 Christina Janko1† Sandra Franz2,3*† Luis E. Munoz1† Stefan Siebig1 Silke Winkler1 Georg Schett1 Kirsten Lauber4 Ahmed Sheriff5 Johan van der Vlag6 Martin Herrmann1
Christina Janko1† Sandra Franz2,3*† Luis E. Munoz1† Stefan Siebig1 Silke Winkler1 Georg Schett1 Kirsten Lauber4 Ahmed Sheriff5 Johan van der Vlag6 Martin Herrmann1
- 1 Department of Internal Medicine 3, Friedrich-Alexander University Erlangen–Nuremberg, Erlangen, Germany
- 2 Department for Dermatology, Venerology and Allergology, University Leipzig, Leipzig, Germany
- 3 MRC Centre for Inflammation and Research, Queen’s Medical Research Institute, University of Edinburgh, Edinburgh, UK
- 4 Department of Radiation Oncology, Ludwig-Maximilians University Munich, Munich, Germany
- 5 Department of Nephrology and Internal Intensive Care Medicine, Charité Berlin, Germany
- 6 Nephrology Research Laboratory, Nijmegen Centre for Molecular Life Sciences, Department of Nephrology, Radboud University Nijmegen Medical Centre, Nijmegen, Netherlands
Systemic lupus erythematosus (SLE) is a chronic inflammatory disease characterized by the production of autoantibodies, formation of immune complexes (IC), and activation of complement that ultimately fuel acute and/or chronic inflammation. Accumulation in blood and tissues of post-apoptotic remnants is considered of etiological and pathological importance for patients with SLE. Besides receptors directly recognizing apoptotic cells, soluble opsonins of the innate immune system bind apoptotic material dependent on the stage of apoptosis. We describe the binding to the surface of secondary necrotic cells (SNEC) of the serum opsonin CRP and further opsonins. We show that anti-dsDNA and anti-CRP autoantibodies bind and sensitize SNEC. Autoantibody-sensitized SNEC were cleared by macrophages in vitro and induced a pro-inflammatory cytokine response. In conclusion, anti-CRP, CRP, and SNEC form a ternary pyrogen endowed with strong pro-inflammatory capabilities which is able to maintain and perpetuate chronic inflammation.
Introduction
Accumulation in blood and tissues of post-apoptotic remnants is discussed to be of etiological and pathological importance for patients with Systemic lupus erythematosus (SLE) since the pathognomonic autoantigens are sequestered inside viable cells. They are, therefore, not accessible to the immune system in healthy persons. These autoantigens are often exposed during cell death. The formation of major complexes containing cellular debris and autoantibodies has been observed in patients with SLE and proposed to be an important pathogen (Munoz et al., 2009). Sensibilization of cellular remnants with autoantibodies shifts the phagocytosis of dead cells from liver and spleen to blood–borne phagocytes, which do not take up unsensitized material. This antibody-dependent pro-inflammatory pathway initiates an amplification loop of inflammation and contributes to the chronification of the autoimmune response (Munoz et al., 2010c).
The recognition by macrophages, of dead cell remnants is based on phosphatidylserine (PS) or on sugar neo-epitopes exposed on the plasma membranes (Schlegel and Williamson, 2001; Bottcher et al., 2006; Franz et al., 2006; Ravichandran and Lorenz, 2007). PS is recognized either directly by receptors as a “nude” lipid, or in combination with soluble proteins working as “adaptor molecules” or “opsonins” that bridge phagocytes and dead cells. These opsonins include milk fat globule protein MFGE8 (Hanayama et al., 2002), growth arrest specific gene product GAS-6 (ligand for the receptor tyrosine kinase MerTK; Scott et al., 2001), β-2-glycoprotein-1 (Balasubramanian and Schroit, 1998), serum-derived protein S (Anderson et al., 2003), annexin I (Arur et al., 2003), annexin A5 (Bondanza et al., 2004), C-reactive protein (CRP; Janko et al., 2009), C1q (Taylor et al., 2000), mannose-binding lectin (Nauta et al., 2004), serum amyloid P-component (Manfredi et al., 2008), the long Pentraxin 3 (Rovere et al., 2000), and further phospholipid or carbohydrate recognizing proteins (Franz et al., 2006; Beer et al., 2008; Sarter et al., 2009). The binding of these opsonins is often dependent on the stage of apoptosis, with PS recognition preceding that of carboneoepitopes (Franz et al., 2007). All these molecules mediate recognition and uptake of dying cells by macrophages and act as bridging opsonins that modulate the inflammatory and immunogenic potential of apoptotic material. Some of them additionally participate in complement activation (Ogden and Elkon, 2006).
C-reactive protein binds to damaged cell membranes via (lyso)phosphatidylcholine and efficiently activates the classical complement pathway avoiding the assembly of the membrane attack complex (MAC). CRP acts as an opsonin supporting ingestion of apoptotic cells by human macrophages and plays a role in the clearance of apoptotic cells, especially during acute phase reactions (Gershov et al., 2000; Vogt et al., 2007). In the case of primary necrosis, the action of nucleases causes an increase in the binding of CRP to necrotic cells that may foster their silent elimination (Janko et al., 2009).
Generally opsonins for dead cells tend to be anti-inflammatory and ameliorate clearance. Their interaction with its cognate receptor of the phagocytic cell often attenuates inflammatory responses (Voll et al., 1997; Aderem and Underhill, 1999). In patients with chronic inflammatory autoimmune diseases these opsonins for dying or dead cells are often targets of autoantibodies (Table 1). Intriguingly, in almost all cases the autoantibodies target only the surface bound opsonins and ignore their circulating counterparts (Shoenfeld et al., 2007; Sjowall et al., 2007; Bigler et al., 2009). The autoimmune response is, therefore, directed against opsonized material and not against the fluid phase proteins (Bell et al., 1998; Shoenfeld et al., 2007; Schaller et al., 2009). The autoantibody-dependent engagement of Fcγ receptors may have devastating consequences since it is prone to shift the process of waste disposal toward inflammation.

Table 1. Opsonins of dead and dying cells as targets of humoral autoimmunity.
Here we describe the exposure of binding sites of anti-dsDNA autoantibodies and of innate opsonins on the surfaces of post-apoptotic remnants and the recognition of these targets by autoantibodies against dsDNA and against surface bound CRP, respectively. The autoantibodies promote the release of inflammatory cytokines by macrophages in both conditions. We postulate that anti-opsonin autoantibodies, opsonins, and (post)apoptotic material form a ternary pyrogen that fuels chronic inflammation in patients with SLE.
Materials and Methods
Patients
Thirty nine patients from our outpatient clinic met the classification criteria of the American College of Rheumatology for SLE were included in this study (Hochberg, 1997). Thirty five NHD served as a control population. An informed consent was obtained from all blood donors and the study received the final approval from the ethics committee of the University Hospital Erlangen. Serum samples were obtained by centrifugation at 1000 g for 15 min of clotted blood, stored at −20°C, thawed once for ELISA, and stored again until phagocytosis assays.
Isolation of CRP
Human CRP was purified from human serum by calcium-dependent affinity chromatography with immobilized phosphorylcholine (Thermo Scientific, Rockford, IL, USA). The monomeric CPR was removed by filtration through Amicon Ultra centrifugation columns (Millipore, MA, USA). The purity of isolated CRP was assured by SDS PAGE and analytical HPLC.
Serological Parameters
Anti-dsDNA autoantibodies were detected by the ability of specific serum antibodies to bind radiolabeled dsDNA employing the Farr method (Wold et al., 1968) adapted by Kredich et al. (1973).
Anti-CRP autoantibodies were detected by ELISA, briefly, NUNC maxisorp 96-well microtiter plates (Nalgene Nunc, New York, NY, USA) were coated overnight at 4°C with 2 μg/well native human CRP in Coating buffer (0.1 M Na2CO3, 0.1 M Na HCo3, pH 9,6) and blocked for 2 h with 1% BSA in PBS. Patient sera were diluted 1:100 in 1% BSA/PBS-0.05% Tween, added in duplicates and incubated for 60 min. After washing with PBS-0.05% Tween, HRP-conjugated rabbit F(ab′)2 anti-human IgG (Southern Biotech) was diluted 1:10000 in PBS-Tween, added to each well and incubated for 60 min. After washing with PBS-Tween, the substrate (1 mg/ml TMB, 0.1 M Na2HPO4, 0.05 M Citric Acid, 0.006% H2O2, pH 5) was added to each well and incubated at room temperature. After 15 min the reaction was stopped with 25% sulfuric acid. Optical densities (OD) were measured at 450 nm. To discriminate positive from negative sera a cutoff was set at the mean value of NHD population plus 2 SD.
Secondary Necrotic Cell-Derived Material
Periphereal blood mononuclear cells (PBMC) served as source for secondary necrotic cell-derived material (SNEC). PBMC were isolated from whole blood by Ficoll density gradient centrifugation using Lymphoflot (Bio-Rad, Dreieich, Germany). Remaining platelets were removed by centrifugation through heat inactivated fetal calf serum (FCS; Invitrogen, Karlsruhe, Germany) and PBMC were subsequently reconstituted in PBS (Invitrogen, Karlsruhe, Germany). Finally SNEC was produced by UVB irradiation (180 mJ/cm2) of PBMC. In order to achieve uniform staining with Trypan blue (0.02%) or propidium iodide (1 μg/ml) in late stages of apoptosis, the lymphocytes were treated with heat (56°C) for 30 min and stored at 4°C in PBS until use. All particles generated by this method showed homogeneous fluorescence staining, scatter, and ligand-binding characteristics detected by flow-cytometry. We further characterized SNEC by analyzing the binding of the following fluorescent labeled dying and dead cell ligands as well as control proteins: BSA, Narcissus pseudonarcissus lectin (Npn), acetylated low density lipoprotein (acLDL), chicken annexin A5 (AxA5), human CRP. Human complement components C1q and C3c, and human IgG were detected employing fluorescent labeled specific antibodies, as control served heat inactivated human serum and NHD serum, respectively. Fluorescence microscopy pictures were done by staining SNEC with propidium iodide and monoclonal antibodies recognizing dsDNA and apoptotic nucleosomes. In the phagocytosis assays SNEC stained with trypan blue was incubated with patients’ sera, respectively for 30 min. Not bound serum proteins were removed by a washing step.
For the evaluation of the role of the opsonin CRP in the formation of ternary complexes, selected anti-dsDNA negative sera from patients with SLE were previously adsorbed 2× against immobilized Phosphorylcholine (Thermo Scientific, Rockford, IL, USA) and SNEC to remove CRP and anti-SNEC antibodies, respectively.
Ligand Specific Interaction Employing Inert Particles
We employed Sephadex beads coated with protein G (Pharmacia) to immobilize the anti-dsDNA monoclonal antibody 33.C9 or normal human IgG. Sephadex beads coated with phosphorylcholine were used to immobilize CRP in the presence or absence of calcium. Coated beads were incubated with fluorescent SNEC for 30 min at room temperature and immediately analyzed by fluorescence microscopy.
Macrophage Preparation
Leukocytes were isolated from heparinized human peripheral blood by density-gradient centrifugation (Ficoll-Paque PLUS, GE Healthcare). Monocytes were enriched by anti-CD14 microbeads (Miltenyi Biotec). Macrophages were generated by culturing CD14+ monocytes for 6 days in RPMI (Biochrome) supplemented with 1% penicillin/streptomycin and 1% glutamine (both Gibco) and 10% heat inactivated FCS (Biochrome), in the presence of 50 U/ml GM-CSF (Behringwerke, Marburg, Germany).
Phagocytosis Assays
Phagocytosis assays were performed in two independent experiments with sera from two patients with SLE of each group (anti-dsDNA−/anti-CRP−, anti-dsDNA+/anti-CRP−, anti-dsDNA+/anti-CRP+, anti-dsDNA−/anti-CRP+) or NHD. Phagocytosis assays were performed as follows; macrophages were rinsed with PBS to wash off serum components of the macrophage differentiation medium. Macrophages were co-cultured with trypan blue-labeled SNEC (non-opsonized or opsonized with different patients sera) in a ratio of 1:10 in serum free medium containing 100 ng/ml LPS at 37°C. After 1 h non-internalized SNEC were removed and macrophages detached with trypsin/EDTA. Phagocytosing macrophages were quantified by flow-cytometry.
Cytokine Quantification
Supernatants from phagocytosis cultures were collected after 18 or 24 h in co-culture with SNEC. Cytokines IL-8, TNF, and IL-10 were quantified by either ELISA (Peprotech) or multiplex bead array technology (Bender Medsystems, Vienna, Austria).
Statistics
Association between parameters were evaluated by a bivariate correlation test. Data are presented as mean ± SD, with n = number of independent experiments. Statistical significance was evaluated using a Student’s t test. All statistic calculations were done with the software SPSS-Statistics 18.0.
Results
Secondary Necrotic Cells-Derived Material Exposes Binding Sites for Several Opsonins
Considering the importance of dsDNA as autoantigen in SLE and the tight interaction between degraded nuclear DNA and CRP, we analyzed its localization in SNEC by flow-cytometry, indirect immunofluorescence, and ligand specific interaction with inert particles. We observed that after execution of apoptosis, peripheral blood lymphocytes contain high amounts of degraded DNA (Figure 1A) and show low binding of C3c, high binding of acLDL, NPn lectin, AxA5, C1q, CRP, and IgG from patients with SLE (Figure 1B, gray curves). Black histograms correspond to BSA–FITC binding as control for AxA5, CRP, acLDL, NPn lectin; to anti-C3c and anti-C1q staining in the presence of decomplemented serum; and to anti-IgG in the presence of NHD serum, respectively. The binding to SNEC of C1q and of autoantibodies from patients with SLE but not that of CRP was abolished by treatment of SNEC with DNase. SNEC show a random distribution of autoantibody (anti-dsDNA and anti-apoptotic nucleosomes) targets on their surfaces that do not overlap with the nuclear chromatin (Figure 1C). To test whether the binding sites for anti-dsDNA and for CRP are not sequestered inside the SNEC but are accessible on their surfaces we employed anti-dsDNA antibodies and CRP immobilized on beads mimicking the curvature of effector phagocytes, respectively. Co-incubation of fluorescent SNEC demonstrates its specific interaction with both immobilized anti-dsDNA and immobilized CRP, respectively. SNEC was captured by beads coated with anti-dsDNA or with CRP but not by those coated with normal human IgG or with CRP-beads in absence of calcium (Figures 1D,E). These experiments reveal that (1) CRP binding sites are accessible for CRP and (2) that both dsDNA and CRP bound to CRP binding sites are accessible for autoantibodies and for professional phagocytes on the surfaces of SNEC.
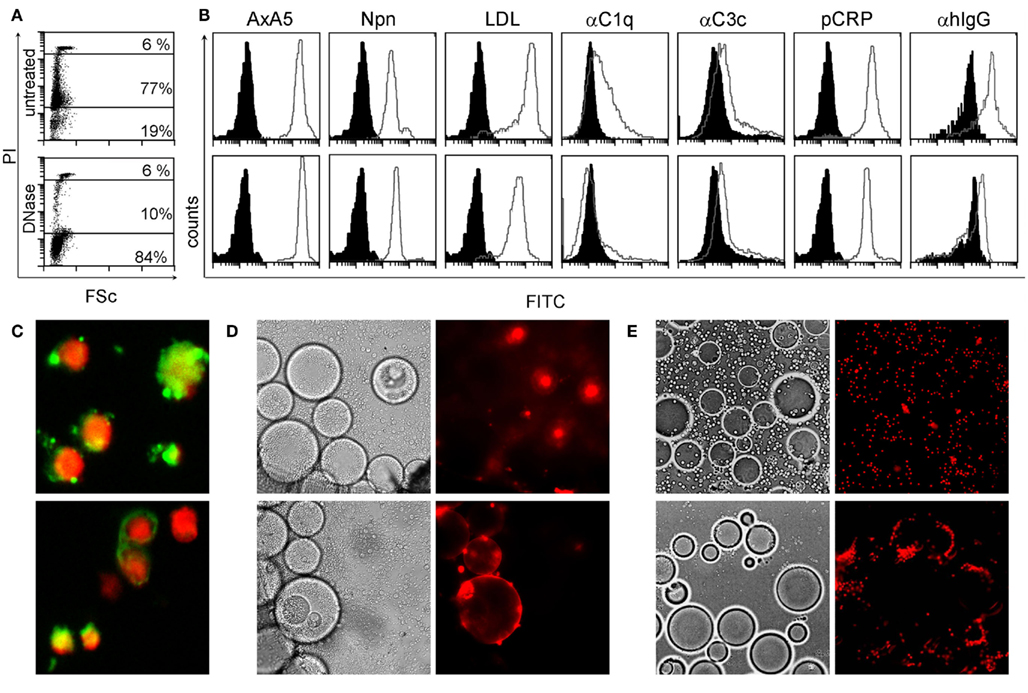
Figure 1. SNEC contain DNA, detected by staining with propidium iodide (PI) (A). SNEC bind AxA5, NPn lectin, acLDL, CRP, serum C1q, and C3c depicted as gray curves as detected by flow-cytometry (B). IgG from patients with SLE sensitize SNEC [(B), right histograms]. BSA–FITC for labeled opsonins, heat inactivated human serum for complement, and NHD serum for SLE serum served as controls, respectively and are shown as black histograms. After treatment with DNAse-1 [(B), lower row], the binding of complement proteins and of IgG from SLE patients is reduced. SNEC expose binding sites for monoclonal antibodies recognizing dsDNA [(C), upper slide] and apoptotic nucleosomes [(C), lower slide]. Binding sites for anti-dsDNA [(D) lower row] and for CRP [(E), lower row] are accessible on the surfaces of SNEC and may, therefore, be employed by phagocytes for uptake. Negative controls (see Materials and Methods) are displayed in the upper rows of D/E.
Autoantibodies against the Dead Cells Opsonin CRP are Frequently Found in Patients with SLE and Do not Correlate with Anti-dsDNA
C-reactive protein interacts with SNEC and may facilitate their phagocytic clearance. SNEC–CRP complexes characterize SLE patients and are seldom found in healthy persons, since apoptotic cells rarely get secondary necrotic in the latter. In the absence of proper clearance bound CRP may itself become a target for the autoimmune response. Therefore, we measured the anti-CRP and anti-dsDNA reactivity in sera from 39 patients with SLE and 35 NHD.
Autoantibodies of the IgG isotype were detected in 61.5 and 2.7% (p < 0.001) of the patients with SLE and of healthy donors, respectively (Figure 2A). Anti-CRP did not correlate with anti-dsDNA (Farr assay; Figure 2B). This allowed us to study the individual contribution of these autoantibodies in the uptake by macrophages of sensitized SNEC and their subsequent cytokine responses.
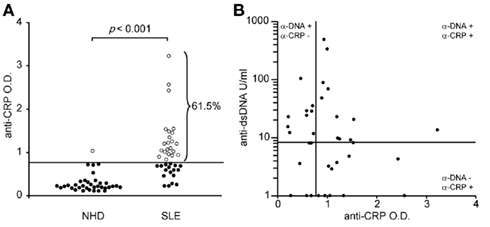
Figure 2. Autoantibodies against CRP are frequently found in sera of patients with SLE (A). Anti-CRP levels do not correlate with anti-dsDNA levels (B).
Sensibilization of SNEC with Autoantibodies Promotes Their Uptake by Macrophages and Fuels an Inflammatory Cytokine Response
Macrophages were co-cultured with (I) pure SNEC or (II) SNEC pre-incubated with serum from healthy donors, or (III) sera from patients with SLE. The latter contained (IIIa) neither anti-dsDNA nor anti-CRP, (IIIb) anti-dsDNA only, (IIIc) anti-CRP only; (IIId) anti-dsDNA; and anti-CRP (Figure 3A). Treatment of SNEC with autoantibody positive sera results in an elevated uptake by macrophages, whereas phagocytosis of SNEC incubated with autoantibody negative serum does not. Sensibilization of SNEC with anti-dsDNA and anti-CRP antibodies promoted the highest phagocytosis index (p < 0.05; Figure 3B).
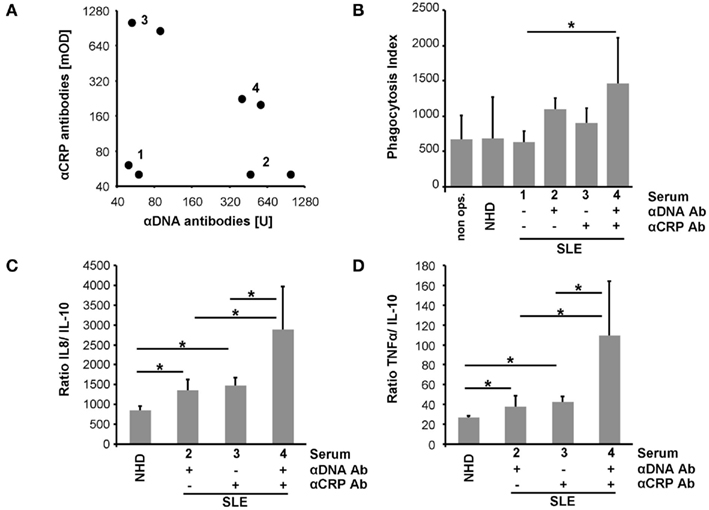
Figure 3. Sensibilization with autoantibodies enhances uptake by macrophages of SNEC and drives an inflammatory cytokine response. Sera selected for phagocytosis assays are shown in (A). SNEC treated with autoantibody positive sera are more efficiently engulfed by macrophages (B). Sensibilization with serum of SNEC induces an elevated macrophage cytokine response upon uptake. In the presence of both anti-dsDNA and anti-CRP phagocytosis of SNEC induces the highest levels of IL-8 and TNF (C,D). This indicates an autoantibody-dependent shift toward an inflammatory cytokine response.
Phagocytosis of apoptotic cells is typically anti-inflammatory. We, therefore, analyzed the cytokine profile of LPS triggered macrophages after the uptake of SNEC in the presence and absence of opsonins. SNEC sensitized with serum of patients with SLE induces an increased production of IL-8 and TNF and a reduced release of IL-10 when compared with SNEC treated with the serum of healthy donors. Figures 3C,D shows the ratios of IL-8 and TNF in relation to IL-10. However, phagocytosis of SNEC targeted with anti-dsDNA and anti-CRP antibodies promoted the strongest inflammatory cytokine response as seen by the highest IL-8 and TNF ratios (Figures 3C,D).
SNEC, the Opsonin CRP, and Anti-CRP Autoantibodies form Ternary Complexes that Shift Clearance of Apoptotic Cells Toward Inflammation
To evaluate the role of CRP, of anti-CRP autoantibodies during the clearance process of SNEC, we depleted CRP and anti-SNEC from anti-dsDNA negative/anti-CRP positive sera. These procedures remove the target structure for anti-CRP antibodies as well as other possible anti-SNEC antibodies. Depleted and reconstituted anti-CRP containing sera were used to sensitize SNEC for phagocytosis. Opsonisation with CRP or sensibilization with anti-CRP antibodies or both does not influence the phagocytosis by macrophages of SNEC (Figure 4A). However, the cytokine profile measured in the culture supernatants showed a significant higher TNF/IL-10 ratio if SNEC was opsonized with CRP and sensitized with anti-CRP autoantibodies, respectively (Figure 4B). The ternary complex of SNEC, CRP, and anti-CRP fosters the pro-inflammatory response of the macrophages.
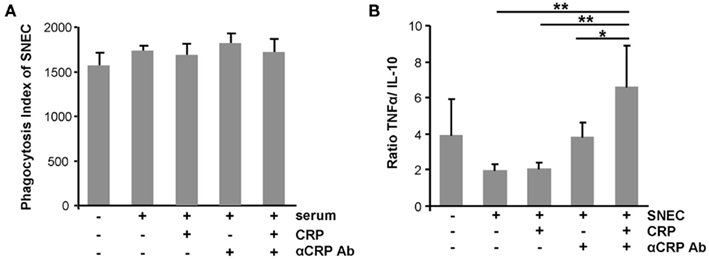
Figure 4. Ternary complexes of autoantibodies and opsonized apoptotic cell remnants provoke inflammation upon uptake by macrophages. The phagocytosis by macrophages of SNEC was not influenced by the opsonisation with CRP or sensibilization with anti-CRP or both when compared to non-opsonized SNEC (A). However, the cytokine profile measured in the culture supernatants showed significantly higher TNF/IL-10 ratio if SNEC was opsonized with CRP and sensitized with anti-CRP (B).
Discussion
In previous work, we have definitely demonstrated that autoantibodies promote phagocytosis of SNEC by blood–borne phagocytes and suggested that shuttling autoantigens into the intracellular milieu of phagocytes is an important trigger of inflammatory cytokine responses in patients with SLE (Munoz et al., 2009). SNEC–IC containing nucleic acids can be considered as a binary pyrogen able to induce much more damage than its single components apart (Munoz et al., 2010b). Since patients with SLE continuously produce SNEC, we proposed SNEC–IC as a lupus “pathogen” playing a role in the chronification of inflammation in patients with SLE (Munoz et al., 2010c).
In the actual work we have demonstrated that not properly cleared SNEC displays not only dsDNA on its surface but binding sites for several further opsonins. Employing inert particles we showed the presence of binding sites for anti-dsDNA and for CRP on the surfaces of SNEC. These are accessible to be recognized by opsonins, autoantibodies, and finally by phagocytes. CRP reportedly binds to SNEC via (lyso)phosphatidylcholine of disturbed membranes (Volanakis and Wirtz, 1979), to nuclear components like histones (Du Clos et al., 1988), and to snRNPs (Jewell et al., 1993). Such DNA-associated proteins become more accessible after DNA degradation during apoptosis and in post-apoptotic cells (Janko et al., 2009).
Anti-CRP autoantibodies have a high prevalence in patients with SLE. This finding has also been reported by several other authors (Bell et al., 1998; Sjowall et al., 2002). The titer of anti-CRP is associated with clinical activity but not with the levels of circulating CRP (Sjowall et al., 2005). It has also been shown that anti-CRP recognizes neo-epitopes of surface bound CRP which displays a monomeric conformation (Bell et al., 1998; Sjowall et al., 2002). The levels of anti-CRP and of anti-dsDNA do not correlate as already been shown by Sjowall et al. (2002). The independency of anti-CRP from anti-dsDNA confers target-bound CRP a role as further autoantigen in autoimmune responses of patients with SLE.
In this work we report the ability of sera to sensitize SNEC for phagocytic clearance by macrophages. Enhanced phagocytosis of SNEC by macrophages is observed in all conditions where anti-dsDNA or anti-CRP, or both are present. We previously reported that anti-dsDNA sensitize SNEC and enhance their uptake by blood–borne phagocytes (Munoz et al., 2009, 2010a). Macrophages play a very important role in the swift engulfment of dying cells (Voll et al., 1997). The involvement of these professional phagocytes in the aberrant clearance process may have additional deleterious consequences for patients with SLE, which often show an impaired clearance capability.
During an acute phase response CRP usually increases dramatically. In patients with SLE CRP is not considered as a marker for inflammation – often CRP levels increase only moderately during flares. This is in striking contrast to other rheumatic diseases with comparable amounts of tissue inflammation such as RA and gout (Becker et al., 1980). High IFNα levels, prototypic for SLE flares, have been shown to suppress IL-6 induced CRP levels by human hepatocytes in vitro (Enocsson et al., 2009). CRP has early been identified as a component of immune complexes (IC) circulating in the plasma of patients with SLE (Maire et al., 1983). CRP bound to circulating or sessile targets may escape detection, resulting in artificially low values of measurable CRP. In a clearance deficiency scenario where post-apoptotic remnants accumulate, CRP can be massively sequestered by this mechanism. We propose that in patients with SLE dead cell bound CRP is an important target for anti-CRP antibodies compromising the normal clearance process.
The complement system and CRP act together as an important backup mechanism for cells that have escaped the early PS-dependent clearance process (Gaipl et al., 2001). After opsonization, CRP activates complement and facilitates silent clearance by macrophages (Gershov et al., 2000). If complement is low, CRP does not enhance the phagocytosis of late apoptotic neutrophils (Hart et al., 2005). To exclude influences of the serum levels of complement of individual sera we performed the phagocytosis assays in the absence of complement. This argues for an Fcγ-receptor involvement during recognition and engulfment by macrophages of sensitized SNEC. Sensibilization of SNEC with either anti-dsDNA or with anti-CRP present in sera from patients with SLE significantly increased the production by macrophages of inflammatory cytokines. We have shown that SNEC exposes binding sites for anti-dsDNA and for CRP on their surfaces and suggest that autoantibodies recognize these targets and thereby form IC that can be taken up by macrophages in an inflammatory fashion. The maximal enhancement of uptake and inflammatory cytokine production was observed employing sera containing both anti-dsDNA and anti-CRP. This observation suggests that concomitant sensibilization with both autoantibody specificities results in a higher density of antibodies bound to the surfaces of the target particles. The spatial vicinity of the Fc portions is critical to trigger Fcγ-receptor mediated phagocytosis (Allen and Aderem, 1996).
Kenyon et al. (2011) recently showed that anti-C3 antibodies in sera from autoimmune mice inhibited uptake of apoptotic cells by blocking C3 recognition by mouse macrophages suggesting that autoantibodies against the dead cell opsonin C3 may contribute to a further suppression of apoptotic cell disposal increasing severity and/or exacerbations in SLE (Kenyon et al., 2011). On the first view this contradicts our results. However, phagocytosis assays crucially depend on experimental details. In contrast to the settings of Kenyon et al. (2011) our phagocytosis assays were performed under serum free conditions. After opsonisation, SNEC was washed twice and resuspended in serum free medium. Therefore, only molecules bound to the target-cells influenced the outcome of phagocytosis.
We employed anti-dsDNA negative sera from patients with SLE and depleted CRP and anti-SNEC, to analyze if circulating CRP is part of a major IC containing SNEC and anti-CRP. Although the indices of SNEC phagocytosis by macrophages were not increased by opsonisation with CRP and/or sensibilization with anti-CRP, the presence of anti-CRP and SNEC-bound CRP induced a strong TNF response of macrophages. The presence of anti-CRP at the phagocytic synapse formed by phagocytes and SNEC might be decisive to start the signaling process that results in cytokine production. Our findings that anti-opsonin antibodies shift the clearance toward inflammation are in agreement with previous findings by Rovere et al. (1999) who reported that anti-beta 2-GP1 antibodies bound to apoptotic cells skew their immunogenicity, enabling DCs to present their antigen with higher efficiency and secrete pro-inflammatory cytokines.
The fact that many opsonins for dead cells are also targeted by IgG autoimmune responses (Table 1) place them in the “crime scene” during the challenge of the tolerance in germinal centers of patients with SLE (Baumann et al., 2002). Anti-CRP in a patient with persistent deficiency for the early anti-inflammatory clearance of apoptotic cells provides the optimal condition to form ternary pro-inflammatory IC composed of anti-CRP, CRP, and SNEC. The Figure 5 shows a schematic representation of the usual and the alternative interactions among SNEC, opsonins, and phagocytes. We conclude that sensibilization by autoantibodies of SNEC, either directly or indirectly, shifts the clearance process by macrophages toward inflammation. The role of the individual receptors involved in this alternative and pathological phagocytosis pathway may differ amongst individual patients.
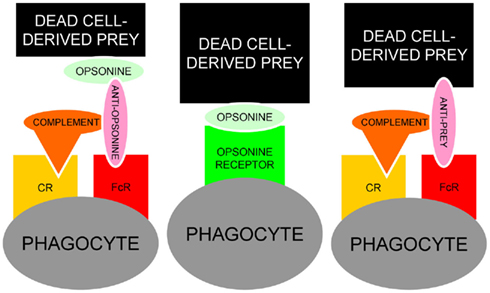
Figure 5. Schematic representation of the usual interaction between SNEC, opsonins and phagocytes (central). In the presence of autoantibodies, alternative models for the recognition by phagocytes of antibody-sensitized SNEC are represented. Anti-opsonin antibodies, e.g., anti-CRP (left) or directly binding anti-SNEC antibodies, e.g., anti-dsDNA (right) may shift the clearance of SNEC toward inflammation engaging the receptors for Fcγ and complement. In addition to autoantibodies against dsDNA and/or opsonins, low complement levels are prone to influence the clearance of post-apoptotic remnants.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was supported by the Masterswitch project of the European Union (EU), by a DFG research fellowship (Sandra Franz), by SFB 643 (Luis E. Munoz), the training Grant GK SFB 643 from the DFG (Christina Janko), an intramural grant of the University Erlangen-Nürnberg (Frauenförderung; Christina Janko), by the IZKF of the University Clinic of Erlangen (Martin Herrmann), and the K. und R. Wucherpfennigstiftung. (Martin Herrmann).
References
Aderem, A., and Underhill, D. M. (1999). Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 17, 593–623.
Allen, L. A., and Aderem, A. (1996). Molecular definition of distinct cytoskeletal structures involved in complement- and Fc receptor-mediated phagocytosis in macrophages. J. Exp. Med. 184, 627–637.
Anderson, H. A., Maylock, C. A., Williams, J. A., Paweletz, C. P., Shu, H., and Shacter, E. (2003). Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat. Immunol. 4, 87–91.
Arur, S., Uche, U. E., Rezaul, K., Fong, M., Scranton, V., Cowan, A. E., Mohler, W., and Han, D. K. (2003). Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev. Cell 4, 587–598.
Balasubramanian, K., and Schroit, A. J. (1998). Characterization of phosphatidylserine-dependent beta2-glycoprotein I macrophage interactions. Implications for apoptotic cell clearance by phagocytes. J. Biol. Chem. 273, 29272–29277.
Bassi, N., Ghirardello, A., Blank, M., Zampieri, S., Sarzi-Puttini, P., Mantovani, A., Shoenfeld, Y., and Doria, A. (2010). IgG anti-pentraxin 3 antibodies in systemic lupus erythematosus. Ann. Rheum. Dis. 69, 1704–1710.
Baumann, I., Kolowos, W., Voll, R. E., Manger, B., Gaipl, U., Neuhuber, W. L., Kirchner, T., Kalden, J. R., and Herrmann, M. (2002). Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 46, 191–201.
Becker, G. J., Waldburger, M., Hughes, G. R., and Pepys, M. B. (1980). Value of serum C-reactive protein measurement in the investigation of fever in systemic lupus erythematosus. Ann. Rheum. Dis. 39, 50–52.
Beer, A., Andre, S., Kaltner, H., Lensch, M., Franz, S., Sarter, K., Schulze, C., Gaipl, U. S., Kern, P., Herrmann, M., and Gabius, H. J. (2008). Human galectins as sensors for apoptosis/necrosis-associated surface changes of granulocytes and lymphocytes. Cytometry A 73, 139–147.
Bell, S. A., Faust, H., Schmid, A., and Meurer, M. (1998). Autoantibodies to C-reactive protein (CRP) and other acute-phase proteins in systemic autoimmune diseases. Clin. Exp. Immunol. 113, 327–332.
Bigler, C., Schaller, M., Perahud, I., Osthoff, M., and Trendelenburg, M. (2009). Autoantibodies against complement C1q specifically target C1q bound on early apoptotic cells. J. Immunol. 183, 3512–3521.
Bondanza, A., Zimmermann, V. S., Rovere-Querini, P., Turnay, J., Dumitriu, I. E., Stach, C. M., Voll, R. E., Gaipl, U. S., Bertling, W., Poschl, E., Kalden, J. R., Manfredi, A. A., and Herrmann, M. (2004). Inhibition of phosphatidylserine recognition heightens the immunogenicity of irradiated lymphoma cells in vivo. J. Exp. Med. 200, 1157–1165.
Bottcher, A., Gaipl, U. S., Furnrohr, B. G., Herrmann, M., Girkontaite, I., Kalden, J. R., and Voll, R. E. (2006). Involvement of phosphatidylserine, alphavbeta3, CD14, CD36, and complement C1q in the phagocytosis of primary necrotic lymphocytes by macrophages. Arthritis Rheum. 54, 927–938.
Du Clos, T. W., Zlock, L. T., and Rubin, R. L. (1988). Analysis of the binding of C-reactive protein to histones and chromatin. J. Immunol. 141, 4266–4270.
Enocsson, H., Sjowall, C., Skogh, T., Eloranta, M. L., Ronnblom, L., and Wettero, J. (2009). Interferon-alpha mediates suppression of C-reactive protein: explanation for muted C-reactive protein response in lupus flares? Arthritis Rheum. 60, 3755–3760.
Franz, S., Frey, B., Sheriff, A., Gaipl, U. S., Beer, A., Voll, R. E., Kalden, J. R., and Herrmann, M. (2006). Lectins detect changes of the glycosylation status of plasma membrane constituents during late apoptosis. Cytometry A 69, 230–239.
Franz, S., Herrmann, K., Fuhrnrohr, B., Sheriff, A., Frey, B., Gaipl, U. S., Voll, R. E., Kalden, J. R., Jack, H. M., and Herrmann, M. (2007). After shrinkage apoptotic cells expose internal membrane-derived epitopes on their plasma membranes. Cell Death Differ. 14, 733–742.
Gaipl, U. S., Kuenkele, S., Voll, R. E., Beyer, T. D., Kolowos, W., Heyder, P., Kalden, J. R., and Herrmann, M. (2001). Complement binding is an early feature of necrotic and a rather late event during apoptotic cell death. Cell Death Differ. 8, 327–334.
Gershov, D., Kim, S., Brot, N., and Elkon, K. B. (2000). C-Reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J. Exp. Med. 192, 1353–1364.
Hanayama, R., Tanaka, M., Miwa, K., Shinohara, A., Iwamatsu, A., and Nagata, S. (2002). Identification of a factor that links apoptotic cells to phagocytes. Nature 417, 182–187.
Hart, S. P., Alexander, K. M., Maccall, S. M., and Dransfield, I. (2005). C-reactive protein does not opsonize early apoptotic human neutrophils, but binds only membrane-permeable late apoptotic cells and has no effect on their phagocytosis by macrophages. J. Inflamm. (Lond.) 2, 5.
Hochberg, M. C. (1997). Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 40, 1725.
Janko, C., Schorn, C., Weidner, D., Sarter, K., Chaurio, R., Sheriff, A., Schett, G., and Munoz, L. E. (2009). Treatment with DNAse I fosters binding to nec PBMC of CRP. Autoimmunity 42, 286–288.
Jewell, W. S., Marnell, L. L., Rokeach, L. A., and Du Clos, T. W. (1993). C-reactive protein (CRP) binding to the Sm-D protein of snRNPS. Identification of a short polypeptide binding region. Mol. Immunol. 30, 701–708.
Kenyon, K. D., Cole, C., Crawford, F., Kappler, J. W., Thurman, J. M., Bratton, D. L., Boackle, S. A., and Henson, P. M. (2011). IgG autoantibodies against deposited C3 inhibit macrophage-mediated apoptotic cell engulfment in systemic autoimmunity. J. Immunol. 187, 2101–2111.
Kredich, N. M., Skyler, J. S., and Foote, L. J. (1973). Antibodies to native DNA in systemic lupus erythematosus. A technique of rapid and quantitative determination. Arch. Intern. Med. 131, 639–644.
Kretz, C. C., Norpo, M., Abeler-Dorner, L., Linke, B., Haust, M., Edler, L., Krammer, P. H., and Kuhn, A. (2010). Anti-annexin 1 antibodies: a new diagnostic marker in the serum of patients with discoid lupus erythematosus. Exp. Dermatol. 19, 919–921.
Maire, M. A., Barnet, M., Carpentier, N., Miescher, P. A., and Lambert, P. H. (1983). Identification of components of IC purified from human sera. I. Immune complexes purified from sera of patients with SLE. Clin. Exp. Immunol. 51, 215–224.
Manfredi, A. A., Rovere, P., Galati, G., Heltai, S., Bozzolo, E., Soldini, L., Davoust, J., Balestrieri, G., Tincani, A., and Sabbadini, M. G. (1998). Apoptotic cell clearance in systemic lupus erythematosus. I. Opsonization by antiphospholipid antibodies. Arthritis Rheum. 41, 205–214.
Manfredi, A. A., Rovere-Querini, P., Bottazzi, B., Garlanda, C., and Mantovani, A. (2008). Pentraxins, humoral innate immunity and tissue injury. Curr. Opin. Immunol. 20, 538–544.
Matsuda, J., Saitoh, N., Gohchi, K., Gotoh, M., and Tsukamoto, M. (1994). Anti-annexin V antibody in systemic lupus erythematosus patients with lupus anticoagulant and/or anticardiolipin antibody. Am. J. Hematol. 47, 56–58.
Munoz, L. E., Janko, C., Chaurio, R. A., Schett, G., Gaipl, U. S., and Herrmann, M. (2010a). IgG opsonized nuclear remnants from dead cells cause systemic inflammation in SLE. Autoimmunity 43, 232–235.
Munoz, L. E., Janko, C., Schulze, C., Schorn, C., Sarter, K., Schett, G., and Herrmann, M. (2010b). Autoimmunity and chronic inflammation – two clearance-related steps in the etiopathogenesis of SLE. Autoimmun. Rev. 10, 38–42.
Munoz, L. E., Lauber, K., Schiller, M., Manfredi, A. A., and Herrmann, M. (2010c). The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat. Rev. Rheumatol. 6, 280–289.
Munoz, L. E., Janko, C., Grossmayer, G. E., Frey, B., Voll, R. E., Kern, P., Kalden, J. R., Schett, G., Fietkau, R., Herrmann, M., and Gaipl, U. S. (2009). Remnants of secondarily necrotic cells fuel inflammation in systemic lupus erythematosus. Arthritis Rheum. 60, 1733–1742.
Nauta, A. J., Castellano, G., Xu, W., Woltman, A. M., Borrias, M. C., Daha, M. R., Van Kooten, C., and Roos, A. (2004). Opsonization with C1q and mannose-binding lectin targets apoptotic cells to dendritic cells. J. Immunol. 173, 3044–3050.
Ogden, C. A., and Elkon, K. B. (2006). Role of complement and other innate immune mechanisms in the removal of apoptotic cells. Curr. Dir. Autoimmun. 9, 120–142.
Oosting, J. D., Derksen, R. H., Bobbink, I. W., Hackeng, T. M., Bouma, B. N., and De Groot, P. G. (1993). Antiphospholipid antibodies directed against a combination of phospholipids with prothrombin, protein C, or protein S: an explanation for their pathogenic mechanism? Blood 81, 2618–2625.
Ravichandran, K. S., and Lorenz, U. (2007). Engulfment of apoptotic cells: signals for a good meal. Nat. Rev. Immunol. 7, 964–974.
Rovere, P., Peri, G., Fazzini, F., Bottazzi, B., Doni, A., Bondanza, A., Zimmermann, V. S., Garlanda, C., Fascio, U., Sabbadini, M. G., Rugarli, C., Mantovani, A., and Manfredi, A. A. (2000). The long pentraxin PTX3 binds to apoptotic cells and regulates their clearance by antigen-presenting dendritic cells. Blood 96, 4300–4306.
Rovere, P., Sabbadini, M. G., Vallinoto, C., Fascio, U., Recigno, M., Crosti, M., Ricciardi-Castagnoli, P., Balestrieri, G., Tincani, A., and Manfredi, A. A. (1999). Dendritic cell presentation of antigens from apoptotic cells in a proinflammatory context: role of opsonizing anti-beta2-glycoprotein I antibodies. Arthritis Rheum. 42, 1412–1420.
Sarter, K., Andre, S., Kaltner, H., Lensch, M., Schulze, C., Urbonaviciute, V., Schett, G., Herrmann, M., and Gabius, H. J. (2009). Detection and chromatographic removal of lipopolysaccharide in preparations of multifunctional galectins. Biochem. Biophys. Res. Commun. 379, 155–159.
Schaller, M., Bigler, C., Danner, D., Ditzel, H. J., and Trendelenburg, M. (2009). Autoantibodies against C1q in systemic lupus erythematosus are antigen-driven. J. Immunol. 183, 8225–8231.
Schlegel, R. A., and Williamson, P. (2001). Phosphatidylserine, a death knell. Cell Death Differ. 8, 551–563.
Scott, R. S., Mcmahon, E. J., Pop, S. M., Reap, E. A., Caricchio, R., Cohen, P. L., Earp, H. S., and Matsushima, G. K. (2001). Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211.
Seelen, M. A., Trouw, L. A., Van Der Hoorn, J. W., Fallaux-Van Den Houten, F. C., Huizinga, T. W., Daha, M. R., and Roos, A. (2003). Autoantibodies against mannose-binding lectin in systemic lupus erythematosus. Clin. Exp. Immunol. 134, 335–343.
Shoenfeld, Y., Szyper-Kravitz, M., Witte, T., Doria, A., Tsutsumi, A., Tatsuya, A., Dayer, J. M., Roux-Lombard, P., Fontao, L., Kallenberg, C. G., Bijl, M., Matthias, T., Fraser, A., Zandman-Goddard, G., Blank, M., Gilburd, B., and Meroni, P. L. (2007). Autoantibodies against protective molecules–C1q, C-reactive protein, serum amyloid P, mannose-binding lectin, and apolipoprotein A1: prevalence in systemic lupus erythematosus. Ann. N. Y. Acad. Sci. 1108, 227–239.
Siegert, C., Daha, M., Westedt, M. L., Van Der Voort, E., and Breedveld, F. (1991). IgG autoantibodies against C1q are correlated with nephritis, hypocomplementemia, and dsDNA antibodies in systemic lupus erythematosus. J. Rheumatol. 18, 230–234.
Sjowall, C., Bengtsson, A. A., Sturfelt, G., and Skogh, T. (2005). Anti-CRP autoantibody levels correlate with disease activity in systemic lupus erythematosus. Semin. Arthritis Rheum. 35, 65. [author reply 66].
Sjowall, C., Eriksson, P., Almer, S., and Skogh, T. (2002). Autoantibodies to C-reactive protein is a common finding in SLE, but not in primary Sjogren’s syndrome, rheumatoid arthritis or inflammatory bowel disease. J. Autoimmun. 19, 155–160.
Sjowall, C., Wettero, J., Bengtsson, T., Askendal, A., Almroth, G., Skogh, T., and Tengvall, P. (2007). Solid-phase classical complement activation by C-reactive protein (CRP) is inhibited by fluid-phase CRP-C1q interaction. Biochem. Biophys. Res. Commun. 352, 251–258.
Taylor, P. R., Carugati, A., Fadok, V. A., Cook, H. T., Andrews, M., Carroll, M. C., Savill, J. S., Henson, P. M., Botto, M., and Walport, M. J. (2000). A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J. Exp. Med. 192, 359–366.
Tincani, A., Spatola, L., Prati, E., Allegri, F., Ferremi, P., Cattaneo, R., Meroni, P., and Balestrieri, G. (1996). The anti-beta2-glycoprotein I activity in human anti-phospholipid syndrome sera is due to monoreactive low-affinity autoantibodies directed to epitopes located on native beta2-glycoprotein I and preserved during species’ evolution. J. Immunol. 157, 5732–5738.
Vogt, B., Fuhrnrohr, B., Muller, R., and Sheriff, A. (2007). CRP and the disposal of dying cells: consequences for systemic lupus erythematosus and rheumatoid arthritis. Autoimmunity 40, 295–298.
Volanakis, J. E., and Wirtz, K. W. (1979). Interaction of C-reactive protein with artificial phosphatidylcholine bilayers. Nature 281, 155–157.
Voll, R. E., Herrmann, M., Roth, E. A., Stach, C., Kalden, J. R., and Girkontaite, I. (1997). Immunosuppressive effects of apoptotic cells. Nature 390, 350–351.
Wold, R. T., Young, F. E., Tan, E. M., and Farr, R. S. (1968). Deoxyribonucleic acid antibody: a method to detect its primary interaction with deoxyribonucleic acid. Science 161, 806–807.
Zandman-Goddard, G., Blank, M., Langevitz, P., Slutsky, L., Pras, M., Levy, Y., Shovman, O., Witte, T., Doria, A., Rovensky, J., and Shoenfeld, Y. (2005). Anti-serum amyloid component P antibodies in patients with systemic lupus erythematosus correlate with disease activity. Ann. Rheum. Dis. 64, 1698–1702.
Keywords: immune complexes, opsonins, CRP, anti-dsDNA, inflammation, SLE
Citation: Janko C, Franz S, Munoz LE, Siebig S, Winkler S, Schett G, Lauber K, Sheriff A, van der Vlag J and Herrmann M (2011) CRP/anti-CRP antibodies assembly on the surfaces of cell remnants switches their phagocytic clearance toward inflammation. Front. Immun. 2:70. doi: 10.3389/fimmu.2011.00070
Received: 15 August 2011;
Paper pending published: 05 October 2011;
Accepted: 14 November 2011;
Published online: 02 December 2011.
Edited by:
Amiram Ariel, University of Haifa, IsraelReviewed by:
Silvano Sozzani, University of Brescia, ItalyPatrizia Rovere Querini, Fondazione Centro San Raffaele del Monte Tabor and Vita-Salute University, Italy
Copyright: © 2011 Janko, Franz, Munoz, Siebig, Winkler, Schett, Lauber, Sheriff, van der Vlag and Herrmann. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Sandra Franz, Department for Dermatology, Venerology and Allergology, Max Bürger Research Centre, University Leipzig, Johannisallee 30, 04103 Leipzig, Germany. e-mail: sandra.franz@medizin.uni-leipzig.de
†Christina Janko Sandra Franz and Luis E. Munoz have contributed equally to this work.