., Ohnuma, N., Obata, K., Ishikawa, R., Yoshikawa, S., Mukai, K., Kawano, Y., Minegishi, Y., Yokozeki, H., Watanabe, N., and Karasuyama, H. (2010). Selective ablation of basophils in mice reveals their nonredundant role in acquired immunity against ticks. J. Clin. Invest. 120, 2867–2875.
Waern, I., Jonasson, S., Hjoberg, J., Bucht, A., Abrink, M., Pejler, G., and Wernersson, S. (2009). Mouse mast cell protease 4 is the major chymase in murine airways and has a protective role in allergic airway inflammation. J. Immunol. 183, 6369–6376.
Wehkamp, J., Stange, E. F., and Fellermann, K. (2009). Defensin-immunology in inflammatory bowel disease. Gastroenterol. Clin. Biol. 33(Suppl. 3), S137–S144.
Weller, K., Foitzik, K., Paus, R., Syska, W., and Maurer, M. (2006). Mast cells are required for normal healing of skin wounds in mice. FASEB J. 20, 2366–2368.
Williams, C. M., and Galli, S. J. (2000). Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J. Exp. Med. 192, 455–462.
Yoo, J., Omori, M., Gyarmati, D., Zhou, B., Aye, T., Brewer, A., Comeau, M. R., Campbell, D. J., and Ziegler, S. F. (2005). Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J. Exp. Med. 202, 541–549.
Yu, M., Tsai, M., Tam, S. Y., Jones, C., Zehnder, J., and Galli, S. J. (2006). Mast cells can promote the development of multiple features of chronic asthma in mice. J. Clin. Invest. 116, 1633–1641.
Keywords: mast cells, inflammation, immunity
Citation: Beghdadi W, Madjene LC, Benhamou M, Charles N, Gautier G, Launay P and Blank U (2011) Mast cells as cellular sensors in inflammation and immunity. Front. Immun. 2:37. doi: 10.3389/fimmu.2011.00037
Received: 15 June 2011; Paper pending published: 07 July 2011;
Accepted: 16 August 2011; Published online: 06 September 2011.
Edited by:
Johan Van Der Vlag, Radboud University Nijmegen Medical Centre, NetherlandsReviewed by:
Martin Herrmann, Universitätsklinikum Erlangen, GermanyJaklien Leemans, Academic Medical Center, Netherlands
Copyright: © 2011 Beghdadi, Madjene, Benhamou, Charles, Gautier, Launay and Blank. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Ulrich Blank, Faculté de Médecine Site Xavier Bichat, INSERM U699, Université Paris-Diderot UMR-S699, 16 Rue Henri Huchard, 75018 Paris, France. e-mail: ulrich.blank@inserm.fr