
- Faculty of Life Sciences, Kyoto Sangyo University, Kyoto, Japan
Over the past two decades, heat shock proteins (HSPs) have been implicated in inflammatory responses and autoimmunity. HSPs were originally believed to maintain protein quality control in the cytosol. However, they also exist extracellularly and appear to act as inflammatory factors. Recently, a growing body of evidence suggested that the other class of stress proteins such as, endoplasmic reticulum (ER) stress proteins, which originally act as protein quality control factors in the secretory pathway and are induced by ER stress in inflammatory lesions, also participate in inflammation and autoimmunity. The immunoglobulin heavy-chain binding protein (Bip)/glucose-regulated protein 78 (GRP78), calnexin, calreticulin, glucose-regulated protein 94 (GRP94)/gp96, oxygen regulated protein 150 (ORP150)/glucose-regulated protein 170 (GRP170), homocysteine-induced ER protein (Herp) and heat shock protein 47 (hsp47)/Serpin H1, which are expressed not only in the ER but also occasionally at the cell surface play pathophysiological roles in autoimmune and inflammatory diseases as pro- or anti-inflammatory factors. Here we describe the accumulating evidence of the participation of ER stress proteins in autoimmunity and inflammation and discuss the critical differences between the two classes of stress proteins.
Introduction
Inflammation is a typical sign of autoimmune diseases. Misdirected immune responses target self-antigens and induce severe inflammatory responses, which sometimes cause death. In addition to numerous components in autoimmune responses, heat shock proteins (HSPs) have been implicated in autoimmune and inflammatory diseases over the past two decades (Hauet-Broere et al., 2006; Van Eden et al., 2007). Many HSPs are so-called molecular chaperones and folding enzymes, which maintain protein folding in a cell (Bukau et al., 2006; Hartl, 2011). Because protein folding is easily impaired by various cytotoxic stresses, such as heat shock, cytotoxic chemicals, hypoxia, and inflammation, prokaryotic to higher eukaryotic organisms have evolved stress responses and HSPs. The transcription of HSPs and their subsequent protein expression are stimulated by cytotoxic stresses, and they immediately restore protein folding and cellular homeostasis to counter toxic stresses (Morimoto, 1998). Thus, HSPs could be postulated to act as intracellular protein homeostasis maintenance factors. However, HSPs have also been reported to be observed in the extracellular fluid (Njemini et al., 2011), and act as pro- and anti-inflammatory factors especially in autoimmune and inflammatory diseases in diverse manners. In inflammatory lesions, HSPs are upregulated by inflammatory stress and are released into the extracellular fluid. Then, the extracellular HSPs specifically induce proinflammatory cytokines and enhance the antigenicity of autoantigens through modulations of antigen presentation (Multhoff, 2006; Yokota and Fujii, 2010). However, the extracellular HSPs can also stimulate anti-inflammatory regulatory T cell responses, thereby inducing the negative feedback control of inflammation (Hauet-Broere et al., 2006; Van Eden et al., 2007). Indeed, immunization with HSP peptides prevents disease development in autoimmune model animals, such as adjuvant arthritis and collagen-induced arthritis (CIA; van Eden et al., 1988; Jorgensen et al., 1998; Wendling et al., 2000). Presently, besides HSPs, another class of stress proteins is also known to exist in eukaryotic cells. Endoplasmic reticulum (ER) stress proteins, the specialized factors involved in protein quality control in the secretory pathway (Hoseki et al., 2010; Araki and Nagata, 2011; Smith et al., 2011), are induced by ER stress, which is very different from cytosolic stress (Mori, 2000; Walter and Ron, 2011). However, similar to HSPs, ER stress proteins are also induced by inflammatory stress (Yoshida, 2007), suggesting that ER stress proteins might also participate in autoimmune and inflammatory responses. Here, we discuss the accumulating evidence that ER stress proteins and their autoantibodies play roles in autoimmune and inflammatory diseases.
Regular Functions of ER Stress Proteins
Before discussing the relevance of ER stress proteins in autoimmune diseases, we have briefly sketched the regular functions of ER stress proteins in a cell. Although the boundary between the two is ambiguous, ER stress proteins can be classified into two groups. The first group consists of molecular chaperones and folding enzymes, which assist the folding and assembly of newly synthesized proteins and prevent misfolding and aggregation of preexisting proteins. The other group consists of protein degradation factors, which mediate the clearance of proteins that should be degraded, such as misfolded proteins. The latter protein clearance pathway is called the ER-associated degradation (ERAD) pathway (McCracken and Brodsky, 1996; Brodsky and McCracken, 1997).
The ER is located at the starting point of the protein secretory pathway. Secretory or membrane proteins are first cotranslationally transported into the ER from cytosolic ribosomes, modified and properly folded, and then translocated to the next step of the secretory pathway (Figures 1A,B; Ni and Lee, 2007; Araki and Nagata, 2011). After the ER, proteins pass through the Golgi apparatus and are finally secreted to the cell surface through a secretory vesicle. In the ER, most proteins are glycosylated and covalently crosslinked with disulfide bonds. Such protein modifications are believed necessary for the structural stabilization of the proteins destined to be secreted into the extracellular environment (Ni and Lee, 2007; Araki and Nagata, 2011). Calnexin and calreticulin are lectin-like chaperones that specifically maintain the folding of glycosylated proteins (Williams, 2006). Protein disulfide isomerase (PDI) is an oxidoreductase that mediates protein disulfide bond formation and isomerization (Tu and Weissman, 2004; Ellgaard and Ruddock, 2005). The immunoglobulin heavy-chain binding protein (Bip)/glucose-regulated protein 78 (GRP78) is a hsp70-type molecular chaperone that maintains protein folding (Ni and Lee, 2007; Araki and Nagata, 2011). Glucose-regulated protein 94 (GRP94)/gp96 and oxygen regulated protein 150 (ORP150) are hsp90 and hsp70 family molecular chaperones, respectively; their functions remain unclear (Ni and Lee, 2007; Araki and Nagata, 2011). Another characteristic stress protein in the ER is heat shock protein 47 (hsp47)/Serpin H1, the only heat shock regulated protein in the ER, is the collagen-specific molecular chaperone (Nagata, 2003; Ishida and Nagata, 2011). Using these molecular chaperones and folding enzymes, secretory and membrane proteins are properly folded in the ER and then transported to the Golgi apparatus. Other chaperones and enzymes were precisely described in a recent article (Araki and Nagata, 2011).
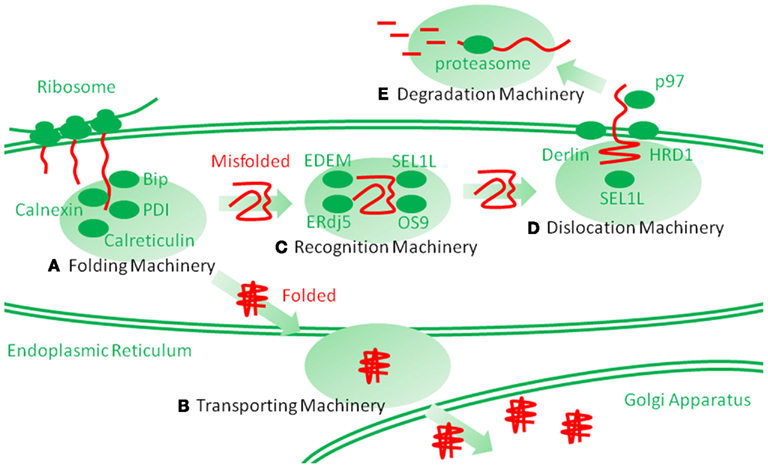
Figure 1. Schematic representation of endoplasmic reticulum (ER) protein folding and ER-associated degradation (ERAD). Secretory and membrane proteins are cotranslationally transported into the ER by cytosolic ribosomes. Newly synthesized unfolded polypeptide are then captured by molecular chaperones and protein folding enzymes (A), which involve immunoglobulin heavy-chain binding protein (Bip), calnexin, calreticulin, protein disulfide isomerase (PDI) and heat shock protein 47 (hsp47), and are properly folded. The folded polypeptides are transported into the Golgi apparatus through transporting machinery (B). However, the proteins that are improperly folded are specifically recognized and isolated by ERAD recognition machinery (C), which involves ERAD-enhancing mannosidase-like protein (EDEM), ERdj5, Bip, SEL1L, osteosarcoma 9 (OS9), and XBP1. The misfolded proteins are then transferred to the dislocation machinery (D), which involves HRD1, Derlin, Herp, and p97. Then, the proteins are dislocated into the cytosol through a putative narrow pore. The dislocated proteins are ubiquitinated by ERAD ubiquitin ligases, such as HRD1, and are finally degraded by huge cytosolic protease complexes, the proteasome (E), into peptides.
Improperly folded proteins are strictly retained in the ER via anchoring by ER chaperones and enter the ERAD pathway (Araki and Nagata, 2011; Smith et al., 2011). ERAD is a multi-step mechanism, which can be divided into the following three steps: (1) the substrate is recognized and isolated, (2) the substrate is dislocated from the ER into the cytosol, and (3) the substrate is then degraded by the ubiquitin proteasome system in the cytosol (Figures 1C–E; Hershko and Ciechanover, 1998). Thereby, the clearance of the proteins from the ER is completed. Among these steps, the first step is mediated by ERAD-enhancing mannosidase-like protein 1 (EDEM-1), ERdj5, Bip, Osteosarcoma 9 (OS9), XTP3 transactivating gene B (XTP3B), and SEL1L (Hosokawa et al., 2001; Ushioda et al., 2008; Hagiwara et al., 2011; Ushioda and Nagata, 2011). The substrate is directly transferred from the calnexin/calreticulin-mediated folding cycle to the EDEM-1-containing degradation recognition complex (Molinari et al., 2003; Oda et al., 2003). The substrate is then transferred from EDEM-1 and SEL1L to the so-called ERAD complex, postulated to form a protein dislocation channel (Mueller et al., 2006, 2008; Christianson et al., 2008). ER stress proteins, such as HRD1/synoviolin, Derlin, and homocysteine-induced ER protein (Herp), are involved in this supramolecular complex (Lilley and Ploegh, 2004, 2005; Oda et al., 2006). The substrate is then dislocated into the cytosol from the ER. During dislocation, the substrate is ubiquitinated by the ER ubiquitin ligase and pulled by p97/VCP from the cytosolic side (Ye et al., 2001, 2003; Tsai et al., 2002). Finally, in the third step, the ubiquitinated substrate is degraded by the cytosolic complex protease, the proteasome. Detailed comprehensive descriptions of the ERAD pathways can be found elsewhere (Araki and Nagata, 2011; Smith et al., 2011).
ER Stress
The stress response for HSPs, called heat shock response, is mediated by transcription factors called heat shock factors (HSFs; Morimoto, 1998). Similarly, the stress response for ER stress proteins, called the unfolded protein response (UPR), is mediated by the unconventional transcription factors, ATF6 and XBP1, and a translation and apoptosis regulating factor, PERK (Figure 2; Mori, 2000; Walter and Ron, 2011). The UPR can be experimentally stimulated by chemical compounds that specifically interfere with protein glycosylation and disulfide bond formation because these protein modifications are necessary for the structural stability of secretory and membrane proteins. In addition, perturbation of calcium storage in the ER causes impairments in the quality of ER proteins because the ER has a high calcium concentration, and many of the ER proteins need calcium ions. Alternatively, the UPR is physiologically activated during plasma cell differentiation in order to expand the protein folding capacity for massive production of immunoglobulins. The activation of the UPR has been observed in various neurodegenerative diseases, inflammatory diseases, and viral infections (Yoshida, 2007). Thus, the UPR can be postulated as a completely independent pathway from cytosolic stresses. Both cytosolic and ER stresses are commonly stimulated by inflammation. Although several studies suggest that ER stress protein levels correlate with inflammatory pathogenesis, the roles of ER stress and ER stress proteins in autoimmune and inflammatory diseases remain unclear. Thus, in addition to the heat shock response, the other axis of the cellular stress response might provide new insights into disease mechanisms and clinical treatments.
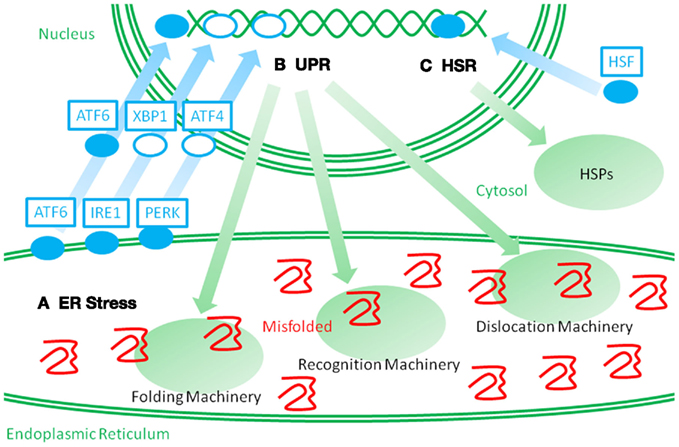
Figure 2. Schematic representation of two distinct stress responses. When misfolded proteins accumulate in the ER, they perturb ER protein homeostasis and organelle functions. Such perturbations are called ER stressors (A). ER stress leads to the activation of membrane spanning stress sensors. ATF6 is a membrane spanning transcription factor that is processed and released from the ER membrane under ER stress, and the stress signal is then transmitted to the nucleus. IRE1 and PERK activate the transcription factors, XBP1 and ATF4, and stimulate a stress response. Such stress responses are called an unfolded protein response (UPR) (B). UPR leads to an upregulation of ER stress proteins, which consist of molecular chaperones and folding enzymes, and ERAD recognition and dislocation factors. Such cross-membrane signal transduction is very different from the heat shock response (HSR) (C). When the cytosolic stresses perturb the cytosolic protein homeostasis, heat shock factor (HSF) is activated and transmits the signal into the nucleus. Then, an upregulation of HSPs is induced. It is a challenge to recover cytosolic protein homeostasis.
Bip/GRP78
The 68-kDa glycoprotein was identified through exploration of autoantigens that predominantly immunoreact with sera from rheumatoid arthritis (RA) patients and thus expected to be associated with RA pathogenesis (Blass et al., 1995, 1997, 1998). Subsequently, the 68-kDa antigen was demonstrated to be identical to the ER resident hsp70 family chaperone Bip (Blass et al., 2001; Corrigall et al., 2001). This crucial ER resident stress protein appears to be expressed at the cell surface and acts as an autoantigen in RA. Indeed, 30–75% of RA patients exhibit Bip overexpression in synovial fluid or autoantibodies to Bip in serum (Blass et al., 2001; Corrigall et al., 2001; Shoda et al., 2011). Although the exact roles of extracellular Bip and anti-Bip autoantibodies in RA pathogenesis remain debatable, three possible functions can be considered. First, extracellular Bip and its antibody may result from inflammatory stress during RA progression, and they might have no pathophysiological roles. Because the UPR is stimulated through inflammation and ER stress proteins, including Bip, are subsequently induced (Yoshida, 2007), Bip could be simply a specific and abundant autoantigen in the inflammatory lesion that is unrelated to RA pathogenesis. Second, the autoantigen might act as an anti-inflammatory factor, similar to HSPs. Several studies suggested that extracellular Bip stimulates the production of the anti-inflammatory cytokines, IL-4 and IL-10, through specific T lymphocytes (Blass et al., 2001; Corrigall et al., 2001, 2004; Bodman-Smith et al., 2003; Brownlie et al., 2006; Panayi and Corrigall, 2006). Furthermore, the pre-administration of Bip protein to mice prevents the in vivo induction of adjuvant arthritis or CIA, both of which are well-known artificial models of autoimmune diseases (Corrigall et al., 2001; Brownlie et al., 2006). Third, the Bip antigen could act as a proinflammatory factor. Bip positively causes immunological responses and inflammation. Recently, Shoda et al. (2011) reported that in addition to the intact Bip antibody, anti-citrullinated Bip (ctBip) antibody is frequently detected in RA patients. The ctBip protein, but not intact Bip, enhances anti-citrullin antibodies and worsens arthritis symptoms in a mouse model of adjuvant arthritis. Citrullination is protein modification in which an arginine residue is converted into a citrulline residue by a specific intracellular enzyme, peptidylarginine deiminase (PAD; Vossenaar et al., 2003). The anti-citrullinated peptide/protein antibody (ACPA) is frequently detected in RA patients (Vincent et al., 2005; Suzuki et al., 2007; van Venrooij et al., 2011). Although the reasons why citrullination is frequently observed and how it participates in RA pathogenesis remains unclear, the relationship between stress proteins and this specific protein modification suggests an undescribed crosstalk between inflammatory stress and disease-specific protein modifications in RA pathogenesis. In addition to RA, the anti-Bip autoantibody is also detected in another autoimmune and inflammatory disease, systemic lupus erythematosus (SLE; Casciola-Rosen et al., 1994; Weber et al., 2010), in which its pathophysiological role remains unknown.
HSP47
Hsp47 is an ER resident molecular chaperone; it is the only HSP in the ER. Hsp47 specifically maintains collagen biosynthesis (Nagata, 2003; Ishida and Nagata, 2011). Its gene disruption in mice causes significant reductions in mature collagens in connective tissues, resulting in embryonic lethality (Nagai et al., 2000; Marutani et al., 2004; Matsuoka et al., 2004). Several studies showed that the levels of anti-hsp47 autoantibody are specifically increased in RA patients (Hattori et al., 1998, 2000, 2001, 2003, 2005). However, little is known about how hsp47 and its autoantibody correlate with RA pathogenesis. In addition to RA, the levels of the autoantibody to hsp47 are also increased in other autoimmune diseases, such as SLE, Sjögren’s syndrome (SjS), mixed connective tissue disease (MCTD), systemic sclerosis (SSc), and non-specific idiopathic pneumonia (Yokota et al., 2003; Fujimoto et al., 2004; Kakugawa et al., 2008). Most of these diseases can be considered connective tissue diseases in which an upregulation of various types of collagen is observed. The expression profiles of collagens and hsp47 are fully consistent in both healthy (Masuda et al., 1998; Yamamura et al., 1998; Hirata et al., 1999; Yasuda et al., 2002) and diseased conditions (Masuda et al., 1994; Naitoh et al., 2001; Sato et al., 2008). Hsp47 might be the protein that stands at the junction of stress, the extracellular matrix (ECM) biogenesis, and autoimmune/connective tissue diseases.
HERP
Lupus nephritis, which is a kidney inflammatory disorder, is one of the manifestations of SLE, a complex autoimmune disease. Among the variety of autoantibodies that are detected in SLE patients, the anti-double-stranded DNA (dsDNA) antibody, which is a type of anti-nuclear antibody (ANA), is most characteristic of SLE and appears to significantly contribute to the pathogenesis of lupus nephritis (Isenberg et al., 2007). Although administration of dsDNA failed to initiate antibody production (Madaio et al., 1984), nucleosome-forming dsDNA elicited the anti-dsDNA antibody production (Rumore and Steinman, 1990; Casciola-Rosen et al., 1994; Voynova et al., 2005), suggesting that proteins like histone can work as an adjuvant for enhancing the antigenicity of dsDNA. Another possibility for anti-dsDNA antibody production is elicitation by cross-reactive protein antigens. Several proteins have been reported to cross-react with the anti-dsDNA antibody (Isenberg et al., 2007). Among them, α-actinin, which is an actin-associated protein, might be a potent candidate for the original antigen, which evokes anti-dsDNA antibody production (Mostoslavsky et al., 2001; Deocharan et al., 2002). However, a dysregulation of the negative selection of B-cell clones, which produce autoantibodies, occurs in these diseases.
Recently, Herp was suggested as a possible cross-reactive antigen of the anti-dsDNA antibody (Hirabayashi et al., 2010). Herp is an ER resident membrane protein that is involved in the ERAD complex and is induced by UPR (Kokame et al., 2000, 2001; Okuda-Shimizu and Hendershot, 2007; Kny et al., 2011; Marutani et al., 2011). Thus, Herp is an ER stress protein, can be induced by inflammatory ER stress. Among the known candidates for the original antigen of the anti-dsDNA antibody including α-actinin, only Herp can be induced by inflammatory stress. Thus, Herp can be the key component that connects inflammatory stress responses and anti-dsDNA antibody production in SLE.
Calreticulin
The anti-Ro/SS-A antibody, which is also considered an ANA, is one of the most studied and crucial markers in many autoimmune diseases, such as SLE, Sjs, SSc, and RA (Anderson et al., 1962; Clark et al., 1969; Alspaugh and Tan, 1975; Schulte-Pelkum et al., 2009; Defendenti et al., 2011). Although it has a strong, well-established association with autoimmune diseases, especially with neonatal lupus, it is unclear how it participates in autoimmune diseases (Schulte-Pelkum et al., 2009; Defendenti et al., 2011). Recently, it was shown that the anti-Ro/SS-A antibody can be categorized into two types, each of which specifically recognizes the 52-kDa (Ro52) and 60-kDa (Ro60) antigens. Ro52 and Ro60 form a Ro ribonucleoprotein (RNP) complex with a couple of other proteins, while the intracellular functions and pathogenic relevancies of those antigens remain unclear (Schulte-Pelkum et al., 2009; Defendenti et al., 2011).
Calreticulin is an essential ER resident lectin-like chaperone, which is induced by ER stress (Yoshida et al., 1998; Mesaeli et al., 1999; Williams, 2006). Calreticulin was first postulated as the Ro/SS-A autoantigen (Collins et al., 1989; McCauliffe et al., 1990). However, it was later demonstrated that calreticulin itself was not an antigen (Lu et al., 1993; Boehm et al., 1994), but it maintained Ro RNP complex formation through its chaperone activity and modulated the antigenicity of the complex (Cheng et al., 1996; Staikou et al., 2003). Although it was shown that calreticulin was not the autoantigen for the Ro/SS-A autoantibody, the anti-calreticulin autoantibody has been independently observed in SLE patients (Boehm et al., 1994), complete congenital heart block (CCHB; Orth et al., 1996), which is one of the manifestations of SLE, and inflammatory bowel disease (Watanabe et al., 2006).
Calnexin, GRP94, and ORP150
Calnexin is an ER resident lectin-like chaperone (Wada et al., 1991, 1997; Hebert et al., 1995; Deprez et al., 2005), which is not induced or is only mildly induced by ER stress (Kamauchi et al., 2005). An autoantibody to calnexin is observed in SLE patients, while its biological relevance remains unknown (Weber et al., 2010).
GRP94 is an essential ER resident hsp90 family chaperone, which is induced by cytosolic and ER stress (Wanderling et al., 2007; Eletto et al., 2010). Anti-GRP94 autoantibodies have been observed in SLE patients (Boehm et al., 1994), RA (Weber et al., 2010), and myasthenia gravis (MG; Suzuki et al., 2011). In addition, cell surface expression of GRP94 itself is detected in patients with type I diabetes (Pagetta et al., 2003) and in GRP94 transgenic mice (Liu et al., 2003). In GRP94 transgenic mice, lupus-like autoimmune disorder and systemic inflammation are induced, suggesting that extracellular GRP94 and its autoantibody have proinflammatory effects in autoimmune diseases.
ORP150 is an essential ER resident hsp70 chaperone that is induced by ER stress and hypoxia (Kitao et al., 2001; Ni and Lee, 2007). An autoantibody to ORP150 is detected in patients with atherosclerosis and type I diabetes (Tsukamoto et al., 1996; Nakatani et al., 2006), but it remains unclear if they have a pathophysiological role in autoimmune responses.
Regulation of ER Stress Protein Distribution
As discussed above, the critical difference between HSPs and ER stress proteins is based on the stress pathway they are induced through. One is through a heat shock response, and the other is through the UPR. However, another striking difference exists, i.e., HSPs are originally distributed in the cytosol. In patients with inflammatory diseases, HSPs are observed in extracellular fluid; this could be due to the destruction of the plasma membrane that accompanies stress-induced apoptosis, necrosis, and phagocytosis or alternative active secretion through exosomes. On the other hand, ER stress proteins are originally located in the secretory pathway. Thus, ER stress proteins can potentially be secreted into the cell surface, after which they escape from the ER retention mechanism. Indeed, some ER stress proteins are expressed on the cell surface without any cell destruction, e.g., calnexin (Okazaki et al., 2000), calreticulin (Jeffery et al., 2011), Bip (Delpino and Castelli, 2002), GRP94 (Altmeyer et al., 1996), and hsp47 (Hebert et al., 1999). How they escape, how they are retained, and whether this leaky expression correlates with autoimmune pathogenesis remains unknown. However, this apparent difference between the two types of stress proteins might cause physiological differences, therefore, this should be examined in autoimmune and inflammatory diseases.
Conclusion
Several studies suggested that ER stress proteins are potent immunomodulating components in autoimmune and inflammatory diseases. Although their pathogenic relevancies remain unclear, some of them appear to participate in disease progression. Their inducibility by inflammatory stress and their original distribution in the secretory pathway could be advantageous for participation in autoimmune and inflammatory responses. Until now, the major evidence of the involvement of ER stress proteins in autoimmune diseases appears to be autoantigens. This situation can be compared to that of HSPs two decades ago. Since then, much has been revealed about the functions of HSPs in addition to being autoantigens. Because HSPs and ER stress proteins share several properties as molecular chaperones, we may be able to expect unidentified and important roles of ER stress proteins in autoimmune response as well as HSPs. Indeed, another issue of the stress proteins in autoimmune responses is their potential immunomodulating abilities. Because chaperones can broadly associate with other proteins, including autoantigens and recruit antigen presentation pathway, they can work as endogenous adjuvants in immune responses. In this review, we focused on the roles of ER stress proteins as autoantigens. However, several other issues that are related to ER stress are also important, e.g., HLA-B27 misfolding in ankylosing spondylitis and anti-apoptotic function of HRD1/synoviolin in synovial cells in RA (Yoshida, 2007; Todd et al., 2008; Yagishita et al., 2008; Colbert et al., 2010). At the moment, the features of ER stress proteins in autoimmunity remain largely unclear. The potential relevance of ER stress proteins in many autoimmune and inflammatory diseases makes them potential clinical targets.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Shinichi Yokota (Sapporo Medical University) for critical reading of the manuscript. This work was supported by Grant-in-Aid for Creative Scientific Research (19G50314; to Kazuhiro Nagata), Grant-in-Aid for Young Scientists (B) (22710193), and Grant-in-Aid for Scientific Research on Innovative Areas (23122520; to Daisuke Morito).
References
Alspaugh, M. A., and Tan, E. M. (1975). Antibodies to cellular antigens in Sjogren’s syndrome. J. Clin. Invest. 55, 1067–1073.
Altmeyer, A., Maki, R. G., Feldweg, A. M., Heike, M., Protopopov, V. P., Masur, S. K., and Srivastava, P. K. (1996). Tumor-specific cell surface expression of the-KDEL containing, endoplasmic reticular heat shock protein gp96. Int. J. Cancer 69, 340–349.
Anderson, J. R., Gray, K. G., Beck, J. S., Buchanan, W. W., and Mc, E. A. (1962). Precipitating auto-antibodies in the connective tissue diseases. Ann. Rheum. Dis. 21, 360–369.
Araki, K., and Nagata, K. (2011). Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 3.
Blass, S., Haferkamp, C., Specker, C., Schwochau, M., Schneider, M., and Schneider, E. M. (1997). Rheumatoid arthritis: autoreactive T cells recognising a novel 68k autoantigen. Ann. Rheum. Dis. 56, 317–322.
Blass, S., Meier, C., Vohr, H. W., Schwochau, M., Specker, C., and Burmester, G. R. (1998). The p68 autoantigen characteristic of rheumatoid arthritis is reactive with carbohydrate epitope specific autoantibodies. Ann. Rheum. Dis. 57, 220–225.
Blass, S., Specker, C., Lakomek, H. J., Schneider, E. M., and Schwochau, M. (1995). Novel 68 kDa autoantigen detected by rheumatoid arthritis specific antibodies. Ann. Rheum. Dis. 54, 355–360.
Blass, S., Union, A., Raymackers, J., Schumann, F., Ungethum, U., Muller-Steinbach, S., De Keyser, F., Engel, J. M., and Burmester, G. R. (2001). The stress protein BiP is overexpressed and is a major B and T cell target in rheumatoid arthritis. Arthritis Rheum. 44, 761–771.
Bodman-Smith, M. D., Corrigall, V. M., Kemeny, D. M., and Panayi, G. S. (2003). BiP, a putative autoantigen in rheumatoid arthritis, stimulates IL-10-producing CD8-positive T cells from normal individuals. Rheumatology (Oxford) 42, 637–644.
Boehm, J., Orth, T., Van Nguyen, P., and Soling, H. D. (1994). Systemic lupus erythematosus is associated with increased auto-antibody titers against calreticulin and grp94, but calreticulin is not the Ro/SS-A antigen. Eur. J. Clin. Invest. 24, 248–257.
Brodsky, J. L., and McCracken, A. A. (1997). ER-associated and proteasomemediated protein degradation: how two topologically restricted events came together. Trends Cell Biol. 7, 151–156.
Brownlie, R. J., Myers, L. K., Wooley, P. H., Corrigall, V. M., Bodman-Smith, M. D., Panayi, G. S., and Thompson, S. J. (2006). Treatment of murine collagen-induced arthritis by the stress protein BiP via interleukin-4-producing regulatory T cells: a novel function for an ancient protein. Arthritis Rheum. 54, 854–863.
Bukau, B., Weissman, J., and Horwich, A. (2006). Molecular chaperones and protein quality control. Cell 125, 443–451.
Casciola-Rosen, L. A., Anhalt, G., and Rosen, A. (1994). Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J. Exp. Med. 179, 1317–1330.
Cheng, S. T., Nguyen, T. Q., Yang, Y. S., Capra, J. D., and Sontheimer, R. D. (1996). Calreticulin binds hYRNA and the 52-kDa polypeptide component of the Ro/SS-A ribonucleoprotein autoantigen. J. Immunol. 156, 4484–4491.
Christianson, J. C., Shaler, T. A., Tyler, R. E., and Kopito, R. R. (2008). OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 10, 272–282.
Clark, G., Reichlin, M., and Tomasi, T. B. Jr. (1969). Characterization of a soluble cytoplasmic antigen reactive with sera from patients with systemic lupus erythmatosus. J. Immunol. 102, 117–122.
Colbert, R. A., Delay, M. L., Klenk, E. I., and Layh-Schmitt, G. (2010). From HLA-B27 to spondyloarthritis: a journey through the ER. Immunol. Rev. 233, 181–202.
Collins, J. H., Xi, Z. J., Alderson-Lang, B. H., Treves, S., and Volpe, P. (1989). Sequence homology of a canine brain calcium-binding protein with calregulin and the human Ro/SS-A antigen. Biochem. Biophys. Res. Commun. 164, 575–579.
Corrigall, V. M., Bodman-Smith, M. D., Brunst, M., Cornell, H., and Panayi, G. S. (2004). Inhibition of antigen-presenting cell function and stimulation of human peripheral blood mononuclear cells to express an antiinflammatory cytokine profile by the stress protein BiP: relevance to the treatment of inflammatory arthritis. Arthritis Rheum. 50, 1164–1171.
Corrigall, V. M., Bodman-Smith, M. D., Fife, M. S., Canas, B., Myers, L. K., Wooley, P., Soh, C., Staines, N. A., Pappin, D. J., Berlo, S. E., Van Eden, W., Van Der Zee, R., Lanchbury, J. S., and Panayi, G. S. (2001). The human endoplasmic reticulum molecular chaperone BiP is an autoantigen for rheumatoid arthritis and prevents the induction of experimental arthritis. J. Immunol. 166, 1492–1498.
Defendenti, C., Atzeni, F., Spina, M. F., Grosso, S., Cereda, A., Guercilena, G., Bollani, S., Saibeni, S., and Puttini, P. S. (2011). Clinical and laboratory aspects of Ro/SSA-52 autoantibodies. Autoimmun. Rev. 10, 150–154.
Delpino, A., and Castelli, M. (2002). The 78 kDa glucose-regulated protein (GRP78/BIP) is expressed on the cell membrane, is released into cell culture medium and is also present in human peripheral circulation. Biosci. Rep. 22, 407–420.
Deocharan, B., Qing, X., Lichauco, J., and Putterman, C. (2002). Alpha-actinin is a cross-reactive renal target for pathogenic anti-DNA antibodies. J. Immunol. 168, 3072–3078.
Deprez, P., Gautschi, M., and Helenius, A. (2005). More than one glycan is needed for ER glucosidase II to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol. Cell 19, 183–195.
Eletto, D., Dersh, D., and Argon, Y. (2010). GRP94 in ER quality control and stress responses. Semin. Cell Dev. Biol. 21, 479–485.
Ellgaard, L., and Ruddock, L. W. (2005). The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 6, 28–32.
Fujimoto, M., Hamaguchi, Y., Yazawa, N., Komura, K., Takehara, K., and Sato, S. (2004). Autoantibodies to a collagen-specific molecular chaperone, heat-shock protein 47, in systemic sclerosis. Clin. Exp. Immunol. 138, 534–539.
Hagiwara, M., Maegawa, K., Suzuki, M., Ushioda, R., Araki, K., Matsumoto, Y., Hoseki, J., Nagata, K., and Inaba, K. (2011). Structural basis of an ERAD pathway mediated by the ER-resident protein disulfide reductase ERdj5. Mol. Cell 41, 432–444.
Hartl, F. U. (2011). Chaperone-assisted protein folding: the path to discovery from a personal perspective. Nat. Med. 17, 1206–1210.
Hattori, T., Fujisawa, T., Sasaki, K., Yutani, Y., Nakanishi, T., Takahashi, K., and Takigawa, M. (1998). Isolation and characterization of a rheumatoid arthritis-specific antigen (RA-A47) from a human chondrocytic cell line (HCS-2/8). Biochem. Biophys. Res. Commun. 245, 679–683.
Hattori, T., Kawaki, H., Kubota, S., Yutani, Y., De Crombrugghe, B., Von Der Mark, K., and Takigawa, M. (2003). Downregulation of a rheumatoid arthritis-related antigen (RA-A47) by ra-a47 antisense oligonucleotides induces inflammatory factors in chondrocytes. J. Cell. Physiol. 197, 94–102.
Hattori, T., Kubota, S., Yutani, Y., Fujisawa, T., Nakanishi, T., Takahashi, K., and Takigawa, M. (2001). Change in cellular localization of a rheumatoid arthritis-related antigen (RA-A47) with downregulation upon stimulation by inflammatory cytokines in chondrocytes. J. Cell. Physiol. 186, 268–281.
Hattori, T., Takahash, K., Yutani, Y., Fujisawa, T., Nakanishi, T., and Takigawa, M. (2000). Rheumatoid arthritis-related antigen 47kDa (RA-A47) is a product of colligin-2 and acts as a human HSP47. J. Bone Miner. Metab. 18, 328–334.
Hattori, T., Von Der Mark, K., Kawaki, H., Yutani, Y., Kubota, S., Nakanishi, T., Eberspaecher, H., De Crombrugghe, B., and Takigawa, M. (2005). Downregulation of rheumatoid arthritis-related antigen RA-A47 (HSP47/colligin-2) in chondrocytic cell lines induces apoptosis and cell-surface expression of RA-A47 in association with CD9. J. Cell. Physiol. 202, 191–204.
Hauet-Broere, F., Wieten, L., Guichelaar, T., Berlo, S., Van Der Zee, R., and Van Eden, W. (2006). Heat shock proteins induce T cell regulation of chronic inflammation. Ann. Rheum. Dis. 65(Suppl. 3), iii65–iii68.
Hebert, C., Norris, K., Della Coletta, R., Reynolds, M., Ordonez, J., and Sauk, J. J. (1999). Cell surface colligin/Hsp47 associates with tetraspanin protein CD9 in epidermoid carcinoma cell lines. J. Cell. Biochem. 73, 248–258.
Hebert, D. N., Foellmer, B., and Helenius, A. (1995). Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 81, 425–433.
Hirabayashi, Y., Oka, Y., Ikeda, T., Fujii, H., Ishii, T., Sasaki, T., and Harigae, H. (2010). The endoplasmic reticulum stress-inducible protein, Herp, is a potential triggering antigen for anti-DNA response. J. Immunol. 184, 3276–3283.
Hirata, H., Yamamura, I., Yasuda, K., Kobayashi, A., Tada, N., Suzuki, M., Hirayoshi, K., Hosokawa, N., and Nagata, K. (1999). Separate cis-acting DNA elements control cell type- and tissue-specific expression of collagen binding molecular chaperone HSP47. J. Biol. Chem. 274, 35703–35710.
Hoseki, J., Ushioda, R., and Nagata, K. (2010). Mechanism and components of endoplasmic reticulum-associated degradation. J. Biochem. 147, 19–25.
Hosokawa, N., Wada, I., Hasegawa, K., Yorihuzi, T., Tremblay, L. O., Herscovics, A., and Nagata, K. (2001). A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2, 415–422.
Isenberg, D. A., Manson, J. J., Ehrenstein, M. R., and Rahman, A. (2007). Fifty years of anti-ds DNA antibodies: are we approaching journey’s end? Rheumatology (Oxford) 46, 1052–1056.
Ishida, Y., and Nagata, K. (2011). Hsp47 as a collagen-specific molecular chaperone. Meth. Enzymol. 499, 167–182.
Jeffery, E., Peters, L. R., and Raghavan, M. (2011). The polypeptide binding conformation of calreticulin facilitates its cell-surface expression under conditions of endoplasmic reticulum stress. J. Biol. Chem. 286, 2402–2415.
Jorgensen, C., Gedon, E., Jaquet, C., and Sany, J. (1998). Gastric administration of recombinant 65 kDa heat shock protein delays the severity of type II collagen induced arthritis in mice. J. Rheumatol. 25, 763–767.
Kakugawa, T., Yokota, S., Mukae, H., Kubota, H., Sakamoto, N., Mizunoe, S., Matsuoka, Y., Kadota, J., Fujii, N., Nagata, K., and Kohno, S. (2008). High serum concentrations of autoantibodies to HSP47 in nonspecific interstitial pneumonia compared with idiopathic pulmonary fibrosis. BMC Pulm. Med. 8, 23. doi:10.1186/1471-2466-8-23
Kamauchi, S., Nakatani, H., Nakano, C., and Urade, R. (2005). Gene expression in response to endoplasmic reticulum stress in Arabidopsis thaliana. FEBS J. 272, 3461–3476.
Kitao, Y., Ozawa, K., Miyazaki, M., Tamatani, M., Kobayashi, T., Yanagi, H., Okabe, M., Ikawa, M., Yamashima, T., Stern, D. M., Hori, O., and Ogawa, S. (2001). Expression of the endoplasmic reticulum molecular chaperone (ORP150) rescues hippocampal neurons from glutamate toxicity. J. Clin. Invest. 108, 1439–1450.
Kny, M., Standera, S., Hartmann-Petersen, R., Kloetzel, P. M., and Seeger, M. (2011). Herp regulates Hrd1-mediated ubiquitylation in a ubiquitin-like domain-dependent manner. J. Biol. Chem. 286, 5151–5156.
Kokame, K., Agarwala, K. L., Kato, H., and Miyata, T. (2000). Herp, a new ubiquitin-like membrane protein induced by endoplasmic reticulum stress. J. Biol. Chem. 275, 32846–32853.
Kokame, K., Kato, H., and Miyata, T. (2001). Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J. Biol. Chem. 276, 9199–9205.
Lilley, B. N., and Ploegh, H. L. (2004). A membrane protein required for dislocation of misfolded proteins from the ER. Nature 429, 834–840.
Lilley, B. N., and Ploegh, H. L. (2005). Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. U.S.A. 102, 14296–14301.
Liu, B., Dai, J., Zheng, H., Stoilova, D., Sun, S., and Li, Z. (2003). Cell surface expression of an endoplasmic reticulum resident heat shock protein gp96 triggers MyD88-dependent systemic autoimmune diseases. Proc. Natl. Acad. Sci. U.S.A. 100, 15824–15829.
Lu, J., Willis, A. C., and Sim, R. B. (1993). A calreticulin-like protein co-purifies with a “60 kD” component of Ro/SSA, but is not recognized by antibodies in Sjogren’s syndrome sera. Clin. Exp. Immunol. 94, 429–434.
Madaio, M. P., Hodder, S., Schwartz, R. S., and Stollar, B. D. (1984). Responsiveness of autoimmune and normal mice to nucleic acid antigens. J. Immunol. 132, 872–876.
Marutani, T., Maeda, T., Tanabe, C., Zou, K., Araki, W., Kokame, K., Michikawa, M., and Komano, H. (2011). ER-stress-inducible Herp, facilitates the degradation of immature nicastrin. Biochim. Biophys. Acta 1810, 790–798.
Marutani, T., Yamamoto, A., Nagai, N., Kubota, H., and Nagata, K. (2004). Accumulation of type IV collagen in dilated ER leads to apoptosis in Hsp47-knockout mouse embryos via induction of CHOP. J. Cell. Sci. 117, 5913–5922.
Masuda, H., Fukumoto, M., Hirayoshi, K., and Nagata, K. (1994). Coexpression of the collagen-binding stress protein HSP47 gene and the alpha 1(I) and alpha 1(III) collagen genes in carbon tetrachloride-induced rat liver fibrosis. J. Clin. Invest. 94, 2481–2488.
Masuda, H., Hosokawa, N., and Nagata, K. (1998). Expression and localization of collagen-binding stress protein Hsp47 in mouse embryo development: comparison with types I and II collagen. Cell Stress Chaperones 3, 256–264.
Matsuoka, Y., Kubota, H., Adachi, E., Nagai, N., Marutani, T., Hosokawa, N., and Nagata, K. (2004). Insufficient folding of type IV collagen and formation of abnormal basement membrane-like structure in embryoid bodies derived from Hsp47-null embryonic stem cells. Mol. Biol. Cell 15, 4467–4475.
McCauliffe, D. P., Zappi, E., Lieu, T. S., Michalak, M., Sontheimer, R. D., and Capra, J. D. (1990). A human Ro/SS-A autoantigen is the homologue of calreticulin and is highly homologous with onchocercal RAL-1 antigen and an aplysia “memory molecule.” J. Clin. Invest. 86, 332–335.
McCracken, A. A., and Brodsky, J. L. (1996). Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J. Cell Biol. 132, 291–298.
Mesaeli, N., Nakamura, K., Zvaritch, E., Dickie, P., Dziak, E., Krause, K. H., Opas, M., Maclennan, D. H., and Michalak, M. (1999). Calreticulin is essential for cardiac development. J. Cell Biol. 144, 857–868.
Molinari, M., Calanca, V., Galli, C., Lucca, P., and Paganetti, P. (2003). Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299, 1397–1400.
Mori, K. (2000). Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 101, 451–454.
Morimoto, R. I. (1998). Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 12, 3788–3796.
Mostoslavsky, G., Fischel, R., Yachimovich, N., Yarkoni, Y., Rosenmann, E., Monestier, M., Baniyash, M., and Eilat, D. (2001). Lupus anti-DNA autoantibodies cross-react with a glomerular structural protein: a case for tissue injury by molecular mimicry. Eur. J. Immunol. 31, 1221–1227.
Mueller, B., Klemm, E. J., Spooner, E., Claessen, J. H., and Ploegh, H. L. (2008). SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. U.S.A. 105, 12325–12330.
Mueller, B., Lilley, B. N., and Ploegh, H. L. (2006). SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J. Cell Biol. 175, 261–270.
Nagai, N., Hosokawa, M., Itohara, S., Adachi, E., Matsushita, T., Hosokawa, N., and Nagata, K. (2000). Embryonic lethality of molecular chaperone hsp47 knockout mice is associated with defects in collagen biosynthesis. J. Cell Biol. 150, 1499–1506.
Nagata, K. (2003). HSP47 as a collagen-specific molecular chaperone: function and expression in normal mouse development. Semin. Cell Dev. Biol. 14, 275–282.
Naitoh, M., Hosokawa, N., Kubota, H., Tanaka, T., Shirane, H., Sawada, M., Nishimura, Y., and Nagata, K. (2001). Upregulation of HSP47 and collagen type III in the dermal fibrotic disease, keloid. Biochem. Biophys. Res. Commun. 280, 1316–1322.
Nakatani, Y., Kaneto, H., Hatazaki, M., Yoshiuchi, K., Kawamori, D., Sakamoto, K., Matsuoka, T., Ogawa, S., Yamasaki, Y., and Matsuhisa, M. (2006). Increased stress protein ORP150 autoantibody production in Type 1 diabetic patients. Diabet. Med. 23, 216–219.
Ni, M., and Lee, A. S. (2007). ER chaperones in mammalian development and human diseases. FEBS Lett. 581, 3641–3651.
Njemini, R., Bautmans, I., Onyema, O. O., Van Puyvelde, K., Demanet, C., and Mets, T. (2011). Circulating heat shock protein 70 in health, aging and disease. BMC Immunol. 12, 24. doi:10.1186/1471-2172-12-24
Oda, Y., Hosokawa, N., Wada, I., and Nagata, K. (2003). EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299, 1394–1397.
Oda, Y., Okada, T., Yoshida, H., Kaufman, R. J., Nagata, K., and Mori, K. (2006). Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J. Cell Biol. 172, 383–393.
Okazaki, Y., Ohno, H., Takase, K., Ochiai, T., and Saito, T. (2000). Cell surface expression of calnexin, a molecular chaperone in the endoplasmic reticulum. J. Biol. Chem. 275, 35751–35758.
Okuda-Shimizu, Y., and Hendershot, L. M. (2007). Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol. Cell 28, 544–554.
Orth, T., Dorner, T., Meyer Zum Buschenfelde, K. H., and Mayet, W. J. (1996). Complete congenital heart block is associated with increased autoantibody titers against calreticulin. Eur. J. Clin. Invest. 26, 205–215.
Pagetta, A., Folda, A., Brunati, A. M., and Finotti, P. (2003). Identification and purification from the plasma of Type 1 diabetic subjects of a proteolytically active Grp94Evidence that Grp94 is entirely responsible for plasma proteolytic activity. Diabetologia 46, 996–1006.
Panayi, G. S., and Corrigall, V. M. (2006). BiP regulates autoimmune inflammation and tissue damage. Autoimmun. Rev. 5, 140–142.
Rumore, P. M., and Steinman, C. R. (1990). Endogenous circulating DNA in systemic lupus erythematosus. Occurrence as multimeric complexes bound to histone. J. Clin. Invest. 86, 69–74.
Sato, Y., Murase, K., Kato, J., Kobune, M., Sato, T., Kawano, Y., Takimoto, R., Takada, K., Miyanishi, K., Matsunaga, T., Takayama, T., and Niitsu, Y. (2008). Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol. 26, 431–442.
Schulte-Pelkum, J., Fritzler, M., and Mahler, M. (2009). Latest update on the Ro/SS-A autoantibody system. Autoimmun. Rev. 8, 632–637.
Shoda, H., Fujio, K., Shibuya, M., Okamura, T., Sumitomo, S., Okamoto, A., Sawada, T., and Yamamoto, K. (2011). Detection of autoantibody to citrullinated BiP in rheumatoid arthritis patients and pro-inflammatory roles of citrullinated BiP in collagen-induced arthritis. Arthritis Res. Ther. 13, R191.
Smith, M. H., Ploegh, H. L., and Weissman, J. S. (2011). Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334, 1086–1090.
Staikou, E. V., Routsias, J. G., Makri, A. A., Terzoglou, A., Sakarellos-Daitsiotis, M., Sakarellos, C., Panayotou, G., Moutsopoulos, H. M., and Tzioufas, A. G. (2003). Calreticulin binds preferentially with B cell linear epitopes of Ro60 kD autoantigen, enhancing recognition by anti-Ro60 kD autoantibodies. Clin. Exp. Immunol. 134, 143–150.
Suzuki, A., Yamada, R., and Yamamoto, K. (2007). Citrullination by peptidylarginine deiminase in rheumatoid arthritis. Ann. N. Y. Acad. Sci. 1108, 323–339.
Suzuki, S., Utsugisawa, K., Iwasa, K., Satoh, T., Nagane, Y., Yoshikawa, H., Kuwana, M., and Suzuki, N. (2011). Autoimmunity to endoplasmic reticulum chaperone GRP94 in myasthenia gravis. J. Neuroimmunol. 237, 87–92.
Todd, D. J., Lee, A. H., and Glimcher, L. H. (2008). The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 8, 663–674.
Tsai, B., Ye, Y., and Rapoport, T. A. (2002). Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 3, 246–255.
Tsukamoto, Y., Kuwabara, K., Hirota, S., Ikeda, J., Stern, D., Yanagi, H., Matsumoto, M., Ogawa, S., and Kitamura, Y. (1996). 150-kD oxygen-regulated protein is expressed in human atherosclerotic plaques and allows mononuclear phagocytes to withstand cellular stress on exposure to hypoxia and modified low density lipoprotein. J. Clin. Invest. 98, 1930–1941.
Tu, B. P., and Weissman, J. S. (2004). Oxidative protein folding in eukaryotes: mechanisms and consequences. J. Cell Biol. 164, 341–346.
Ushioda, R., Hoseki, J., Araki, K., Jansen, G., Thomas, D. Y., and Nagata, K. (2008). ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science 321, 569–572.
Ushioda, R., and Nagata, K. (2011). The endoplasmic reticulum-associated degradation and disulfide reductase ERdj5. Meth. Enzymol. 490, 235–258.
van Eden, W., Thole, J. E., Van Der Zee, R., Noordzij, A., Van Embden, J. D., Hensen, E. J., and Cohen, I. R. (1988). Cloning of the mycobacterial epitope recognized by T lymphocytes in adjuvant arthritis. Nature 331, 171–173.
Van Eden, W., Wick, G., Albani, S., and Cohen, I. (2007). Stress, heat shock proteins, and autoimmunity: how immune responses to heat shock proteins are to be used for the control of chronic inflammatory diseases. Ann. N. Y. Acad. Sci. 1113, 217–237.
van Venrooij, W. J., Van Beers, J. J., and Pruijn, G. J. (2011). Anti-CCP antibodies: the past, the present and the future. Nat. Rev. Rheumatol. 7, 391–398.
Vincent, C., Nogueira, L., Clavel, C., Sebbag, M., and Serre, G. (2005). Autoantibodies to citrullinated proteins: ACPA. Autoimmunity 38, 17–24.
Vossenaar, E. R., Zendman, A. J., Van Venrooij, W. J., and Pruijn, G. J. (2003). PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays 25, 1106–1118.
Voynova, E. N., Tchorbanov, A. I., Todorov, T. A., and Vassilev, T. L. (2005). Breaking of tolerance to native DNA in nonautoimmune mice by immunization with natural protein/DNA complexes. Lupus 14, 543–550.
Wada, I., Kai, M., Imai, S., Sakane, F., and Kanoh, H. (1997). Promotion of transferrin folding by cyclic interactions with calnexin and calreticulin. EMBO J. 16, 5420–5432.
Wada, I., Rindress, D., Cameron, P. H., Ou, W. J., Doherty, J. J. II, Louvard, D., Bell, A. W., Dignard, D., Thomas, D. Y., and Bergeron, J. J. (1991). SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J. Biol. Chem. 266, 19599–19610.
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086.
Wanderling, S., Simen, B. B., Ostrovsky, O., Ahmed, N. T., Vogen, S. M., Gidalevitz, T., and Argon, Y. (2007). GRP94 is essential for mesoderm induction and muscle development because it regulates insulin-like growth factor secretion. Mol. Biol. Cell 18, 3764–3775.
Watanabe, K., Ohira, H., Orikasa, H., Saito, K., Kanno, K., Shioya, Y., Obara, K., and Sato, Y. (2006). Anti-calreticulin antibodies in patients with inflammatory bowel disease. Fukushima J. Med. Sci. 52, 1–11.
Weber, C. K., Haslbeck, M., Englbrecht, M., Sehnert, B., Mielenz, D., Graef, D., Distler, J. H., Mueller, R. B., Burkhardt, H., Schett, G., Voll, R. E., and Furnrohr, B. G. (2010). Antibodies to the endoplasmic reticulum-resident chaperones calnexin, BiP and Grp94 in patients with rheumatoid arthritis and systemic lupus erythematosus. Rheumatology (Oxford) 49, 2255–2263.
Wendling, U., Paul, L., Van Der Zee, R., Prakken, B., Singh, M., and Van Eden, W. (2000). A conserved mycobacterial heat shock protein (hsp) 70 sequence prevents adjuvant arthritis upon nasal administration and induces IL-10-producing T cells that cross-react with the mammalian self-hsp70 homologue. J. Immunol. 164, 2711–2717.
Williams, D. B. (2006). Beyond lectins: the calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell. Sci. 119, 615–623.
Yagishita, N., Yamasaki, S., Nishioka, K., and Nakajima, T. (2008). Synoviolin, protein folding and the maintenance of joint homeostasis. Nat. Clin. Pract. Rheumatol. 4, 91–97.
Yamamura, I., Hirata, H., Hosokawa, N., and Nagata, K. (1998). Transcriptional activation of the mouse HSP47 gene in mouse osteoblast MC3T3-E1 cells by TGF-beta 1. Biochem. Biophys. Res. Commun. 244, 68–74.
Yasuda, K., Hirayoshi, K., Hirata, H., Kubota, H., Hosokawa, N., and Nagata, K. (2002). The Kruppel-like factor Zf9 and proteins in the Sp1 family regulate the expression of HSP47, a collagen-specific molecular chaperone. J. Biol. Chem. 277, 44613–44622.
Ye, Y., Meyer, H. H., and Rapoport, T. A. (2001). The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656.
Ye, Y., Meyer, H. H., and Rapoport, T. A. (2003). Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 162, 71–84.
Yokota, S., and Fujii, N. (2010). Immunomodulatory activity of extracellular heat shock proteins and their autoantibodies. Microbiol. Immunol. 54, 299–307.
Yokota, S., Kubota, H., Matsuoka, Y., Naitoh, M., Hirata, D., Minota, S., Takahashi, H., Fujii, N., and Nagata, K. (2003). Prevalence of HSP47 antigen and autoantibodies to HSP47 in the sera of patients with mixed connective tissue disease. Biochem. Biophys. Res. Commun. 303, 413–418.
Yoshida, H., Haze, K., Yanagi, H., Yura, T., and Mori, K. (1998). Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273, 33741–33749.
Keywords: autoimmunity, inflammation, ER stress, ERAD, molecular chaperone
Citation: Morito D and Nagata K (2012) ER stress proteins in autoimmune and inflammatory diseases. Front. Immun. 3:48. doi: 10.3389/fimmu.2012.00048
Received: 15 December 2011; Paper pending published: 10 January 2012;
Accepted: 28 February 2012; Published online: 15 March 2012.
Edited by:
Willem Van Eden, Utrecht University, NetherlandsReviewed by:
Rik Lories, Katholieke Universiteit Leuven, BelgiumCristina Bonorino, Pontificia Universidade Catolica do Rio Grande do Sul, Brazil
Ruurd Van Der Zee, Universiteit Utrecht, Netherlands
Copyright: © 2012 Morito and Nagata. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Kazuhiro Nagata, Laboratory of Molecular and Cellular Biology, Faculty of Life Sciences, Kyoto Sangyo University, Kamigamo Motoyama, Kyoto, Kyoto 603-8555, Japan. e-mail: nagata@cc.kyoto-su.ac.jp