 C. A. Thomson1† Y. Wang1† L. M. Jackson1 M. Olson1 W. Wang1 A. Liavonchanka2,3 L. Keleta4,5 V. Silva6 S. Diederich6 R. B. Jones7 J. Gubbay8,9 J. Pasick10 M. Petric11 François Jean6 V. G. Allen8,9 E. G. Brown4,5 J. M. Rini2,3 J. W. Schrader1*
C. A. Thomson1† Y. Wang1† L. M. Jackson1 M. Olson1 W. Wang1 A. Liavonchanka2,3 L. Keleta4,5 V. Silva6 S. Diederich6 R. B. Jones7 J. Gubbay8,9 J. Pasick10 M. Petric11 François Jean6 V. G. Allen8,9 E. G. Brown4,5 J. M. Rini2,3 J. W. Schrader1*- 1 The Biomedical Research Centre, University of British Columbia, Vancouver, BC, Canada
- 2 Department of Molecular Genetics, University of Toronto, Toronto, ON, Canada
- 3 Department of Biochemistry, University of Toronto, Toronto, ON, Canada
- 4 Department of Biochemistry, Microbiology and Immunology, Faculty of Medicine, University of Ottawa, Ottawa, ON, Canada
- 5 Emerging Pathogens Research Centre, University of Ottawa, Ottawa, ON, Canada
- 6 Department of Microbiology and Immunology, University of British Columbia, Vancouver, BC, Canada
- 7 Department of Immunology, University of Toronto, Toronto, ON, Canada
- 8 Ontario Agency for Health Protection and Promotion, Toronto, ON, Canada
- 9 Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada
- 10 Canadian Food Inspection Agency, National Centre for Foreign Animal Disease, Winnipeg, MB, Canada
- 11 BC Centre for Disease Control, Vancouver, BC, Canada
Most monoclonal antibodies (mAbs) generated from humans infected or vaccinated with the 2009 pandemic H1N1 (pdmH1N1) influenza virus targeted the hemagglutinin (HA) stem. These anti-HA stem mAbs mostly used IGHV1-69 and bound readily to epitopes on the conventional seasonal influenza and pdmH1N1 vaccines. The anti-HA stem mAbs neutralized pdmH1N1, seasonal influenza H1N1 and avian H5N1 influenza viruses by inhibiting HA-mediated fusion of membranes and protected against and treated heterologous lethal infections in mice with H5N1 influenza virus. This demonstrated that therapeutic mAbs could be generated a few months after the new virus emerged. Human immunization with the pdmH1N1 vaccine induced circulating antibodies that when passively transferred, protected mice from lethal, heterologous H5N1 influenza infections. We observed that the dominant heterosubtypic antibody response against the HA stem correlated with the relative absence of memory B cells against the HA head of pdmH1N1, thus enabling the rare heterosubtypic memory B cells induced by seasonal influenza and specific for conserved sites on the HA stem to compete for T-cell help. These results support the notion that broadly protective antibodies against influenza would be induced by successive vaccination with conventional influenza vaccines based on subtypes of HA in viruses not circulating in humans.
Introduction
Each year seasonal influenza causes serious illnesses in 3–5 million humans and 200,000–500,000 deaths (Chen and Subbarao, 2009). Protection against influenza is due to high-avidity antibodies against the hemagglutinin (HA) protein, which protrudes from the viral envelope and attaches the virion to the sialic acid receptor on the cells of the host (Wiley and Skehel, 1987). The hemagglutinin protein has a globular head which mediates the attachment of the virus to the host cells, and a stem, which mediates the fusion of the viral membrane to the host cell membrane, enabling the viral genome to enter the cells (Wiley and Skehel, 1987). If antibodies against the head of the hemagglutinin are of sufficient affinity/avidity and sterically inhibit the receptor-binding site, they block attachment of the virus to the host cell (Knossow et al., 2002). These protective antibodies are very specific for a given isolate/subtype of influenza virus because the replication of the influenza virus genome is error-prone. Thus influenza viruses are rapidly selected with mutants of the HA that decrease the affinity of the protective antibodies so that they no longer block attachment of the mutant virus. This variation in the influenza virus is called “antigenic drift” and three dominant drifted influenza viruses are incorporated into updated seasonal influenza vaccines to induce protective antibodies to these variants.
Occasionally a more drastic change in the antigenicity of the HA termed “antigenic shift” occurs. Consequently humans have no protective antibodies against the “antigenically shifted” influenza virus. If an antigen-shifted influenza virus transmits readily between humans, it can cause an influenza pandemic. The 1918 “Spanish” H1N1 influenza pandemic was probably caused by an influenza virus that had come from avian species (Yen and Webster, 2009). The HA of the 1918 H1N1, 1957 H2N2, 1968 H3N2, and 2009 pandemic H1N1 influenza viruses ultimately originated from avian influenza viruses (Yen and Webster, 2009). The 2009 pandemic influenza A H1N1 virus (pdmH1N1) was generated by reassortment of the eight RNA molecules of the influenza genome between two swine influenza viruses, both with genes from avian, swine, and human influenza viruses (Ding et al., 2009; Garten et al., 2009). In the HA of the 2009 pandemic H1N1 influenza, 21% of the amino-acids of the ectodomain were non-identical with the corresponding residues in seasonal H1N1 virus and ∼50% in the key epitopes on the HA head were non-identical (Xu et al., 2010). Infection of humans with influenza viruses not circulating in humans can cause serious human illness, the highly pathogenic avian H5N1 influenza having a mortality rate in humans of over 60% (Yen and Webster, 2009).
In contrast to the HA head, mutations in the HA stem are not well-tolerated for viral infectivity because, after the virus is endocytosed, the low pH in the endosome induces a complex conformational change in the HA stem (Wiley and Skehel, 1987). This conformational change exposes a peptide that mediates fusion of the viral and the endosomal membranes, enabling the viral genome to access the cytoplasm. Antibodies against the HA stem can block this conformational change and membrane fusion (Okuno et al., 1993) and thus infectivity. Artificial phage-display mAbs using the human immunoglobulin heavy (H) chain and light (L) chain genes that were selected to bind the H5 HA neutralize the infectivity of multiple subtypes of influenza viruses by targeting a conserved site on the HA stem (Throsby et al., 2008; Sui et al., 2009). These mAbs preferentially used the H chain variable region gene IGHV1-69. Crystallographic studies of two of these artificial mAbs in complex with HA demonstrated that the germline IGHV1-69 encodes key residues for a binding site for an epitope on the stem of the HA (Ekiert et al., 2009; Sui et al., 2009). The L chain did not contact the HA stem.
Techniques that enable copying of natural human monoclonal antibodies (mAbs) binding to HA offers great advantages in dissecting the distribution of protective antibodies in terms of their affinity, epitope, cross-reactivity, heterosubtypic protection, V-gene usage, clonotypic dominance, and numbers of somatic mutations. Wrammert et al. (2008) generated copies of natural mAbs from newly formed plasmablasts shortly after seasonal influenza vaccination and found that none of the monoclonal antibodies were heterosubtypic and all targeted the hemagglutinin head (Wrammert et al., 2011). Likewise, memory B cells making heterosubtypic antibodies against the HA from H5N1 were undetectable in normal humans (Corti et al., 2010). However, after seasonal influenza vaccination, heterosubtypic memory B cells (mainly against the HA stem and using IGHV1-69) could be detected in some individuals, although the frequency was variable and 26- to 200-fold less than that of memory B cells making antibodies specific for the seasonal influenza vaccine (Corti et al., 2010). A small amount of heterosubtypic antibody in the serum was detected but was insufficient to neutralize the H5N1 influenza virus (Corti et al., 2010), consistent with immunity against influenza being very isolate-specific (Wiley and Skehel, 1987). As the gene used for most heterosubtypic antibodies against the HA stem, IGHV1-69, is present in most humans and the structural data indicated that the H chain without the help of the L chain made the critical contacts with the HA stem by germline IGHV1-69 encoded side-chains, these observations raised the question of why effective levels of cross-protective heterosubtypic antibodies are not induced by infections or vaccinations with seasonal influenza (Throsby et al., 2008; Sui et al., 2009).
Wrammert et al. (2011) reported that 5 of the 15 mAbs against HA generated from three out of four people infected with pdmH1N1 (Garten et al., 2009) were against the HA stem and four used IGHV1-69. These five anti-HA stem mAbs neutralized pdmH1N1 and seasonal H1N1 influenza viruses. We made more extensive observations and have demonstrated that the anti-HA stem mAbs induced by pdmH1N1 infection neutralized another subtype of influenza A virus, the highly pathogenic avian H5N1 influenza. They also protected potently against and effectively treated lethal infections in mice with H5N1. These published observations (Wrammert et al., 2011) and our observations in the winter of 2009–2010 suggested that infection with pdmH1N1 induces a high proportion of PB making heterosubtypic antibodies and raised important questions. Would circulating levels of heterosubtypic antibodies reach protective levels? Would the pdmH1N1 vaccine similarly induce a protective heterosubtypic antibody response? What was different about infection with pdmH1N1 and why were there not a high frequency of anti-HA stem mAbs induced by infection with seasonal influenza? Here we provide answers to these questions.
Results
Anti-HA Stem mAbs Dominate in both Infected and Vaccinated Subjects
When the 2009 pandemic emerged, we set out to generate rapidly therapeutic truly human monoclonal antibodies (mAbs) against the HA (Mozdzanowska et al., 1997; Luke et al., 2006; Simmons et al., 2007; Zhou et al., 2007; Hung et al., 2011) of the pdmH1N1 virus (pdmHA). We used three approaches to generate human monoclonal antibodies (mAbs) that reacted with HA of the pdmH1N1 virus. First, we randomly cloned antibodies from newly generated plasmablasts (PB) circulating in the blood of recently infected patients. This approach was based upon observations that ∼7 days after vaccination, PB that secrete antibodies specific for the vaccine appear in the blood (Heilmann et al., 1987; Barington et al., 1990) and form a significant fraction of the total PB (Odendahl et al., 2005; Wrammert et al., 2008). It also exploited techniques that we had previously used to generate monoclonal antibodies from blood-borne PB by RT-PCR and cloning and expression of the DNA encoding the antigen-binding site from single PB (Babcook et al., 1996). In the absence of data on the kinetics of entry of infection-specific PB into the blood during infections, we collected blood from subjects with laboratory-confirmed pdmH1N1 infections ∼7 days after the onset of symptoms. We found many PB secreting antibodies against pdmHA in the blood after infection (Figure 1). This demonstrates that, during an infection, at least some newly generated PB enter the blood and do not all remain in lymphoid tissues near the site of infection, consistent with data published afterward with human infection with Respiratory Syncytial Virus (Lee et al., 2010). We obtained our first mAb (I4-128) against pdmHA in August 2009, only months after the pdmH1N1 virus was identified. The mAb used IGHV1-69 and bound readily to the HA of the highly pathogenic avian influenza A/Hong Kong/156/197 (H5N1) virus (Figures 1C,D). As a second strategy we also purified PB cells that bound fluorochrome-labeled pdmHA and used RT-PCR and cloning to generate additional mAbs from subjects infected or vaccinated with pdmH1N1 (Table 1). We also used fluorescent cell sorting to purify individual class-switched memory B cells that bound to fluorochrome-labeled pdmHA, expanded the B cells into clones and assayed the supernatants after a week for the presence of antibodies to pdmHA and used RT-PCR and cloning to generate mAbs against pdmHA.
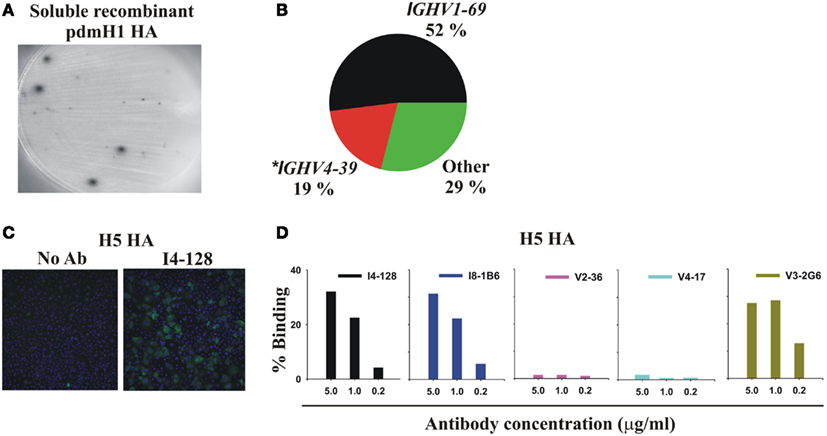
Figure 1. Infection or vaccination with pdmH1N1 induces a dominant antibody response that preferentially uses IGHV1-69 and that cross-reacts with the HA of the highly pathogenic avian H5N1 influenza virus. (A) Elispot assay showing spots of anti-pdmHA antibodies that had been secreted by individual PB. (B) Pie chart representing all 48 mAbs from Table 1. The 95% confidence intervals of IGHV1-69 usage were 38–66%. *8 of 9 mAbs using IGHV4-39 were from a single subject, V2. (C,D) Cross-reactivity of selected mAbs with the HA of influenza A/Hong Kong/156/197 (H5N1; Clade 0) expressed by the A549 human cell-line. Binding was quantified by indirect immunofluorescence and the Cellomics instrument. (C) Cellomics images of cells stained with either secondary antibodies alone or first with mAb I4-128 (5 μg/ml). (D) Percentage of cells stained with varying concentrations of the indicated mAbs. Note that anti-stem HA mAbs that did not use IGHV1-69, like V3-1E8 and I5-52 also bound to the H5 HA in this assay (data not shown). KE5, an human anti-HCMV monoclonal antibody (McLean et al., 2005), was used as a negative control with no significant binding observed (data not shown).
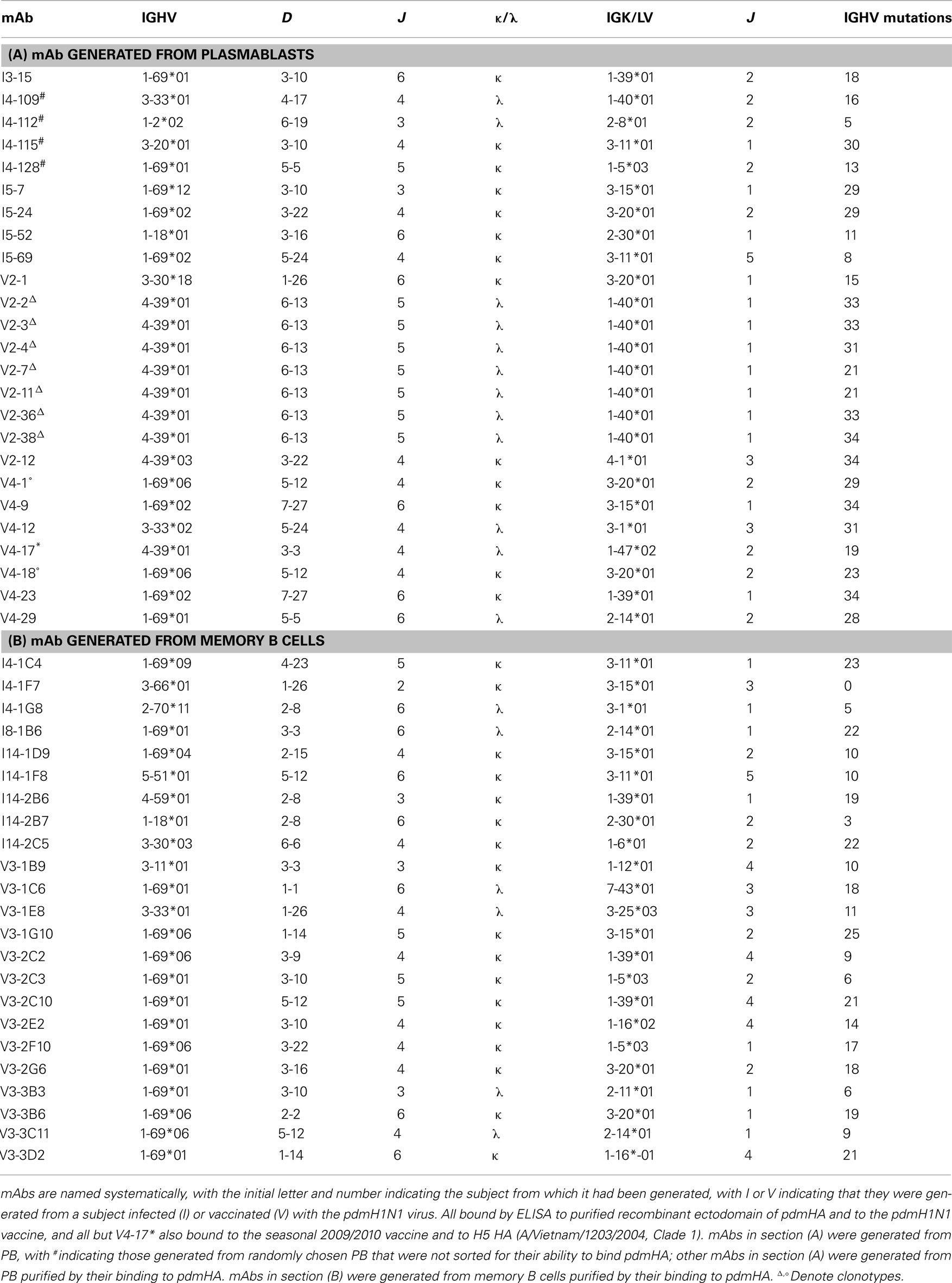
Table 1. Truly human monoclonal antibodies binding to pdmHA.
In total, from five infected and three vaccinated subjects, we generated 48 mAbs against pdmHA (Table 1). The sequences of the 10 best-binding mAbs to pdmH1N1, 3 against the HA head (V2-36, V2-7, and V4-17) and 7 against the HA stem (I4-128, I5-24, I8-1B6, V3-1G10, V3-2C3, V3-2G6, and V3-3D2), are listed in Table 2. Strikingly, 52% of the mAbs used the heavy-chain immunoglobulin variable region gene IGHV1-69 (Table 1; Figure 1B) which is used by only 3.6% of random B cells from blood (De Wildt et al., 1999). The high frequency of usage of IGHV1-69 in mAbs against pdmHA was seen whether the mAbs were generated from subjects infected (44%) or vaccinated (57%) with pdmH1N1, or from either PB (40%) or memory B cells (65%).

Table 2. Nucleotide sequences of the variable regions of 10 monoclonal neutralizing antibodies that had the highest avidity for pdmH1N1 hemagglutinin and that were studied in detail.
A minority of mAbs (7 of 38 mAbs tested) inhibited hemagglutination by pdmH1N1 (Figure 2A) and most were of one clonotype (Table 1) and bound to the HA head. We found like others, that select mAbs using IGHV1-69 and other IGHV genes bound to a site on the HA stem as judged by inhibition of the binding of C179, a cross-protective anti-HA stem mAb (Okuno et al., 1993; Figure 2B). We noted, however, that this assay was not specific for antibodies against the HA stem. Thus a mAb (V2-36) that was clearly directed against the HA head because it inhibited hemagglutination (Figure 2A), also inhibited binding of the anti-HA stem mAb C179 (Figure 2B). However, we confirmed that many mAbs that inhibited the binding of C179 to pdmHA, bound to the HA stem by showing that they did not inhibit hemagglutination by pdmH1N1 and that their binding was sensitive to pre-treatment of HA at pH 5 (Figure 2C), which altered the conformation of the HA stem (Wiley and Skehel, 1987; Throsby et al., 2008). The binding by the anti-HA head mAb, V2-36 was not affected by this treatment (Figure 2C). By these three criteria, failure to inhibit hemagglutination by influenza virus, competitive inhibition of C179 binding to HA, and inhibition of binding to the mAb by pre-treatment of the HA at pH 5, another 6% of the anti-pdmHA mAbs not using IGHV1-69 mapped to the HA stem and probably bound to subtly different epitopes (e.g., the mAb I5-52, which uses IGHV1-18 Figures 2A–C). We tested 20 out of 25 IGHV1-69-using mAbs for inhibition of hemagglutination with pdmH1N1 and none proved positive at 20 μg/ml. We tested 24 of the IGHV1-69-using mAbs for binding to pdmHA treated at pH 5. We found that the binding of 20 of the mAbs was dramatically reduced. Throsby et al. (2008), found a heterogeneity in the binding to low pH treated HA with different IGHV1-69-using mAbs. However, we are still assessing the proportion of mAbs against the HA stem. The most conclusive way to prove that they are against the HA stem, is by making escape mutants, which has proven difficult (Throsby et al., 2008; Sui et al., 2009; Wang et al., 2010). As can be seen in Figure 2D, the anti-HA stem mAbs also competed with biotinylated I5-24 (which used IGHV1-69) for binding to pdmHA. Of note, V2-36, which clearly was against the HA head (Figure 2A), competed with biotinylated I5-24 for binding to pdmHA (Figure 2D). Moreover 3 out of 3 IGHV1-69-using mAbs bound to the HA of highly pathogenic avian influenza A/Hong Kong/156/197 (H5N1) virus (Figure 1D). Only two mAbs using IGHV1-69 (from subject V4) were members of the same clonotype. Notably, in the subject with the highest proportion of mAbs using IGHV1-69, V3 (86%), all of the mAbs were from different clonotypes.
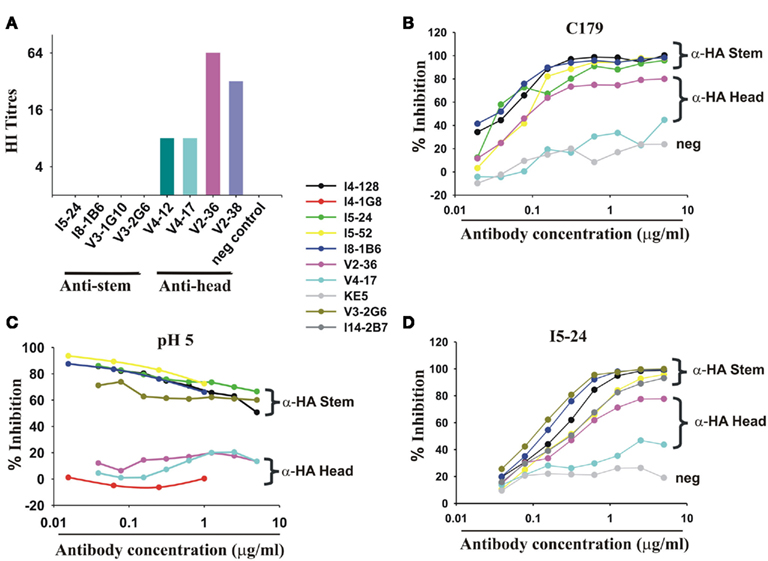
Figure 2. Infection or vaccination with pdmH1N1 induces a dominant antibody response targeting the HA stem. (A) mAbs to the HA stem fail to inhibit hemagglutination but a minority of mAbs inhibit hemagglutination by pdmH1N1. All mAbs were at 40 μg/ml and diluted 50% with virus in the first well. No detectable hemagglutination means that hemagglutination was not inhibited at 20 μg/ml. (B) Anti-HA stem mAbs inhibit binding of a mouse mAb (C179 (Okuno et al., 1993)) to the HA stem in a competition ELISA. Note that mAb I5-52, not using IGHV1-69, also inhibited C179 binding. (C) pH 5 pre-treatment of HA selectively decreases binding of anti-HA stem mAbs whether encoded by IGHV1-69 or other IGHV genes (like I5-52) but not anti-HA head mAbs like V2-36. (D) ELISA showing competitive inhibition of the indicated mAbs of the binding of biotinylated I5-24, which uses IGHV1-69 and binds to the HA stem. Note that V2-36 also inhibits binding of I5-24. Also shown is competitive inhibition of I5-24 binding by I14-2B7, which uses IGHV1-18 and IGKV2-30. KE5, an human anti-HCMV monoclonal antibody (McLean et al., 2005), was used as a negative control.
Anti-HA Stem mAbs Recognized Cross-Reactive Epitopes on Conventional Seasonal and PdmH1N1 Vaccines
With one exception, the anti-HA head mAb V4-17, all mAbs including both anti-HA stem and head mAbs, bound to the 2009/2010 seasonal influenza vaccine and the pdmH1N1 vaccine (Figure 3; Table 1) and all bound to the purified ectodomain of H5 HA of an H5N1 avian influenza virus A/Vietnam 1203/2004 (Clade 1; Table 1; Figure 3). The binding of the anti-HA stem mAbs to the seasonal influenza and pdmH1N1 vaccine was comparable to the binding of the anti-HA head mAbs (e.g., binding of V2-36 to the seasonal influenza and pdmH1N1 vaccine and binding of V4-17 to the pdmH1N1 vaccine, Figure 3). Thus, the two conventionally manufactured vaccines were a good source of the conserved epitopes on the HA stem and thus the failure of the seasonal influenza vaccine to induce protective levels of heterosubtypic anti-HA stem antibodies (Wrammert et al., 2008; Corti et al., 2010) was not due to the lack of the conserved epitopes in the vaccine.
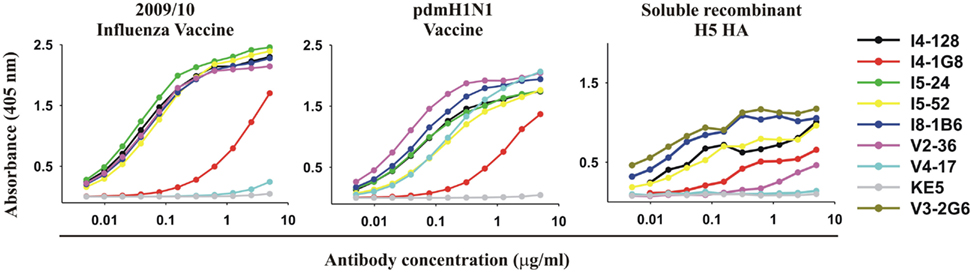
Figure 3. Vaccines for seasonal influenza or pdmH1N1 and purified recombinant HA from highly pathogenic avian H5N1 influenza exhibit the epitope targeted by anti-HA stem mAbs. ELISA reactivity with the 2009/2010 seasonal influenza vaccine, the pdmH1N1 vaccine, and purified recombinant ectodomain of the HA of an H5N1 avian influenza virus A/Vietnam 1203/2004 (Clade 1) of 5 anti-HA stem mAbs (V3-2G6, I4-128, I5-24, I8-1B6 using IGHV1-69 and I5-52 using IGHV1-18), with for comparison, three anti-HA head antibodies (I4-1G8, V2-36, and V4-17).
Neutralization of pdmH1N1 and Avian H5N1 Influenza Virus Infectivity by Anti-HA Stem Antibodies
None of our anti-HA stem mAbs neutralized the pdmH1N1 or current seasonal H1N1 influenza viruses in the standard WHO microneutralization assay, performed in two experienced public health laboratories. However, the minority of mAbs that inhibited hemagglutination with the respective influenza virus, neutralized. The WHO neutralization assay effectively detects antibodies that block viral attachment, as the mixture of virus and antibody is incubated with the cells for only 3 h and then washed off. Subsequently the cells are cultured for 3–4 days to allow the influenza virus to replicate and produce a cytopathic effect.
We reasoned that, in the standard assay, the mixture of the viruses and mAb will only be in contact with the host cells for 3 h and, if the mAb targets the HA stem rather than the head, the viruses will successfully attach to the host cells. After the virus and the mAb are washed away, some of the mAb will dissociate from viruses that are not endocytosed but still attached to the cells. When the virus is finally endocytosed there may be insufficient mAb bound to the virus to inhibit membrane fusion. Thus the virus will replicate and can infect other cells, unimpeded by the mAb as it is no longer present.
The WHO assay was designed to detect neutralizing antibodies in serum. The mixture of virus and sera was washed away after 3 h incubation with the host cells because the human serum contained trypsin inhibitors. The inclusion of trypsin in the assay was essential because enzymatic action of trypsin was necessary to cleave the hemagglutinin on the virions in order to see a cytopathic effect. This assay worked well with seasonal influenza where the neutralizing antibodies were directed at the HA head and blocked viral attachment. We modified the assay by leaving the virus and mAbs in the cultures for the duration of the assay, mimicking the constant presence of antibodies in an infection in vivo. With this simple modification, all of the anti-HA stem mAbs tested then completely neutralized pdmH1N1 at concentrations ranging from 78 to 1250 ng/ml (Figure 4A).
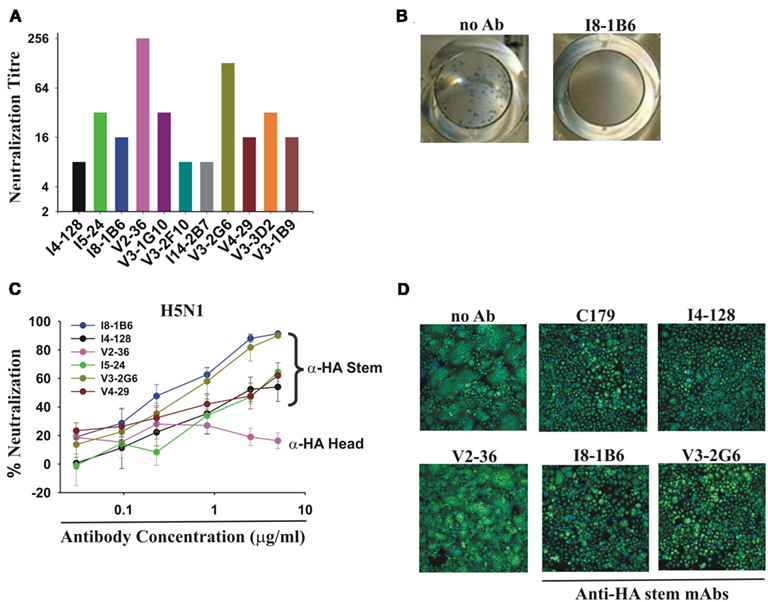
Figure 4. Neutralization of pdmH1N1 and the highly pathogenic avian H5N1 influenza A viruses by anti-HA stem mAbs. (A) Neutralization of pdmH1N1 by mAbs present for the entire assay. Titrations commenced at a concentration of 5 μg/ml of each mAb. The most potent mAbs at neutralization were V2-36 (against the HA head) and V3-2G6 (against the HA stem), which inhibited infectivity completely at ∼40 ng/ml and ∼80 ng/ml. Anti-HA stem mAbs not using IGHV1-69 like I14-2B7, using IGHV1-18, and V3-1B9, using IGHV3-11, also completely neutralized pdmH1N1. Neutralizing titers against influenza virus A/Brisbane/59/07(H1N1) were 2- to 32-fold lower in all cases (data not shown). No neutralization activity against Brisbane influenza A/Brisbane/10/2007 (H3N2) by any mAb tested was observed (data not shown), consistent with previous observations on mAbs using IGHV1-69 (Throsby et al., 2008; Sui et al., 2009). (B,C) Neutralization of infectivity of the highly pathogenic Influenza A/Goose/Ger/R1400/07 (H5N1) avian virus. (B) Shown are representative images of the plaque reduction assay performed with influenza A virus A/Goose/Ger/R1400/07 (H5N1) and mAb I8-1B6 (5 μg/ml). (C) Only V2-36 (which failed to bind to H5 HA, Figure 1) failed to neutralize infectivity. (D) Anti-HA stem mAbs encoded by IGHV1-69 inhibit H5 HA-mediated fusion, correlating with low binding to the HA of highly pathogenic avian influenza A/Hong Kong/156/197 (H5N1) virus (Figure 1). Note the decreased syncytia formation in wells treated by the anti-stem mAb, C179, or the IGHV1-69-using mAbs, I4-128, I8-1B6, and V3-2G6, in contrast to the control with no antibody added (no Ab) and the anti-HA head mAb, V2-36, which did not bind to this H5 HA.
Anti-HA stem mAbs that neutralized pdmH1N1 effectively also neutralized infectivity of the highly pathogenic avian influenza virus A/Goose/Ger/R1400/07 (H5N1; Clade 2.2; Figures 4B,C). We demonstrated that the mechanism of neutralization of infectivity was inhibition of H5 HA-mediated fusion (Throsby et al., 2008; Ekiert et al., 2009; Sui et al., 2009; Figure 4D). These anti-HA stem mAbs had no neutralization activity against the A/Ck/Ger/R28/03 (H7N7) influenza virus (data not shown) consistent with the failure of most engineered mAbs using IGHV1-69 to bind to Group 2 subtypes of HA (Throsby et al., 2008; Sui et al., 2009).
Therapeutic Effects of mAbs Against the Head and Stem of pdmH1N1 on Lethal in vivo Infections with a Human Isolate of pdmH1N1
We then tested selected mAbs which we had generated against the head and the stem of the pdmHA, for their therapeutic efficacy in mice with lethal infections of a human isolate of pdmH1N1. We treated groups of five mice with each of six anti-HA stem mAbs 24 h after lethal intranasal doses of a human isolate of pdmH1N1. With a single intraperitoneal injection of 300 μg, we saw significant increases in survival rates (Figure 5A). Sui et al. (2009), pointed out that IGHV1-69 is the only germline V-gene to encode a Phe at the top of the CDRH2 loop and that the Phe makes several critical interactions with H5 HA. Six alleles of IGHV1-69 (*02, *04, *08, *09, *10, and *11) have a Leu in place of a Phe in this position. Our data on I5-24 show that an allele of IGHV1-69 that has a Leu can bind strongly to the HA stem and can treat a lethal infection with pdmH1N1 (Figures 2 and 5A). In addition other mAbs using IGHV1-69*02 or IGHV1-69*04 bound to the HA stem (Table 1). It is interesting that the ancestral gene probably encoded a Leu as the only IGHV1-69 genes from the Chimpanzees, the Gorilla, and the Orangutan encode a Leu at that position and these great apes have respective common ancestors with humans 6, 10, and 12–16 million years ago (Scally et al., 2012). Treatment with an anti-HA head mAb, V2-36, with a dose of 180 μg (∼9 mg/kg) also was successful in treating mice infected with pdmH1N1 (Figure 5B).
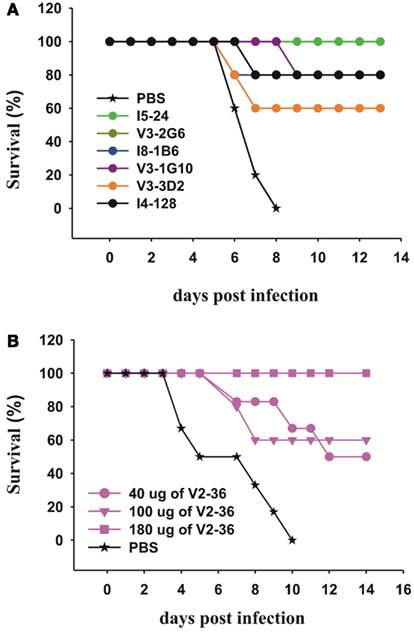
Figure 5. Therapeutic effects of mAbs on lethal infections with a human isolate of pdmH1N1. (A,B) Groups of 5 CD-1 mice were infected intranasally with 2 × 105 PFU of A/Halifax/210/2009 (pdmH1N1). Twenty-four hours post-infection, the mice were treated IP with (A) 300 μg of the indicated anti-HA stem mAbs (the survival curves for V3-2G6 and I8-1B6 are superimposed and mainly superimposed with the curve of I4-128) or (B) with the indicated doses of the anti-HA head mAb, V2-36.
Many of the mAbs Were Derived from Memory B Cells Induced by Seasonal Influenza but Some may Have been Derived from Naïve B Cells
The PB that entered the blood ∼7–10 days after infection or vaccination with pdmH1N1 were likely induced by cross-activation by pdmHA of memory B cells, as the median number of somatic mutations in the IGHV gene was 29 versus 13.6 ± 4.8 in memory B cells or germinal center B cells (Wrammert et al., 2008; Figure 6). All but one of the mAbs, V4-17, cross-reacted with the 2009/2010 seasonal influenza vaccine (Figure 3). Thus it is likely that these memory B cells, including those against the HA stem, were elicited by previous contact with seasonal influenza (Corti et al., 2010). A minority of the mAbs (12.5%) exhibited equal to or less than six somatic mutations (Table 1) and thus were likely to be ultimately derived from naïve B cells activated by pdmHA (Smith et al., 1997). Four mAbs were against the HA stem and two used IGHV1-69. Given that these mAbs were from a different clonotype (Table 1), this observation suggests that a primary response against influenza, includes heterosubtypic antibodies against the HA stem.
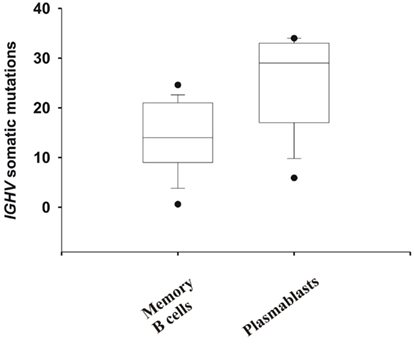
Figure 6. Evidence that most mAbs originated from memory B cells that were activated by pdmH1N1. Shown are the number of somatic mutations in IGHV genes of mAbs generated from PB and memory B cells. Whiskers show the 5/95th percentile. Mann–Whitney Rank Sum Test of the number of somatic mutations in IGHV genes of mAbs generated from memory B cells (N = 23) versus PB (N = 25) demonstrates a significant difference (p < 0.001). Note that more than 50% of the mAbs generated from PB (which all but one cross-reacted with seasonal influenza vaccine) exhibited IGHV genes with more than 10% of their nucleotides mutated (>28 mutations), with the median number of mutations being 29.
Analysis of the Response of Subject V2
We analyzed the frequency of anti-HA stem mAbs generated from each subject (Table 3). We found at least one anti-HA stem mAb from each subject with one exception, V2 who was healthy and was vaccinated with pdmH1N1 vaccine and expressed at least one IGHV1-69 gene (data not shown). If we assumed that the frequency of anti-HA stem mAbs using IGHV1-69 in responses to pdmH1N1 is 50%, the probability of obtaining the observed result from V2 by chance alone was 0.002 and thus, unlikely (Table 3).
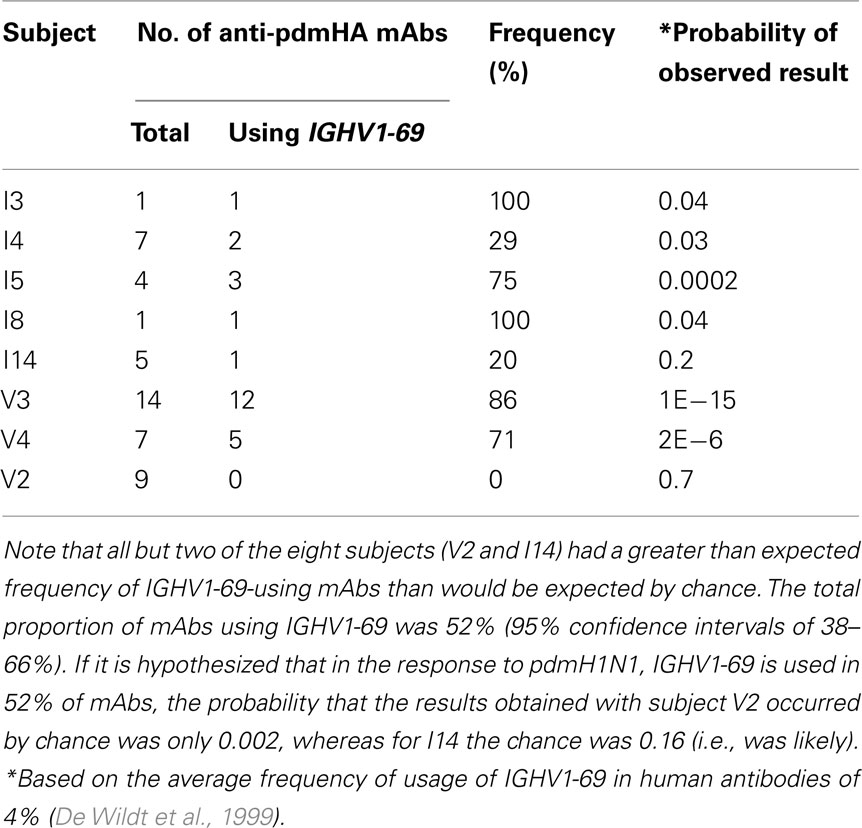
Table 3. Expected versus observed IGHV1-69 usage per subject.
We sought a cause for this and we noted that the antibody response of V2 to pdmH1N1 vaccine had two similarities to the antibody response of humans to the seasonal influenza vaccine. Firstly, eight of nine mAbs from V2 blocked hemagglutination and were therefore directed against the HA head and used IGHV4-39. Second, seven of these belonged to the same clonotype (Figure 7) and the other, V2-12, also used IGHV4-39 but used different IGHD and IGHJ genes, and used an IGKV rather than IGHL gene. We know that the dominant clonotypic response against the HA head in V2 was similar to results obtained after vaccination with seasonal influenza (Wrammert et al., 2008). The dominant clonotype in V2 had a high number of somatic mutations in IGHV4-39 ranging from 21 to 34 (Table 1) indicating they were derived from memory B cells that had been activated to differentiate to PB by the HA of pdmH1N1. That all of the mAbs from this subject, V2, bound to seasonal influenza vaccine (Table 1), suggested that the memory B cells had been induced by a seasonal H1N1 influenza virus or a related virus. Consistent with this, V2 was aged over 60, and had likely been in contact with an influenza virus related to the pdmH1N1 (Hancock et al., 2009). Thus, in V2, the lack of a prominent response to the pdmH1N1 vaccine of antibodies using IGHV1-69, correlated with a dominant antibody response against the HA head, similar to that which occurs in seasonal influenza (Wrammert et al., 2008, 2011).
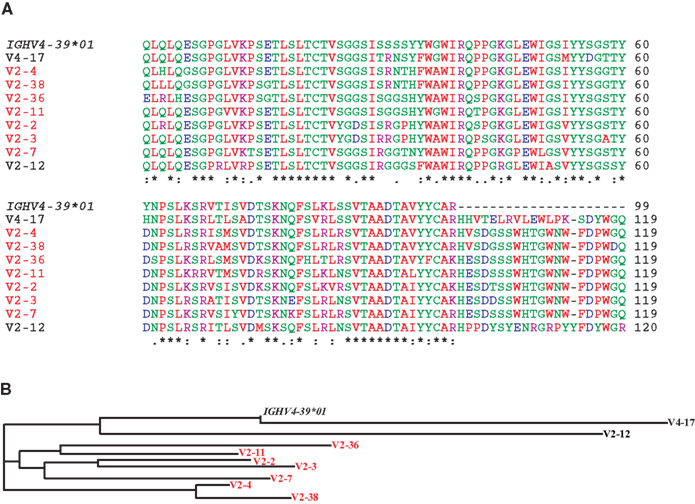
Figure 7. The dominant clonotype of mAbs against the HA head generated from subject V2. (A) Shown are alignments of the amino acid sequences of the variable region of the H chain of the dominant clonotype from V2 using the IGHV4-39, another mAb against the HA head from V2 also using IGHV4-39, and a mAb against the HA head from V4 also using IGHV4-39. We also included the amino acid sequence encoded by the germline IGHV4-39*01 allele to indicate somatic mutations. Note that the mAbs in red belong to the same clonotype as seen by the common IGHD-6-13-, IGHJ-5-encoded residues (and the similar light chains, Table 1). Shown in (B) is a phylogram using the Clustal-W website.
Evidence that Human Vaccination with PdmH1N1 Provides Passive Immunity that Protects Mice against Lethal Infection with H5N1 Influenza Virus
We next asked whether the dominant cross-protective antibody response in memory B cells induced by vaccination with the pdmH1N1 vaccine correlated with circulating cross-protective antibodies in human plasma. To test the protective effect of the human plasma from a subject vaccinated with pdmH1N1 vaccine, we passively transferred human plasma to mice and subsequently gave them a lethal intranasal dose of a heterologous H5N1 influenza virus. We tested the ability of plasma from donor V3 taken 14 days and 1 year after vaccination to protect mice against a lethal infection with H5N1 influenza (Figure 8A). We used as a control, plasma from a young adult donor taken in 2006, to ensure the subject could not have been in contact with the 2009 pandemic H1N1 virus. We observed that 400 μl of plasma from V3 taken 14 days after vaccination completely protected the mice from a lethal infection with H5N1 influenza. We also observed that 400 μl of plasma from V3 collected a year after the vaccination protected mice against death from a lethal infection with H5N1 influenza. The protection seemed to wane after a year after the vaccination, as although all of the mice treated with this plasma survived, the infection with H5N1 produced a small weight loss. However three times the dose of human plasma from V3 taken 1 year after vaccination (which gave much lower concentrations of human immunoglobulins in the mice than in the donor’s plasma) protected the mice against significant weight loss (Figure 8A).
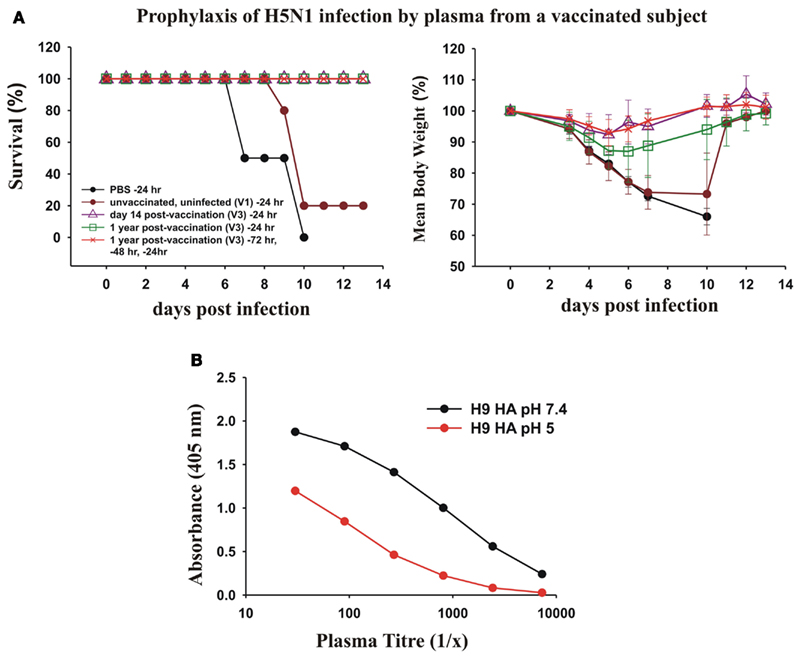
Figure 8. Prophylactic effects of human plasma from a vaccinated subject against lethal infections with H5N1 avian influenza virus. (A) Twenty-four hours before intranasal infection of BALB/c mice with 2 × 105 PFU of A/Hong Kong/213/2003 (H5N1) virus, groups of five mice were treated with a 400 μl intraperitoneal injection of PBS as a control, or 400 μl of plasma collected from subject V3 either 14 days after pdmH1N1 vaccination or 1 year after pdmH1N1 vaccination, and as a control, plasma from a young individual unvaccinated and uninfected with pdmH1N1, collected in 2006. Another group of five mice were pre-treated 72, 48, and 24 h prior to infection with 400 μl of plasma from V3 collected 1 year after pdmH1N1 vaccination. (B) pH 5 treatment of H9 HA drastically decreases the binding of V3 sera compared with binding of H9 HA treated at pH 7.4.
The cross-protective, heterosubtypic antibodies were probably against a conserved site on the HA stem. This was supported by the results of ELISA assay of the V3 plasma against untreated and pH 5-treated HA of H9. The titer of the plasma against pH 5-treated H9 HA was drastically reduced, consistent with most of the heterosubtypic antibodies being against the conserved site on the HA stem (Figure 8B).
Evidence that a MAb Generated from a Human Vaccinated with PdmH1N1 Protects against and is an Effective Therapeutic for Mice with a Lethal Infection with a Heterologous H5N1 Influenza Virus
We took our most potent mAb anti-HA stem mAb generated from a subject vaccinated with pdmH1N1 and tested the dose that completely protected against a lethal infection with H5N1 influenza. As can be seen (Figure 9A), very low doses (5 μg i.p.) completely protected mice from death. A higher dose (15 μg i.p.) completely protected against weight loss. This established that antibodies against the HA stem that were induced by pdmH1N1 vaccination mediated protection against a heterologous H5N1 influenza.
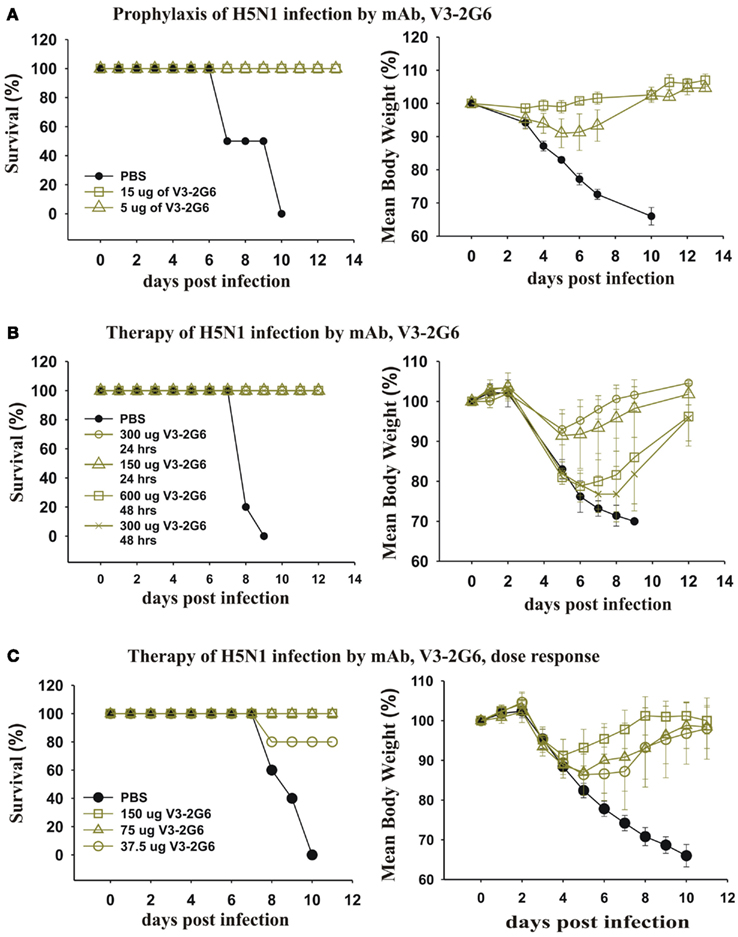
Figure 9. Therapeutic and prophylactic effects of purified mAb against lethal infections with H5N1 avian influenza virus. (A) Twenty-four hours before intranasal infection with 2 × 105 PFU of A/Hong Kong/213/2003 (H5N1) virus, three groups (five mice each) of BALB/c mice were injected intraperitoneally with PBS as a control, or 15 or 5 μg of V3-2G6 generated from a subject vaccinated with the pdmH1N1 vaccine. The data shows survival rates and average weight at over 14 days. (B,C) Groups of five mice were infected intranasally with 2 × 105 PFU of A/Hong Kong/213/2003 (H5N1) virus and (B) treated after 24 h with 150 or 300 μg of V3-2G6 or after 48 h with 300 or 600 μg of V3-2G6 and (C) treated after 24 h with 150, 75 or 37.5 μg of V3-2G6.
We tested whether the same mAb had therapeutic effects on a lethal infection with a heterologous H5N1 influenza (Figure 9B). As can be seen 24 h after infection with H5N1 influenza virus, when the mice were obviously severely ill and had loss ∼10% of their weight, administration of V3-2G6 at either 150 or 300 μg completely cured the mice. We also delayed therapy for an additional 24 h, at 48 h after infection with H5N1 influenza virus, when the mice had lost more than 20% of their weight. The administration of V3-2G6 at either 150 or 300 μg completely cured the mice. Shown in Figure 9C, is a dose–response experiment which shows that 75 μg cured all of the mice from a lethal infection with H5N1 influenza virus.
Discussion
Our data show that vaccination with the pdmH1N1 vaccine or infection with the virus, induced in many humans a dominant cross-protective, heterosubtypic antibody response against a conserved site on the HA stem. The majority of the anti-HA stem mAbs were encoded by IGHV1-69 but a minority were encoded by different immunoglobulin variable region genes, which may bind to different, overlapping epitopes. This was consistent with the fact that the pandemic vaccine exhibited the conserved epitopes bound by anti-HA stem mAbs (Figure 3). In contrast, comparable experiments examining the frequency of anti-HA stem mAbs in humans after seasonal influenza vaccination, found only antibodies against the HA head and no mAbs against the HA stem (Wrammert et al., 2008) or found, in some individuals, very low frequencies of memory B cells making heterosubtypic, cross-protective mAbs (Corti et al., 2010). This was despite the fact that, as we showed, the seasonal influenza vaccine exhibited abundantly the conserved epitopes recognized by mAbs against the HA stem (Figure 3). Unlike the seasonal influenza vaccine, the pdmH1N1 vaccine, in our experiments, was administered with the AS03 adjuvant system (Morel et al., 2011). However, we generated two mAbs against pdmHA from a subject vaccinated with unadjuvanted pdmH1N1 vaccine and one of the mAbs used IGHV1-69 (unpublished data). Thus it seems that the adjuvant is not responsible for the predominance of the heterosubtypic antibodies induced by the pdmH1N1 vaccine, but this will need to be investigated further.
What was the difference between the pdmH1N1 vaccine, which induces a dominant cross-protective antibody response against the conserved HA stem, and seasonal influenza vaccines, which induce a dominant, isolate-specific response against the mutable HA head? It is unlikely that the intrinsic nature of the pdmH1N1 antigen or its presentation to the immune system (Wei et al., 2010) was a factor in the dominance of the anti-HA stem antibody response to pdmH1N1, as this occurred in subjects with infections and vaccinations with pdmH1N1. Infections and vaccinations involve many differences in the presentation and nature of the antigen (in infections it was produced by human cells, in vaccines, in hens’ eggs), the site of the immune response, and the associated inflammatory responses. The probable mechanism through which the seasonal influenza vaccine fails to induce an effective cross-protective anti-HA stem antibody response and only induces an undetectable (Wrammert et al., 2011) or ineffectual (Corti et al., 2010) heterosubtypic response, was elucidated by the detailed dissection of the antibody response of subject V2. This subject responded to the pdmH1N1 vaccine like a seasonal influenza vaccine, with a dominant clonotypic antibody response against the HA head, which inhibited hemagglutination (Figure 2A). That there was an inverse correlation with the frequency of antibodies against the HA stem with the activation of memory B cells against the HA head in V2, is most plausibly explained by elegant experiments that have documented the competition between B cells for presenting antigen to T-cell helpers (Schwickert et al., 2011). The data show that, in V2, the pdmH1N1 vaccine activated the dominant clonotype of memory B cells against the HA head which differentiated into PB. These activated B cells would have physically outcompeted (Schwickert et al., 2011) those rare memory B cells against the HA stem in presenting cross-reactive peptides shared between virus subtypes (Russell and Liew, 1979; Doherty and Kelso, 2008) to helper T cells (Schwickert et al., 2011). The same thing would happen in normal humans vaccinated with the seasonal influenza vaccine, where it has been demonstrated that the vaccine activates pauci-clonal memory B cells against the HA head (Wrammert et al., 2008). In contrast, in most subjects infected or vaccinated with pdmH1N1, there was a paucity of memory B cells that were activated by the pdmHA head. Even if a few memory B cells had low affinity for the pdmHA, they would not compete for T-cell help with memory B cells making higher affinity antibodies against the HA stem (Schwickert et al., 2011). We also showed evidence that probable naïve B cells which have a diminished ability to interact with T cells than memory B cells (Tangye and Tarlinton, 2009), were activated by the HA stem of pdmH1N1 and collaborated with helper T cells. This is probably because, in the absence of memory B cells that were activated by the HA head of the pdmH1N1, the naïve B cells could competitively present antigens to the memory T cells induced by shared peptides between virion proteins (Russell and Liew, 1979; Doherty and Kelso, 2008).
Wrammert et al. (2011), observed that the sole mAb against pdmHA that they had isolated from a critically ill donor was specific for only pdmH1N1 and not for other H1N1 viruses; they speculate that the reason this donor may have had more serious illness was that they had fewer cross-reactive memory B cells. Our results are consistent with this notion. Subject I14 spent 2 weeks in an intensive care unit and only two out of five of the anti-pdmHA mAbs generated from this donor were against the HA stem. One mAb used IGHV1-69 with 10 mutations, and another used IGHV1-18 with 3 mutations. Thus, our results and those of Wrammert et al. (2011), suggest that the number of heterosubtypic memory B cells correlates with protection against serious influenza infection. This supports the protection afforded by these heterosubtypic antibodies and highlights the utility of inducing them using vaccines.
Our results show that a conventionally prepared influenza vaccine can induce in humans, broadly cross-protective antibodies against the HA stem. Our results suggest that the unusual dominance of the cross-protective antibodies against conserved sites on the HA stem induced by pdmH1N1, was due to the absence of memory B cells which are activated by the HA head of the pdmH1N1. The one subject, V2, that had a high frequency of memory B cells making hemagglutination inhibitory antibodies against the HA head of pdmH1N1, did not make an IGHV1-69-using mAb against the HA stem. This suggests a novel vaccination strategy, namely to vaccinate with conventional vaccines against influenza A viruses with HA that have not been circulating in humans. Based on our results with the pdmH1N1 vaccine, this strategy, with the appropriate choice of vaccine strains from Group 1 and Group 2 of the HA not circulating in humans, should induce antibodies against all conserved epitopes on the HA stem including those against epitopes shared by various Group 1 HA subtypes targeted by IGHV1-69 encoded mAbs (Ekiert et al., 2009; Sui et al., 2009), epitopes shared by various Group 2 HA subtypes (Ekiert et al., 2011), and epitopes shared by all 16 Group 1 and 2 HA subtypes (Corti et al., 2011). Additionally there may be many others yet to be defined, for example, targeted by the non-IGHV1-69 anti-HA stem mAbs we have documented here, or on the HA head (Yoshida et al., 2009). However all of the reported epitopes are accessible on purified HA and also on infectious viruses, as mAbs against them neutralize infectivity (Sui et al., 2009; Corti et al., 2011; Ekiert et al., 2011). It would be interesting to see whether the broadly cross-protective mAbs that bind to Group 2 HA subtypes (Ekiert et al., 2009) or all 16 Group 1 and 2 HA subtypes (Corti et al., 2011) bind to conventional vaccines. We predict that they will. The mAb that neutralized influenza viruses from all 16 HA subtypes was generated from a subject infected with pdmH1N1 (Corti et al., 2011), supporting the present demonstration that immunization with an “antigen-shifted” HA of pdmH1N1 results in the activation of rare heterosubtypic memory B cells. Also supporting our vaccination strategy is that this mAb that neutralized all 16 subtypes of HA, evolved from an antibody that neutralized only Group 1, but later acquired mutations that also neutralized Group 2 subtypes of viruses (Corti et al., 2011). Our vaccination strategy should be suitable for all ages; our demonstration that the primary antibody response to a “new” antigenic HA includes heterosubtypic antibodies probably originating from naive B cells, indicates that young children should respond to a series of these vaccinations.
Kaur et al. (2011), reviewed advances in inducing broadly neutralizing antibodies against influenza. One strategy that induced broadly neutralizing antibodies against the HA stem was an initial DNA vaccination with an HA in naïve mice, ferrets or non-human primates followed by immunization with an identical HA using the seasonal influenza vaccine or an adenovirus vector (Wei et al., 2010). The DNA vaccination was thought to act by increasing the number of helper T cells. Our results suggest that it will fail in adult humans, due to the plethora of memory B cells present in humans against the HA head induced by previous contact with related influenza viruses. However, as foreshadowed by Wei et al. (2010), DNA vaccination with HA may be useful to increase T helper cells in children and memory B cells against the HA stem, in combination with our strategy of using conventional vaccines including HA from influenza viruses not circulating in humans. The second prime-boost strategy reviewed by Kaur et al. (2011), was fundamentally different from Wei et al. (2010), because it was based on immunizing sequentially with different antigenically drifted H3 HA spanning the past 40 years since H3 appeared in humans (Wang et al., 2010). The frequency of broadly neutralizing mAbs in their experiments was low (3 out of 120 mAbs or 2.5% and it was not clear whether the 3 mAbs were a clonotype; Wang et al., 2010) and potency of their best mAb was >2 logs less potent at preventing disease in mice than our best human mAb induced by vaccination with pdmH1N1 vaccine. This is probably because relatively few anti-HA stem mAbs were induced in the mice because they immunized in the chronological order that these drifted influenza strains arose, which mimicks the human experience with H3N2. Each subsequent immunization would activate low affinity memory B cells against the HA head and these memory B cells would out-compete for T-cell help the memory B cells against conserved sites on the HA stem. However this was an important paper because it is the first demonstration of broadly neutralizing mAbs against the HA stem of Group 2 HA subtype influenza viruses. With subsequent examples of human mAbs broadly neutralizing against Group 2 HA subtypes of influenza viruses (Ekiert et al., 2011) and all 16 Group 1 and 2 HA subtypes (Corti et al., 2011), there is hope that sequential vaccination with Group 1 and 2 subtypes of HA that are not circulating in humans and have not been exposed to the subject will induce broadly cross-protective antibodies. Kaur et al. (2011), were enthusiastic about a minimalist approach of designing new immunogens that had the conserved protective epitopes and excluded the variable regions. Although progress has been made, our demonstration that a conventional vaccine that was developed and registered in less than 6 months induces broadly neutralizing antibodies may lead the way to a faster and cheaper universal influenza vaccine rather than developing a new immunogen which will require extensive testing and investment.
Further experimental and epidemiological studies need to be undertaken on cross-protective antibodies in the sera after the first pdmH1N1 vaccination and vaccination with other strains of influenza virus not circulating in humans, like H5N1. Moreover, there is a need to develop a simple and reliable serum test that can predict heterosubtypic protection. The WHO standard neutralization assay and the hemagglutination inhibition assay are inadequate to monitor the degree of protection in serum induced by this new vaccination strategy. Although we demonstrated that circulating antibodies in a human after vaccination with pdmH1N1 can provide passive protection to mice from a lethal infection with H5N1 influenza virus, this is not a practical assay. Binding assays, for example with an ELISA against a heterologous HA, are poor at discriminating between high levels of low affinity antibodies and low levels of high affinity antibodies and cannot predict protection. As well, our results show that competitive ELISA’s with an anti-HA stem mAb (Corti et al., 2010; Wei et al., 2010) have to be cautiously interpreted due to false positives resulting from steric interference from antibodies against the HA head. Pica et al. (2012) recently developed chimeric hemagglutinin constructs and demonstrated that patients infected with the pdmH1N1 virus had significant increases in their titers of anti-stem antibodies, supporting our conclusions.
The demonstration that a conventional vaccine given safely to millions of humans induces a dominant, cross-protective antibody response against the HA stem given by us and others (Pica et al., 2012), points to a readily testable vaccination strategy for inducing broad-spectrum protective antibodies against influenza by vaccinating successively with conventional vaccines or live attenuated viruses based on influenza viruses with an “antigen-shifted” HA that humans have not been exposed to.
Materials and Methods
Collection of Blood Samples and PBMC Isolation
Twenty to 50 ml samples of venous blood were collected from individuals confirmed to have had pdmH1N1 infections (verified by RT-PCR of nasopharyngeal swabs) or from individuals vaccinated with the pdmH1N1 vaccine. Blood samples were collected for generation of mAbs from PB at 7–10 days after vaccination or 7 days after onset of clinical symptoms with infection with pdmH1N1, or for generation of mAbs from memory B cells 14 days after vaccination or 2–8 weeks after infection. Mononuclear cells (PBMCs) and plasma samples were prepared using Ficoll–Hypaque were aliquoted and frozen. These studies were approved by the Research Ethics Boards of the University of British Columbia and the University of Toronto. Infected subjects I4 and I5 were male and I3, I8, and I14 were female and were respectively aged 23, 23, 49, 55, and 41 years. I4 and I5 were managed as outpatients and only I14 was seriously ill and was in the intensive care unit for 2 weeks. Vaccinated subjects had been immunized with pdmH1N1 with Arepanrix™ H1N1 AS03-Adjuvanted H1N1 Pandemic Influenza Vaccine. V2, V3, and V4 were all male and aged 63, 26, and 42. Samples were collected between May and December 2009 and all subjects gave informed consent. All subjects had been previously vaccinated with the seasonal influenza vaccine but none more recently than the preceding winter.
Generation of MAbs
We collected blood from convalescent patients with confirmed infections with pdmH1N1 and used RT-PCR to amplify and clone the cDNA encoding the antibodies made by individual, newly generated plasmablasts (PB) in the blood, and expressed them as mAbs (Babcook et al., 1996). However, because it was not known that the PB entered the blood after an infection rather than remaining in the site of infection, we also generated mAbs from memory B cells in blood. When the pdmH1N1 vaccine became available in November 2009, we also collected blood from vaccinated subjects.
Flow Cytometry
PBMCs were thawed, washed in PBS containing 2% BSA and held for 30 min on ice with labeled antibodies. For PB isolation, the antibodies used were against the following antigens conjugated with the following fluorochromes: CD19-APC-H7, CD3-FITC, IgM-FITC, CD20-FITC, CD27-PE, and CD38-PE–Cy7. For memory B cell isolation, CD19-APC-H7, CD3-FITC, IgM-FITC, and CD27-PE were used. All antibodies were from BD Biosciences, with the exception of the IgM-FITC (Beckman Coulter). For the isolation of pdmHA-binding PB, purified pdmHA labeled with Alexa-647 was added to the antibodies. For the isolation of pdmHA-binding memory B cells, pdmHA labeled with Alexa-647 and biotin-conjugated e-pdmH1 HA were mixed with the antibodies and streptavidin-PE–Cy7 (Invitrogen) was subsequently added following a wash step. To purify PB, PBMCs were first gated on CD19pos/CD20neg/CD3neg/IgMneg cells, then on CD27high/CD38high cells according to the known phenotype of PB (Medina et al., 2002; Odendahl et al., 2005) and in the indicated cases pdmHA-binding PB were isolated. The PB were individually deposited into PCR tubes containing RT-lysis buffer and frozen at −80°C. Memory cells were isolated by first gating on CD19pos/CD3neg/IgMneg cells, and then on CD27high cells, and then on cells double-labeled with Alexa-647-conjugated and biotin-conjugated pdmHA, the latter identified by binding of streptavidin-PE–Cy7. Single pdmHA-binding B memory cells were sorted into individual wells on 96-well plates. Each well contained 50,000 irradiated EL4 cells supplemented with 5% T-cell supernatant to induce expansion and differentiation into plasmablasts as described (Wen et al., 1987). After 1 week wells were screened for the production of pdmHA-specific antibodies by ELISA.
Amplification, Cloning, Expression, and Purification of MAbs
Individual PB were just lysed, but RNA was purified using the Absolutely RNA Microprep Kit (Stratagene) from clones that were generated from individual memory B cells. cDNA from PB or clones was synthesized using constant region primers with SuperScript III Reverse Transcriptase (Invitrogen) followed by PCR-amplification using Platinum pfx and Platinum Taq DNA Polymerase (Invitrogen). Sequences were determined on at least three clones and immunoglobulin V-gene usage and somatic mutations were determined by use of the IMGT website (Lefranc et al., 1999). Restriction sites for cloning into the heavy- and light-chain expression vectors (PHC and PLC) were included in the PCR primers as previously described (McLean et al., 2005). The heavy- and light-chain expression plasmids were co-transfected into BOSC 293 cells and the antibodies purified using Protein A Sepharose as before (McLean et al., 2005).
Production, Purification and Labeling of Trimeric Soluble pdmHA (pdmHA)
The H1N1 influenza virus (A/California/04/2009, accession number GQ117044) hemagglutinin (H1 HA) cDNA was kindly provided by Dr. Ruben Donis of the CDC (Atlanta, GA, USA). A fusion protein containing the extramembranous region of the HA (residues 18-520) followed by the bacteriophage T4 fibritin foldon domain and the 3xFLAG tag was secreted from HEK293T cells. The purified trimeric fusion protein was desialylated with Sialidase A (Prozyme, USA) and then labeled with either DyLight 649 NHS ester or Sulfo-NHS-LC biotin (Pierce, USA), followed by limited trypsin (Sigma, USA) cleavage to give a mixture of both uncleaved (HA0) and cleaved (HA1/HA2) soluble trimers.
ELISA
Monoclonal antibodies were screened by evaluation of binding to recombinant pdmHA in an ELISA. Ninety-six well plates were coated with 50 μl of purified recombinant pdmHA (6 μg/ml) or purified recombinant ectodomain of the HA of an H5N1 avian influenza virus A/Vietnam 1203/2004 (Clade 1) from Protein Sciences Corporation (1 μg/ml). Primary antibodies, either in transfected cell supernatants or purified IgG, were added for 1.5 h at 37°C, washed, and detected using goat-anti-human IgG-AP (SouthernBiotech). To evaluate the binding of the identified antibodies against the HA subtypes present in recent influenza vaccines, 96-well plates were coated with either the pdmH1N1 vaccine (Arepanrix™ H1N1, but without the adjuvant) at 2 μg/ml, or the 09/10 seasonal influenza vaccine “Influvac™” (Solvay Pharma) containing three different HA subtypes (A/Brisbane/59/2007 (H1N1)-like strain, A/Brisbane/10/2007 (H3N2)-like strain, B/Brisbane/60/2008-like strain at 6 μg/ml.
Elispot Assays
Wells of 96-well plates (MultiScreen-IP filter Plate, PVDF, Millipore) were coated with 1 μg of pdmHA overnight at 4°C, and blocked with fetal calf serum. Fifty microliters of culture medium containing the cell populations to be assayed, or as a control medium alone, were added to each well, and the plate was incubated for overnight at 37°C to allow secretion of antibodies. Antibodies against pdmHA were captured in the immediate vicinity of the secreting cell. The spots of anti-HA antibodies that had been secreted by individual PB were visualized by incubation with a mixture of goat antibodies against the human Kappa or Lambda chains conjugated with alkaline phosphatase (SouthernBiotech), developed using BCIP/NBT as a substrate.
Competition ELISA with C179 or I5-24
The indicated competing antibodies were pre-incubated on the pdmH1N1 vaccine-coated plate for 1 h before addition of C179 or biotinylated I5-24 at a final concentration of 0.2 μg/ml. The binding of C179 or biotinylated I5-24 was assayed respectively by goat anti-mouse immunoglobulins or streptavidin conjugated with alkaline phosphatase.
Low PH-Treatment of HA Bound to ELISA Plates
Pandemic H1N1 vaccine (2 μg/ml), or H9 HA (2 μg/ml; BEI Resources) was coated to ELISA plates overnight and wells were treated by adding fusion buffer (10 mM HEPES, 10 mM MES, pH 5), or as a control PBS at pH 7.4, and incubated for 1 h at 37°C. Plates were then washed with PBS and the indicated mAbs were titrated and assayed in the normal way.
Microneutralization of PdmH1N1 Assay
The assay was performed first following the WHO Manual on Animal Influenza Diagnosis and Surveillance (www.wpro.who.int/internet/resources.ashx/CSR/Publications/manual+on+animal+ai+diagnosis+and+surveillance.pdf). Briefly, the monoclonal antibody was subjected to twofold serial dilutions in a microtiter plate beginning at 1:2 and an equal volume of pdmH1N1 virus containing 100 TCID50 were added to each dilution and incubated for 2 h at 37°C. The mixtures were added to respective wells of a microtiter plate containing monolayers of MDCK cells in serum-free Megavir medium containing TCPK-treated trypsin and incubated for 3 h after which the medium was replaced by fresh Megavir medium containing TCPK-treated trypsin. The monolayers were monitored on days 3, 4, and 5 for the development of cytopathic effect (CPE). The reciprocal of the highest dilution of the antibody that inhibited the development of viral CPE was designated as the titer. A modified form of the assay consisted of eliminating the step of omitting the removal of the virus-antibody mixtures after 3 h of incubation and allowing the mAb and the virus to remain in the medium for the duration of the assay.
Binding to Cell-Expressed H5 HA
Recombinant adenoviruses expressing influenza A HA (AdHA) from A/Hong Kong/156/97 (H5N1; Hoelscher et al., 2008) were provided by Dr. Suryaprakash Sambhara (Centers for Disease Control and Prevention, Atlanta, GA, USA). The AdHA proprotein convertase (AdHA-PC) containing the HA multi-basic cleavage site was utilized. The Adenovirus empty (AdEmpty) was used as a negative control (Viraquest Inc.). To promote HA stability, NH4Cl (10 mM) was added to the culture medium during AdHA infection. Monolayers of A549 cells in 96-well plates were infected with AdHA at a MOI of 500 and incubated for 40 h prior to fixation with 4% formaldehyde. Then, cells were incubated with the indicated concentrations of mAb and treated with FITC-labeled anti-human immunoglobulin antibodies. Nuclei were counterstained with Hoechst. Images were recorded and analyzed/counted using the Cellomics instrument (Thermo Scientific). Data shown are the average of duplicate measurements from a representative experiment.
Assay for Inhibition of HA-Mediated Fusion
A549 cells were seeded and infected with AdHA as described in the method for the binding assay. At 40 h post-infection, cells were washed with PBS, and incubated with the indicated antibodies (20 μg/ml) for 30 min at 37°C. Then, cells were washed again and treated with fusion buffer (10 mM HEPES, 10 mM MES, pH 5) for 5 min at room temperature. Media was replaced with normal cell culture media and cells were incubated at 37°C for a 5-h period to allow for syncytia formation. To monitor syncytia formation, cells were labeled with 10 μM Cell Tracker Green CMFDA (Molecular Probes) for 30 min at 37°C, followed by further incubation for 30 min in fresh media before samples were fixed with 4% formaldehyde. Nuclei were counterstained with Hoechst dye (1 μg/ml). Images were analyzed using the Cellomics system.
H5N1 Plaque Reduction Assay
Approximately 250 plaque forming units (PFU) of influenza viruses A/Goose/Ger/R1400/07 (H5N1) and A/Ck/Ger/R28/03 (H7N7) were incubated in triplicate with the mAbs at twice the indicated final concentrations at 37°C for 1 h at a final volume of 80 μl. 40 μl of the virus-antibody mixtures were transferred onto MDCK cell monolayers in 96-well plates and incubated for 1 h at 37°C. Then the cells were overlaid with 40 μl of 1.5% carboxy-methyl cellulose. 30 h later, plaques were visualized by immunostaining using a mouse monoclonal anti-NP antibody (F26NP9-2-1). Plaques were counted and % neutralization was calculated by setting the infection without mAb as 0% neutralization. Data shown are the mean of triplicate measurements with SD.
Therapeutic and Prophylactic Testing of Antibodies against Fatal PdmH1N1 and H5N1 Pneumonia in Mice
The human clinical isolate of pandemic H1N1 influenza, A/Halifax/210/2009 or the H5N1 vaccine strain, A/Hong Kong/213/2003 on the PR8/34 backbone were used to induce fatal viral pneumonia in CD-1 mice or BALB/c mice respectively. Groups of 5 CD-1 or BALB/c mice (19–21 Gm, females) were infected under halothane anesthesia (3.5% in O2) with 50 μl of 2 × 105 pfu of either H5N1 or pmdH1N1 virus in PBS. Mice were either treated with an intraperitoneal injection of 0.5 ml of purified monoclonal antibodies 1 day before or 1–2 days after infection to monitor protective or therapeutic effects. Controls were treated with only PBS. Survival and weight loss was monitored for 12–14 days to assess the effects against fatal pandemic H1N1 or H5N1 infections in mice. Similarly, mice were either treated with an intraperitoneal injection of 400 μl of human plasma 1 day before or 400 μl of human plasma for each of 3, 2, or 1 days before infection with H5N1 to monitor protective effects.
Ethics statement
Animal studies were performed under the supervision of a veterinarian (DVM) and carried out in compliance with the guidelines of the Canadian Council on Animal Care (CCAC) as outlined in the Care and Use of Experimental Animals, Vol. 1, 2nd Edn. (1993). The animal care protocol was approved by the University of Ottawa Animal Care Committee (Protocol Number: BMI-85) and all efforts were made to minimize suffering and mice were euthanized at humane end-points, if infection resulted in greater than 30% body weight loss plus respiratory distress.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Andy Johnson and Jason Wong for FACS, Dewi Schrader for statistical analyses, Lenka Allan and Fabio Rossi for advice with FACS and microscopy, Rama Kandiah for neutralization assays, and Martine Boutin, Heather Braybrook, Emma-Kate Loveday, and Yohannes Berhane for advice and reagents, Sherie Duncan and Kasmintan Schrader for discussion, and Jeremy Carver, Denis Ferkany and Dale Cummins for their help. Supported by grants from the Canadian Institutes for Health Research to J. W. Schrader (POR-100131 and IBF-103106), J. M. Rini and F. Jean (TPA-90195) and from the International Consortium for Anti-Virals to J. W. Schrader and J. M. Rini, and by the Michael Smith Foundation for Health Research. J. W. Schrader has relevant patent applications.
References
Babcook, J. S., Leslie, K. B., Olsen, O. A., Salmon, R. A., and Schrader, J. W. (1996). A novel strategy for generating monoclonal antibodies from single, isolated lymphocytes producing antibodies of defined specificities. Proc. Natl. Acad. Sci. U.S.A. 93, 7843–7848.
Barington, T., Heilmann, C., and Andersen, V. (1990). Quantitation of antibody-secreting cells in the blood after vaccination with Haemophilus influenzae type b conjugate vaccine. Scand. J. Immunol. 31, 515–522. [Published erratum appears in Scand. J. Immunol. 32, 59].
Chen, G. L., and Subbarao, K. (2009). Attacking the flu: neutralizing antibodies may lead to “universal” vaccine. Nat. Med. 15, 1251–1252.
Corti, D., Suguitan, A. L., Pinna, D., Silacci, C., Fernandez-Rodriguez, B. M., Vanzetta, F., Santos, C., Luke, C. J., Torres-Velez, F. J., Temperton, N. J., Weiss, R. A., Sallusto, F., Subbarao, K., and Lanzavecchia, A. (2010). Heterosubtypic neutralizing antibodies are produced by individuals immunized with a seasonal influenza vaccine. J. Clin. Invest. 120, 1663–1673.
Corti, D., Voss, J., Gamblin, S. J., Codoni, G., Macagno, A., Jarrossay, D., Vachieri, S. G., Pinna, D., Minola, A., Vanzetta, F., Silacci, C., Fernandez-Rodriguez, B. M., Agatic, G., Bianchi, S., Giacchetto-Sasselli, I., Calder, L., Sallusto, F., Collins, P., Haire, L. F., Temperton, N., Langedijk, J. P. M., Skehel, J. J., and Lanzavecchia, A. (2011). A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza a hemagglutinins. Science 333, 850–856.
De Wildt, R. M., Hoet, R. M., Van Venrooij, W. J., Tomlinson, I. M., and Winter, G. (1999). Analysis of heavy and light chain pairings indicates that receptor editing shapes the human antibody repertoire. J. Mol. Biol. 285, 895–901.
Ding, N., Wu, N., Xu, Q., Chen, K., and Zhang, C. (2009). Molecular evolution of novel swine-origin A/H1N1 influenza viruses among and before human. Virus Genes 39, 293–300.
Doherty, P. C., and Kelso, A. (2008). Toward a broadly protective influenza vaccine. J. Clin. Invest. 118, 3273–3275.
Ekiert, D. C., Bhabha, G., Elsliger, M. A., Friesen, R. H., Jongeneelen, M., Throsby, M., Goudsmit, J., and Wilson, I. A. (2009). Antibody recognition of a highly conserved influenza virus epitope. Science 324, 246–251.
Ekiert, D. C., Friesen, R. H. E., Bhabha, G., Kwaks, T., Jongeneelen, M., Yu, W., Ophorst, C., Cox, F., Korse, H. J. W. M., Brandenburg, B., Vogels, R., Brakenhoff, J. P. J., Kompier, R., Koldijk, M. H., Cornelissen, L. A. H. M., Poon, L. L. M., Peiris, M., Koudstaal, W., Wilson, I. A., and Goudsmit, J. (2011). A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 333, 843–850.
Garten, R. J., Davis, C. T., Russell, C. A., Shu, B., Lindstrom, S., Balish, A., Sessions, W. M., Xu, X., Skepner, E., Deyde, V., Okomo-Adhiambo, M., Gubareva, L., Barnes, J., Smith, C. B., Emery, S. L., Hillman, M. J., Rivailler, P., Smagala, J., De Graaf, M., Burke, D. F., Fouchier, R. A., Pappas, C., Alpuche-Aranda, C. M., Lopez-Gatell, H., Olivera, H., Lopez, I., Myers, C. A., Faix, D., Blair, P. J., Yu, C., Keene, K. M., Dotson, P. D. Jr., Boxrud, D., Sambol, A. R., Abid, S. H., St George, K., Bannerman, T., Moore, A. L., Stringer, D. J., Blevins, P., Demmler-Harrison, G. J., Ginsberg, M., Kriner, P., Waterman, S., Smole, S., Guevara, H. F., Belongia, E. A., Clark, P. A., Beatrice, S. T., Donis, R., Katz, J., Finelli, L., Bridges, C. B., Shaw, M., Jernigan, D. B., Uyeki, T. M., Smith, D. J., Klimov, A. I., and Cox, N. J. (2009). Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325, 197–201.
Hancock, K., Veguilla, V., Lu, X., Zhong, W., Butler, E. N., Sun, H., Liu, F., Dong, L., Devos, J. R., Gargiullo, P. M., Brammer, T. L., Cox, N. J., Tumpey, T. M., and Katz, J. M. (2009). Cross-reactive antibody responses to the 2009 pandemic H1N1 influenza virus. N. Engl. J. Med. 361, 1945–1952.
Heilmann, C., Henrichsen, J., and Pedersen, F. K. (1987). Vaccination-induced circulation of human B cells secreting type-specific antibodies against pneumococcal polysaccharides. Scand. J. Immunol. 25, 61–67.
Hoelscher, M. A., Singh, N., Garg, S., Jayashankar, L., Veguilla, V., Pandey, A., Matsuoka, Y., Katz, J. M., Donis, R., Mittal, S. K., and Sambhara, S. (2008). A broadly protective vaccine against globally dispersed clade 1 and clade 2 H5N1 influenza viruses. J. Infect. Dis. 197, 1185–1188.
Hung, I. F., To, K. K., Lee, C.-K., Lee, K.-L., Chan, K., Yan, W.-W., Liu, R., Watt, C.-L., Chan, W.-M., Lai, K.-Y., Koo, C.-K., Buckley, T., Chow, F.-L., Wong, K.-K., Chan, H.-S., Ching, C.-K., Tang, B. S., Lau, C. C., Li, I. W., Liu, S.-H., Chan, K.-H., Lin, C.-K., and Yuen, K.-Y. (2011). Convalescent plasma treatment reduced mortality in patients with severe pandemic influenza A (H1N1) 2009 virus infection. Clin. Infect. Dis. 52, 447–456.
Kaur, K., Sullivan, M., and Wilson, P. C. (2011). Targeting B cell responses in universal influenza vaccine design. Trends Immunol. 32, 524–531.
Knossow, M., Gaudier, M., Douglas, A., Barrere, B., Bizebard, T., Barbey, C., Gigant, B., and Skehel, J. J. (2002). Mechanism of neutralization of influenza virus infectivity by antibodies. Virology 302, 294–298.
Lee, F. E.-H., Falsey, A. R., Halliley, J. L., Sanz, I., and Walsh, E. E. (2010). Circulating antibody-secreting cells during acute respiratory syncytial virus infection in adults. J. Infect. Dis. 202, 1659–1666.
Lefranc, M. P., Giudicelli, V., Ginestoux, C., Bodmer, J., Muller, W., Bontrop, R., Lemaitre, M., Malik, A., Barbie, V., and Chaume, D. (1999). IMGT, the international ImMunoGeneTics database. Nucleic Acids Res. 27, 209–212.
Luke, T. C., Kilbane, E. M., Jackson, J. L., and Hoffman, S. L. (2006). Meta-analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Ann. Intern. Med. 145, 599–609.
McLean, G. R., Olsen, O. A., Watt, I. N., Rathanaswami, P., Leslie, K. B., Babcook, J. S., and Schrader, J. W. (2005). Recognition of human cytomegalovirus by human primary immunoglobulins identifies an innate foundation to an adaptive immune response. J. Immunol. 174, 4768–4778.
Medina, F., Segundo, C., Campos-Caro, A., Gonzalez-Garcia, I., and Brieva, J. A. (2002). The heterogeneity shown by human plasma cells from tonsil, blood, and bone marrow reveals graded stages of increasing maturity, but local profiles of adhesion molecule expression. Blood 99, 2154–2161.
Morel, S., Didierlaurent, A., Bourguignon, P., Delhaye, S., Baras, B., Jacob, V. R., Planty, C., Elouahabi, A., Harvengt, P., Carlsen, H., Kielland, A., Chomez, P., Garçon, N., and Van Mechelen, M. (2011). Adjuvant system AS03 containing α-tocopherol modulates innate immune response and leads to improved adaptive immunity. Vaccine 29, 2461–2473.
Mozdzanowska, K., Furchner, M., Washko, G., Mozdzanowski, J., and Gerhard, W. (1997). A pulmonary influenza virus infection in SCID mice can be cured by treatment with hemagglutinin-specific antibodies that display very low virus-neutralizing activity in vitro. J. Virol. 71, 4347–4355.
Odendahl, M., Mei, H., Hoyer, B. F., Jacobi, A. M., Hansen, A., Muehlinghaus, G., Berek, C., Hiepe, F., Manz, R., Radbruch, A., and Dorner, T. (2005). Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood 105, 1614–1621.
Okuno, Y., Isegawa, Y., Sasao, F., and Ueda, S. (1993). A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J. Virol. 67, 2552–2558.
Pica, N., Hai, R., Krammer, F., Wang, T. T., Maamary, J., Eggink, D., Tan, G. S., Krause, J. C., Moran, T., Stein, C. R., Banach, D., Wrammert, J., Belshe, R. B., Garcia-Sastre, A., and Palese, P. (2012). Hemagglutinin stalk antibodies elicited by the 2009 pandemic influenza virus as a mechanism for the extinction of seasonal H1N1 viruses. Proc. Natl. Acad. Sci. U.S.A. 109, 2573–2578.
Russell, S. M., and Liew, F. Y. (1979). T cells primed by influenza virion internal components can cooperate in the antibody response to haemagglutinin. Nature 280, 147–148.
Scally, A., Dutheil, J. Y., Hillier, L. W., Jordan, G. E., Goodhead, I., Herrero, J., Hobolth, A., Lappalainen, T., Mailund, T., Marques-Bonet, T., McCarthy, S., Montgomery, S. H., Schwalie, P. C., Tang, Y. A., Ward, M. C., Xue, Y., Yngvadottir, B., Alkan, C., Andersen, L. N., Ayub, Q., Ball, E. V., Beal, K., Bradley, B. J., Chen, Y., Clee, C. M., Fitzgerald, S., Graves, T. A., Gu, Y., Heath, P., Heger, A., Karakoc, E., Kolb-Kokocinski, A., Laird, G. K., Lunter, G., Meader, S., Mort, M., Mullikin, J. C., Munch, K., O‚ Äôconnor, T. D., Phillips, A. D., Prado-Martinez, J., Rogers, A. S., Sajjadian, S., Schmidt, D., Shaw, K., Simpson, J. T., Stenson, P. D., Turner, D. J., Vigilant, L., Vilella, A. J., Whitener, W., Zhu, B., Cooper, D. N., Jong, P. D., Dermitzakis, E. T., Eichler, E. E., Flicek, P., Goldman, N., Mundy, N. I., Ning, Z., Odom, D. T., Ponting, C. P., Quail, M. A., Ryder, O. A., Searle, S. M., Warren, W. C., Wilson, R. K., Schierup, M. H., Rogers, J., Tyler-Smith, C., and Durbin, R. (2012). Insights into hominid evolution from the gorilla genome sequence. Nature 483, 169–175.
Schwickert, T. A., Victora, G. D., Fooksman, D. R., Kamphorst, A. O., Mugnier, M. R., Gitlin, A. D., Dustin, M. L., and Nussenzweig, M. C. (2011). A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J. Exp. Med. 208, 1243–1252.
Simmons, C. P., Bernasconi, N. L., Suguitan, A. L., Mills, K., Ward, J. M., Chau, N. V., Hien, T. T., Sallusto, F., Ha Do, Q., Farrar, J., De Jong, M. D., Lanzavecchia, A., and Subbarao, K. (2007). Prophylactic and therapeutic efficacy of human monoclonal antibodies against H5N1 influenza. PLoS Med. 4, e178. doi:10.1371/journal.pmed.0040178
Smith, K. G., Light, A., Nossal, G. J., and Tarlinton, D. M. (1997). The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. 16, 2996–3006.
Sui, J., Hwang, W. C., Perez, S., Wei, G., Aird, D., Chen, L. M., Santelli, E., Stec, B., Cadwell, G., Ali, M., Wan, H., Murakami, A., Yammanuru, A., Han, T., Cox, N. J., Bankston, L. A., Donis, R. O., Liddington, R. C., and Marasco, W. A. (2009). Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 16, 265–273.
Tangye, S. G., and Tarlinton, D. M. (2009). Memory B cells: effectors of long-lived immune responses. Eur. J. Immunol. 39, 2065–2075.
Throsby, M., Van Den Brink, E., Jongeneelen, M., Poon, L. L., Alard, P., Cornelissen, L., Bakker, A., Cox, F., Van Deventer, E., Guan, Y., Cinatl, J., Ter Meulen, J., Lasters, I., Carsetti, R., Peiris, M., De Kruif, J., and Goudsmit, J. (2008). Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS ONE 3, e3942. doi:10.1371/journal.pone.0003942
Wang, T. T., Tan, G. S., Hai, R., Pica, N., Petersen, E., Moran, T. M., and Palese, P. (2010). Broadly protective monoclonal antibodies against H3 influenza viruses following sequential immunization with different hemagglutinins. PLoS Pathog. 6, e1000796. doi:10.1371/journal.ppat.1000796
Wei, C.-J., Boyington, J. C., McTamney, P. M., Kong, W.-P., Pearce, M. B., Xu, L., Andersen, H., Rao, S., Tumpey, T. M., Yang, Z.-Y., and Nabel, G. J. (2010). Induction of broadly neutralizing H1N1 influenza antibodies by vaccination. Science 329, 1060–1064.
Wen, L., Hanvanich, M., Werner-Favre, C., Brouwers, N., Perrin, L. H., and Zubler, R. H. (1987). Limiting dilution assay for human B cells based on their activation by mutant EL4 thymoma cells: total and antimalaria responder B cell frequencies. Eur. J. Immunol. 17, 887–892.
Wiley, D. C., and Skehel, J. J. (1987). The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 56, 365–394.
Wrammert, J., Koutsonanos, D., Li, G.-M., Edupuganti, S., Sui, J., Morrissey, M., McCausland, M., Skountzou, I., Hornig, M., Lipkin, W. I., Mehta, A., Razavi, B., Del Rio, C., Zheng, N.-Y., Lee, J.-H., Huang, M., Ali, Z., Kaur, K., Andrews, S., Amara, R. R., Wang, Y., Das, S. R., O’Donnell, C. D., Yewdell, J. W., Subbarao, K., Marasco, W. A., Mulligan, M. J., Compans, R., Ahmed, R., and Wilson, P. C. (2011). Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med. 208, 181–193.
Wrammert, J., Smith, K., Miller, J., Langley, W. A., Kokko, K., Larsen, C., Zheng, N. Y., Mays, I., Garman, L., Helms, C., James, J., Air, G. M., Capra, J. D., Ahmed, R., and Wilson, P. C. (2008). Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671.
Xu, R., Ekiert, D. C., Krause, J. C., Hai, R., Crowe, J. E., and Wilson, I. A. (2010). Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 328, 357–360.
Yen, H.-L., and Webster, R. G. (2009). Pandemic influenza as a current threat. Curr. Top. Microbiol. Immunol. 333, 3–24.
Yoshida, R., Igarashi, M., Ozaki, H., Kishida, N., Tomabechi, D., Kida, H., Ito, K., and Takada, A. (2009). Cross-protective potential of a novel monoclonal antibody directed against antigenic site B of the hemagglutinin of influenza A viruses. PLoS Pathog. 5, e1000350. doi:10.1371/journal.ppat.1000350
Keywords: pandemic H1N1 influenza, vaccines, cross-protective antibodies, heterosubtypic, plasmablasts, memory B cells, competition for T-cell help, hemagglutinin
Citation: Thomson CA, Wang Y, Jackson LM, Olson M, Wang W, Liavonchanka A, Keleta L, Silva V, Diederich S, Jones RB, Gubbay J, Pasick J, Petric M, Jean F, Allen VG, Brown EG, Rini JM and Schrader JW (2012) Pandemic H1N1 influenza infection and vaccination in humans induces cross-protective antibodies that target the hemagglutinin stem. Front. Immun. 3:87. doi: 10.3389/fimmu.2012.00087
Received: 16 February 2012; Paper pending published: 06 March 2012;
Accepted: 04 April 2012; Published online: 08 May 2012.
Edited by:
Harry W. Schroeder, University of Alabama at Birmingham, USAReviewed by:
Patrick C. Wilson, University of Chicago, USANina Luning Prak, Trustees of the University of Pennsylvania, USA
Copyright: © 2012 Thomson, Wang, Jackson, Olson, Wang, Liavonchanka, Keleta, Silva, Diederich, Jones, Gubbay, Pasick, Petric, Jean, Allen, Brown, Rini and Schrader. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: J. W. Schrader, The Biomedical Research Centre, University of British Columbia, 2222 Health Sciences Mall, Vancouver, BC, Canada V6T 1Z3. e-mail: john@brc.ubc.ca
†C. A. Thomson and Y. Wang have contributed equally to this work.