
- 1 Centre for Infection, Immunity and Disease Mechanisms, School of Health Sciences and Social Care, Brunel University, London, UK
- 2 Centre for Biotechnology and Bioinformatics, School of Life Sciences, Jawaharlal Nehru Institute for Advanced Studies, Secunderabad, Andhra Pradesh, India
Surfactant proteins SP-A and SP-D are hydrophilic, collagen-containing calcium-dependent lectins, which appear to have a range of innate immune functions at pulmonary as well as extrapulmonary sites. These proteins bind to target ligands on pathogens, allergens, and apoptotic cells, via C-terminal homotrimeric carbohydrate recognition domains, while the collagen region brings about the effector functions via its interaction with cell surface receptors. SP-A and SP-D deal with various pathogens, using a range of innate immune mechanisms such as agglutination/aggregation, enhancement of phagocytosis, and killing mechanisms by phagocytic cells and direct growth inhibition. SP-A and SP-D have also been shown to be involved in the control of pulmonary inflammation including allergy and asthma. Emerging evidence suggest that SP-A and SP-D are capable of linking innate immunity with adaptive immunity that includes modulation of dendritic cell function and helper T cell polarization. This review enumerates immunological properties of SP-A and SP-D inside and outside lungs and discusses their importance in human health and disease.
Introduction
Surfactant proteins, SP-A and SP-D, are collagen-containing C-type (calcium-dependent) lectins called collectins, which were originally identified as surfactant-associated proteins involved in pulmonary surfactant homeostasis (Weaver and Whitsett, 1991). Subsequent studies involving structural and functional characterization of these proteins have suggested that SP-A and SP-D are potent innate immune molecules in the lungs that are involved in viral neutralization, clearance of bacteria, fungi, apoptotic and necrotic cells, down regulation of allergic reaction and resolution of inflammation (Kishore et al., 2001, 2006; Wright, 2005). Their primary structures include an N-terminal, triple-helical collagen region, and a C-terminal homotrimeric C-type lectin or carbohydrate recognition domain (CRD). The trimeric CRDs can recognize carbohydrate or charge patterns on microbes, allergens, and dying cells acting as a soluble pattern recognition receptors (PRRs) while the collagen region can interact with receptor molecules present on a variety of immune cells in order to initiate clearance mechanisms (Kishore et al., 2006).
SP-A and SP-D are large hydrophilic proteins, as opposed to SP-B and SP-C, the other two hydrophobic surfactant proteins found in the lungs. Their primary structure is organized into a cysteine-containing N-terminal region, a triple-helical collagen region composed of repeating Gly-X-Y triplets, an α-helical coiled-coil neck region, and a C-terminus comprising a C-type lectin or CRD region (reviewed in Kishore et al., 2006). SP-A and SP-D are large oligomeric structures, each assembled from multiple copies of either one or two polypeptide chains. SP-A has a hexameric structure in which six structural subunits associate to form a molecule of 630 kDa made up of 18 chains. SP-D is composed of oligomers of a 130 kDa subunit comprising three identical polypeptide chains of 43 kDa. Human SP-D is assembled into a 520 kDa tetrameric structure with four of the homotrimeric subunits linked via their N-terminal regions, but multimers, trimers, dimers and monomers are also possible (Holmskov et al., 2003; Kishore et al., 2006; Figures 1A,B).
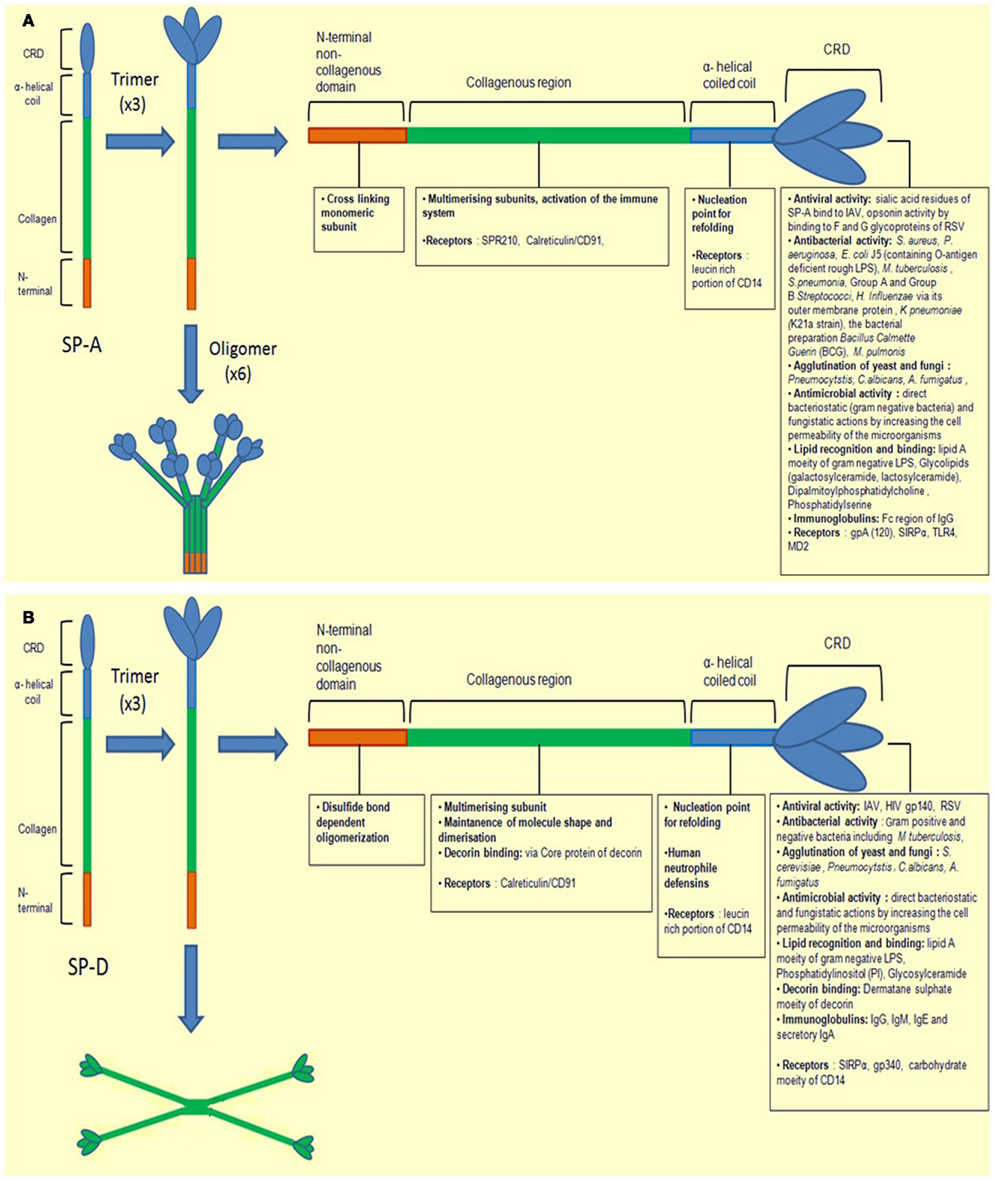
Figure 1. Illustration of a molecule of (A) SP-A and (B) SP-D depicting different regions. The molecules are first shown as monomers and trimers. They are divided into four subunits, the N-terminal non-collagenous domain, collagenous region, helical neck, and C-terminal carbohydrate recognition domain. Each sub-unit has different ligand binding affinities.
SP-A and SP-D can bind various self and non-self ligands, mostly via CRDs on the target surface in a carbohydrate and calcium-dependent manner, while the collagen region can recruit and activate the immune cells for the clearance of pathogens and apoptotic/necrotic cells (Kishore et al., 2006) (Figure 2). In addition to anti-microbial activities, SP-A and SP-D also play an important role in the control of inflammation triggered by self, non-self and altered self cells and molecules. Thus, they have a pivotal role in the clearance of apoptotic and necrotic cells, dampening of allergic reactions, maintenance of pregnancy (by virtue of their presence in the amniotic fluid), and modulating dendritic cell (DC) and T cell properties (Figure 2). The major immune-related functions of SP-A and SP-D are being reviewed here.
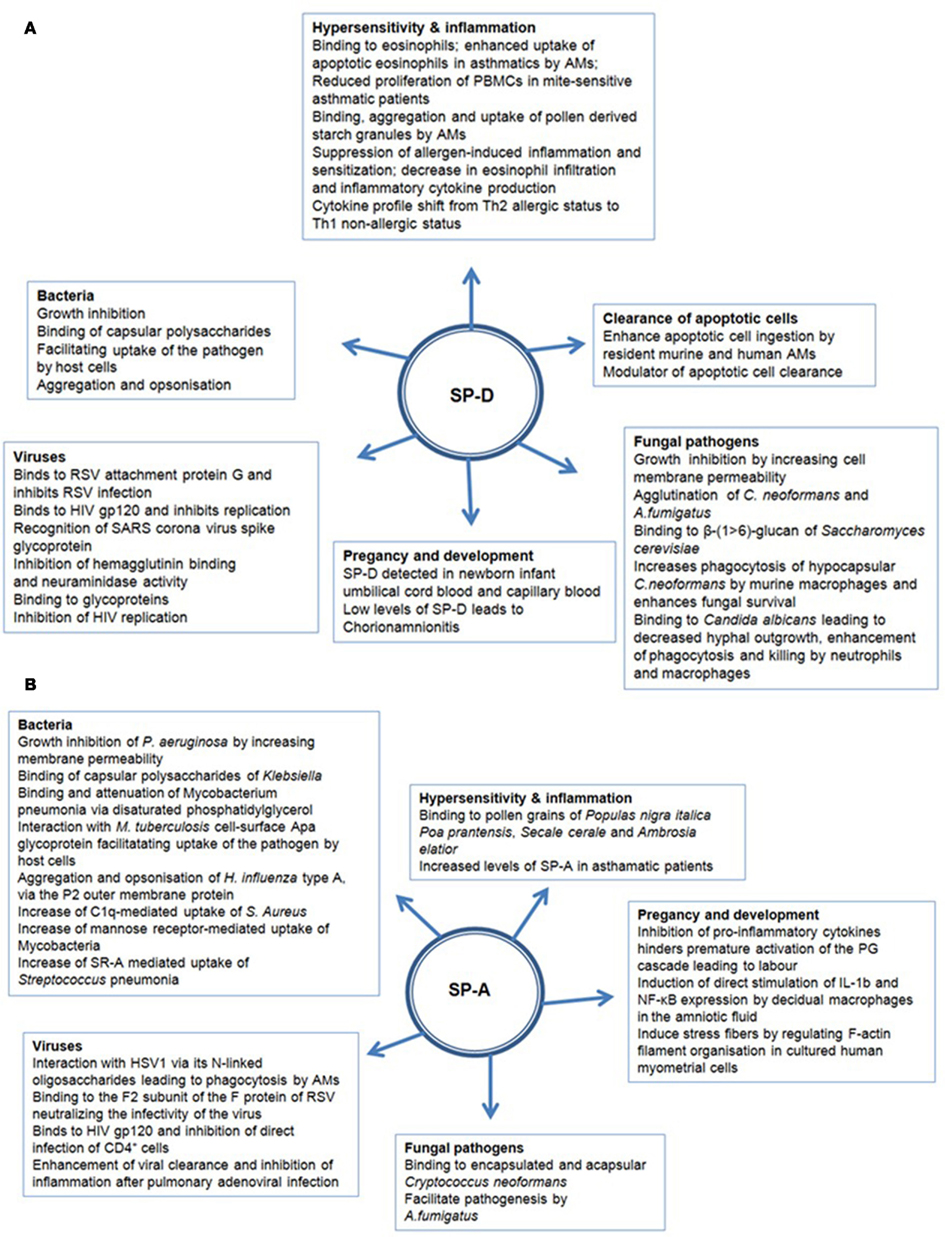
Figure 2. Multiple functions of (A) SP-D and (B) SP-A in human health and disease.
Anti-Microbial Properties of SP-A and SP-D
SP-A and SP-D play a central role in the protection of the lungs against respiratory infection. SP-A and SP-D are able to bind to a variety of microbes (viruses, bacteria, yeast, and fungi), thus acting as opsonins and having direct and indirect biological effects (Crouch, 2000; Lawson and Reid, 2000; Crouch and Wright, 2001; Shepherd, 2002; Tables 1 and 2). There is one report of SP-D binding to the parasite Schistosoma mansoni (van de Wetering et al., 2004b). SP-A and SP-D bind and/or agglutinate a wide range of microbes that enhance their attachment to phagocytic cells and can lead to killing and enhanced clearance. SP-A and SP-D deal with various pathogens via a number of innate immune mechanisms that include agglutination/aggregation, enhanced phagocytosis, and superoxidative burst by phagocytes, chemotaxis, growth inhibition, and modulation of toll-like receptor (TLR) functions (Kishore et al., 2006).
Interaction with Bacteria
One of the first studies showing that surfactant proteins may be involved in pulmonary immunity against infection was conducted 40 years ago and involved isolating total surfactant by centrifugation and showed enhanced killing, but not phagocytosis of Staphylococcus aureus by alveolar macrophages (AMs), in vitro (LaForce et al., 1973). Subsequently, SP-A was shown to enhance uptake of sensitized erythrocytes through Fc and complement receptor 1 (CR1) present on AMs (Tenner et al., 1989; van Iwaarden et al., 1992).
SP-A and SP-D can bind to Gram-negative and Gram-positive bacteria. Lipopolysaccharide (LPS) is an established ligand for surfactant proteins and a major component of the cell wall of Gram-negative bacteria. There are essentially two types of LPS. Firstly, smooth LPS which is comprised of the O-specific antigen, complete core oligosaccharides and the endotoxin-principal region, lipid A. Secondly, rough LPS serotypes which lacks the O-specific antigen, but has the lipid A region and increasingly shorter core oligosaccharides in some bacterial mutants (Silhavy et al., 2010).
SP-D was first shown to bind to rough LPS via CRDs and agglutinate Escherichia coli (Kuan et al., 1992). SP-A and SP-D can also bind to rough serotypes of LPS, via contrasting mechanisms. SP-A binds to the lipid A region of LPS (van Iwaarden et al., 1994; Kalina et al., 1995). SP-D has been shown to bind via its trimeric CRDs only rough LPS derived from a select species of Gram-negative bacteria such as Klebsiella pneumoniae and Pseudomonas aeruginosa (Lim et al., 1994; Kishore et al., 1996). SP-D binds strongly to LPS from E. coli and Salmonella, but not to lipid A, LPS from oligosaccharide deficient strains or smooth LPS, suggesting that core terminal sugar residues (e.g., heptose and glucose) may be important in binding SP-D to LPS (Kuan et al., 1992). As with SP-D, SP-A only seems to bind rough LPS (van Iwaarden et al., 1994), often mediating subsequent phagocytosis and killing of the bacterium (Pikaar et al., 1995). SP-A binds also to lipid A and rough serotypes of LPS lacking O-specific antigen, but not to smooth LPS. SP-A has also been demonstrated to interact with the capsular polysaccharides of Klebsiella species (Kabha et al., 1997).
SP-A and SP-D can directly inhibit the growth of Gram-negative bacteria by increasing membrane permeability (Wu et al., 2003). SP-A inhibits growth of P. aeruginosa by increasing membrane permeability (van Iwaarden et al., 1994). However, a recent study has shown that through quorum-sensing, P. aeruginosa is able to resist SP-A activities, via a flagellum-mediated mechanism producing exoproteases that degrade SP-A (Kuang et al., 2011).
SP-A is not able to interact with peptidoglycan isolated from Gram-positive bacteria (Murakami et al., 2002) or zymosan isolated from yeast (Sato et al., 2003). In contrast, H. influenza type A, the major outer membrane protein, is a ligand for SP-A (McNeely and Coonrod, 1994) whilst in Klebsiella, SP-A interacts with a capsular polysaccharide (Kabha et al., 1997). However, SP-D interacts with Gram-positive bacteria via binding to the cell wall components lipoteichoic acid and peptidoglycan via CRDs (van de Wetering et al., 2001). SP-D is also able to bind lipoarabinomannan (LAM) from M. tuberculosis and M. avium (Ferguson et al., 1999; Kudo et al., 2004) and to cell surface lipids on Mycoplasma pneumoniae (Chiba et al., 2002). SP-A has been shown to bind M. pneumonia via disaturated phosphatidylglycerol and is able to attenuate its growth (Piboonpocanun et al., 2005). SP-A can aggregate and opsonize H. influenza type A, but not type B, via the P2 outer membrane protein (McNeely and Coonrod, 1994). SP-A interacts with the M. tuberculosis cell surface Apa glycoprotein, which is a potential adhesion molecule, and may facilitate uptake of the pathogen by host cells (Ragas et al., 2007).
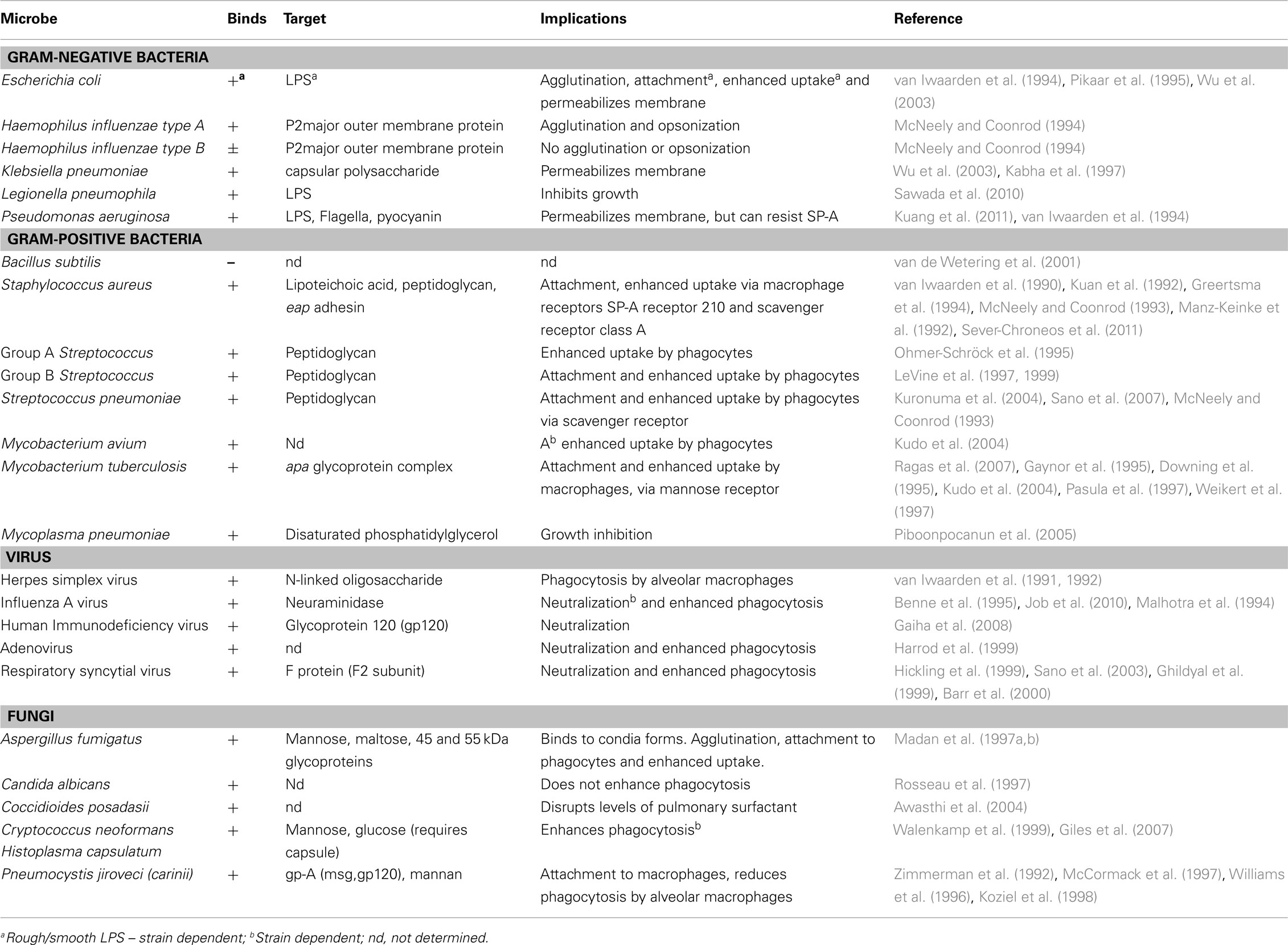
Table 1. Interactions with SP-A and various microbes.
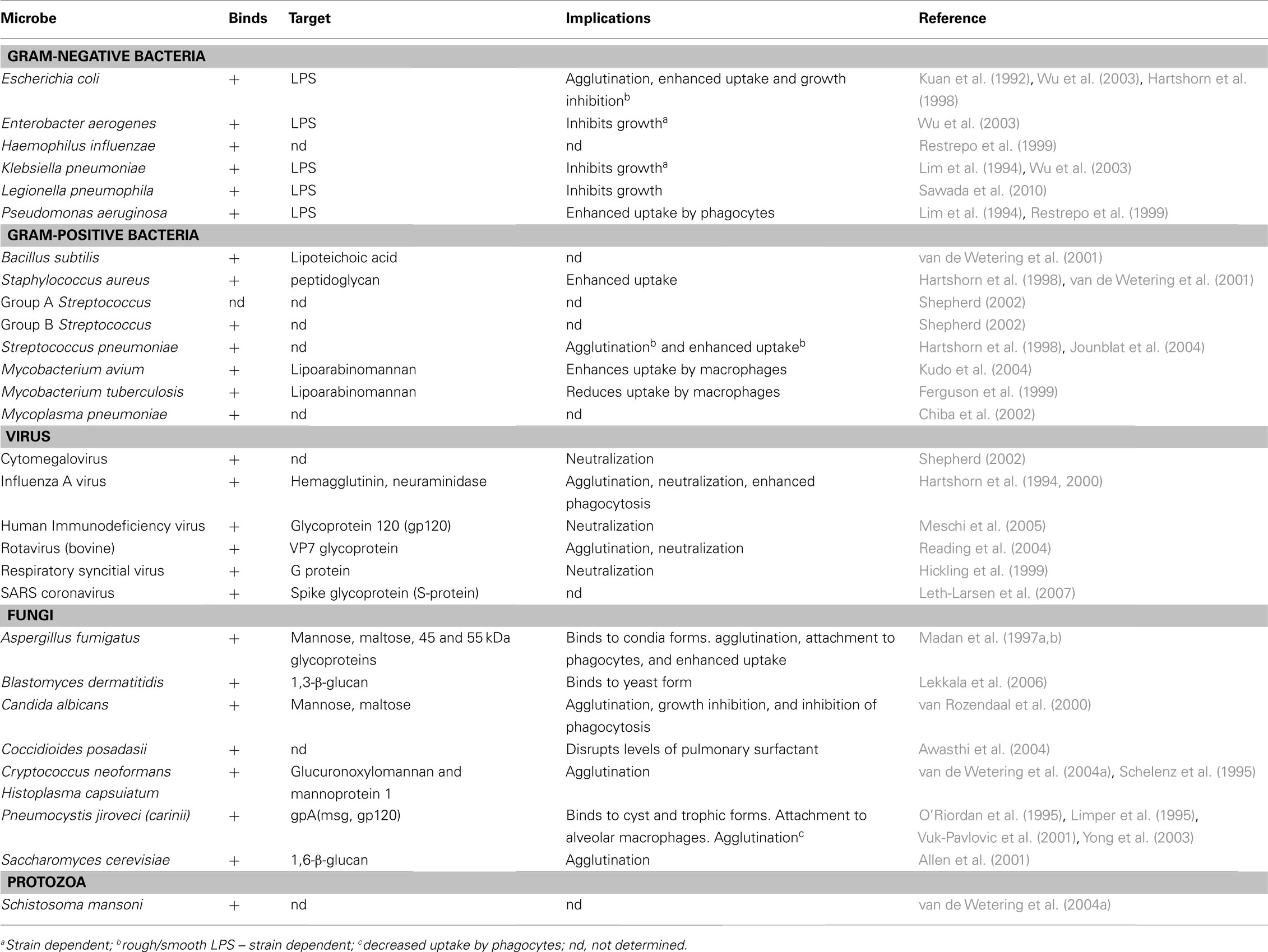
Table 2. Interactions with SP-D and various microbes.
To date, a number of in vitro studies have shown that SP-A and SP-D increase receptor-mediated uptake, independent of serum, of a variety of microbes including bacteria (Lawson and Reid, 2000). SP-A increases C1q-mediated uptake of S. aureus (Greertsma et al., 1994). For Mycobacteria, SP-A increases mannose receptor-mediated uptake (Gaynor et al., 1995; Kudo et al., 2004), and for Streptococcus pneumoniae, SP-A increases scavenger receptor A (SR-A)-mediated uptake of the bacterium (Kuronuma et al., 2004). Curiously, SP-D has been found to reduce uptake of M. tuberculosis (Ferguson et al., 1999), but enhances phagocytosis of Mycobacterium avium (Kudo et al., 2004). These opposing effects of SP-A and SP-D on the uptake of M. tuberculosis suggest different roles for SP-A and SP-D in the clearance of bacterial infection. Thus, interaction of SP-A and SP-D with the pathogen can either enhance or suppress their interactions with phagocytes. SP-D reduces the uptake of M. tuberculosis by macrophages by interacting directly with the bacterium (Ferguson et al., 1999) and this effect does not involve bacterial agglutination (Ferguson et al., 2002). In addition to enhancing phagocytosis via opsonization, SP-A and SP-D can also directly stimulate phagocytosis, by the upregulation of cell surface phagocytic receptors in macrophages, without the need for microbial binding. This has been shown for the mannose receptor on monocyte-derived macrophages, where both SP-A and SP-D have been shown to up-regulate its activity (Beharka et al., 2002; Kudo et al., 2004). Furthermore, in SP-A gene-deficient (SP-A-/-) mice, expression of mannose receptor is decreased, suggesting that SP-A plays a central role in modulating the activity of this receptor (Beharka et al., 2002). It has also been found that SP-A enhances the phagocytosis of S. pneumoniae by AMs, via significant upregulation in the expression of cell surface scavenger receptor A (SR-A; Kuronuma et al., 2004). The mechanisms (e.g., signaling pathways) by which collectin phagocytic receptors actually trigger phagocytosis are not well understood. A receptor for SP-A has been identified called SPR-210, which is a 210 kDa cell surface protein from a macrophage like cell line U973 (Chroneos et al., 1996) and is also found on type II Pneumocytes and AMs. Antibodies generated against SPR-210, inhibited the enhancement by SP-A on the phagocytosis of Mycobacterium bovis BCG by macrophages (Weikert et al., 2000). Subsequently, another receptor called gp-340 was also found that binds SP-A and SP-D (Holmskov et al., 1997; Tino and Wright, 1999).
SP-A and SP-D also play important roles modulating reactive oxygen and nitrogen intermediates in the phagocytes during the killing of intracellular bacteria. A number of studies have shown that SP-A modulates this process in Mycobacterium-infected cells. For example, SP-A enhances macrophage killing of M. bovis BCG (Bacillus Calmette-Guérin) by increasing the level of NO (Weikert et al., 2000). However, in IFN-γ stimulated macrophages, SP-A decreases nitric oxide levels in response to M. tuberculosis and M. avium (Pasula et al., 1999; Hussain et al., 2003). NO production induced by IFN-γ is TNF-α dependant and it is thought that SP-A decreases NO levels, by inhibiting TNF-α production and activation of the transcription factor NF-κB (Hussain et al., 2003). SP-A is also found to enhance the killing of Mycoplasma pulmonis, by increasing the levels of NO (Hickman-Davis et al., 1998).
Bacterial cell wall components such as LPS and peptidoglycan (pathogen-associated molecular patterns; PAMPs) are potent stimulators of inflammation and can also interact with PRRs such as CD14 or TLR, and activate downstream intracellular signaling. SP-A and SP-D can bind to PRRs and directly influence the inflammatory response. For example, SP-A binds to CD14 on AMs, preventing the binding of smooth LPS to CD14, and reducing TNF-α production (Sano et al., 1999). These findings are supported by studies showing that TNF-α, which is induced by smooth LPS, is significantly increased in SP-A−/− compared to wild-type mice (Borron et al., 2000). Equally, SP-A has also been shown to inhibit the peptidoglycan-induced secretion of TNF-α by directly binding to TLR-2 (Murakami et al., 2002). Therefore, the presence of SP-A significantly reduces peptidoglycan-induced pro-inflammatory cytokine responses and NF-κB activation. However, SP-A enhances the pro-inflammatory response induced by rough LPS. It is interesting that rough LPS is a ligand for SP-A, whereas peptidoglycan is not. SP-A is thought to interact with CD14 via its neck region, but binds with rough LPS via the CRD domain, possibly resulting in a trimolecular complex (Sano et al., 1999).
Interaction of SP-A and SP-D with Viruses
Direct interaction of SP-A and SP-D with a number of viruses more often results in viral neutralization and enhanced phagocytosis. For example, both SP-A and SP-D inhibit hemagglutinin binding activity of influenza A virus (IAV) (Malhotra et al., 1993; Hartshorn et al., 1994; Benne et al., 1995), whilst SP-D also decreases neuraminidase activity (Reading et al., 1997). Moreover, SP-D exhibits strong anti- IAV activity via its CRD region binding to carbohydrates (mannosylated, N-linked) on neuraminidase and hemagglutinin of the virus (Hartshorn et al., 1994, 2000). It appears that some Pandemic H1N1 IAV strains are resistant to the neutralization by SP-A because of differences in the N-glycosylation of viral hemagglutinin (Job et al., 2010). SP-A can also interact with herpes simplex virus type 1 (HSV-1) via its N-linked oligosaccharides that results in SP-A-mediated phagocytosis of HSV-1 by AMs (van Iwaarden et al., 1991, 1992). SP-A can interact with respiratory syncytial virus (RSV) (Hickling et al., 1999; Sano et al., 1999) and binds to the F2 subunit of the F protein of RSV and neutralizes the infectivity of the virus (Ghildyal et al., 1999; Sano et al., 2003). Additionally, SP-D binds to RSV attachment protein G and inhibits RSV infection in vivo and in vitro (Hickling et al., 1999). A recent study has shown that RSV-infected cells (a bronchiolar epithelial cell line) have a reduction in SP-A protein levels via a mechanism that affects SP-A mRNA translation efficiency (Bruce et al., 2009).
SP-A has been found to bind to Human Immunodeficiency Virus (HIV) gp120 and this inhibits direct infection of CD4+ cells, but also enhances the transfer of infection to CD4+ T cells mediated by DCs (Gaiha et al., 2008). SP-D binds to HIV envelope protein gp120 and inhibits HIV replication (Meschi et al., 2005). SP-D recognizes the SARS corona virus spike glycoprotein and can activate macrophages (Leth-Larsen et al., 2007). SP-D has also been found to bind bovine strains of the non-enveloped rotavirus via the VP7 glycoprotein (Reading et al., 2004). There is also a report showing that SP-A can enhance viral clearance and inhibit inflammation after pulmonary adenoviral infection (Harrod et al., 1999).
Interaction of SP-A and SP-D with Primary and Opportunistic Fungal Pathogens
SP-A and SP-D have been shown to bind to a variety of fungi, mostly opportunistic fungal pathogens, resulting in direct inhibition of growth and enhancement phagocytosis. However, the downstream immune response elicited by surfactant proteins can also contribute to fungal pathogenesis.
SP-A and SP-D inhibit the growth of Histoplasma capsulatum directly by increasing the permeability of the cell membrane (McCormack et al., 2003). SP-D has also been found to agglutinate C. neoformans and Aspergillus fumigatus (Schelenz et al., 1995; Madan et al., 1997a). SP-A and SP-D bind to A. fumigatus conidia and enhance phagocytosis and killing by neutrophils and AMs (Madan et al., 1997a). In a murine model of invasive pulmonary aspergillosis (IPA), SP-D, but not SP-A, was found to protect immunosuppressed mice from an otherwise fatal challenge with A. fumigatus conidia (Madan et al., 2001). In a further study, therapeutic protection by native full-length SP-D (and a recombinant fragment of human SP-D containing homotrimeric neck and CRD domains, designated rhSP-D) was associated with a reduction in the growth of fungal hyphae in the murine lungs, and increased levels of TNF-α and IFN-γ (Singh et al., 2009). SP-D gene-deficient (SP-D−/−) mice are also more susceptible to IPA, while SP-A gene-deficient (SP-A−/−) mice acquire resistance to IPA (Madan et al., 2010), suggesting that SP-A may facilitate pathogenesis by A. fumigatus. A. fumigatus-challenged SP-A-/- mice showed less mortality (40%) than the wild-type mice (100%) and increased mortality (60%) following administration of SP-A with decreased TNF-α and IFN-γ to IL-4 ratio than SP-A-/- IPA mice. The SP-D-/- IPA mice (57.14%) showed similar mortality as wild-type mice (60%). However, the SP-D-/- IPA mice (42.86% mortality on day 2) died earlier than the WT-IPA mice (20% mortality on day 2), showed a higher hyphal density and tissue injury in lungs. Treatment with SP-D or rhSP-D fragment reduced the mortality to 50 and 33%, respectively, consistent with higher IFN-γ to IL-4 ratios in treated SP-D-/- mice, compared to untreated control group. The results showed that SP-D-/- mice are more susceptible to IPA while SP-A gene-deficient mice acquire resistance to IPA (Madan et al., 2010).
The fungal ligands recognized by SP-A include the 45and 55 kDa glycoproteins secreted by A. fumigatus (Madan et al., 1997b). SP-A also binds to a 120 kDa surface glycoprotein of Pneumocystis jirovecii (formally P. carinii) (Zimmerman et al., 1992) via its CRD region (McCormack et al., 1997). SP-D, but not SP-A, binds to Saccharomyces cerevisiae cells, and the ligand for SP-D was determined to be the yeast glucan, β-(1 > 6)-glucan (Allen et al., 2001). SP-A binds to both encapsulated and acapsular Cryptococcus neoformans, but does not appear to enhance acapsular C. neoformans phagocytosis (Walenkamp et al., 1999). In contrast, SP-D increases phagocytosis of hypocapsular C. neoformans by murine macrophages and enhances fungal survival (Geunes-Boyer et al., 2009). In a study involving SP-A−/− and wild-type mice and an intranasal C. neoformans infection model, it was found that disease progression was not influenced by SP-A (Giles et al., 2007). Both encapsulated and acapsular Cryptococci can bind SP-D, but SP-D showed the highest affinity and aggregation for acapsular C. neoformans (van de Wetering et al., 2004a). The ligands for SP-D were identified as capsular components glucuronoxylomannan (GXM) and mannoprotein 1 (MP1) (van de Wetering et al., 2004a).
SP-D also binds to Candida albicans, resulting in agglutination, growth inhibition, decreased hyphal outgrowth and inhibition of phagocytosis by AMs (van Rozendaal et al., 2000). Recent data has suggested that SP-D binds to Blastomyces dermatitidis via β-glucan molecule (Lekkala et al., 2006).
Recently, the roles of SP-A and SP-D have also been examined in Coccidioides infection. Both SP-A and SP-D bind to Coccidiodal antigens, but it has been shown that Coccidioides infection also disrupts the expression of pulmonary SP-A and SP-D, potentially aiding pathogenesis (Awasthi et al., 2004).
Role of SP-A and SP-D in the Control of Lung Hypersensitivity and Inflammation
SP-A was first shown to bind to pollen grains of Populas nigra italica (Lombardy poplar), Poa prantensis (Kentucky blue grass), Secale cerale (cultivated rye), and Ambrosia elatior (short ragweed) in a calcium and carbohydrate (mannose) dependant manner (Malhotra et al., 1993). SP-A, SP-D, and rhSP-D bind to 3-week culture filtrate (3wcf) and two immunodominant glycoprotein allergens, gp45 and gp55, of A. fumigatus in a calcium, dose-dependent and sugar-specific manner (Madan et al., 1997b), as well as allergens derived from Dermatophagoides pteronyssinus (house dust mite) (Wang et al., 1996, 1998). SP-A, SP-D, and rhSP-D can inhibit specific IgE binding to the allergens of A. fumigatus and block allergen-induced histamine release by basophils (Madan et al., 1997b). Consistent with binding of SP-A, SP-D, and rhSP-D binding to mite allergens and competitive inhibition of IgE-allergen interaction, SP-D reduces the proliferation of peripheral blood mononuclear cells (PBMC) isolated from mite-sensitive asthmatic children (Wang et al., 1998). Dodecameric form of SP-D has been shown to induce aggregation of pollen derived starch granules (PSG) derived from Dactylis glomerata and Phleum pratense and increase their binding and uptake by AMs. Pre-treatment of these PSG with SP-D resulted in inhibiting the release of β-hexosaminidase in PSG induced IgE dependent mast cell degranulation (Erpenbeck et al., 2005).
In view of the ability of SP-A and SP-D to inhibit IgE-mediated histamine release by basophils, a number of allergic murine models have been used to examine in vivo effects of SP-A and SP-D following allergenic challenge. Intranasal administration of SP-D and rhSP-D in a murine model of pulmonary hypersensitivity induced by A. fumigatus allergens and antigens suppressed specific IgG and IgE levels in serum, reduced peripheral and pulmonary eosinophilia, and caused helper T cell (Th) polarization from allergic Th2 to a protective Th1 phenotype (i.e., lowering the levels of IL-4 and IL-5 and elevating IFN-γ level in the spleen supernatant; Madan et al., 2001). The rhSP-D, when given intranasal to Derp mice (a murine model of lung allergy induced by house dust mite allergens) decreased Derp specific IgE levels, peripheral blood eosinophilia and pulmonary infiltration, in addition to causing Th1 polarization (Singh et al., 2003).
The susceptibility of SP-A−/− or SP-D−/− mice to the A. fumigatus allergen challenge, as compared with the wild-type mice, has also been examined (Madan et al., 2005). Both SP-A−/− and SP-D−/− mice show intrinsic hypereosinophilia and several-fold increase in levels of IL-5 and IL-13, and lower IFN-γ to IL-4 ratio in the lungs, suggesting a Th2 bias of immune response. This Th2 polarization was reversible by treating SP-A−/− or SP-D−/− mice with exogenous intranasal SP-A or SP-D delivery, respectively. SP-D−/− mice were found more susceptible than wild-type mice to pulmonary hypersensitivity induced by A. fumigatus allergens whilst SP-A−/− mice were found to be nearly resistant to sensitization. Intranasal treatment with SP-D or rhSP-D rescued the sensitized SP-D−/− mice, while SP-A-treated and sensitized.
SP-A−/− mice showed several-fold elevated levels of IL-13 and IL-5, resulting in increased pulmonary eosinophilia and damaged lung tissue. These data validated important roles for SP-A and SP-D in offering resistance to pulmonary allergenic challenge (Erpenbeck et al., 2006).
In an interesting study, Haczku et al. (2006) have shown that mice sensitized and challenged with either A. fumigatus have increased SP-D levels in their lung that gets exaggerated further in response to IL-4 or IL-13 treatment. STAT-6-deficient mice as well as in IL-4/IL-13 double knockout mice do not appear to have upregulation of SP-D expression following A. fumigatus allergen challenge. It is likely that SP-D protein levels, which is dependent on IL-4 and IL-13, interferes with T cell sensitization, thus protecting lungs from airway inflammation. Consistent with these in vivo studies, when cultured alveolar type II cells are exposed to IL-13 or IFN-γ, the expression of surfactant proteins are altered in vitro (Ito and Mason, 2010). IL-13 decreases SP-D in levels, whereas IFN-γ has the opposite effect. Thus, over-expression of IL-13 can lead to SP-D deficiency in the lungs.
SP-A and SP-D have been shown to modulate DCs and eosinophils. The SP-D mediated binding and uptake of E. coli by bone marrow derived mouse DCs has been shown to increase antigen presentation of E. coli expressed proteins to a T cell hybridoma (Brinker et al., 2001). Curiously, pre-treatment of immature DCs with SP-A (and C1q) has been shown to inhibit LPS-mediated surface expression of maturation markers: MHC class II and CD86. Stimulation of immature DCs by SP-A also inhibits the allostimulation of T cells, enhances dextran endocytosis, and alters DCs chemotaxis toward RANTES (Brinker et al., 2003).
In view of therapeutic effects of rhSP-D in murine models of lung allergy (Madan et al., 2001; Singh et al., 2003) and hypereosinophilic SP-D gene-deficient mice (Madan et al., 2005), direct interaction of rhSP-D with human eosinophils derived from allergic patients and healthy donors and its consequences have been examined (Mahajan et al., 2008). The rhSP-D shows a sugar- and calcium-dependent binding to human eosinophils, suggesting binding via CRD region. Compared to eosinophils derived from non-allergic healthy donors, eosinophils derived from allergic patients show a significant increase in apoptosis, oxidative burst and CD69 expression in the presence of rhSP-D. However, eosinophils from healthy donors, when primed with IL-5, show an increase in apoptosis on incubation with rhSP-D. In addition, the uptake of apoptotic eosinophils by macrophage cell line J774A.1 is significantly enhanced by rhSP-D. These two processes may account for reduction in peripheral and pulmonary eosinophilia in the mouse models following therapeutic treatment with rhSP-D.
Clearance of Apoptotic Cells by SP-A and SP-D
Apoptosis (programmed cell death) and the non-inflammatory removal of dying cells are the important features in embryonic normal development, maintenance of tissue homeostasis, and resolution of inflammation (Wyllie et al., 1980; Haslett et al., 1994). Ineffective clearance of apoptotic cells can lead to the accumulation of late apoptotic cells or secondary necrotic cells in the lungs (Vandivier et al., 2002; Bianchi et al., 2008; Vandivier et al., 2002; Litvack and Palaniyar, 2010). An inflammatory immune response may occur to reduce the accumulation of these dying cells (Clark and Reid, 2002; Palaniyar et al., 2005). SP-A and SP-D have been shown to enhance apoptotic cell ingestion by resident murine and human AMs in vitro (Vandivier et al., 2002). SP-D is a potent modulator of apoptotic cell clearance in a naïve lung when compared to SP-A. SP-A and SP-D bind to apoptotic cells and enhance apoptotic cell uptake by phagocytes through a mechanism dependent on calreticulin and CD91, similar to that of C1q, suggesting that the entire collectin family of innate immune proteins (including C1q) works through a common receptor complex to enhance removal of apoptotic cells, and that collectins are integral, organ-specific components of the clearance machinery (Vandivier et al., 2002). In resting, non-inflamed lung, lung collectins suppress AM phagocytic function through interacting with SIRPα and enhance apoptotic cell removal by opsonizing apoptotic cells and helping their removal through CD91 (Gardai et al., 2003; Janssen et al., 2008).
Extrapulmonary Existence and Functions of SP-A and SP-D
Several immunolocalization studies provide evidence of the expression of the SP-A and SP-D at extrapulmonary sites. A significant level of SP-D has been detected immunohistochemically in human trachea, brain, testis, salivary gland, heart, prostate, kidneys, small intestine, pancreas, and placenta. Lower levels of expression have also been detected in spleen, adrenal gland, uterus, and mammary glands (Fisher and Mason, 1995; Madsen et al., 2000; Herias et al., 2007). SP-D immunoreactivity has also been detected in epithelial cells of large and small ducts of the parotid gland, sweat and lacrimal glands, epithelial cells of gall bladder and intra-hepatic bile ducts, exocrine pancreatic ducts, epidermal cells, esophagus and small intestine, and in the urinary tract, including the collecting ducts of the kidney (Madsen et al., 2000; Brauer et al., 2007).
SP-A, on the other hand, is found to be at low levels in the human and rat small and large intestines (Lin et al., 2001), mesentery, and colon (Rubio et al., 1995; Chailley-Heu et al., 1997; Madsen et al., 2003). SP-A is found in human prostate, thymus, amniotic fluid and epithelium, and salivary glands in very low quantities (Madsen et al., 2003). SP-A has also been detected in the entire human lacrimal apparatus including lacrimal gland, nasolacrimal ducts, conjunctiva, cornea, and human tear fluid (Brauer et al., 2007). Both SP-A and SP-D have been immunohistochemically localized in the fetal membranes (amniotic epithelium and chorionic membrane) and the choriodecidual layer of the late pregnant uterus (Miyamura et al., 1994). SP-A and SP-D, along with SP-B and SP-C, can be immunohistochemically localized on the human skin as well as human skin-derived cell lines (Mo et al., 2007).
The presence of SP-A and/or SP-D in the extrapulmonary environment, especially in organs and tissues, which are most exposed to pathogens, supports their roles as important proteins of the innate immune system even outside the lungs. The gastrointestinal tract was the first site to be examined for extrapulmonary presence of the surfactant proteins (Fisher and Mason, 1995; Rubio et al., 1995). Following immunolocalization, SP-A was found to be expressed in areas distal to the stomach such as a fine lining of the luminal surface of villi and on some jejunal cells (Rubio et al., 1995). On the other hand, SP-D is localized mainly to the stomach and some parts of the large and small intestine (Fisher and Mason, 1995; Madsen et al., 2000). The presence of SP-D in stomach is interesting as it is an organ that is potentially in direct contact with pathogens. SP-D as a major opsonin is likely to facilitate phagocytosis by macrophages of pathogens introduced via the stomach. In the case of SP-A, it has been known to be involved in surfactant homeostasis in the lung (Kuroki and Akino, 1991; Weaver and Whitsett, 1991). The hydrophobic gut lining bears a strong resemblance to the lung surfactant with phosphotidylcholine being the major phospholipid (Lichtenberger et al., 1983). Hence, SP-A might be involved in the maintenance of the gut surfactant function.
Consistent with the observation that SP-A and SP-D are mostly found in tissues susceptible to infection, recent studies have reported their expression in human paranasal sinus mucosa, as confirmed by immunolocalization (Woodworth et al., 2006). SP-A and SP-D are also found in Eustachian tube epithelium (Paananen et al., 2001). Thus, the potential roles of SP-A and SP-D in mucosal immunity are likely to be an emerging area of research.
Association of SP-A and SP-D in Human Health and Diseases
A fine-tuned activity of SP-A and SP-D is vital for first line host defense and immune regulation within pulmonary and extrapulmonary sites. Varying serum levels of SP-A and SP-D have been associated with a range of diseases including idiopathic pulmonary fibrosis, sarcoidosis, pulmonary alveolar proteinosis, hypersensitivity pneumonitis, acute lung injury (ALI)/acute respiratory distress syndrome (ARDS), chronic obstructive pulmonary disease (COPD), interstitial lung disease (ILD), rheumatoid arthritis (RA), and systemic scleroderma (Table 3).
The mean normal serum concentration of SP-D ranges between 48.7 and 109 ng/ml (Kuroki et al., 1998) and mean BAL concentration can be 880 ng/ml (Kuroki et al., 1998). The physiological levels of SP-A in serum is 25 ng/ml (Greene et al., 1999). The physiological levels of these proteins undergo changes when challenged by external stimuli such as allergens during asthma. Measurement of the altered expression of these immunomodulatory proteins in serum during health as well as disease has emerged as important biomarkers for a range of pathological conditions.
SP-A and SP-D in Pulmonary Disorders
In lung diseases such as pulmonary fibrosis and idiopathic interstitial pneumonia, the serum levels of the surfactant proteins (especially SP-D) are inversely related to their respective levels in the BAL. The widely accepted view is that the underlying disease mechanism increases the alveolar-capillary permeability and also affects the basement membrane. These changes lead to the leakage of the surfactant proteins into the blood vessels and, hence increase in the overall serum level measurement. For example, in idiopathic interstitial pneumonia, the basement membranes of the alveoli and vessels are injured (Kawanami et al., 1982; Wells et al., 1993). This, in turn, facilitates the leaking of SP-A from the alveolar space into the blood vessels (Kuroki et al., 1993). This finding supports previous studies in guinea pigs, where exposure to cigarette smoke increases the alveolar permeability (Simani et al., 1974). Thus, along with being markers of an underlying lung disease and its progression, the measurement of surfactant proteins also acts as a good marker for alveolar integrity. An exception to the hypothesis is asthma and allergen-induced airway inflammation. In these cases, levels of SP-A and SP-D increase in serum (Koopmans et al., 2004) as well as BALF (Erpenbeck et al., 2006).
Other conditions linking the levels of the SP-A and SP-D with pathology are smoking and radiotherapeutic pneumonitis. Serum levels of the surfactant proteins are seen to be upregulated in smokers when compared to non-smokers (Nomori et al., 1998; Mazur et al., 2011). Consistent with notion of the high serum levels and the alveolar-vascular translocation, the BAL levels of SP-D was the lowest in smokers (12–19 μg/ml) when compared to the normal levels of BAL SP-D (51 μg/ml). The decrease in BAL SP-D might be contributing to the host defense imbalance within the lungs, thus explaining the increase in the incidence of infections in smokers (More et al., 2010). An increase in serum SP-D and decrease of BAL SP-D levels not dependent on the occurrence of COPD in the same individual. In addition to the elevated levels of SP-D, smoking has also been recently reported to affect the multimeric structure of the SP-D molecule, forming lower molecular weight SP-D molecules (Winkler et al., 2011).
In cystic fibrosis (CF), the BAL levels of SP-A and SP-D vary according to the organ involved and severity of infection. For example, SP-A and SP-D gene expression has been reported to be increased in the sino-mucosal tissue in CF patients (Skinner et al., 2007; Woodworth et al., 2007b). However, during bacterial infections, especially Pseudomonas aeruginosa, the SP-A and SP-D levels in the BALF decrease dramatically (Noah et al., 2003). The possible reason for this decrease might be due to impaired production of SP-A and SP-D by the respiratory epithelial cells or increased consumption of SP-A and SP-D due to a demand for increased phagocytosis of the pathogen involved. Proteolytic degradation of SP-D by P. aeruginosa elastase and protease IV has been previously reported, which can lead to hampering of immune functions of SP-D in the lungs (Alcorn and Wright, 2004a; Malloy et al., 2005).
The level of SP-A and SP-D in serum are likely to be good biomarkers for the clinical outcomes of certain disorders. For instance, a decline in the systemic SP-D in COPD has linked with a better clinical outcome (Sin et al., 2008). ALI and ARDS also exhibit similar association of serum level of SP-D and mortality (Eisner et al., 2003). Post-mortem subjects in a study showed extremely high levels of circulating SP-D when compared to early-stage ALI/ARDS patients (Eisner et al., 2003).
Inflammation of the lungs as an adverse affect of radiation therapy is termed as radiation pneumonitis. This occurs in 5–15% of patients undergoing radiation therapy as a treatment of cancers affecting lungs, breasts, lymphomas, and other cancers. An increased production of SP-A and SP-D that is correlated to the intensity of radiation administered has been noticed in lungs afflicted by radiation pneumonitis (Sasaki et al., 2001). About 50–60 Gy of radiation led to 68–74% increase of SP-D levels when compared to SP-A (21–26%). Thus monitoring of the SP-A and SP-D levels can potentially be good biomarkers to ascertain side effects of radiation.
SP-A and SP-D and Extrapulmonary Diseases
SP-A and SP-D found at extrapulmonary sites also exhibit a varying pattern of expression in response to stimulus. Chronic rhinosinusitis involves inflammation of the sinonasal cavity, which can be triggered by infection, allergy, or other autoimmune conditions. During episodes of chronic rhinosinusitis, the levels of both SP-A and SP-D have been reported to be upregulated. For example, chronic rhino sinusitis due to A. fumigatus exhibited upregulated levels of SP-D when compared to normal sinonasal tissue (Ooi et al., 2007). In two separate studies, increased levels of SP-A were observed during chronic rhinosinusitis as well as allergic rhinitis (Lee et al., 2006; Wootten et al., 2006).
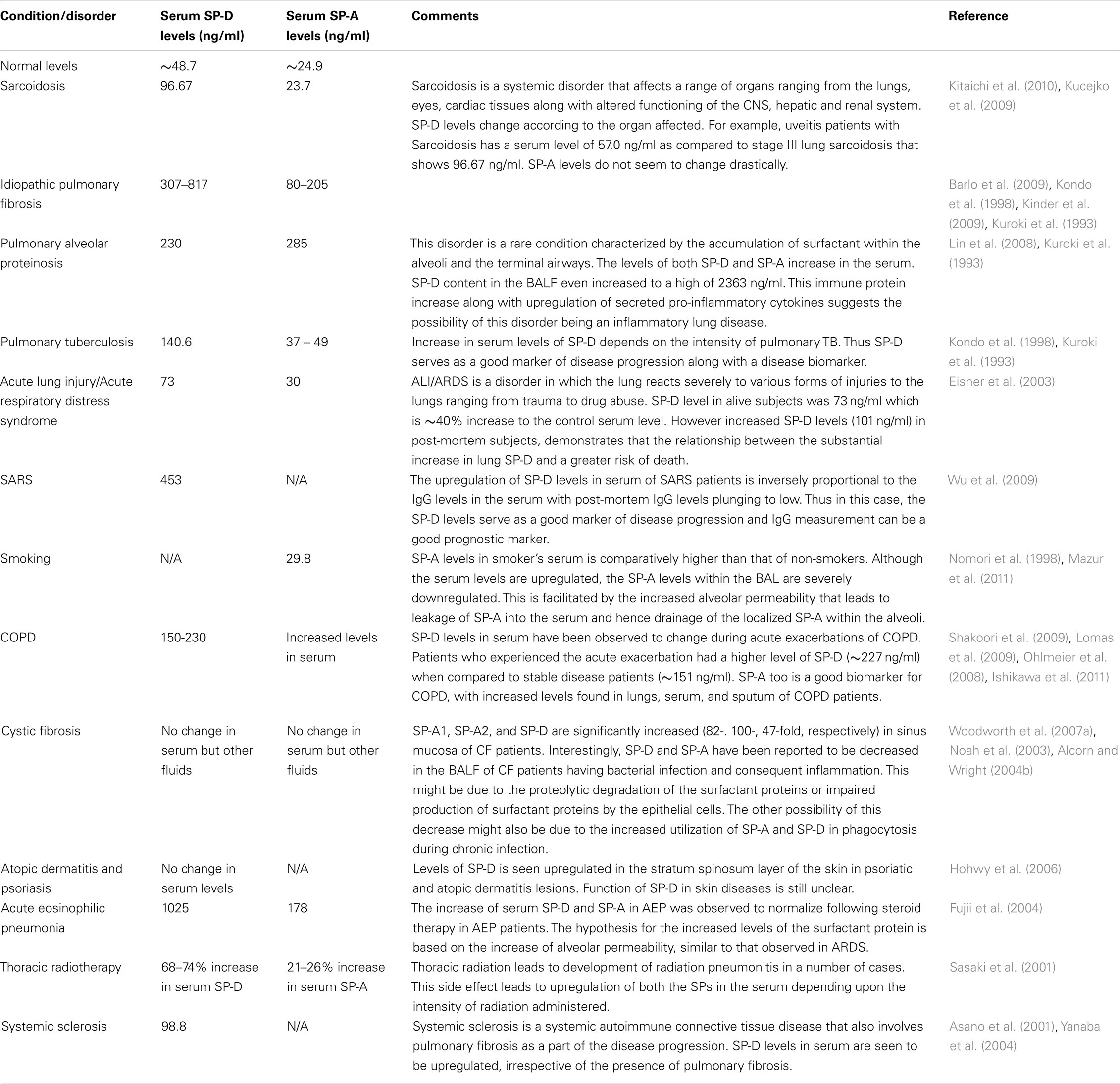
Table 3. Serum levels of SP-D and SP-A in various pulmonary and extra-pulmonary diseases.
Rheumatoid arthritis is a chronic inflammatory systemic disorder that affects a range of tissues and organs, but predominantly affects the synovial joints. SP-A and SP-D have been detected in the synovial fluid as part of the lubricating surfactant (Kankavi, 2006). Consistent with the established surfactant-related functions of SP-A and SP-D in the lungs, it has been speculated that SP-A would also facilitate the adsorption of surface-active phospholipid to the articular surface, whereas and SP-D might regulate the phospholipid homeostasis within the joint (Kankavi, 2006). Lamellar bodies are specialized intracellular organelles of epithelial cells that are responsible for the packaging and secretion of surfactant (Dobbie et al., 1995). These lamellar bodies found in synoviocytes in joints of patients afflicted with RA have been associated with expression of SP-A (Dobbie, 1996). The lamellar bodies are known to secrete the endocytosed SP-A. Thus, a 6.1 and 3.5 fold increased levels of SP-D and SP-A, respectively, have been reported in RA synovial fluid (Kankavi, 2006). Considerable antigenic cross-reactivity was observed between SP-A and a mycobacterial heat shock protein (hsp) 65 (Dobbie et al., 1994). The possibility of SP-A being a potential autoantigen in RA led to the identification of autoantibodies directed against SP-A within the synovial fluid (Trinder et al., 2000). On the other hand, recent studies report the decreased levels of circulating SP-D in RA patients that is inversely associated with disease activity (Hoegh et al., 2008; Christensen et al., 2010). Decreased levels of SP-D in RA could be due to the increased clearance of SP-D from the serum by deposition in the inflamed joint or by complex formation with cellular waste. SP-D as a scavenger molecule enhancing the clearance of DNA and apoptotic cells via macrophages is likely to reduce the anti-double-stranded DNA autoantibody production (Palaniyar et al., 2005). This mechanism is supported by the inverse association between SP-D and disease activity measurements and gradual increase of SP-D levels following treatment. SP-D is accumulated within the synovial fluid through diffusion into the joint cavity from the circulation (Kushner and Somerville, 1971; Pejovic et al., 1995). Interestingly, trimeric forms (130 kDa subunit of SP-D) of SP-D are abundant in the synovial fluid. Although the functions of trimeric SP-D are not well documented, it has been demonstrated to exhibit both pro-inflammatory (via calreticulin-CD91) and anti-inflammatory (via SIRPα) functions (Gardai et al., 2003; Guo et al., 2008). Furthermore recent studies on the disruption of multimeric SP-D during inflammation has shown that this degradation of the multimer into nitrosylated trimeric SP-D exhibits a pro-inflammatory response (Guo et al., 2008). This response is brought about by the CD91/calreticulin receptor interaction and consequent p38 MAPK pathway (Gardai et al., 2003; Guo et al., 2008). Consistent with these findings, increased inflammatory response is associated with an increase in p38 phosphorylation, an important determinant of synovitis severity (Schett et al., 2008). Thus, low levels of SP-D in serum and increased expression of SP-D in the synovial fluid may contribute to the persistent low-grade subclinical joint inflammation seen in RA patients (Brown et al., 2006). This can also be a good biomarker of inflammation and disease activity in the RA patients.
SP-D synthesis has been demonstrated to be localized to vascular endothelial cells in both mice and human (Madsen et al., 2000; Sorensen et al., 2006). The study was carried out in SP-D−/− and control mice fed on an atherogenic diet. Interestingly, the SP-D−/− mice was protected against the development of atherosclerotic lesions in the aortic root with very small foamy cell lesions when compared to the mature cholesterol-laden collagenous atherosclerotic plaques in the control mice. Nevertheless, there was a presence of foamy macrophages within the lung due to lipid engorgement, a typical feature due to SP-D deficiency. When treated with rhSP-D fragment, the anti-inflammatory Th2 response in the blood of the SP-D−/− mice shifted to a Th1 response with the upregulation of the pro-inflammatory cytokine TNFα. Also observed was the reduction of plasma lipid concentrations including HDL and LDL. Thus the Th2 → Th1 shift along with increased susceptibility to development of atherosclerosis suggests the pro-atherogenic potential of SP-D (Sorensen et al., 2006).
SP-D has also been recently reported to be involved in skin diseases such as psoriasis and atopic dermatitis (Hohwy et al., 2006). When immunohistochemically stained, affected skin biopsies from psoriasis and atopic dermatitis patients showed no difference in SP-D immunoreactivity to control healthy skin biopsies on the basal keratinocyte layer level (Hohwy et al., 2006). However, in the stratum spinosum, where keratinization is initialized, SP-D showed higher intensity of immunostaining. Interestingly, the serum level of SP-D was not altered in either condition suggesting that unlike ILDs in which there is a spill-over due to increased alveolar permeability, there was no leakage of SP-D from the psoriatic lesions (Hohwy et al., 2006). The same study also reported that production of SP-D transcripts was not upregulated, and hence, the increase in infiltrating inflammatory cells might be the source of SP-D in the lesions. This study is supported by the finding of yet another study that reports the strong surfactant protein (SP-A, B, C, and D) expression in the lesions of patients affected with psoriasis, atopic dermatitis, lichen planus, and Behcet’s disease (Akman et al., 2008). Thus, further studies are required to elucidate the exact role and regulation of SP-D in inflammatory skin diseases.
SP-A and SP-D in Reproductive Tissues
The ubiquitous roles of SP-A and SP-D are extended to the human reproductive system. Alongside being involved in the respiratory function of the newborn lung during the transition from the intrauterine aqueous environment to extrauterine oxygen, the surfactant proteins are also involved in the process of parturition and innate immune mechanisms during pregnancy.
The localization of SP-A and SP-D in the female reproductive tract suggests the role of these proteins in the process of reproduction. SP-D has been localized in non-pregnant uterus, ovaries, and oviduct of humans (Oberley et al., 2004). SP-D protein and mRNA has been detected in the villous and extra villous trophoblast of the human placenta (Oberley et al., 2004). SP-D mRNA and protein have been detected in the vagina, uterus, ovary, cervix, and oviduct in mouse (Akiyama et al., 2002). Both SP-A and SP-D have been reported to be present in the genital tract of the mare (Kankavi, 2007).
SP-D mRNA in mouse uterus was reported to be hormonally regulated, with peak levels present at estrus and the lowest levels at diesterus (Oberley et al., 2007). Human SP-A has been localized in both pre- and post-menopausal vaginal stratified squamous epithelium as well as in vaginal lavage fluid (MacNeill et al., 2004). Both SP-A and SP-D can be detected in human amniotic fluid as early as 26 weeks of gestation. However, by 40 weeks of gestation, the level of SP-A rises dramatically from 3 to 24 μg/ml with a less pronounced rise of SP-D levels. Thus, the 1:1 ratio of SP-A and SP-D during 26–34 weeks of gestation rises to a striking 6:1 ratio by end of term (Miyamura et al., 1994).
Role of SP-A and SP-D in Maintenance of Pregnancy
The immunoregulatory function of SP-A and SP-D are also seen in the control of cytokine production within the uterine environment. During human labor, the intrauterine tissue macrophages, especially of the decidua, releases excess of prostaglandin F2α (PGF2α; Norwitz et al., 1992a). Along with the release of PGF2α, there is a concomitant release of other macrophage-associated products such as pro-inflammatory cytokine TNF-α (Norwitz et al., 1992b). Inhibition of TNF-α production by SP-A in lung macrophages can hinder the premature activation of the PG cascade, and hence, when this inhibition of pro-inflammatory cytokines takes place during pregnancy, it leads to regulation of the onset of labor (Alcorn and Wright, 2004b). In support of this observation, the down regulation of pro-inflammatory cytokines and chemokines (IL1β, CXCL2, CXCL5) was observed when human amnion explants were incubated with SP-A (Lee et al., 2010). SP-A and SP-D are also involved in clearance of apoptotic cells and necrotic cells (Schagat et al., 2001; Jakel et al., 2010) and this ability to clear dead cells from the uterine tissues can decrease the chances of inflammation due to the presence of dead cells in the uterine environment (Vandivier et al., 2002).
Role of SP-A and SP-D in Parturition
Control of parturition depends on genetic, hormonal, mechanical and/or environmental factors. Research into the control of parturition is of utmost importance as spontaneous pre-term delivery, i.e., delivery before 37 weeks of gestation, is the major cause of neonatal mortality. Signals for parturition might arise from either the maternal side or the fetal side or both (Challis et al., 2005). The process of parturition is associated with an increase in the release of PGs into the uterine environment (Keirse, 1978; Gibb, 1998). The precursor for PG synthesis is arachidonic acid/arachidonate which involves cleaving of the arachidonate from the phospholipid by a phospholipase, which in human amnion is, amniocyte diacyl phosphatidylethanolamine by phospholipase A2 and phosphotidylinositol by phospholipase C and diacylglycerol lipase (Okita et al., 1982). During the development of the fetal lung, the surfactant is synthesized and released to the surrounding amniotic fluid. The process of parturition and fetal lung maturation has been closely associated since survival of the neonate is closely linked with development of surfactant within in the fetal lung (Pryhuber et al., 1991). Respiratory failure due to abnormal lung development is responsible for majority of neonatal death observed in pre-term babies. The link between the fetal lung surfactant protein levels and the initiation of pre-term labor is supported by the observation that the fetal surfactant has also been associated with contributing arachidonate for PG synthesis by amnion cells (Bernal and Phizackerley, 2000). In full-term labor, the secretion and accumulation of fetal surfactant into the amniotic fluid in the third trimester of pregnancy might lead to the gradual increase of PG that induces normal birth. However, in pre-term labor, retarded lung maturation might be leading to an excessive secretion of surfactant, and thus of arachidonate, which contributes to early enhancement of PG production, that in turn, induces labor and leads to pre-term birth. Recent studies have shown that the surfactant proteins, especially SP-A, is directly linked to the induction of parturition in mice (Condon et al., 2004).
Initiation of labor is associated with an inflammatory response characterized by an increased production of IL-1β and NF-κB by intrauterine macrophages. Intra-amniotic injection of SP-A in mice caused pre-term delivery within 6–24 h and intra-amniotic administration of an SP-A antibody or NF-κB inhibitor delays labor by 24 h (Condon et al., 2004), thus facing a pro-inflammatory action at the end of gestation. SP-A has been reported to induce direct stimulation of IL-1β and NF-κB expression by decidual macrophages in the amniotic fluid (Condon et al., 2004). This increase of pro-inflammatory cytokines leads to the PG cascade which in turn leads to the onset of labor. Thus, SP-A appears to be a potential initiator of parturition. Yet another study that supports the pro-parturition effects of SP-A is the potential ability of SP-A to induce stress fibers by regulating F-actin filament organization in cultured human myometrial cells (Breuiller-Fouche et al., 2010). Stress fibers are dynamic structures that aid in maintaining the endothelial cell integrity during the exertion of tensile stress on the vessel wall during growth of the uterine body (Sago et al., 1993). Thus, SP-A may be a vital pro-parturition, immunoregulatory molecule during pregnancy where a delicate balance between stringent host defense and protection of allogenic fetus is required. However, the levels of SP-A is observed to decrease rapidly in spontaneous human parturition at full-term (Chaiworapongsa et al., 2008). This is in comparison to subjects at full-term but not in labor. Further studies into the effects caused by the sudden withdrawal of expression of SP-A in the amniotic fluid can show the association of surfactant proteins at term and how it modulates the process of parturition.
SP-A and SP-D in Fetal and Neonatal Organs
Expression of SP-D has been detected in newborn infant umbilical cord blood and capillary blood. The varied levels in the capillary blood reflected the mode of delivery, with the highest levels being detected in newborns being delivered through cesarean section (Dahl et al., 2005). This study showed that the perinatal conditions played an important role in regulation of the levels of SP-D in the newborn circulation. High levels of SP-D in the cord blood are likely to be influenced by mother’s smoking habits (Dahl et al., 2005). Smoking during pregnancy also decreases the levels of SP-D in the capillary blood of the newborn (Dahl et al., 2006). Smoking may lead to decrease in trophoblast proliferation and reduction of the length of villous capillaries in the placenta leading to abnormal gas and nutrient exchange (Larsen et al., 2002).
The human fetal lung expresses SP-A and SP-D and the production of SP-D has been detected in bronchiolar and terminal epithelium at 16–21 weeks of gestation by hybridization and immunohistochemical analysis (Dulkerian et al., 1996; Mori et al., 2002). Several studies have reported that after the production of SP-D, it begins to peak gradually during gestation and then peaks down during later stages of gestation (Dulkerian et al., 1996; Stahlman et al., 2002). This is evident from the observation that umbilical cord blood SP-D levels were twice as high in premature infants when compared to the full-term babies (Dahl et al., 2006). The same study also reported that neonate with high levels of SP-D in the umbilical blood and capillary blood later developed sepsis. This increase has been speculated to be due to a capillary leak leading the decrease of SP-D in the neonatal lung. This, in turn, will increase the susceptibility of the newborn to infections or due to pro-inflammatory effects of SP-D such as stimulation of TNF-α production induced by LPS, as evident in studies on premature newborn baboons (Awasthi et al., 2001). The higher levels of SP-D in umbilical cord blood observed in Dahl et al., 2006 has been speculated to be due to the impairment of alveolar-vascular integrity during development and that the leakage of proteins into circulation might have taken place even before birth. Regulation of inflammation in newborns by SP-D has also been shown in animal studies. When surfactant mixed with SP-D was administered to premature newborn lambs under ventilation for resuscitation, the SP-D was seen to inhibit lung inflammation caused by the ventilation with downregulated levels of pro-inflammatory cytokines (Sato et al., 2010).
SP-A, apparently an important player in pregnancy and parturition, is also involved in neonatal immunity. In mice studies, progeny of SP-A−/− mice bred in bacteria-laden environment showed a high rate of mortality (P < 0.001) compared to wild-type mice (P < 0.05; George et al., 2008). Interestingly, the progeny of SP-A−/− showed significant gastrointestinal tract pathology (Bacillus sp. and Enterococcus sp. Peritonitis) and blood congestion in lung tissue due to systemic inflammation and consequent upregulation of pro-inflammatory cytokines. The GI tract pathology may arise due to ingestion of bacteria during nursing. However, when the SP-A−/− pups were administered SP-A orally, there was an increase in the rate of survival. Thus, SP-A is seen to play a critical role in immunoprotection of newborn mice (George et al., 2008).
SP-A and SP-D are also involved in dealing with intrauterine infections. Infection within the sterile intrauterine environment can lead to the initiation of inflammation which might lead to pre-term labor (Kim et al., 2009). This would include an exaggerated pro-inflammatory cytokine response that in turn would lead to release of PGs that can induce parturition by pre-term cervical softening, contractions and rupture of the protective fetal membranes. The localization of the SP-A and SP-D in fetal membranes and uterine tissue (Miyamura et al., 1994) supports their role in active host defense mechanisms, including during intrauterine infections.
Chorioamnionitis is the inflammation of the fetal membranes due to a bacterial infection. Bacterial infection in this context would mean an influx of PGs, cytokines, and other mediators of inflammation. Pregnancy demonstrates a strong Th2 character at the feto-maternal interface in which there is a predominance of immunosuppressive activity, including elevated levels of anti-inflammatory mediators such as IL-10, and lowered levels of pro-inflammatory cytokines, such as IL-1β and TNF-α. However, the presence of bacterial products such as LPS can switch on pro-inflammatory cytokine production and lead to unwanted expulsion of the fetus from the uterus. Chorioamnionitis is commonly developed at 26–32 weeks of gestation when SP-A (Wang et al., 1996) and SP-D levels are low (Lahra and Jeffery, 2004). The association of the low levels of SP-A and SP-D and the consequent susceptibility of the fetal membranes to bacterial challenge demonstrate the importance of SP-A and SP-D in immunoregulation and primary host defense against pathogens during pregnancy.
Conclusions
Studies involving gene knockout mice, murine models of lung hypersensitivity and infection, and functional characterization of cell surface receptors have revealed the diverse roles of SP-A and SP-D in the control of lung infection and inflammation. Studies that demonstrate how a range of immune cells, cytokines/chemokines, and surface receptors are modulated by SP-A and SP-D in response to pathogens and allergens in a given microenvironment are crucial in understanding how these proteins link innate and adaptive immunity. The therapeutic effects of rhSP-D in allergic and invasive aspergillosis in murine models are worth investigating as a follow-up clinical trial as an adjunct therapy especially in IPA. Clearly, SP-A and SP-D levels and SNPs in various forms of respiratory diseases are likely to find their translational applications. A recently proposed model based on studies with the calreticulin-CD91 complex as a receptor for SP-A and SP-D has suggested an anti-inflammatory role for SP-A and SP-D in naïve lungs which would help minimize the potential damage that continual low level exposure to pathogens, allergens and apoptosis can cause. However, when the lungs are overwhelmed with exogenous insults, SP-A and SP-D can assume pro-inflammatory roles in order to complement pulmonary innate and adaptive immunity (Gardai et al., 2003). However, the in vitro, in vivo, and ex vivo effects of rhSP-D, which is composed of homotrimeric neck and CRD region, appear to suggest the existence of additional, crucial SP-D receptor(s) that is specific to the lectin domain. Identifying extrapulmonary functions of SP-A and SP-D at mucosal surfaces remain an attractive area of research. Thus, how SP-A and SP-D are involved in the maintenance of pregnancy and subsequent parturition need further investigation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ALI, acute lung injury; AM, alveolar macrophages; ARDS, acute respiratory distress syndrome; BAL, bronchoalveolar lavage; BALF, bronchoalveolar lavage fluid; COPD, chronic obstructive pulmonary disease; CRD, carbohydrate recognition domain; DC, dendritic cells; IFN, interferon; IL, interleukin; LAM, lipoarabinomannan; LPS, lipopolysaccharides; MMP, matrix metalloproteinases; NO, nitric oxide; PAP, pulmonary alveolar proteinosis; PBMCs, peripheral blood mononuclear cell; PG, prostaglandin; RA, rheumatoid arthritis; RSV, respiratory syncytial virus; SP-A, surfactant protein A; SP-D, surfactant protein D; SR-A, scavenger receptor A; TLR, toll-like receptor; TNF, tumor necrosis factor.
References
Akiyama, J., Hoffman, A., Brown, C., Allen, L., Edmondson, J., Poulain, F., and Hawgood, S. (2002). Tissue distribution of surfactant proteins A and D in the mouse. J. Histochem. Cytochem. 2002, 993–996.
Akman, A., Kankavi, O., Ciftcioglu, M., and Alpsoy, E. (2008). Surfactant proteins in inflammatory skin diseases: controlled study. Arch. Dermatol. Res. 300, 353–356.
Alcorn, J., and Wright, J. (2004a). Degradation of pulmonary surfactant protein D by Pseudomonas aeruginosa elastase abrogates innate immune function. J. Biol. Chem. 279, 30871–30879.
Alcorn, J., and Wright, J. (2004b). Surfactant protein A inhibits alveolar macrophage cytokine production by CD14-independent pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 286, L129–36.
Allen, M., Voelker, D., and Mason, R. (2001). Interactions of surfactant proteins A and D with Saccharomyces cerevisiae and Aspergillus fumigatus. Infect. Immun. 69, 2037–2044.
Asano, Y., Ihn, H., Yamane, K., Yazawa, N., Kubo, M., Fujimoto, M., and Tamaki, K. (2001). Clinical significance of surfactant protein D as a serum marker for evaluating pulmonary fibrosis in patients with systemic sclerosis. Arthritis Rheum. 44, 1363–1369.
Awasthi, S., Coalson, J., Yoder, B., Crouch, E., and King, R. (2001). Deficiencies in lung surfactant proteins A and D are associated with lung infection in very premature neonatal baboons. Am. J. Respir. Crit. Care Med. 163, 389–397.
Awasthi, S., Magee, D., and Coalson, J. (2004). Coccidioides posadasii infection alters the expression of pulmonary surfactant proteins (SP)-A and SP-D. Respir. Res. 5, 28–33.
Barlo, N., van Moorsel, C., Ruven, H., Zanen, P., van den Bosch, J., and Grutters, J. (2009). Surfactant protein-D predicts survival in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffuse Lung Dis. 26, 155–161.
Barr, F. E., Pedigo, H., Johnson, T. R., and Shepherd, V. L. (2000). Surfactant protein-A enhances uptake of respiratory syncytial virus by monocytes and U937 macrophages. Am. J. Respir. Cell Mol. Biol. 23, 586–592.
Beharka, A., Gaynor, C., Kang, B., Voelker, D., McCormack, F., and Schlesinger, L. (2002). Pulmonary surfactant protein A upregulates activity of the mannose receptor, a pattern recognition receptor expressed on human macrophages. J. Immunol. 169, 3565–3573.
Benne, C., Kraaijeveld, C., van Strijp, J., Brouwer, E., Harmsen, M., Verhoef, J., van Golde, L. M., and van Iwaarden, J. F. (1995). Interactions of surfactant protein A with influenza A viruses: binding and neutralization. J. Infect. Dis. 171, 335–341.
Bernal, A., and Phizackerley, P. (2000). Fetal surfactant as a source of arachidonate in human amniotic fluid. Prostaglandins Other Lipid Mediat. 60, 59–70.
Bianchi, S. M., Prince, L. R., McPhillips, K., Allen, L., Marriott, H. M., and Taylor, G. W. (2008). Impairment of apoptotic cell engulfment by pyocyanin, a toxic metabolite of Pseudomonas aeruginosa. Am. J. Respir. Crit. Care Med. 177, 35–43.
Borron, P., McIntosh, J., Korfhagen, T., Whitsett, J., Taylor, J., and Wright, J. (2000). Surfactant-associated protein A inhibits LPS-induced cytokine and nitric oxide production in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 278, L840–L847.
Brauer, L., Kindler, C., Jäger, K., Sel, S., Nölle, B., Pleyer, U., Ochs, M., and Paulsen, F. P. (2007). Detection of surfactant proteins A and D in human tear fluid and the human lacrimal system. Invest. Ophthalmol. Vis. Sci. 48, 3945–3953.
Breuiller-Fouche, M., Dubois, O., Sediki, M., Garcia-Verdugo, I., Palaniyar, N., Tanfin, Z., Chissey, A., Cabrol, D., Charpigny, G., and Mehats, C. (2010). Secreted surfactant protein A from fetal membranes induces stress fibers in cultured human myometrial cells. Am. J. Physiol. Endocrinol. Metab. 298, E1188–E1197.
Brinker, K. G., Garner, H., and Wright, J. R. (2003). Surfactant protein A modulates the differentiation of murine bone marrow-derived dendritic cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 284, L232–L241.
Brinker, K. G., Martin, E., Borron, P., Mostaghel, E., Doyle, C., Harding, C. V., and Wright, J. R. (2001). Surfactant protein D enhances bacterial antigen presentation by bone marrow-derived dendritic cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L1453–L1463.
Brown, A., Quinn, M., Karim, Z., Conaghan, P., Peterfy, C., Hensor, E., Wakefield, R. J., O’Connor, P. J., and Emery, P. (2006). Presence of significant synovitis in rheumatoid arthritis patients with disease-modifying antirheumatic drug-induced clinical remission: evidence from an imaging study may explain structural progression. Arthritis Rheum. 54, 3761–3773.
Bruce, S. R., Atkins, C. L., Colasurdo, G. N., and Alcorn, J. L. (2009). Respiratory syncytial virus infection alters surfactant protein A expression in human pulmonary epithelial cells by reducing translation efficiency. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L559–L567.
Chailley-Heu, B., Rubio, S., Rougier, J. P., Ducroc, R., Barlier-Mur, A. M., Ronco, P., and Bourbon, J. R. (1997). Expression of hydrophilic surfactant proteins by mesentery cells in rat and man. Biochem. J. 328, 251–256.
Chaiworapongsa, T., Hong, J., Hull, W., Kim, C., Gomez, R., Mazor, M., Romero, R., and Whitsett, J. A. (2008). The concentration of surfactant protein-A in amniotic fluid decreases in spontaneous human parturition at term. J. Matern. Fetal Neonatal Med. 2008, 652–659.
Challis, J., Bloomfield, F., Bocking, A., Casciani, V., Chisaka, H., Connor, K., Dong, X., Gluckman, P., Harding, J. E., Johnstone, J., Li, W., Lye, S., Okamura, K., and Premyslova, M. (2005). Fetal signals and parturition. J. Obstet. Gynaecol. Res. 31, 492–499.
Chiba, H., Pattanajitvilai, S., Evans, A., Harbeck, R., and Voelker, D. (2002). Human surfactant protein D (SP-D) binds Mycoplasma pneumonia by high affinity interactions with lipids. J. Biol. Chem. 277, 20379–20385.
Christensen, A., Sørensen, G., Hørslev-Petersen, K., Holmskov, U., Lindegaard, H. M., Junker, K., Hetland, M. L., Stengaard-Pedersen, K., Jacobsen, S., Lottenburger, T., Ellingsen, T., Andersen, L. S., Hansen, I., Skjødt, H., Pedersen, J. K., Lauridsen, U. B., Svendsen, A., Tarp, U., Pødenphant, J., Vestergaard, A., Jurik, A. G., Østergaard, M., and Pand, J. (2010). Circulating surfactant protein -D is low and correlates negatively with systemic inflammation in early, untreated rheumatoid arthritis. Arthritis Res. Ther. 12, R39.
Chroneos, Z., Abdolrasulnia, R., Whitsett, J., Rice, W., and Shepherd, V. (1996). Purification of a cell-surface receptor for surfactant protein A. J. Biol. Chem. 271, 16375–16383.
Clark, H., and Reid, K. (2002). Structural requirements for SP-D function in vitro and in vivo: therapeutic potential of recombinant SP-D. Immunobiology 205, 619–631.
Condon, J., Jeyasuria, P., Faust, J., and Mendelson, C. (2004). Surfactant protein secreted by the maturing mouse fetal lung acts as a hormone that signals the initiation of parturition. Proc. Natl. Acad. Sci. U.S.A. 101, 4978–4983.
Crouch, E., and Wright, J. R. (2001). Surfactant proteins a and d and pulmonary host defense. Annu. Rev. Physiol. 63, 521–554.
Dahl, M., Holmskov, U., Husby, S., and Juvonen, P. (2006). Surfactant protein D levels in umbilical cord blood and capillary blood of premature infants. The influence of perinatal factors. Pediatr. Res. 59, 806–810.
Dahl, M., Juvonen, P., Holmskov, U., and Husby, S. (2005). Surfactant protein D in newborn infants: factors influencing surfactant protein D levels in umbilical cord blood and capillary blood. Pediatr. Res. 58, 908–912.
Dobbie, J., Hind, C., Meijers, P., Bodart, C., Tasiaux, N., Perret, J., and Anderson, J. D. (1995). Lamellar body secretion: ultrastructural analysis of an unexplored function of synoviocytes. Br. J. Rheumatol. 34, 13–23.
Dobbie, J. W. (1996). Surfactant protein A and lamellar bodies: a homologous secretory function of peritoneum, synovium, and lung. Perit. Dial. Int. 16, 574–581.
Dobbie, J. W., Tasiaux, N., Meijers, P., Anderson, J. D., Bodart, C., Hind, C., Bourguet, C., and Perret, J. (1994). Lamellar bodies in synoviocytes, mesothelium and specific epithelia as possible site of auto-antigen in rheumatoid disease. Br. J. Rheumatol. 6, 508–519.
Downing, J. F., Pasula, R., Wright, J. R., Twigg, H. L., and Martin, W. J. (1995). Surfactant protein a promotes attachment of Mycobacterium tuberculosis to alveolar macrophages during infection with human immunodeficiency virus. Proc. Natl. Acad. Sci. U. S. A. 92, 4848–4852.
Dulkerian, S., Gonzales, L., Ning, Y., and Ballard, P. (1996). Regulation of surfactant protein D in human fetal lung. Am. J. Respir. Cell Mol. Biol. 15, 781–786.
Eisner, M., Parsons, P., Matthay, M., Ware, L., and Greene, K. (2003). Plasma surfactant protein levels and clinical outcomes in patients with acute lung injury. Thorax 58, 983–988.
Erpenbeck, V., Malherbe, D., Sommer, S., Schmiedl, A., Steinhilber, W., Ghio, A. J., Krug, N., Wright, J. R., and Hohlfeld, J. M. (2005). Surfactant protein D increases phagocytosis and aggregation of pollen-allergen starch granules. Am. J. Physiol. Lung Cell. Mol. Physiol. 288, L692–L698.
Erpenbeck, V., Ziegert, M., Cavalet-Blanco, D., Martin, C., Baelder, R., Glaab, T., Braun, A., Steinhilber, W., Luettig, B., Uhlig, S., Hoymann, H. G., Krug, N., and Hohlfeld, J. M. (2006). Surfactant protein D inhibits early airway response in Aspergillus fumigatus-sensitized mice. Clin. Exp. Allergy 36, 930–940.
Ferguson, J., Voelker, D., McCormack, F., and Schlesinger, L. (1999). Surfactant protein D binds to Mycobacterium tuberculosis bacilli and lipoarabinomannan via carbohydrate–lectin interactions resulting in reduced phagocytosis of the bacteria by macrophages. J. Immunol. 163, 312–321.
Ferguson, J., Voelker, D., Ufnar, J., Dawson, A., and Schlesinger, L. (2002). Surfactant protein D inhibition of human macrophage uptake of Mycobacterium tuberculosis is independent of bacterial agglutination. J. Immunol. 168, 1309–1314.
Fisher, J., and Mason, R. (1995). Expression of pulmonary surfactant protein D in rat gastric mucosa. Am. J. Respir. Cell Mol. Biol. 12, 13–18.
Fujii, M., Tanaka, H., Kameda, M., Fujii, M., Tanaka, S., Ohashi, K., Chiba, H., Takahashi, H., and Abe, S. (2004). Elevated serum surfactant protein A and D in a case of acute eosinophilic pneumonia. Intern. Med. 43, 423–426.
Gaiha, G. D., Dong, T., Palaniyar, N., Mitchell, D. A., Reid, K. B., and Clark, H. W. (2008). Surfactant protein A binds to HIV and inhibits direct infection of CD4 + cells, but enhances dendritic cell-mediated viral transfer. J. Immunol. 181, 601–609.
Gardai, S., Xiao, Y., Dickinson, M., Nick, J., Voelker, D., Greene, K., and Henson, P. (2003). By binding SIRPα or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 115, 13–23.
Gaynor, C., McCormack, F., Voelker, D., McGowan, S., and Schlesinger, L. (1995). Pulmonary surfactant protein A mediates enhanced phagocytosis of Mycobacterium tuberculosis by a direct interaction with human macrophages. J. Immunol. 155, 5343–5351.
George, C., Goss, K., Meyerholz, D., Lamb, F., and Snyder, J. (2008). Surfactant-associated protein A provides critical immunoprotection in neonatal mice. Infect. Immun. 76, 380–389.
Geunes-Boyer, S., Oliver, T. N., Janbon, G., Lodge, J. K., and Heitman, J. (2009). Surfactant protein D increases phagocytosis of hypocapsular Cryptococcus neoformans by murine macrophages and enhances fungal survival. Infect. Immun. 7, 2783–2794.
Ghildyal, R., Hartley, C., Varrasso, A., Meanger, J., Voelker, D. R., Anders, E. M., and Mills, J. (1999). Surfactant protein A binds to the fusion glycoprotein of respiratory syncytial virus and neutralizes virion infectivity. J. Infect. Dis. 180, 2009–2013.
Giles, S., Zaas, A., Reidy, M., Perfect, J., and Wright, J. (2007). Cryptococcus neoformans is resistant to surfactant protein A mediated host defense mechanisms. PLoS ONE 2, e1370.
Greene, K. E., Wright, J. R., Steinberg, K. P., Ruzinski, J. T., Caldwell, E., Wong, W. B., Hull, W., Whitsett, J. A., Akino, T., Kuroki, Y., Nagae, H., Hudson, L. D., and Martin, T. R. (1999). Serial changes in surfactant-associated proteins in lung and serum before and after onset of ARDS. Am. J. Respir. Crit. Care Med. 160, 1843–1850.
Greertsma, M. F., Nibbering, P. H., Haagsman, H. P., Daha, M. R., and van Furth, R. (1994). Binding of surfactant protein A to C1q receptors mediates phagocytosis of Staphylococcus aureus by monocytes. Am. J. Physiol. 267, L578–L584.
Guo, C., Atochina-Vasserman, E., Abramova, E., Foley, J., Zaman, A., Crouch, E., Beers, M. F., Savani, R. C., and Gow, A. J. (2008). S-nitrosylation of surfactant protein-D controls inflammatory function. PLoS Biol. 6, e266.
Haczku, A., Cao, Y., Vass, G., Kierstein, S., Nath, P., Atochina-Vasserman, E. N., Scanlon, S. T., and Li, L. (2006). IL-4 and IL-13 form a negative feedback circuit with surfactant protein-D in the allergic airway response. J. Immunol. 176, 3557–3565.
Harrod, K. S., Trapnell, B. C., Otake, K., Korfhagen, T. R., and Whitsett, J. A. (1999). SP-A enhances viral clearance and inhibits inflammation after pulmonary adenoviral infection. Am. J. Physiol. 277(3 Pt 1), L580–L588.
Hartshorn, K., Crouch, E., White, M., Eggleton, P., Tauber, A., Chang, D., and Sastry, K. (1994). Evidence for a protective role of pulmonary surfactant protein D (SP-D) against influenza A viruses. J. Clin. Invest. 94, 311–319.
Hartshorn, K. L., Crouch, E., White, M. R., Colamussi, M. L., Kakkanatt, A., Tauber, B., Shepherd, V., and Sastry, K. N. (1998). Pulmonary surfactant proteins A and D enhance neutrophil uptake of bacteria. Am. J. Physiol. 274, L958–L969.
Hartshorn, K., White, M., Voelker, D., Coburn, J., Zaner, K., and Crouch, E. (2000). Mechanism of binding of surfactant protein D to influenza A viruses: importance of binding to haemagglutinin to antiviral activity. Biochem. J. 351(Pt 2), 449–458.
Haslett, C., Savill, J. S., Whyte, M. K., Stern, M., Dransfield, I., and Meagher, L. C. (1994). Granulocyte apoptosis and the control of inflammation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 345, 327–333.
Herias, M., Hogenkamp, A., van Asten, A., Tersteeg, M., van Eijk, M., and Haagsman, H. (2007). Expression sites of the collectin SP-D suggest its importance in first line host defence: power of combining in situ hybridisation, RT-PCR and immunohistochemistry. Mol. Immunol. 44, 3324–3332.
Hickling, T. P., Bright, H., Wing, K., Gower, D., Martin, S. L., Sim, R. B., and Malhotra, R. (1999). A recombinant trimeric surfactant protein D carbohydrate recognition domain inhibits respiratory syncytial virus infection in vitro and in vivo. Eur. J. Immunol. 29, 3478–3484.
Hickman-Davis, J., Lindsey, J., Zhu, S., and Matalon, S. (1998). Surfactant protein A mediates mycoplasmacidal activity of alveolar macrophages. Am. J. Physiol. 274, L270–L277.
Hoegh, S., Lindegaard, H., Sorensen, G., Høj, A., Bendixen, C., Junker, P., and Holmskov, U. (2008). Circulating surfactant protein D is decreased in early rheumatoid arthritis: a 1-year prospective study. Scand. J. Immunol. 67, 71–76.
Hohwy, T., Otkjaer, K., Madsen, J., Søerensen, G., Nielsen, O., Vestergaard, C., Steiniche, T., Holmskov, U., and Lomholt, H. (2006). Surfactant protein D in atopic dermatitis and psoriasis. Exp. Dermatol. 15, 168–174.
Holmskov, U., Lawson, P., Teisner, B., Tornoel, I., Willis, A., Morgan, C., Koch, C., and Reid, K. B. (1997). Isolation and characterization of a new member of the scavenger receptor superfamily, glycoprotein-340(gp-340), as a lung surfactant protein-D binding molecules. J. Biol. Chem. 272, 13743–13749.
Holmskov, U., Thiel, S., and Jensenius, J. C. (2003). Collections and ficolins: humoral lectins of the innate immune defense. Annu. Rev. Immunol. 547–578.
Hussain, S., Wright, J., and Martin, W. II. (2003). Surfactant protein A decreases nitric oxide production by macrophages in a tumor necrosis factor-α-dependent mechanism. Am. J. Respir. Cell Mol. Biol. 28, 520–527.
Ishikawa, N., Hattori, N., Tanaka, S., Horimasu, Y., Haruta, Y., Yokoyama, A., Kohno, N., and Kinnula, V. L. (2011). Levels of surfactant proteins A and D and KL-6 are elevated in the induced sputum of chronic obstructive pulmonary disease patients: a sequential sputum analysis. Respiration. 82, 10–18.
Ito, Y., and Mason, R. J. (2010). The effect of interleukin-13 (IL-13) and interferon-γ (IFN-γ) on expression of surfactant proteins in adult human alveolar type II cells in vitro. Respir. Res. 157.
Jakel, A., Clark, H., Reid, K., and Sim, R. (2010). The human lung surfactant proteins A (SP-A) and D (SP-D) interact with apoptotic target cells by different binding mechanisms. Immunobiology 215, 551–558.
Janssen, W. J., McPhillips, K. A., Dickinson, M. G., Linderman, D. J., Morimoto, K., Xiao, Y. Q., and Oldham, K. M. (2008). Surfactant proteins A and D suppress alveolar macrophage phagocytosis via interaction with SIRP alpha. Am. J. Respir. Crit. Care Med. 178, 158–167.
Job, E., Deng, Y., Tate, M., Bottazzi, B., Crouch, E., Dean, M., Mantovani, A., Brooks, A. G., and Reading, P. C. (2010). Pandemic H1N1 influenza A viruses are resistant to the antiviral activities of innate immune proteins of the collectin and pentraxin superfamilies. J. Immunol. 185, 4284–4291.
Jounblat, R., Kadioglu, A., Iannelli, F., Pozzi, G., Eggleton, P., and Andrew, P. W. (2004). Binding and agglutination of Streptococcus pneumoniae by human surfactant protein D (SP-D) vary between strains, but SP-D fails to enhance killing by neutrophils. Infect. Immun. 72, 709–716.
Kabha, K., Schmegner, J., Keisari, Y., Parolis, H., Schlepper-Schaefer, J., and Ofek, I. (1997). SP-A enhances phagocytosis of Klebsiella by interaction with capsular polysaccharides and alveolar macrophages. Am. J. Physiol. 272, L344–L352.
Kalina, M., Blau, H., Riklis, S., and Kravtsov, V. (1995). Interaction of surfactant protein A with bacterial lipopolysaccharide may affect some biological functions. Am. J. Physiol. 268, L144–L151.
Kankavi, O. (2006). Increased expression of surfactant protein A and D in rheumatoid arthritic synovial fluid (RASF). Croat. Med. J. 47, 155–161.
Kankavi, O., Ata, A., and Gungor, O. (2007). Surfactant proteins A and D in the genital tract of mares. Anim. Reprod. Sci. 98, 259–270.
Kawanami, O., Ferrans, V., and Crystal, R. (1982). Structure of alveolar epithelial cells in patients with fibrotic lung disorders. Lab. Invest. 46, 39–53.
Keirse, M. (1978). Biosynthesis and metabolism of prostaglandins in the pregnant human uterus. Adv. Prostaglandin Thromboxane Res. 4, 87–102.
Kim, M., Romero, R., Gervasi, M., Kim, J., Yoo, W., Lee, D., Mittal, P., Erez, O., Kusanovic, J. P., Hassan, S. S., and Kim, C. J. (2009). Widespread microbial invasion of the chorioamniotic membranes is a consequence and not a cause of intra-amniotic infection. Lab. Invest. 89, 924–936.
Kinder, B., Brown, K., McCormack, F., Ix, J., Kervitsky, A., Schwarz, M., and King, T. E. Jr. (2009). Serum surfactant protein-A is a strong predictor of early mortality in idiopathic pulmonary fibrosis. Chest 135, 1557–1563.
Kishore, U., Greenhough, T., Waters, P., Shrive, A. K., Ghai, R., Kamran, M. F., Bernal, A. L., Reid, K. B., Madan, T., and Chakraborty, T. (2006). Surfactant proteins SP-A and SP-D: structure, function and receptors. Mol. Immunol. 43, 1293–1315.
Kishore, U., Strong, P., Perdikoulis, M. V., and Reid, K. B. (2001). A recombinant homotrimer, composed of the alpha helical neck region of human surfactant protein D and C1q B chain globular domain, is an inhibitor of the classical complement pathway. J. Immunol. 166, 559–565.
Kishore, U., Wang, J. Y., Hoppe, H. J., and Reid, K. B. (1996). The alpha-helical neck region of human lung surfactant protein D is essential for the binding of the carbohydrate recognition domains to lipopolysaccharides and phospholipids. Biochem. J. 318(Pt 2), 505–511.
Kitaichi, N., Kitamura, M., Namba, K., Ishida, S., and Ohno, S. (2010). Elevation of surfactant protein D, a pulmonary disease biomarker, in the sera of uveitis patients with sarcoidosis. Jpn. J. Ophthalmol. 54, 81–84.
Kondo, A., Oketani, N., Maruyama, M., Taguchi, Y., Yamaguchi, Y., Miyao, H., and Mashima, I. (1998). Significance of serum surfactant protein-D (SP-D) level in patients with pulmonary tuberculosis. Kekkaku 73, 585–590.
Koopmans, J. G., van der Zee, J. S., Krop, E. J., Lopuhaä, C. E., Jansen, H. M., and Batenburg, J. J. (2004). Serum surfactant protein D is elevated in allergic patients. Clin. Exp. Allergy 34, 1827–1833.
Koziel, H., Phelps, D. S., Fishman, J. A., Armstrong, M. Y., Richards, F. F., and Rose, R. M. (1998). Surfactant protein-A reduces binding and phagocytosis of pneumocystis carinii by human alveolar macrophages in vitro. Am. J. Respir. Cell Mol. Biol. 18, 834–843.
Kuan, S., Rust, K., and Crouch, E. (1992). Interactions of surfactant protein D with bacterial lipopolysaccharides. Surfactant protein D is an Escherichia coli-binding protein in bronchoalveolar lavage. J. Clin. Invest. 90, 97–106.
Kuang, Z., Hao, Y., Hwang, S., Zhang, S., Kim, E., Akinbi, H., Schurr, M. J., Irvin, R. T., Hassett, D. J., and Lau, G. W. (2011). The Pseudomonas aeruginosa flagellum confers resistance to pulmonary surfactant protein-A by impacting the production of exoproteases through quorum-sensing. Mol. Microbiol. 79, 1220–1235.
Kucejko, W., Chyczewska, E., Naumnik, W., and Ossolinska, M. (2009). Concentration of surfactant protein D, Clara cell protein CC-16 and IL-10 in bronchoalveolar lavage (BAL) in patients with sarcoidosis, hypersensivity pneumonitis and idiopathic pulmonary fibrosis. Folia Histochem. Cytobiol. 47, 225–230.
Kudo, K., Sano, H., Takahashi, H., Kuronuma, K., Yokota, S., Fujii, N., Shimada, K., Yano, I., Kumazawa, Y., Voelker, D. R., Abe, S., and Kuroki, Y. (2004). Pulmonary collectins enhance phagocytosis of Mycobacterium avium through increased activity of mannose receptor. J. Immunol. 172, 7592–7602.
Kuroki, Y., and Akino, T. (1991). Pulmonary surfactant protein A (SP-A) specifically binds dipalmitoylphosphatidylcholine. J. Biol. Chem. 266, 3068–3073.
Kuroki, Y., Takahashi, H., Chiba, H., and Akino, T. (1998). Surfactant proteins A and D: disease markers. Biochim. Biophys. Acta. 1408, 334–345.
Kuroki, Y., Tsutahara, S., Shijubo, N., Takahashi, H., Shiratori, M., Hattori, A., Honda, Y., Abe, S., and Akino, T. (1993). Elevated levels of lung surfactant protein A in sera from patients with idiopathic pulmonary fibrosis and pulmonary alveolar proteinosis. Am. Rev. Respir. Dis. 147, 723–729.
Kuronuma, K., Sano, H., Kato, K., Kudo, K., Hyakushima, N., and Yokota, S. (2004). Pulmonary surfactant protein A augments the phagocytosis of Streptococcus pneumonia by alveolar macrophages through a casein kinase 2-dependent increase of cell surface localization of scavenger receptor A. J. Biol. Chem. 279, 21421–21430.
Kushner, I., and Somerville, J. (1971). Permeability of human synovial membrane to plasma proteins. Relationship to molecular size and inflammation. Arthritis Rheum. 14, 560–570.
LaForce, F., Kelly, W., and Huber, G. (1973). Inactivation of Staphylococci by alveolar macrophages with preliminary observations on the importance of alveolar lining material. Am. Rev. Respir. Dis. 108, 784–790.
Lahra, M., and Jeffery, H. (2004). A fetal response to chorioamnionitis is associated with early survival after preterm birth. Am. J. Obstet. Gynecol. 190, 147–151.
Larsen, L., Clausen, H., and Jonsson, L. (2002). Stereologic examination of placentas from mothers who smoke during pregnancy. Am. J. Obstet. Gynecol. 186, 531–537.
Lawson, P., and Reid, K. (2000). The roles of surfactant proteins A and D in innate immunity. Immunol. Rev. 173, 66–78.
Lee, D., Romero, R., Kim, C., Chaiworapongsa, T., Tarca, A., Lee, J., Suh, Y. L., Mazaki-Tovi, S., Vaisbuch, E., Mittal, P., Draghici, S., Erez, O., Kusanovic, J. P., Hassan, S. S., and Kim, J. S. (2010). Surfactant protein-A as an anti-inflammatory component in the amnion: implications for human pregnancy. J. Immunol. 184, 6479–6491.
Lee, H., Kang, H., Woo, J., Chae, S., Lee, S., and Hwang, S. (2006). Upregulation of surfactant protein A in chronic rhinosinusitis. Laryngoscope 116, 328–330.
Lekkala, M., LeVine, A., Linke, M., Crouch, E., Linders, B., Brummer, E., and Stevens, D. A. (2006). Effect of lung surfactant collectins on bronchoalveolar macrophage interaction with Blastomyces dermatitidis: inhibition of tumor necrosis factor alpha production by surfactant protein D. Infect. Immun. 74, 4549–4556.
LeVine, A. M., Bruno, M. D., Huelsman, K. M., Ross, G. F., Whitsett, J. A., and Korfhagen, T. R. (1997). Surfactant protein A-deficient mice are susceptible to group B streptococcal infection. J. Immunol. 158, 4336–4340.
LeVine, A. M., Kurak, K. E., Wright, J. R., Watford, W. T., Bruno, M. D., Ross, G. F., Whitsett, J. A., and Korfhagen, T. R. (1999). Surfactant protein-A binds group B streptococcus enhancing phagocytosis and clearance from lungs of surfactant protein-A-deficient mice. Am. J. Respir. Cell Mol. Biol. 20, 279–286.
Leth-Larsen, R., Zhong, F., Chow, V. T., Holmskov, U., and Lu, J. (2007). The SARS coronavirus spike glycoprotein is selectively recognized by lung surfactant protein D and activates macrophages. Immunobiology 212, 201–211.
Lichtenberger, L. M., Graziani, L. A., Dial, E. J., and Butler, B. D. (1983). Role of surface-active phospholipids in gastric cytoprotection. Science 219, 1327–1329.
Lim, B., Wang, J., Holmskov, U., Hoppe, H., and Reid, K. (1994). Expression of the carbohydrate recognition domain of lung surfactant protein D and demonstration of its binding to lipopolysaccharides of gram-negative bacteria. Biochem. Biophys. Res. Commun. 202, 1674–1680.
Limper, A. H., Crouch, E. C., O’Riordan, D. M., Chang, D., Vuk-Pavlovic, Z., Standing, J. E., Kwon, K. Y., and Adlakha, A. (1995). Surfactant protein-D modulates interaction of Pneumocystis carinii with alveolar macrophages. J. Lab. Clin. Med. 126, 416–422.
Lin, F., Chen, Y., and Chang, S. (2008). Clinical importance of bronchoalveolar lavage fluid and blood cytokines, surfactant protein D, and Kerbs von Lungren 6 antigen in idiopathic pulmonary alveolar proteinosis. Mayo Clin. Proc. 83, 1344–1349.
Lin, Z., deMello, D., Phelps, D., Koltun, W., Page, M., and Floros, J. (2001). Both human SP-A1 and Sp-A2 genes are expressed in small and large intestine. Pediatr. Pathol. Mol. Med. 5, 367–386.
Litvack, M., and Palaniyar, N. (2010). Review: soluble innate immune pattern-recognition proteins for clearing dying cells and cellular components: implications on exacerbating or resolving inflammation. Innate Immun. 16, 191–200.
Lomas, D., Silverman, E., Edwards, L., Locantore, N., Miller, B., Horstman, D., and Tal-Singer, R. (2009). Serum surfactant protein D is steroid sensitive and associated with exacerbations of COPD. Eur. Respir. J. 34, 95–102.
MacNeill, C., Umstead, T., Phelps, D., Lin, Z., Floros, J., Shearer, D., Shearer, D. A., and Weisz, J. (2004). Surfactant protein A, an innate immune factor, is expressed in the vaginal mucosa and is present in vaginal lavage fluid. Immunology 111, 91–99.
Madan, T., Eggleton, P., Kishore, U., Strong, P., Aggrawal, S., Sarma, P., and Reid, K. B. (1997a). Binding of pulmonary surfactant proteins A and D to Aspergillus fumigatus conidia enhances phagocytosis and killing by human neutrophils and alveolar macrophages. Infect. Immun. 65, 3171–3179.
Madan, T., Kishore, U., Shah, A., Eggleton, P., Strong, P., Wang, J. Y., Aggrawal, S. S., Sarma, P. U., and Reid, K. B. (1997b). Lung surfactant proteins A and D can inhibit specific IgE binding to the allergens of Aspergillus fumigatus and block allergen-induced histamine release from human basophils. Clin. Exp. Immunol. 110, 241–249.
Madan, T., Kishore, U., Singh, M., Strong, P., Hussain, E., Reid, K., and Sarma, P. U. (2001). Protective role of lung surfactant protein D in a murine model of invasive pulmonary aspergillosis. Infect. Immun. 69, 2728–2731.
Madan, T., Reid, K. B., Clark, H., Singh, M., Nayak, A., Sarma, P. U., Hawgood, S., and Kishore, U. (2010). Susceptibility of mice genetically deficient in SP-A or SP-D gene to invasive pulmonary aspergillosis. Mol. Immunol. 47, 1923–1930.
Madan, T., Reid, K. B., Singh, M., Sarma, P. U., and Kishore, U. (2005). Susceptibility of mice genetically deficient in the surfactant protein (SP)-A or SP-D gene to pulmonary hypersensitivity induced by antigens and allergens of Aspergillus fumigatus. J. Immunol. 174, 6943–6954.
Madsen, J., Kliem, A., Tornoe, I., Skjodt, K., Koch, C., and Holmskov, U. (2000). Localization of lung surfactant protein D on mucosal surfaces in human tissues. J. Immunol. 164, 5866–5870.
Madsen, J., Tornoe, I., Nielsen, O., Koch, C., Steinhilber, W., and Holmskov, U. (2003). Expression and localization of lung surfactant protein A in human tissues. Am. J. Respir. Cell Mol. Biol. 29, 591–597.
Mahajan, L., Madan, T., Kamal, N., Singh, V., Sim, R., Telang, S., Ramchand, C. N., Waters, P., Kishore, U., and Sarma, P. U. (2008). Recombinant surfactant protein-D selectively increases apoptosis in eosinophils of allergic asthmatics and enhances uptake of apoptotic eosinophils by macrophages. Int. Immunol. 20, 993–1007.
Malhotra, R., Haurum, J., Thiel, S., Jensenius, J., and Sim, R. (1993). Pollen grains bind to lung alveolar type II cells (A549) via lung surfactant protein A (SP-A). Biosci. Rep. 13, 79–90.
Malhotra, R., Haurum, J. S., Thiel, S., and Sim, R. B. (1994). Binding of human collectins (SP-A and MBP) to influenza virus. Biochem. J. 304, 455–461.
Malloy, J., Veldhuizen, R., Thibodeaux, B., O’Callaghan, R., and Wright, J. (2005). Pseudomonas aeruginosa protease IV degrades surfactant proteins and inhibits surfactant host defense and biophysical functions. Am. J. Physiol. Lung Cell. Mol. Physiol. 288, L409–L418.
Manz-Keinke, H., Plattner, H., and Schlepper-Schäfer, J. (1992). Lung surfactant protein A (SP-A) enhances serum-independent phagocytosis of bacteria by alveolar macrophages. Eur. J. Cell Biol. 57, 95–100.
Mazur, W., Toljamo, T., Ohlmeier, S., Vuopala, K., Nieminen, P., Kobayashi, H., and Kinnula, V. L. (2011). Elevation of surfactant protein A in plasma and sputum in cigarette smokers. Eur. Respir. J. 38, 277–284.
McCormack, F., Festa, A., Andrews, R., Linke, M., and Walzer, P. (1997). The carbohydrate recognition domain of surfactant protein A mediates binding to the major surface glycoprotein of Pneumocystis carinii. Biochemistry 36, 8092–8099.
McCormack, F., Gibbons, R., Ward, S., Kuzmenko, A., Wu, H., and Deepe, G. (2003). Macrophage-independent fungicidal action of the pulmonary collectins. J. Biol. Chem. 278, 36250–36256.
McNeely, T. B., and Coonrod, J. D. (1993). Comparison of the opsonic activity of human surfactant protein A for Staphylococcus aureus and Streptococcus pneumoniae with rabbit and human macrophages. J. Infect. Dis. 167, 91–97.
McNeely, T., and Coonrod, J. (1994). Aggregation and opsonization of type A but not type B Haemophilus influenza by surfactant protein A. Am. J. Respir. Cell Mol. Biol. 11, 114–122.
Meschi, J. C., Crouch, E. C., Skolnik, P., Yahya, K., Holmskov, U., Leth-Larsen, R., and Tornoe, I. (2005). Surfactant protein D binds to human immunodeficiency virus (HIV) envelope protein gp120 and inhibits HIV replication. J. Gen. Virol. 86(Pt 11), 3097–3107.
Miyamura, K., Malhotra, R., Hoppe, H., Reid, K., Phizackerley, P., Macpherson, P., and López Bernal, A. (1994). Surfactant proteins A (SP-A) and D (SP-D): levels in human amniotic fluid and localization in the fetal membranes. Biochim. Biophys. Acta 1210, 303–307.
Mo, Y., Kankavi, O., Masci, P., Mellick, G., Whitehouse, M., Boyle, G., Parsons, P. G., Roberts, M. S., and Cross, S. E. (2007). Surfactant protein expression in human skin: evidence and implications. J. Invest. Dermatol. 127, 381–386.
More, J., Voelker, D., Silveira, L., Edwards, M., Chan, E., and Bowler, R. (2010). Smoking reduces surfactant protein D and phospholipids in patients with and without chronic obstructive pulmonary disease. BMC Pulm. Med. 10, 53.
Mori, K., Kurihara, N., Hayashida, S., Tanaka, M., and Ikeda, K. (2002). The intrauterine expression of surfactant protein D in the terminal airways of human fetuses compared with surfactant protein A. Eur. J. Pediatr. 161, 431–434.
Murakami, S., Iwaki, D., Mitsuzawa, H., Sano, H., Takahashi, H., Voelker, D., Mellick, G. D., Whitehouse, M. W., Boyle, G. M., Parsons, P. G., Roberts, M. S., and Cross, S. E. (2002). Surfactant protein A inhibits peptidoglycan-induced tumor necrosis factor- secretion in U937 cells and alveolar macrophages by direct interaction with Toll-like receptor 2. J. Biol. Chem. 277, 6830–6837.
Noah, T., Murphy, P., Alink, J., Leigh, M., Hull, W., Stahlman, M., and Whitsett, J. A. (2003). Bronchoalveolar lavage fluid surfactant protein-A and surfactant protein-D are inversely related to inflammation in early cystic fibrosis. Am. J. Respir. Crit. Care Med. 168, 685–691.
Nomori, H., Horio, H., Fuyuno, G., Kobayashi, R., Morinaga, S., and Suemasu, K. (1998). Serum surfactant protein A levels in healthy individuals are increased in smokers. Lung 176, 355–361.
Norwitz, E., López Bernal, A., and Starkey, P. (1992a). Tumor necrosis factor-alpha selectively stimulates prostaglandin F2 alpha production by macrophages in human term decidua. Am. J. Obstet. Gynecol. 167, 815–820.
Norwitz, E., Starkey, P., and López Bernal, A. (1992b). Prostaglandin D2 production by term human decidua: cellular origins defined using flow cytometry. Obstet. Gynecol. 80, 440–445.
Oberley, R., Goss, K., Ault, K., Crouch, E., and Snyder, J. (2004). Surfactant protein D is present in the human female reproductive tract and inhibits Chlamydia trachomatis infection. Mol. Hum. Reprod. 10, 861–870.
Oberley, R. E., Goss, K. L., Hoffmann, D. S., Ault, K. A., Neff, T. L., Ramsey, K. H., Ramsey, K. H., and Snyder, J. M. (2007). Regulation of surfactant protein D in the mouse female reproductive tract in vivo. Mol. Hum. Reprod. 13, 863–868.
Ohlmeier, S., Vuolanto, M., Toljamo, T., Vuopala, K., Salmenkivi, K., Myllärniemi, M., and Kinnula, V. L. (2008). Proteomics of human lung tissue identifies surfactant protein A as a marker of chronic obstructive pulmonary disease. J. Proteome Res. 7, 5125–5132.
Ohmer-Schröck, D., Schlatterer, C., Plattner, H., and Schlepper-Schäfer, J. (1995). Lung surfactant protein A (SP-A) activates a phosphoinositide/calcium signaling pathway in alveolar macrophages. J. Cell Sci. 108, 3695–3702.
Okita, J., MacDonald, P., and Johnston, J. (1982). Mobilization of arachidonic acid from specific glycerophospholipids of human fetal membranes during early labor. J. Biol. Chem. 257, 14029–14034.
Ooi, E., Wormald, P., Carney, A., James, C., and Tan, L. (2007). Surfactant protein d expression in chronic rhinosinusitis patients and immune responses in vitro to Aspergillus and alternaria in a nasal explant model. Laryngoscope 117, 51–57.
O’Riordan, D. M., Standing, J. E., Kwon, K. Y., Chang, D., Crouch, E. C., and Limper, A. H. (1995). Surfactant protein D interacts with Pneumocystis carinii and mediates organism adherence to alveolar macrophages. J. Clin. Invest. 95, 2699–2710.
Paananen, R., Sormunen, R., Glumoff, V., van Eijk, M., and Hallman, M. (2001). Surfactant proteins A and D in Eustachian tube epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L660–L667.
Palaniyar, N., Clark, H., Nadesalingam, J., Shih, M. J., Hawgood, S., and Reid, K. B. (2005). Innate immune collectin surfactant protein D enhances the clearance of DNA by macrophages and minimizes anti-DNA antibody generation. J. Immunol. 174, 7352–7358.
Pasula, R., Downing, J. F., Wright, J. R., Kachel, D. L., Davis, T. E. Jr., and Martin, W. J. (1997). Surfactant protein A (SP-A) mediates attachment of Mycobacterium tuberculosis to murine alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 17, 209–217.
Pasula, R., Wright, J., Kachel, D., and Martin, W. II. (1999). Surfactant protein A suppresses reactive nitrogen intermediates by alveolar macrophages in response to Mycobacterium tuberculosis. J. Clin. Invest. 103, 483–490.
Pejovic, M., Stankovic, A., and Mitrovic, D. (1995). Determination of the apparent synovial permeability in the knee joint of patients suffering from osteoarthritis and rheumatoid arthritis. Br. J. Rheumatol. 34, 520–524.
Piboonpocanun, S., Mitsuzawa, H., Martin, W., Murphy, R., Harbeck, R., and Voelker, D. (2005). Surfactant protein A binds Mycoplasma pneumoniae with high affinity and attenuates its growth by recognition of disaturated phosphatidylglycerol. J. Biol. Chem. 280, 9–17.
Pikaar, J., Voorhout, W., van Golde, L., Verhoef, J., Van Strijp, J., and van Iwaarden, J. (1995). Opsonic activities of surfactant proteins A and D in phagocytosis of gram-negative bacteria by alveolar macrophages. J. Infect. Dis. 172, 481–489.
Pryhuber, G., Hull, W., Fink, I., McMahan, M., and Whitsett, J. (1991). Ontogeny of surfactant proteins A and B in human amniotic fluid as indices of fetal lung maturity. Pediatr. Res. 30, 597–605.
Ragas, A., Roussel, L., Puzo, G., and Rivière, M. (2007). The Mycobacterium tuberculosis cell-surface glycoprotein apa as a potential adhesin to colonize target cells via the innate immune system pulmonary C-type lectin surfactant protein A. Biol. Chem. 282, 5133–5142.
Reading, P. C., Holmskov, U., and Anders, E. M. (2004). Antiviral activity of bovine collectins against rotaviruses. J. Gen. Virol. 79(Pt 9), 2255–2263.
Reading, P., Morey, L., Crouch, E., and Anders, E. (1997). Collectin-mediated antiviral host defense of the lung: evidence from influenza virus infection of mice. J. Virol. 71, 8204–8212.
Restrepo, C. I., Dong, Q., Savov, J., Mariencheck, W. I., and Wright, J. R. (1999). Surfactant protein D stimulates phagocytosis of Pseudomonas aeruginosa by alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 21, 576–585.
Rosseau, S., Guenther, A., Seeger, W., and Lohmeyer, J. (1997). Phagocytosis of viable Candida albicans by alveolar macrophages: lack of opsonin function of surfactant protein A. J. Infect. Dis. 175, 421–428.
Rubio, S., Lacaze-Masmonteil, T., Chailley-Heu, B., Kahn, A., Bourbon, J., and Ducroc, R. (1995). Pulmonary surfactant protein A (SP-A) is expressed by epithelial cells of small and large intestine. J. Biol. Chem. 270, 12162–12169.
Sago, H., Sugimoto, K., Fujii, S., Iinuma, K., Yamashita, K., Kitagawa, M., and Terashima, Y. (1993). Dynamic changes in stress fiber expression in rat uterine vein endothelial cells associated with pregnancy. Microvasc. Res. 46, 178–189.
Sano, H., Kuronuma, K., Kudo, K., Mitsuzawa, H., Sato, M., Murakami, S., and Kuroki, Y. (2007). Regulation of inflammation and bacterial clearance by lung collectins. Respirology 11, S46–S50.
Sano, H., Nagai, K., Tsutsumi, H., and Kuroki, Y. (2003). Lactoferrin and surfactant protein A exhibit distinct binding specificity to F protein and differently modulate respiratory syncytial virus infection. Eur. J. Immunol. 33, 2894–2902.
Sano, H., Sohma, H., Muta, T., Nomura, S., Voelker, D., and Kuroki, Y. (1999). Pulmonary surfactant protein A modulates the cellular response to smooth and rough lipopolysaccharides by interaction with CD14. J. Immunol. 163, 387–395.
Sasaki, R., Soejima, T., Matsumoto, A., Maruta, T., Yamada, K., Ota, Y., Kawabe, T., Nishimura, H., Sakai, E., Ejima, Y., and Sugimura, K. (2001). Clinical significance of serum pulmonary surfactant proteins a and d for the early detection of radiation pneumonitis. Int. J. Radiat. Oncol. Biol. Phys. 50, 301–307.
Sato, A., Whitsett, J., Scheule, R., and Ikegami, M. (2010). Surfactant protein-d inhibits lung inflammation caused by ventilation in premature newborn lambs. Am. J. Respir. Crit. Care Med. 181, 1098–1105.
Sato, M., Sano, H., Iwaki, D., Kudo, K., Konishi, M., Takahashi, H., Takahashi, T., Imaizumi, H., Asai, Y., and Kuroki, Y. (2003). Direct binding of Toll-like receptor 2 to zymosan, and zymosan-induced NF- B activation and TNF- secretion are down-regulated by lung collectin surfactant protein A. J. Immunol. 171, 417–425.
Sawada, K., Ariki, S., Kojima, T., Saito, A., Yamazoe, M., Nishitani, C., Shimizu, T., Takahashi, M., Mitsuzawa, H., Yokota, S., Sawada, N., Fujii, N., Takahashi, H., and Kuroki, Y. (2010). Pulmonary collectins protect macrophages against pore-forming activity of Legionella pneumophila and suppress its intracellular growth. J. Biol. Chem., 285, 8434–8443.
Schagat, T., Wofford, J., and Wright, J. (2001). Surfactant protein A enhances alveolar macrophage phagocytosis of apoptotic neutrophils. J. Immunol. 166, 2727–2733.
Schelenz, S., Malhotra, R., Sim, R. B., Holmskov, U., and Bancroft, G. J. (1995). Binding of host collectins to the pathogenic yeast Cryptococcus neoformans: human surfactant protein D acts as an agglutinin for acapsular yeast cells. Infect. Immun. 63, 3360–3366.
Schett, G., Zwerina, J., and Firestein, G. (2008). The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann. Rheum. Dis. 67, 909–916.
Sever-Chroneos, Z., Krupa, A., Davis, J., Hasan, M., Yang, C. H., Szeliga, J., Herrmann, M., Hussain, M., Geisbrecht, B. V., Kobzik, L., and Chroneos, Z. C. (2011). Surfactant protein A (SP-A)-mediated clearance of Staphylococcus aureus involves binding of SP-A to the staphylococcal adhesin eap and the macrophage receptors SP-A receptor 210 and scavenger receptor class A. J. Biol. Chem. 286, 4854–4870.
Shakoori, T., Sin, D., Ghafoor, F., Bashir, S., and Bokhari, S. (2009). Serum surfactant protein D during acute exacerbations of chronic obstructive pulmonary disease. Dis. Markers 27, 287–294.
Shepherd, V. L. (2002). Pulmonary surfactant protein D: a novel link between innate and adaptive immunity. Am. J. Physiol. Lung Cell. Mol. Physiol. 282, L516–L517.
Silhavy, T. J., Kahne, D., and Walker, S. (2010). The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2, a000414.
Simani, A., Inoue, S., and Hogg, J. (1974). Penetration of the respiratory epithelium of guinea pigs following exposure to cigarette smoke. Lab. Invest. 31, 75–81.
Sin, D., Man, S., Marciniuk, D., Ford, G., FitzGerald, M., Wong, E., York, E., Mainra, R. R., Ramesh, W., Melenka, L. S., Wilde, E., Cowie, R. L., Williams, D., Gan, W. Q., and Rousseau, R. (2008). The effects of fluticasone with or without salmeterol on systemic biomarkers of inflammation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 177, 1207–1201.
Singh, M., Madan, T., Waters, P., Parida, S., Sarma, P., and Kishore, U. (2003). Protective effects of a recombinant fragment of human surfactant protein D in a murine model of pulmonary hypersensitivity induced by dust mite allergens. Immunol. Lett. 86, 297–307.
Singh, M., Madan, T., Waters, P., Sonar, S., Singh, S. K., Kamran, M. F., and Bernal, A. L. (2009). Therapeutic effects of recombinant forms of full-length and truncated human surfactant protein D in a murine model of invasive pulmonary aspergillosis. Mol. Immunol. 46, 2363–2369.
Skinner, M., Schlosser, R., Lathers, D., Neal, J., Woodworth, B., Hall, J., Newton, D. A., and Baatz, J. E. (2007). Innate and adaptive mediators in cystic fibrosis and allergic fungal rhinosinusitis. Am. J. Rhinol. 2007, 538–541.
Sorensen, G., Madsen, J., Kejling, K., Tornoe, I., Nielsen, O., Townsend, P., Poulain, F., Nielsen, C. H., Reid, K. B., Hawgood, S., Falk, E., and Holmskov, U. (2006). Surfactant protein D is proatherogenic in mice. Am. J. Physiol. Heart Circ. Physiol. 290, H2286–H2294.
Stahlman, M., Gray, M., Hull, W., and Whitsett, J. (2002). Immunolocalization of surfactant protein-D (SP-D) in human fetal, newborn, and adult tissues. J. Histochem. Cytochem. 50, 651–660.
Tenner, A., Robinson, S., Borchelt, J., and Wright, J. (1989). Human pulmonary surfactant protein (SP-A), a protein structurally homologous to C1q, can enhance FcR- and CR1-mediated phagocytosis. J. Biol. Chem. 264, 13923–13928.
Tino, M., and Wright, J. (1999). Glycoprotein-340 binds surfactant protein-A (SP-A) and stimulates alveolar macrophage migration in an SP-A-independent manner. Am. J. Respir. Cell Mol. Biol. 20, 759–768.
Trinder, P., Hickling, T., Sim, R., Brackertz, D., Loos, M., and Maeurer, M. (2000). Humoral autoreactivity directed against surfactant protein-A (SP-A) in rheumatoid arthritis synovial fluids. Clin. Exp. Immunol. 120, 183–187.
van de Wetering, J., van Eijk, M., van Golde, L., Hartung, T., van Strijp, J., and Batenburg, J. (2001). Characteristics of surfactant protein A and D binding to lipoteichoic acid and peptidoglycan, 2 major cell wall components of gram-positive bacteria. J. Infect. Dis. 184, 1143–1151.
van de Wetering, J. K., van Remoortere, A., Vaandrager, A. B., Batenburg, J. J., van Golde, L. M., Hokke, C. H., and van Hellemond, J. J. (2004a). Surfactant protein D binding to terminal alpha1-3-linked fucose residues and to Schistosoma mansoni. Am. J. Respir. Cell Mol. Biol. 31, 565–572.
van de Wetering, J. K., Coenjaerts, F. E., Vaandrager, A. B., van Golde, L. M., and Batenburg, J. J. (2004b). Aggregation of Cryptococcus neoformans by surfactant protein D is inhibited by its capsular component glucuronoxylomannan. Infect Immun. 72, 145–153.
van Iwaarden, F., Welmers, B., Verhoef, J., Haagsman, H. P., and van Golde, L. M. (1990). Pulmonary surfactant protein A enhances the host-defense mechanism of rat alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 2, 91–98.
van Iwaarden, J., Pikaar, J., Storm, J., Brouwer, E., Verhoef, J., Oosting, R., van Golde, L. M., and van Strijp, J. A. (1994). Binding of surfactant protein A to the lipid A moiety of bacterial lipopolysaccharides. Biochem. J. 303, 407–411.
van Iwaarden, J., van Strijp, J., Ebskamp, M., Welmers, A., Verhoef, J., and van Golde, L. (1991). Surfactant protein A is opsonin in phagocytosis of herpes simplex virus type 1 by rat alveolar macrophages. Am. J. Physiol. 261(2 Pt 1), L204–L209.
van Iwaarden, J., van Strijp, J., Visser, H., Haagsman, H., Verhoef, J., and van Golde, L. (1992). Binding of surfactant protein A (SPA) to herpes simplex virus type 1-infected cells is mediated by the carbohydrate moiety of SP-A. J. Biol. Chem. 267, 25039–25043.
van Rozendaal, B., van Spriel, A., van De Winkel, J., and Haagsman, H. (2000). Role of pulmonary surfactant protein D in innate defense against Candida albicans. J. Infect. Dis. 182, 917–922.
Vandivier, R., Ogden, C., Fadok, V. A., Hoffmann, P., Brown, K., Botto, M., Walport, M. J., Fisher, J. H., Henson, P. M., and Greene, K. E. (2002). Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J. Immunol. 169, 3978–3986.
Vuk-Pavlovic, Z., Standing, J. E., Crouch, E. C., and Limper, A. H. (2001). Carbohydrate recognition domain of surfactant protein D mediates interactions with Pneumocystis carinii glycoprotein A. Am. J. Respir. Cell Mol. Biol. 24, 475–484.
Walenkamp, A., Verheul, A., Scharringa, J., and Hoepelman, I. (1999). Pulmonary surfactant protein A binds to Cryptococcus neoformans without promoting phagocytosis. Eur. J. Clin. Invest. 29, 83–92.
Wang, J., Shieh, C., You, P., Lei, H., and Reid, K. (1998). Inhibitory effect of pulmonary surfactant proteins A and D on allergen-induced lymphocyte proliferation and histamine release in children with asthma. Am. J. Respir. Crit. Care Med. 158, 510–518.
Wang, J. Y., Kishore, U., Lim, B. L., Strong, P., and Reid, K. B. (1996). Interaction of human lung surfactant proteins A and D with mite (Dermatophagoides pteronyssinus) allergens. Clin. Exp. Immunol. 106, 367–373.
Weaver, T., and Whitsett, J. (1991). Function and regulation of expression of pulmonary surfactant-associated proteins. Biochem. J. 273, 249–264.
Weikert, L. F., Edwards, K., Chroneos, Z. C., Hager, C., Hoffman, L., and Shepherd, V. L. (1997). SP-A enhances uptake of bacillus Calmette-Guérin by macrophages through a specific SP-A receptor. Am. J. Physiol. 272, L989–L995.
Weikert, L., Lopez, J., Abdolrasulnia, R., Chroneos, Z., and Shepherd, V. (2000). Surfactant protein A enhances mycobacterial killing by rat macrophages through a nitric oxide-dependent pathway. Am. J. Physiol. Lung Cell. Mol. Physiol. 279, L216–L223.
Wells, A., Hansell, D., Harrison, N., Lawrence, R., Black, C., and du Bois, R. (1993). Clearance of inhaled 99mTc-DTPA predicts the clinical course of fibrosing alveolitis. Eur. Respir. J. 6, 797–802.
Williams, M. D., Wright, J. R., March, K. L., and Martin, W. J. (1996). Human surfactant protein A enhances attachment of Pneumocystis carinii to rat alveolar macrophages. Am. J. Respir. Cell Mol. Biol. 14, 232–238.
Winkler, C., Atochina-Vasserman, E., Holz, O., Beers, M., Erpenbeck, V., Krug, N., Roepcke, S., Lauer, G., Elmlinger, M., and Hohlfeld, J. M. (2011). Comprehensive characterisation of pulmonary and serum surfactant protein D in COPD. Respir. Res. 12, 12–29.
Woodworth, B., Lathers, D., Neal, J., Skinner, M., Richardson, M., Young, M. R., and Schlosser, R. J. (2006). Immunolocalization of surfactant protein A and D in sinonasal mucosa. Am. J. Rhinol. 20, 461–465.
Woodworth, B. A., Neal, J. G., Newton, D., Joseph, K., Kaplan, A. P., Baatz, J. E., and Schlosser, R. J. (2007a). Surfactant protein A and D in human sinus mucosa: a preliminary report. ORL J. Otorhinolaryngol. Relat. Spec. 69, 57–60.
Woodworth, B., Wood, R., Baatz, J., and Schlosser, R. (2007b). Sinonasal surfactant protein A1, A2, and D gene expression in cystic fibrosis: a preliminary report. Otolaryngol. Head Neck Surg. 137, 34–38.
Wootten, C., Labadie, R., Chen, A., and Lane, K. (2006). Differential expression of surfactant protein A in the nasal mucosa of patients with allergy symptoms. Arch. Otolaryngol. Head Neck Surg. 132, 1001–1007.
Wright, J. R. (2005). Immunoregulatory functions of surfactant proteins. Nat. Rev. Immunol. 1, 58–68.
Wu, H., Kuzmenko, A., Wan, S., Schaffer, L., Weiss, A., Fisher, J., Kim, K. S., and McCormack, F. X. (2003). Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J. Clin. Invest. 111, 1589–1602.
Wu, Y., Liu, Z., Wei, R., Pan, S., Mao, N., Chen, B., Han, J. J., Zhang, F. S., Holmskov, U., Xia, Z. L., de Groot, P. G., Reid, K. B., Xu, W. B., and Sorensen, G. L. (2009). Elevated plasma surfactant protein D (SP-D) levels and a direct correlation with anti-severe acute respiratory syndrome coronavirus-specific IgG antibody in SARS patients. Scand. J. Immunol. 69, 508–515.
Wyllie, A. H., Kerr, J. F., and Currie, A. R. (1980). Cell death: the significance of apoptosis. Int. Rev. Cytol. 68, 251–306.
Yanaba, K., Hasegawa, M., Takehara, K., and Sato, S. (2004). Comparative study of serum surfactant protein-D and KL-6 concentrations in patients with systemic sclerosis as markers for monitoring the activity of pulmonary fibrosis. J. Rheumatol. 31, 1112–1120.
Yong, S. J., Vuk-Pavlovic, Z., Standing, J. E., Crouch, E. C., and Limper, A. H. (2003). Surfactant protein D-mediated aggregation of Pneumocystis carinii impairs phagocytosis by alveolar macrophages. Infect. Immun. 71, 1662–1671.
Keywords: surfactant, innate immunity, hypersensitivity, infection, macrophage, pattern recognition receptor
Citation: Nayak A, Dodagatta-Marri E, Tsolaki AG and Kishore U (2012) an insight into the diverse roles of surfactant proteins, SP-A and SP-D in innate and adaptive immunity. Front. Immun. 3:131. doi: 10.3389/fimmu.2012.00131
Received: 05 September 2011; Accepted: 07 May 2012;
Published online: 07 June 2012.
Edited by:
Mohamed Daha, Leiden University Medical Center, NetherlandsReviewed by:
Mohamed Daha, Leiden University Medical Center, NetherlandsDan Anthony Mitchell, University of Warwick, UK
Copyright: © 2012 Nayak, Dodagatta-Marri, Tsolaki and Kishore. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Uday Kishore, Centre for Infection, Immunity and Disease Mechanisms, School of Health Sciences and Social Care, Brunel University, Heinz Wolff Building, UB8 3PH London, UK. e-mail: uday.kishore@brunel.ac.uk; ukishore@hotmail.com