
- Department of Rheumatology, Mayo Clinic, Rochester, MN, USA
The characteristic serologic feature of systemic lupus erythematosus (SLE) is autoantibodies against one’s own nucleic acid or nucleic acid-binding proteins – DNA and RNA-binding nuclear proteins. Circulating autoantibodies can deposit in the tissue, causing inflammation and production of cytokines such as type 1 interferon (IFN). Investigations in human patients and animal models have implicated environmental as well as genetic factors in the biology of the SLE autoimmune response. Viral/Bacterial nucleic acid is a potent stimulant of innate immunity by both toll-like receptor (TLR) and non-TLR signaling cascades. Additionally, foreign DNA may act as an immunogen to drive an antigen-specific antibody response. Self nucleic acid is normally restricted to the nucleus or the mitochondria, away from the DNA/RNA sensors, and mechanisms exist to differentiate between foreign and self nucleic acid. In normal immunity, a diverse range of DNA and RNA sensors in different cell types form a dynamic and integrated molecular network to prevent viral infection. In SLE, pathologic activation of these sensors occurs via immune complexes consisting of autoantibodies bound to DNA or to nucleic acid-protein complexes. In this review, we will discuss recent studies outlining how mismanaged nucleic acid sensing networks promote autoimmunity and result in the over-production of type I IFN. This information is critical for improving therapeutic strategies for SLE disease.
Introduction
The normal immune system strikes a delicate balance between defense against foreign invasion and the prevention of misdirected responses against self-antigens. Sometimes, this intricate balance becomes faulty due to genetic, environmental, or other factors leading to breakdown of self-tolerance and the onset of an autoimmune disorder. Systemic Lupus Erythematosus (SLE) is a prototype autoimmune disease that affects the skin, kidney, musculoskeletal, and hematologic systems and is characterized by presence of various autoantibodies against self-components, especially double-stranded DNA (dsDNA) and RNA-binding nuclear proteins. Amongst SLE patients, the female to male ratio is 9:1, suggesting that sex-related factors are important in the development of the disease (1, 2). Many genetic factors have been strongly associated with disease susceptibility (3, 4). Exposure to several viruses and bacterial infections, and also UV light are known to trigger SLE (5). Thus, it is considered that SLE occurs when an environmental trigger acts on a genetically predisposed individual, leading to a loss of tolerance toward native proteins (6). Multiple immune system abnormalities contribute to the pathogenesis of SLE, including abnormal clearance of apoptotic cells and immune complexes, over-production of type I interferon (IFN), reduced thresholds for B and T lymphocyte activation, and production of autoantibodies against self-antigens (7). These autoantibodies are directed against nucleic acids and RNA-binding proteins such as Ro, La, and Sm (8). Tissue damage is mediated in part by deposition of immune complexes in the affected organs, followed by activation of downstream inflammatory pathways mediated by complement and FcR engagement of innate immune cells (9). Viruses such as Cytomegalovirus (CMV), Epstein–Barr (EBV), and Parvovirus B19 are frequently involved as environmental triggers in lupus. Hypomethylated bacterial and viral DNA are potent inducers of immune responses through TLR signaling cascade finally leading to type 1 IFN over-expression, B cell activation, production of autoantibodies, and interleukin (IL)-6 (10).
Many patients with SLE have high circulating levels of type I IFN (11). Some individuals treated with IFN-α for chronic viral infections developed de novo SLE that was resolved when IFN-α was withdrawn (12, 13). Additionally, within SLE families abnormally high IFN-α levels have been found clustered (14). A recent genome-wide association study has identified additional novel genetic loci associated with high serum IFN-α in SLE patients (15, 16). Taken together, these data support the idea that genetically determined endogenous elevations in IFN-α predispose to human SLE.
How Does Lupus Start?
The etiology of lupus is considered to be multifactorial involving multiple genes and environmental factors such as infections, hormones, and drugs (Figure 1) (17). It is considered that unrestrained immune response to apoptotic cells and decreased disposal of apoptotic material are important initiators of the autoimmune response in SLE. Genomic DNA is not accessible to the immune system under standard conditions as it is safely sequestered in the nucleus or in mitochondria under the tight control of DNA damage and repair response systems. However, when cells die through apoptosis, apoptotic bodies containing fragmented cellular material and abnormal surface antigens, circulate in the body enabling the immune system to access new epitopes (18). Under normal conditions cellular mechanisms exist to ensure that apoptotic debris is not immunogenic to self, but these mechanisms can fail. It seems likely that defective clearance of apoptotic material and modifications to DNA such as hypomethylation can promote SLE (19). Recent reports suggest that neutrophil extracellular traps (NETs) are a potent stimulus for type 1 IFN release by plasmacytoid dendritic cells (DCs), and play an important role in propagation of the lupus phenotype (20–23). Neutrophils are specialized immune cells that are rapidly recruited to sites of inflammation in response to microbial infections. One of the mechanisms of neutrophil action is the formation of “NETs” (24). NETs are made of processed chromatin bound to granular and selected cytoplasmic proteins. NETs are released by neutrophils to control microbial infections (24). This release of chromatin is the result of a unique form of cell death, called “NETosis.” Material derived from NETosis can contribute to SLE by serving as source of autoantigen, propagating inflammation, and tissue damage (21, 23, 25, 26). In an interesting recent study, Sangaletti et al. suggested that NETs may provide antigens to DCs and in this way promote immune responses against neutrophil antigens in the autoimmune disease small vessel vasculitis, which is characterized by antibodies against cytoplasmic proteins in neutrophils (23). It is possible that NETs may provide nuclear antigens to immune cells in a similar way in SLE.
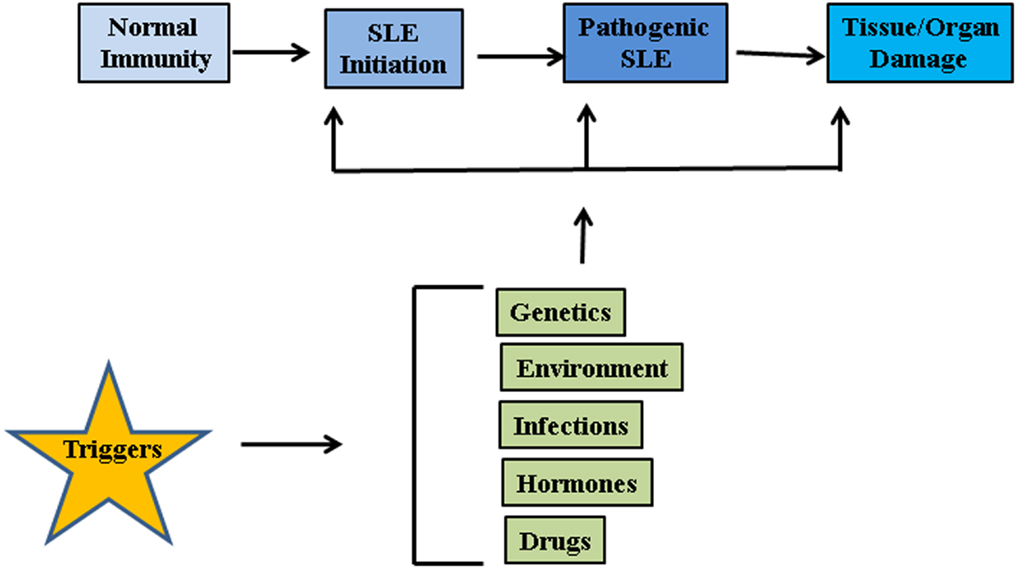
Figure 1. Factors associated with SLE pathogenesis. Genetic factors, environmental influences such as radiations, repeated infections, hormonal imbalances, and certain drugs may act on innate immune system and disrupt the intricate balance between protection against foreign invasion and self-defense.
Pathways through which our own nuclear material is able to induce pro-inflammatory responses are a topic of active research. At least three distinct types of nucleic acid recognition receptors are recognized: (1) the toll-like receptors (TLRs), which recognize nucleic acids on the plasma membranes and endosomes; (2) the nucleotide binding and oligomerization domain (NOD) receptors (NLRs), which monitor the cytosolic compartment and also interact with TLR pathways; and (3) the retinoid acid inducible gene (RIG)-I-like receptors that recognize RNA or DNA in the cytoplasm (RLRs). Many of these receptors may directly or indirectly participate in the pathogenesis of SLE (27).
Toll-Like Receptor Mediated Signaling in Lupus
Toll-like receptors are major components of the innate immune system that activate multiple inflammatory pathways and coordinate systemic defense against microbial pathogens. Data from animal models and human patients suggest that improper engagement of TLR pathways by endogenous or exogenous ligands may lead to the initiation of autoimmune responses and tissue injury (28). Endosomal TLRs (TLR-3, -7, -8, and -9) are potent activators of DCs and B cells. TLR-3 is specific for double-stranded RNA (dsRNA), TLRs-7 and -8 for single-stranded RNA (ssRNA), and TLR-9 is specific for dsDNA (29, 30). TLRs are expressed predominantly in DCs, B cells, macrophages, monocytes, and neutrophils. Cell surface receptors, such as the B cell receptor (BCR) and FcγRIIa, facilitate the endocytosis of nucleic acid containing material or immune complexes (31, 32). Chromatin-containing immune complexes can stimulate B cells up to 100-fold more effectively than complexes without nucleic acids apparently due to collective engagement of BCR and TLR (31–34). Thus, dual engagement of the BCR and the TLR can induce abnormal activation of B cells and break immune tolerance. In human lupus, an increased proportion of B cells and monocytes expressed TLR-9 among patients with active SLE compared to patients with inactive disease (35). TLR activation in combination with T cell derived IL-21 markedly increased B cell differentiation into plasma cells (36).
All TLR family members, including TLRs-7, -8, -9 are type I membrane proteins composed of a ligand-binding ectodomain containing 18–25 tandem copies of leucine-rich repeats (LRRs), a transmembrane domain, and a conserved cytoplasmic toll/interleukin-1 receptor (TIR) domain. Ligand-induced dimerization and conformational rearrangement of the TIR domains leads to the creation of two symmetry-related sites which allow binding of the cognate signaling adaptor molecules (37, 38). Two main adaptors are utilized by TLRs, namely Myeloid Differentiation Factor-88 (MyD88) (TLR-7, -8, and -9) and TIR domain-containing adaptor inducing IFN-β (TRIF) (TLR-3). These adaptors mediate the recruitment of a series of kinases that lead to the formation of specific macromolecular signaling platforms for inflammatory reactions. IL-1 receptor-associated kinase 4 (IRAK-4) is recruited to MyD88 and is activated after recruitment (38). IRAK-4, in turn, activates IL-1 receptor-associated kinase 1 (IRAK 1) via phosphorylation (39, 40). These activated kinases recruit tumor necrosis factor receptor-associated factor 6 (TRAF-6), which is an E3 ubiquitin ligase required for activation of NFκB by freeing it from its inhibitor, I kappa B (IκB) (41). In addition to this, interferon regulatory factors (IRFs) IRF5 and IRF7 are recruited to the MyD88/IRAK/TRAF6 complex, where they become phosphorylated and activated (42, 43). Ultimately, the transcription factors NFκB and IRF5 and IRF7 are activated and translocate into the nucleus where they initiate gene transcription and production of pro-inflammatory cytokines and type I IFN (Figure 2) (43–45). Unlike TLR-7, -8, and -9, TLR-3 signaling is MyD88-independent and utilizes adaptor protein TRIF (46). TRIF also recruits additional proteins necessary for downstream signaling, including TRAF-family member-associated NFκB-activator-binding kinase 1 (TBK1), TRAF3, and receptor-interacting protein 1 (RIP1) (40). TRIF interaction with TBK1 is necessary for the activation of IRF-3, which is a transcription factor involved in the production of interferon beta (IFNβ). (47). TLR-3 can also activate NFκB by the interaction of TRIF with TRAF-6 or RIP1 (40, 48) leading to up-regulated IFNα production and secretion of other pro-inflammatory cytokines.
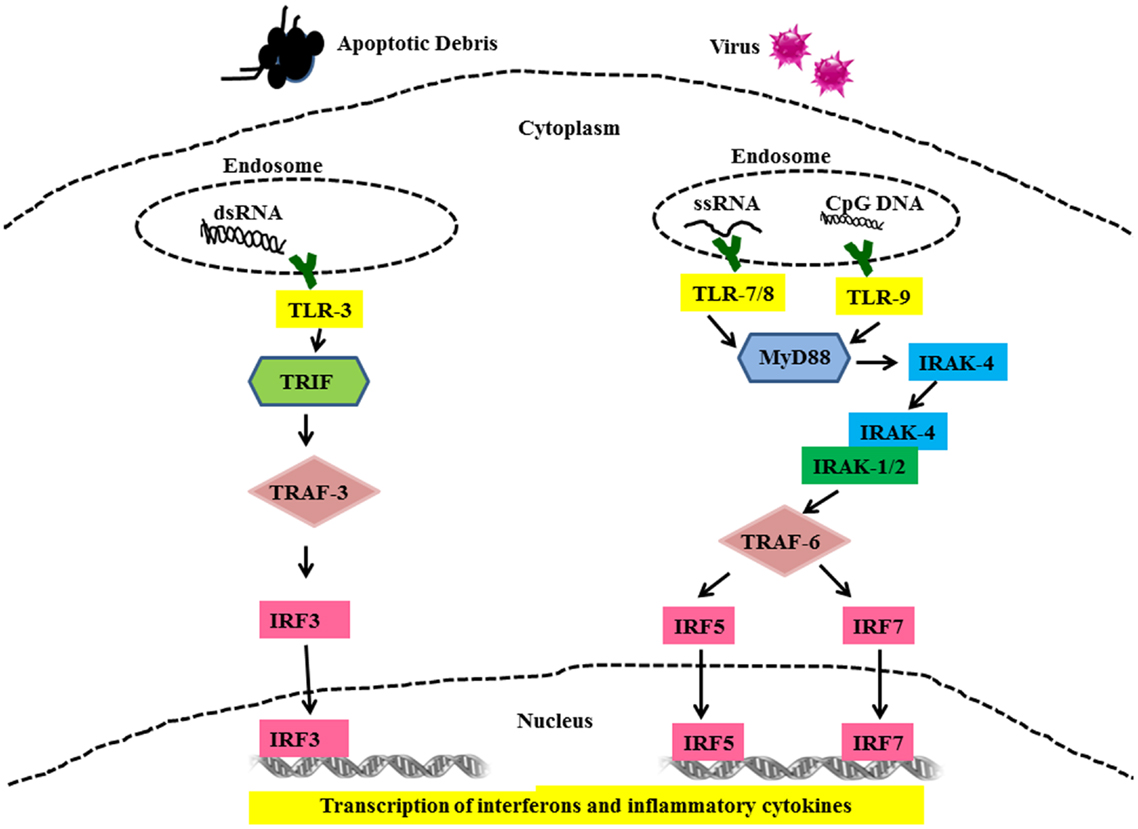
Figure 2. Toll-like receptor mediated signaling in SLE. Cells use TLRs as sensors to detect the presence of viruses and apoptotic debris via TLR-3, -7, -8, and -9. Nuclear material is trafficked to the endosome triggering TLRs signaling. Binding of cognate ligands to these TLRs recruits MyD88, a main signaling intermediate involved in TLR-7, -8, and -9 signaling. MyD88 recruits interleukin-1 receptor-associated kinase (IRAK)-4. IRAK-4 binds and phosphorylates IRAK-1, which in turn recruits Tumor necrosis factor (TNF) receptor-associated factor (TRAF) 6. IRF5 and IRF7 are then shuttled to the nucleus and these events set the stage for the transcription of IFN-α and other pro-inflammatory cytokines. TLR-3 signaling is MyD88-independent and utilizes TRIF and TRAF3 as signaling intermediates finally leading to activation of IRF3 and production of IFN-α and other pro-inflammatory cytokines.
Genetic Factors Associated with TLR-Dependent IFNα Pathway in Lupus
One of the most striking immune system abnormalities in SLE patients is the frequent up-regulation of the type I IFN pathway (49, 50). IFNα is critical player in SLE progression and severity, and has been shown to induce the production of autoantibodies when administered to non-SLE patients (12, 51). An interesting report describes remission of SLE in a patient which was attributed to unresponsiveness to both TLR-7 and -9 stimulation after development of common variable immunodeficiency – (CVID-) like disease (52). Genetic variations in many of the components of the TLR signaling pathway have been associated with SLE, such as TLR-7, IRF5, IRF7, IRF8, IRAK1, and TNFAIP3 (53–59). Three of the nine genes in the IRF family have been genetically associated with SLE (60). Additionally, some of these genetic polymorphisms have been associated with increased type I IFN in SLE patients, supporting the idea that these genetic variations modulate the output of the TLR pathway (42, 60–64). The implication of these genes in SLE strongly supports the primary relevance of the TLR and IFNα pathway in the disease phenotype (63, 65). Additionally, many of these genetic polymorphisms in the TLR pathway are associated with the formation of autoantibodies (62–64, 66), supporting the concept of a feed-forward loop in which genetic variations in the TLR pathway enhance autoantibody production, and then the autoantibodies form immune complexes which stimulate the TLR pathway and result in increased type I IFN production in the setting of the same genetic variations. The TLR pathways are important in B cell maturation, and it is possible that genetically programed TLR pathway over-activity could promote autoantibody formation in B cells. Then after immune complexes are formed, these stimulate the TLR pathway in DCs and macrophages, and the same polymorphisms promote increased cytokine output from these cells.
Toll-Independent Signaling in Lupus
Signaling through RIG-1 Like Receptors in Lupus
After viruses enter the cytoplasm and start replicating, infected host cells can sense and activate anti-viral responses in response to viral nucleic acids. This sensing occurs in the cytoplasm, and is independent of the cell surface and endosomal TLRs. Thus far, three cytosolic RNA helicases have been identified, RIG-I (retinoic acid – inducible gene I), MDA5 (melanoma differentiation – associated gene 5), and LGP2 (laboratory of genetics and physiology 2) that act as RNA sensors to mediate TLR-independent IFN-α/β induction in the presence of replicating RNA viruses (37, 67). Unlike membrane-bound TLRs, RLRs reside in the cytoplasm and sense cytoplasmic RNA. RIG-I contains tandem caspase recruitment domain (CARD)-like regions at its N-terminus and the central DExD/H helicase domain which has an ATP-binding motif and a C-terminal repressor domain which binds to RNA (68, 69). MDA5 contains tandem CARD-like regions and a DExD/H helicase domain, but it is unknown whether the C-terminal region of MDA5 really functions as repressor domain. LGP2 contains a DExD/H helicase domain and a repressor domain, but lacks the CARD-like region. LGP2 was suggested to be a negative regulator of RNA virus-induced responses, because the LGP2 repressor domain binds to that of RIG-I and suppresses signaling by interfering with the self-association of RIG-I (70, 71). Findings suggest that RIG-I and MDA5 have specificities in their detection of RNA viruses, through recognition of distinct viral RNA structures. RIG-I can recognize ssRNA bearing a 5′-triphosphate moiety (72, 73). In the case of self-RNA, 5′-triphosphate structures are removed or masked by a cap structure, which suggests a discrimination mechanism between self- and non-self RNA. RIG-I and MDA5 can distinguish dsRNA by size; RIG-I can bind short dsRNA whereas MDA5 can bind long dsRNA (74). Although LGP2 was considered a negative regulator, LGP2-deficient mice exhibited complicated phenotypes (75) and higher levels of type I IFN in response to polyinosinic: polycytidylic acid (Poly I:C) and vesicular stomatitis virus (VSV), but decreased type I IFN following encephalomyocarditis virus (EMCV) infection, suggesting that LGP2 can negatively or positively regulate RIG-I and MDA5 responses depending on the type of RNA virus (75).
Ligand binding to RLRs induces conformational changes leading to association with mitochondrial-associated IFN-β promoter stimulator 1 (IPS-1) through card-card domain interactions (76–79). IPS-1 then recruits TRAF3, which activates TANK-binding kinase 1 (TBK1) and IκB kinase (IKK) – related kinases IKKϵ (80). This leads to the phosphorylation and nuclear translocation of IRF-3 and -7 resulting in the transcription of IFN type 1 genes (81, 82) (Figure 3). IPS-1 also interacts with FAS-associated death domain protein (FADD) and receptor-interacting protein 1 (RIP-1) (76), which activate caspase-8 and caspase-10, resulting in NF-κB activation and production of inflammatory cytokines (83, 84). Genetic studies in SLE have strongly implicated the RLR pathways in SLE susceptibility. Variants in both MDA5 and IPS-1 have been associated with SLE susceptibility and with altered activation of the type I IFN pathway in SLE patients in vivo (85, 86). This again supports the idea that multiple nucleic acid recognition pathways are involved in SLE pathogenesis.
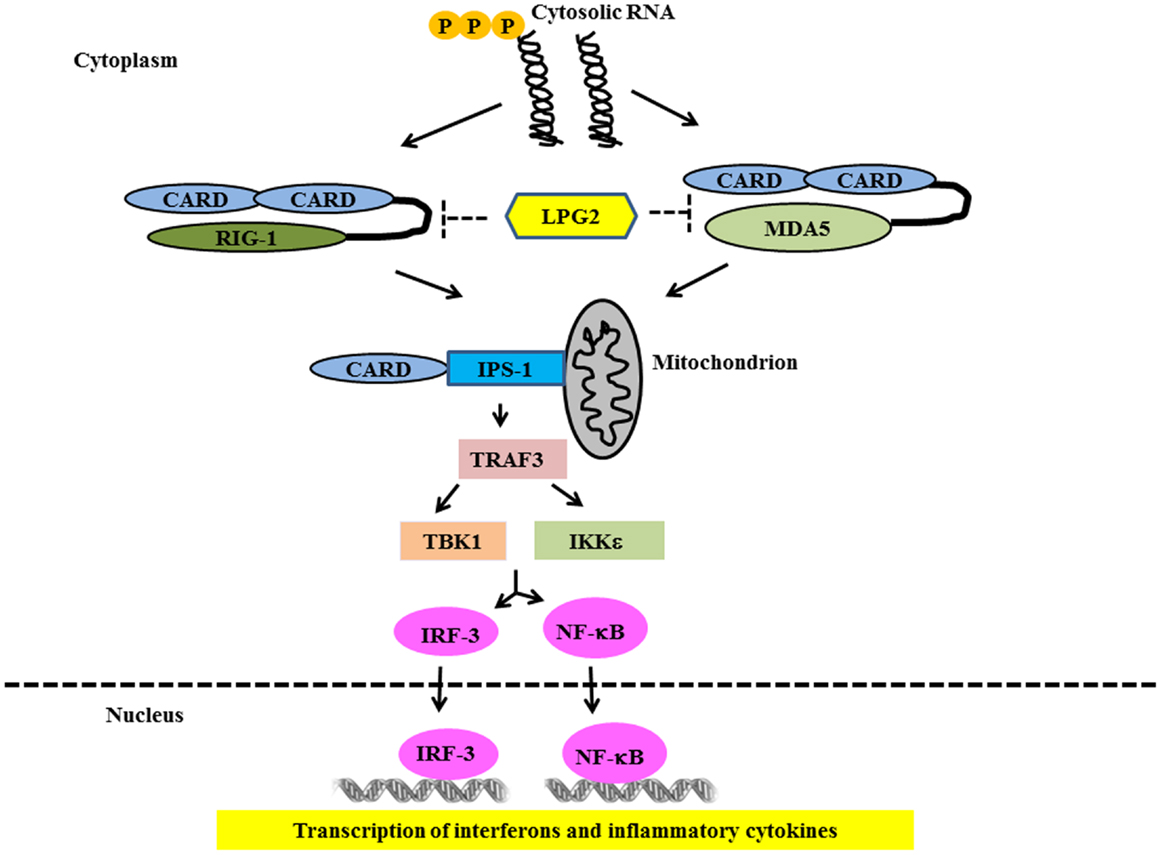
Figure 3. Signaling through RIG-1 like Receptors in lupus. Following recognition of the cytosolic RNA, RIG-I, and MDA5 associate with the adapter IPS-1 via CARD-like domains. IPS-1 is localized to the mitochondrion and initiates signaling leading to activation of IRF3 and NFκB that finally lead of over-production of type 1 IFN and other inflammatory cytokines.
Signaling through Nucleotide Binding and Oligomerization Domain (NLR) Receptors in Lupus
The NOD (NLR) family of receptors are key molecules that drive inflammatory responses by forming a multi-protein complex called “inflammasome.” The inflammasome drives the processing and release of cytokines such as the pro-inflammatory cytokines IL-1β and IL-18. Several inflammasome complexes have been identified in recent years. Of the known inflammasomes, NLRP3, absent in melanoma 2 (AIM2), and IFN inducible protein 16 (IFI16) inflammasomes have been linked to immune responses to intracellular DNA, as well as bacterial and viral infections (87). IL-1β is important in activating neutrophils, macrophages, DCs, and T cells, whereas IL-18 is crucial for IFN-γ production by NK cells and T cells (88). IL-1β and IL-18 are regulated at both transcriptional and post-translational levels. Upon transcriptional induction by TLRs and other sensor systems, IL-1β and IL-18 are synthesized as inactive precursor proteins, which are subsequently processed by the cysteine protease caspase-1 (IL-1β converting enzyme) (89). Conversion of procaspase-1 into an enzymatically active form, caspase-1, occurs upon formation of a multi-protein inflammasome complex (89). Previous reports have suggested that the NLRP3 inflammasome is involved in mediating the inflammatory responses to both DNA and RNA viruses (90, 91). In human SLE macrophages, NETs induce robust activation of the NLRP3 inflammasome (92).
Several groups independently identified AIM2 as a receptor for cytosolic DNA that leads to caspase-1 activation and IL-1β secretion (93, 94). AIM2 binds cytosolic DNA of self and non-self origin, including bacterial, viral, and mammalian DNA, in a sequence-independent manner (95). Recent evidence indicates that the AIM2-related protein IFI16 also forms an inflammasome complex following Kaposi sarcoma – associated herpes virus infection of endothelial cells (96). Several groups independently identified STING as a key component of the DNA-sensing pathway (97, 98). STING/MITA translocates to perinuclear regions where it interacts with TBK1 to relay downstream signals to IRF3 (Figure 4). STING deficiency in macrophages or DCs leads to a markedly impaired type I IFN response to B-DNA and immunostimulatory DNA or to infection with DNA viruses, including HSV-1, human CMV, and vaccinia virus (97, 98). Initial studies showed that STING also interacted with components of the RNA-recognition machinery, such as RIG-I, where it was linked to type I IFN induction in response to VSV, a negative-strand RNA virus (97, 99). Murine models support the relevance of AIM2 in susceptibility to lupus-like disease in the NZB × NZW mouse (100).
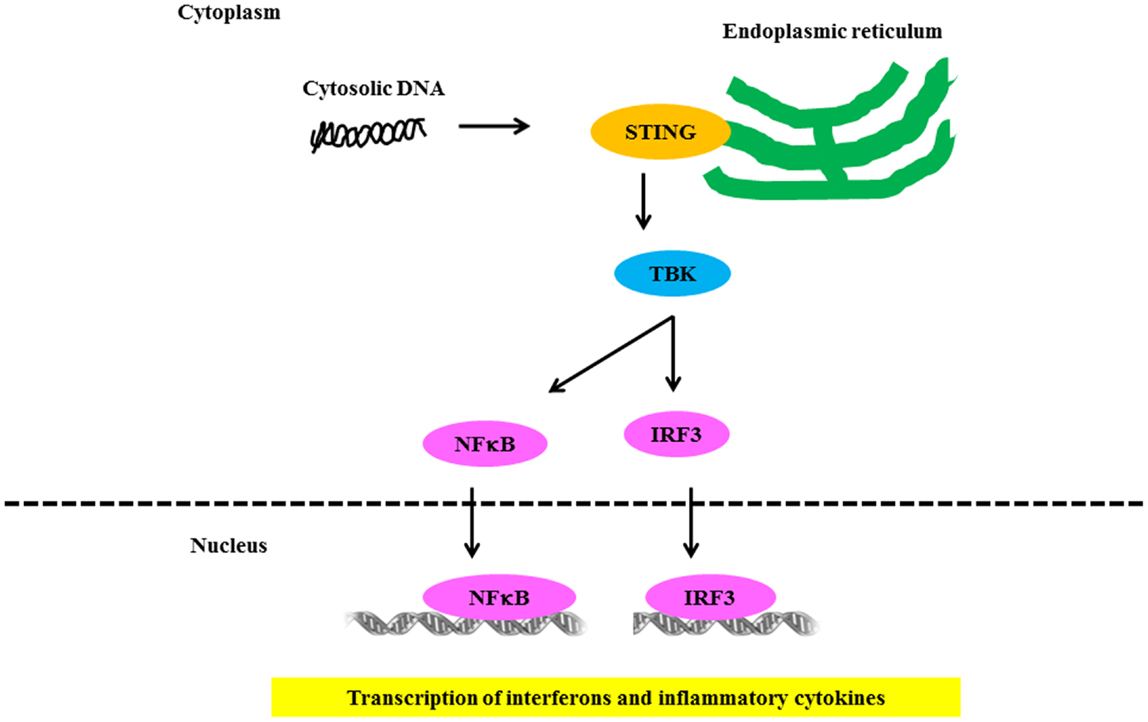
Figure 4. Signaling through NLR receptors in lupus. Intracellular DNA following microbial infection or phagocytosis of immune complexes can potentially trigger the assembly of NLRs. The nucleic acid-induced signaling pathway converges on the adaptor STING and the kinase TBK1, which phosphorylates IRF3 to mediate downstream signaling events leading to transcriptional induction of type 1 IFN and other inflammatory cytokines.
Other Cytosolic Nucleic Acid Sensors
DNAse-I, II, and III
Production of type I IFN and inflammatory cytokines are important for protecting the host against infections; however overstimulation of innate immune pathways can induce autoimmune disease (101). Normally, host nucleic acid is limited to the nucleus and mitochondria whereas; host cellular DNA/RNA sensors are localized in the cytoplasmic compartment. Thus, accidental activation of inflammatory cytokine pathways by host defense sensors is largely averted. However, faulty clearance of self-nuclear material from apoptotic/necrotic bodies can cause improper activation of cytokines including type I IFN production.
One level of self-defense is provided by cellular endonucleases, such as Dnase-I, Dnase-II, and Dnase-III/Trex-1, which are involved in the clearance of extracellular, lysosomal, and cytosolic DNA, respectively. Genetic deficiencies of Dnase-I have been identified in SLE patients (102), and Dnase I – deficient mice develop a lupus-like syndrome (103). Dnase-I defects lead to the accumulation of extracellular DNA produced by apoptotic and necrotic cells, which is immunogenic and can lead to type I IFN production (101, 104). Dnase-II is expressed in lysosomes, where it degrades DNA from engulfed apoptotic/necrotic cells (105). Dnase-II knockout mice are embryonically lethal. However, they are viable on the IFNR1 knockout background, indicating that type I IFN mediates the lethality of Dnase-II genetic deficiency (101, 106). This finding supports the concept that inefficient nucleic acid degradation promotes type I IFN excess and subsequent SLE disease. Dnase-III is another nuclease that is normally involved in the clearance of cell-intrinsic ssDNA (107, 108). DNAse-III is 3′-5′ exonuclease and is localized to the endoplasmic reticulum. In the absence of DNAse-III, there is an accumulation ∼60-bp ssDNA, believed to be produced during replication, which leads to the activation of ATM-dependent DNA-damage associated checkpoint pathways (109). Stetson et al. (110) revealed a role for DNAse-III in preventing cell-intrinsic initiation of autoimmunity. Trex-1 substrates are ssDNA, which are either the by-products of replication and/or reverse transcribed from endogenous retroelements. Loss of function mutations in the human DNAse-III gene cause Aicardi–Goutieres Syndrome (AGS) (111, 112). Different rare DNAse-III mutations also cause monogenic chilblain lupus, and common genetic variations in DNAse-III have also been associated with risk of SLE, suggesting that a common mechanism may underlie these disorders (113–115).
Other DNA and RNA Sensors
DNA-dependent activator of IRFs (DAI) is another cytoplasmic DNA sensor capable of activating IRF-3 and NF-κB, resulting in type I IFN production. DAI interacts directly with dsDNA in vitro and this interaction in turn enhances DAI association with IRF-3. DAI-induced IRF-3 phosphorylation is dependent on TBK1 (47, 116). Recently, Zhang et al. (117) reported that DAI expression is predominantly increased in SLE patients as well as in activated lymphocyte-derived self-apoptotic DNA (ALD-DNA)-induced lupus mice. ALD-DNA could induce the dimerization/oligomerization of DAI and activate DAI signaling pathways via regulating calcium signaling, thus resulting in aberrant macrophage activation and lupus nephritis, implying the possible mechanisms for the recognition and regulation of ALD-DNA-induced pathological macrophage activation in the context of SLE disease (117).
Recently, Kondo et al. (118) identified MRE11 as a sensor for exogenous dsDNA, which is required for STING trafficking and type I IFN induction. The report reveals that MRE11 contributes to recognition of a broad spectrum of dsDNA and MRE11-mediated intracellular DNA recognition is to respond to damaged host cells, rather than defense against foreign pathogens (118). DDX41 is another DExD/H-box helicase that can interact with synthetic dsDNA through the DEAD domain in vitro and DDX41 is required for DNA-dependent induction of type I IFN in myeloid DCs through a pathway dependent on STING and TBK1 (119).
Found in the cytoplasm, RNA polymerase III is known to transcribe AT-rich DNA into dsRNA transcripts characterized by uncapped 5′-triphosphate moieties. This can act as a ligand for RIG-I. Subsequently, RIG-I signals via IPS-1 to induce the expression of type I IFN and other cytokines (72, 120). Ku80 is an abundant nuclear protein that is known to bind dsDNA with high affinity.
A recent study (121) identified Ku70, as the newest member of the cytosolic DNA-sensing machinery with in IFN production. Ku70 was identified as a DNA-binding protein in HEK-293 cells by DNA-affinity purification followed by mass spectrometry. Notably, Ku70 is involved in the production of type III IFN (λ1), but not type I IFN (α or β) in response to a variety of transfected DNA (>500 bp) in HEK-293 (121). It seems likely that we will continue to identify additional DNA and RNA sensors, and that some of these novel mediators will also play a role in SLE pathogenesis.
Conclusion
In recent years, there has been tremendous progress in understanding how cells recognize and respond to microbial threats. Many DNA and RNA sensors have been identified that are dedicated to detection and elimination of microbial infection and clearing cellular damage. Sometimes these beneficial immune responses lose their fidelity and thus contribute to pathogenesis of autoimmune diseases. It is striking that many of the classical components of these pathways have been genetically associated with risk of SLE. This emphasizes the primary importance of nucleic acid handling and innate immune sensors in the pathogenesis of SLE. In SLE, it seems likely that stimulation of these pathways occurs via the combined contribution of microbial nucleic acids as well as self-tissue-derived stimuli. Work from our group and others supports a model in which immune complexes containing nucleic acid and free nucleic acid are a micro-environmental factor that cooperates with genetic variation in the nucleic acid sensing pathways to produce immune system dysregulation and risk of SLE (62). Understanding the molecular mechanisms of how the innate nucleic acid recognition system is dsyregulated in SLE will suggest new therapeutic avenues directed toward the inhibition of nucleic acid recognition by their sensors, downstream signaling events, and inhibition of end-stage mediators. This will lead to the new era of molecular medicine for the treatment of intractable autoimmune diseases like SLE.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Timothy B. Niewold – Research grants from the NIH (R01 AR060861, K08 AI083790, and Clinical Research Loan Repayment AI071651).
References
1. Schwartzman-Morris J, Putterman C. Gender differences in the pathogenesis and outcome of lupus and of lupus nephritis. Clin Dev Immunol (2012) 2012:604892. doi: 10.1155/2012/604892
2. Weckerle CE, Niewold TB. The unexplained female predominance of systemic lupus erythematosus: clues from genetic and cytokine studies. Clin Rev Allergy Immunol (2011) 40(1):42–9. doi:10.1007/s12016-009-8192-4
3. Moser KL, Kelly JA, Lessard CJ, Harley JB. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun (2009) 10(5):373–9. doi:10.1038/gene.2009.39
4. Kariuki SN, Niewold TB. Genetic regulation of serum cytokines in systemic lupus erythematosus. Transl Res (2010) 155(3):109–17. doi:10.1016/j.trsl.2009.08.012
5. Doria A, Canova M, Tonon M, Zen M, Rampudda E, Bassi N, et al. Infections as triggers and complications of systemic lupus erythematosus. Autoimmun Rev (2008) 8(1):24–8. doi:10.1016/j.autrev.2008.07.019
6. Zandman-Goddard G, Shoenfeld Y. Infections and SLE. Autoimmunity (2005) 38(7):473–85. doi:10.1080/08916930500285352
7. Niewold TB. Interferon alpha as a primary pathogenic factor in human lupus. J Interferon Cytokine Res (2011) 31(12):887–92. doi:10.1089/jir.2011.0071
8. Tan EM. Antinuclear antibodies: diagnostic markers for autoimmune diseases and probes for cell biology. Adv Immunol (1989) 44:93–151. doi:10.1016/S0065-2776(08)60641-0
9. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med (2008) 358(9):929–39. doi:10.1056/NEJMra071297
10. Poole BD, Scofield RH, Harley JB, James JA. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity (2006) 39(1):63–70. doi:10.1080/08916930500484849
11. Weckerle CE, Franek BS, Kelly JA, Kumabe M, Mikolaitis RA, Green SL, et al. Network analysis of associations between serum interferon-alpha activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum (2011) 63(4):1044–53. doi:10.1002/art.30187
12. Niewold TB, Swedler WI. Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin Rheumatol (2005) 24(2):178–81. doi:10.1007/s10067-004-1024-2
13. Ronnblom LE, Alm GV, Oberg KE. Possible induction of systemic lupus erythematosus by interferon-alpha treatment in a patient with a malignant carcinoid tumour. J Intern Med (1990) 227(3):207–10. doi:10.1111/j.1365-2796.1990.tb00144.x
14. Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun (2007) 8:492–502. doi:10.1038/sj.gene.6364408
15. Kariuki SN, Franek BS, Kumar AA, Arrington J, Mikolaitis RA, Utset TO, et al. Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Res Ther (2010) 12(4):R151. doi:10.1186/ar3101
16. Koldobskaya Y, Ko K, Kumar AA, Agik S, Arrington J, Kariuki SN, et al. Gene-expression-guided selection of candidate loci and molecular phenotype analyses enhance genetic discovery in systemic lupus erythematosus. Clin Dev Immunol (2012) 2012:682018. doi:10.1155/2012/682018
17. Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med (2003) 349(16):1526–33. doi:10.1056/NEJMoa021933
18. Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med (1994) 179(4):1317–30. doi:10.1084/jem.179.4.1317
19. Crow MK. Developments in the clinical understanding of lupus. Arthritis Res Ther (2009) 11(5):245. doi:10.1186/ar2762
20. Bouts YM, Wolthuis DF, Dirkx MF, Pieterse E, Simons EM, van Boekel AM, et al. Apoptosis and NET formation in the pathogenesis of SLE. Autoimmunity (2012) 45(8):597–601. doi:10.3109/08916934.2012.719953
21. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3(73):73ra20. doi:10.1126/scitranslmed.3001201
22. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med (2011) 3(73):73ra19. doi:10.1126/scitranslmed.3001180
23. Sangaletti S, Tripodo C, Chiodoni C, Guarnotta C, Cappetti B, Casalini P, et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood (2012) 120(15):3007–18. doi:10.1182/blood-2012-03-416156
24. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med (2009) 15(6):623–5. doi:10.1038/nm.1959
25. Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol (2012) 188(7):3522–31. doi:10.4049/jimmunol.1102404
26. Liu CL, Tangsombatvisit S, Rosenberg JM, Mandelbaum G, Gillespie EC, Gozani OP, et al. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res Ther (2012) 14(1):R25. doi:10.1186/ar3933
27. Kontaki E, Boumpas DT. Innate immunity in systemic lupus erythematosus: sensing endogenous nucleic acids. J Autoimmun (2010) 35(3):206–11. doi:10.1016/j.jaut.2010.06.009
28. Papadimitraki ED, Bertsias GK, Boumpas DT. Toll like receptors and autoimmunity: a critical appraisal. J Autoimmun (2007) 29(4):310–8. doi:10.1016/j.jaut.2007.09.001
29. Horton CG, Pan ZJ, Farris AD. Targeting Toll-like receptors for treatment of SLE. Mediators Inflamm (2010) 2010:498980. doi:10.1155/2010/498980
30. Moresco EM, LaVine D, Beutler B. Toll-like receptors. Curr Biol (2011) 21(13):R488–93. doi:10.1016/j.cub.2011.05.039
31. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature (2002) 416(6881):603–7. doi:10.1038/416603a
32. Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, et al. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci U S A (2009) 106(29):12061–6. doi:10.1073/pnas.0905441106
33. Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol (2007) 8(5):487–96. doi:10.1038/ni1457
34. Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med (2005) 202(9):1171–7. doi:10.1084/jem.20050630
35. Papadimitraki ED, Choulaki C, Koutala E, Bertsias G, Tsatsanis C, Gergianaki I, et al. Expansion of toll-like receptor 9-expressing B cells in active systemic lupus erythematosus: implications for the induction and maintenance of the autoimmune process. Arthritis Rheum (2006) 54(11):3601–11. doi:10.1002/art.22197
36. Nakou M, Knowlton N, Frank MB, Bertsias G, Osban J, Sandel CE, et al. Gene expression in systemic lupus erythematosus: bone marrow analysis differentiates active from inactive disease and reveals apoptosis and granulopoiesis signatures. Arthritis Rheum (2008) 58(11):3541–9. doi:10.1002/art.23961
37. Kawai T, Akira S. TLR signaling. Cell Death Differ (2006) 13(5):816–25. doi:10.1038/sj.cdd.4401850
38. Kenny EF, O’Neill LA. Signalling adaptors used by Toll-like receptors: an update. Cytokine (2008) 43(3):342–9. doi:10.1016/j.cyto.2008.07.010
39. Kawasaki T, Kawai T, Akira S. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity. Immunol Rev (2011) 243(1):61–73. doi:10.1111/j.1600-065X.2011.01048.x
40. O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol (2007) 7(5):353–64. doi:10.1038/nri2079
41. Markin CJ, Saltibus LF, Spyracopoulos L. Dynamics of the RING domain from human TRAF6 by 15N NMR spectroscopy: implications for biological function. Biochemistry (2008) 47(38):10010–7. doi:10.1021/bi800252x
42. Cham CM, Ko K, Niewold TB. Interferon regulatory factor 5 in the pathogenesis of systemic lupus erythematosus. Clin Dev Immunol (2012) 2012:780436. doi:10.1155/2012/780436
43. Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol (2004) 5(10):1061–8. doi:10.1038/ni1118
44. Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol (2001) 19:623–55. doi:10.1146/annurev.immunol.19.1.623
45. Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature (2005) 434(7034):772–7. doi:10.1038/nature03464
46. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science (2003) 301(5633):640–3. doi:10.1126/science.1087262
47. McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci U S A (2004) 101(1):233–8. doi:10.1073/pnas.2237236100
48. Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci (2008) 1143:1–20. doi:10.1196/annals.1443.020
49. Ko K, Franek BS, Marion M, Kaufman KM, Langefeld CD, Harley JB, et al. Genetic ancestry, serum interferon-alpha activity, and autoantibodies in systemic lupus erythematosus. J Rheumatol (2012) 39(6):1238–40. doi:10.3899/jrheum.111467
50. Niewold TB, Clark DN, Salloum R, Poole BD. Interferon alpha in systemic lupus erythematosus. J Biomed Biotechnol (2010) 2010:948364. doi:10.1155/2010/948364
51. Ioannou Y, Isenberg DA. Current evidence for the induction of autoimmune rheumatic manifestations by cytokine therapy. Arthritis Rheum (2000) 43(7):1431–42. doi:10.1002/1529-0131(200007)43:7<1431::AID-ANR3>3.0.CO;2-E
52. Visentini M, Conti V, Cagliuso M, Tinti F, Siciliano G, Trombetta AC, et al. Regression of systemic lupus erythematosus after development of an acquired toll-like receptor signaling defect and antibody deficiency. Arthritis Rheum (2009) 60(9):2767–71. doi:10.1002/art.24760
53. Deng Y, Zhao J, Sakurai D, Kaufman KM, Edberg JC, Kimberly RP, et al. MicroRNA-3148 modulates allelic expression of toll-like receptor 7 variant associated with systemic lupus erythematosus. PLoS Genet (2013) 9(2):e1003336. doi:10.1371/journal.pgen.1003336
54. Sigurdsson S, Nordmark G, Göring HH, Lindroos K, Wiman AC, Sturfelt G, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet (2005) 76(3):528–37. doi:10.1086/428480
55. International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN)Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet (2008) 40(2):204–10. doi:10.1038/ng.81
56. Kaufman KM, Zhao J, Kelly JA, Hughes T, Adler A, Sanchez E, et al. Fine mapping of Xq28: both MECP2 and IRAK1 contribute to risk for systemic lupus erythematosus in multiple ancestral groups. Ann Rheum Dis (2013) 72(3):437–44. doi:10.1136/annrheumdis-2012-201851
57. Lodolce JP, Kolodziej LE, Rhee L, Kariuki SN, Franek BS, McGreal NM, et al. African-derived genetic polymorphisms in TNFAIP3 mediate risk for autoimmunity. J Immunol (2010) 184(12):7001–9. doi:10.4049/jimmunol.1000324
58. Adrianto I, Wen F, Templeton A, Wiley G, King JB, Lessard CJ, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet (2011) 43(3):253–8. doi:10.1038/ng.766
59. Lessard CJ, Adrianto I, Ice JA, Wiley GB, Kelly JA, Glenn SB, et al. Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. Am J Hum Genet (2012) 90(4):648–60. doi:10.1016/j.ajhg.2012.02.023
60. Salloum R, Niewold TB. Interferon regulatory factors in human lupus pathogenesis. Transl Res (2011) 157(6):326–31. doi:10.1016/j.trsl.2011.01.006
61. Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum (2008) 58(8):2481–7. doi:10.1002/art.23613
62. Niewold TB, Kelly JA, Kariuki SN, Franek BS, Kumar AA, Kaufman KM, et al. IRF5 haplotypes demonstrate diverse serological associations which predict serum interferon alpha activity and explain the majority of the genetic association with systemic lupus erythematosus. Ann Rheum Dis (2012) 71(3):463–8. doi:10.1136/annrheumdis-2011-200463
63. Salloum R, Franek BS, Kariuki SN, Rhee L, Mikolaitis RA, Jolly M, et al. Genetic variation at the IRF7/PHRF1 locus is associated with autoantibody profile and serum interferon-alpha activity in lupus patients. Arthritis Rheum (2010) 62(2):553–61. doi:10.1002/art.27182
64. Chrabot BS, Kariuki SN, Zervou MI, Feng X, Arrington J, Jolly M, et al. Genetic variation near IRF8 is associated with serologic and cytokine profiles in systemic lupus erythematosus and multiple sclerosis. Genes Immun (2013). doi:10.1038/gene.2013.42. [Epub ahead of print].
65. Niewold TB. Interferon alpha-induced lupus: proof of principle. J Clin Rheumatol (2008) 14(3):131–2. doi:10.1097/RHU.0b013e318177627d
66. Cherian TS, Kariuki SN, Franek BS, Buyon JP, Clancy RM, Niewold TB. Brief Report: IRF5 systemic lupus erythematosus risk haplotype is associated with asymptomatic serologic autoimmunity and progression to clinical autoimmunity in mothers of children with neonatal lupus. Arthritis Rheum (2012) 64(10):3383–7. doi:10.1002/art.34571
67. Meylan E, Tschopp J. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol Cell (2006) 22(5):561–9. doi:10.1016/j.molcel.2006.05.012
68. Cui S, Eisenächer K, Kirchhofer A, Brzózka K, Lammens A, Lammens K, et al. The C-terminal regulatory domain is the RNA 5’-triphosphate sensor of RIG-I. Mol Cell (2008) 29(2):169–79. doi:10.1016/j.molcel.2007.10.032
69. Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, et al. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol Cell (2008) 29(4):428–40. doi:10.1016/j.molcel.2007.11.028
70. Yoneyama M, Fujita T. Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity (2008) 29(2):178–81. doi:10.1016/j.immuni.2008.07.009
71. Yoneyama M, Fujita T. [Non-self RNA-sensing mechanism of RIG-I RNA helicase]. Seikagaku (2008) 80(9):838–43.
72. Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H, et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science (2006) 314(5801):994–7. doi:10.1126/science.1132505
73. Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science (2006) 314(5801):997–1001. doi:10.1126/science.1132998
74. Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med (2008) 205(7):1601–10. doi:10.1084/jem.20080091
75. Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, et al. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol (2007) 178(10):6444–55.
76. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol (2005) 6(10):981–8. doi:10.1038/ni1243
77. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell (2005) 122(5):669–82. doi:10.1016/j.cell.2005.08.012
78. Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med (2006) 203(7):1795–803. doi:10.1084/jem.20060792
79. Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell (2005) 19(6):727–40. doi:10.1016/j.molcel.2005.08.014
80. Saha SK, Pietras EM, He JQ, Kang JR, Liu SY, Oganesyan G, et al. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J (2006) 25(14):3257–63. doi:10.1038/sj.emboj.7601220
81. Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science (2003) 300(5622):1148–51. doi:10.1126/science.1081315
82. Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, et al. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med (2003) 198(7):1043–55. doi:10.1084/jem.20031023
83. Balachandran S, Thomas E, Barber GN. A FADD-dependent innate immune mechanism in mammalian cells. Nature (2004) 432(7015):401–5. doi:10.1038/nature03124
84. Takahashi K, Kawai T, Kumar H, Sato S, Yonehara S, Akira S. Roles of caspase-8 and caspase-10 in innate immune responses to double-stranded RNA. J Immunol (2006) 176(8):4520–4.
85. Molineros JE, Maiti AK, Sun C, Looger LL, Han S, Kim-Howard X, et al. Admixture mapping in lupus identifies multiple functional variants within IFIH1 associated with apoptosis, inflammation, and autoantibody production. PLoS Genet (2013) 9(2):e1003222. doi:10.1371/journal.pgen.1003222
86. Pothlichet J, Niewold TB, Vitour D, Solhonne B, Crow MK, Si-Tahar M. A loss-of-function variant of the antiviral molecule MAVS is associated with a subset of systemic lupus patients. EMBO Mol Med (2011) 3(3):142–52. doi:10.1002/emmm.201000120
87. Atianand MK, Fitzgerald KA. Molecular basis of DNA recognition in the immune system. J Immunol (2013) 190(5):1911–8. doi:10.4049/jimmunol.1203162
88. Gracie JA, Robertson SE, McInnes IB. Interleukin-18. J Leukoc Biol (2003) 73(2):213–24. doi:10.1189/jlb.0602313
89. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell (2002) 10(2):417–26. doi:10.1016/S1097-2765(02)00599-3
90. Barlan AU, Danthi P, Wiethoff CM. Lysosomal localization and mechanism of membrane penetration influence nonenveloped virus activation of the NLRP3 inflammasome. Virology (2011) 412(2):306–14. doi:10.1016/j.virol.2011.01.019
91. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med (2009) 206(1):79–87. doi:10.1084/jem.20081667
92. Kahlenberg JM, Carmona-Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol (2013) 190(3):1217–26. doi:10.4049/jimmunol.1202388
93. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458(7237):514–8. doi:10.1038/nature07725
94. Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol (2010) 11(5):395–402. doi:10.1038/ni.1864
95. Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol (2010) 11(5):385–93. doi:10.1038/ni.1859
96. Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe (2011) 9(5):363–75. doi:10.1016/j.chom.2011.04.008
97. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455(7213):674–8. doi:10.1038/nature07317
98. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity (2008) 29(4):538–50. doi:10.1016/j.immuni.2008.09.003
99. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature (2009) 461(7265):788–92. doi:10.1038/nature08476
100. Choubey D. Interferon-inducible Ifi200-family genes as modifiers of lupus susceptibility. Immunol Lett (2012) 147(1–2):10–7. doi:10.1016/j.imlet.2012.07.003
101. Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol (2005) 6(1):49–56. doi:10.1038/ni1146
102. Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, et al. Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet (2001) 28(4):313–4. doi:10.1038/91070
103. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Möröy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet (2000) 25(2):177–81. doi:10.1038/76032
104. Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature (2006) 443(7114):998–1002. doi:10.1038/nature05245
105. Evans CJ, Aguilera RJ. DNase II: genes, enzymes and function. Gene (2003) 322:1–15. doi:10.1016/j.gene.2003.08.022
106. Kawane K, Fukuyama H, Yoshida H, Nagase H, Ohsawa Y, Uchiyama Y, et al. Impaired thymic development in mouse embryos deficient in apoptotic DNA degradation. Nat Immunol (2003) 4(2):138–44. doi:10.1038/ni881
107. Mazur DJ, Perrino FW. Excision of 3’ termini by the Trex1 and TREX2 3’ →5’ exonucleases. Characterization of the recombinant proteins. J Biol Chem (2001a) 276(20):17022–9.
108. Mazur DJ, Perrino FW. Structure and expression of the TREX1 and TREX2 3’– >5’ exonuclease genes. J Biol Chem (2001b) 276(18):14718–27. doi:10.1074/jbc.M010051200
109. Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell (2007) 131(5):873–86. doi:10.1016/j.cell.2007.10.017
110. Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell (2008) 134(4):587–98. doi:10.1016/j.cell.2008.06.032
111. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet (2006) 38(8):917–20. doi:10.1038/ng1845
112. Crow YJ, Rehwinkel J. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum Mol Genet (2009) 18(R2):R130–6. doi:10.1093/hmg/ddp293
113. Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet (2007) 39(9):1065–7. doi:10.1038/ng2091
114. Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet (2007) 80(4):811–5. doi:10.1086/513443
115. Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun (2011) 12(4):270–9. doi:10.1038/gene.2010.73
116. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature (2007) 448(7152):501–5. doi:10.1038/nature06013
117. Zhang W, Zhou Q, Xu W, Cai Y, Yin Z, Gao X, et al. DNA-dependent activator of interferon-regulatory factors (DAI) promotes lupus nephritis by activating the calcium pathway. J Biol Chem (2013) 288(19):13534–50. doi:10.1074/jbc.M113.457218
118. Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, Barber GN, et al. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc Natl Acad Sci U S A (2013) 110(8):2969–74. doi:10.1073/pnas.1222694110
119. Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol (2011) 12(10):959–65. doi:10.1038/ni.2091
120. Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell (2009) 138(3):576–91. doi:10.1016/j.cell.2009.06.015
Keywords: systemic lupus erythematosus, nucleic acid sensor, type 1 interferon, TLR, DNA, RNA
Citation: Shrivastav M and Niewold TB (2013) Nucleic acid sensors and type I interferon production in systemic lupus erythematosus. Front. Immunol. 4:319. doi: 10.3389/fimmu.2013.00319
Received: 22 August 2013; Accepted: 20 September 2013;
Published online: 07 October 2013.
Edited by:
Fabrizio Mattei, Istituto Superiore di Sanità, ItalyCopyright: © 2013 Shrivastav and Niewold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timothy B. Niewold, Department of Immunology, Division of Rheumatology, Mayo Clinic, 200 1st Street SW, Guggenheim Building 3-42, Rochester, MN 55905, USA e-mail: niewold.timothy@mayo.edu