
- Department of Molecular Biology and Biochemistry, The Institute for Immunology, University of California Irvine, Irvine, CA, USA
As a vital second messenger in the activation of lymphocytes, the divalent cation Ca2+ plays numerous roles in adaptive immune responses. Importantly, Ca2+ signaling is essential for T cell activation, tolerance of self-antigens, and homeostasis. Supporting the essential role of Ca2+ signaling in T cell biology, the Ca2+ regulated protein phosphatase calcineurin is a key target of pharmacologic inhibition for preventing allograft rejection and for autoimmune therapy. Recent studies have highlighted the unique role of Stim1 and Orai1/2 proteins in the regulation of store-operated/calcium release activated calcium (CRAC) channels in the context of T cells. While Ca2+ is known to modulate T cell activation via effects on calcineurin and its target, nuclear factor of activated T cells (NFAT), this second messenger also regulates other pathways, including protein kinase C, calmodulin kinases, and cytoskeletal proteins. Ca2+ also modulates the unique metabolic changes that occur during in distinct T cell stages and subsets. Herein, we discuss the means by which Ca2+ mobilization modulates cellular metabolism following T cell receptor ligation. Further, we highlight the crosstalk between mitochondrial metabolism, reactive oxygen species (ROS) generation, and CRAC channel activity. As a target of mitochondrial ROS and Ca2+ regulation, we describe the involvement of the serine/threonine kinase DRAK2 in the context of these processes. Given the important roles for Ca2+ dependent signaling and cellular metabolism in adaptive immune responses, the crosstalk between these pathways is likely to be important for the regulation of T cell activation, tolerance, and homeostasis.
Introduction
The adaptive immune system exhibits a wide potential for responding to infectious agents encountered throughout a lifetime. Notably, T cells play a critical role in mounting an appropriate response through the activation of their T cell receptor (TCR) upon the recognition of specific antigen presented through the major histocompatibility complex (MHC). This foreign peptide: MHC complex activates a T cell with specificity for the foreign antigen, culminating in proliferation, and differentiation into effector cells. In order to stimulate naïve T cells, ligation of the TCR by antigen must be accompanied by a costimulatory signal. Costimulation serves as a mechanism to modulate the strength of the TCR signal and promote higher gene expression of immunomodulatory cytokines like interleukin-2, as well as other factors that facilitate T cell proliferation and differentiation (1, 2). When a naïve T cell becomes activated, in the context of proper costimulation, it executes developmental reprograming characterized by rapid growth, proliferation, and acquisition of specialized effector functions. Given that this is an energetically demanding process, T cells undergo biochemical and biophysical reprograming to meet these requirements (3).
A key mediator of T cell signaling is the divalent cation calcium (Ca2+). Indeed, this second messenger is so important to T cell biology that its downstream signaling serves as a vital pharmacological target for the treatment of autoimmune disease and the prevention of chronic allograft rejection (4). As with many cell types, ligation of surface molecules that induce phospholipase C leads to mobilization of intracellular calcium, with depletion of Ca2+ from the endoplasmic reticulum (ER). However, it has long been known that for sustained signaling downstream of Ca2+ release, T cells bear special plasma membrane Ca2+ channels that promote the influx of extracellular Ca2+ (5). While beyond the scope of the present review, recent work in several laboratories has revealed that T cells and a select few other cell types, express plasma membrane channels that are activated in response to depletion of intracellular stores of Ca2+ (6). These calcium release activated calcium (CRAC) channels are activated by Stim1, an ER membrane protein that itself is induced upon depletion of Ca2+ stores in ER. Major efforts are currently underway to understand the biophysics of CRAC activation, and importantly, to determine if manipulation of CRAC signaling may be of therapeutic benefit.
In the present review, we examine the link between cellular metabolism and calcium signaling in the context of T cells. As described, Ca2+ modulates a variety of signaling cascades downstream of the TCR and costimulatory receptors, many of which impinge upon bioenergetic and biosynthetic pathways needed for T cell clonal expansion, differentiation, and homeostasis. First, we briefly outline several important control points that orchestrate cellular metabolic changes following TCR stimulation. Next, we discuss the crosstalk between mitochondrial metabolism and CRAC activity, describing the influence of the metabolic status of a given T cell on Ca2+ mobilization and signaling. Finally, the impact of the release of mitochondrial reactive oxygen species (ROS) on Ca2+ signaling is considered, with a focus on the immunoregulatory serine/threonine kinase DRAK2 in this process.
Modulation of Cellular Metabolism Following T Cell Activation
Although one might consider metabolic regulation a mundane housekeeping function of cells, it is becoming apparent that specific metabolic programs are induced in distinct T cell subsets and developmental stages. For example, the metabolic status of naïve T cells is significantly different from their activated counterparts. Naïve T cells actively maintain a quiescent state through engagement of both intracellular signaling pathways and cell extrinsic signals (7), resulting in efficient use of available energy sources (8). These quiescent lymphocytes maintain a catabolic state and do not actively take up nutrients, nor do they secrete effector cytokines (9). Once committed to a T cell fate in the thymus, cellular metabolism plays a vital role in the development and proliferation of double negative (DN) thymocyte precursors (11). In DN thymocytes, both interleukin-7 (IL-7) and Notch prevent cell death and promote pre-T cell development via activation of glycolytic metabolism and the Akt signaling pathway (12). Following release from the thymus, antigenic stimulation of mature T cells facilitates metabolic changes that support various bioenergetically dependent processes needed for rapid clonal expansion (9). It is proposed that T cells must shift from catabolic to anabolic metabolism in order to rapidly proliferate, likely allowing them to respond to microbial infection [(10) p. 2313]. Indeed, CD8 T cells have the capacity to divide once every 4–6 h (13), a process that is highly energy dependent. Naïve T cells appear to favor energetically efficient processes such as the tricarboxylic acid (TCA) cycle linked to the generation of ATP via oxidative phosphorylation (OXPHOS), which results in roughly 30–32 ATP units per molecule of glucose. In contrast, antigenically stimulated T cells rapidly shift to a dependence on aerobic glycolysis, a less efficient process that yields only two ATP units per molecule of glucose (14, 15). Activated T cells that fail to switch metabolic processes are rendered anergic or undergo apoptosis (16), likely a consequence of failing to accommodate the specific bioenergetic demands of proliferation and differentiation (17, 18). Thus, it is clear that the metabolic status must match the needs of distinct T cell subsets and developmental stages in order for appropriate immune responses to be generated.
With regard to the intracellular signaling involved in metabolic regulation, it has long been appreciated that the mechanistic target of rapamycin (mTOR) has a critical role in T cell activation and metabolism (19). In the context of metabolism, mTOR serves to integrate nutrient and immune signals, including the availability of amino acids and oxygen, as well as the presence of extracellular growth factors. mTOR then acts as an effector to modulate downstream cellular metabolic processes needed to meet the demands of the cell upon stimulation (20). Such processes include protein translational initiation via phosphorylation of S6K1 and eIF4E (21), and lipid biosynthesis through the transcription factor SREBP1 and the nuclear protein receptor PPARγ (22). It is crucial, however, that mTOR does not become prematurely activated until T cells are antigenically stimulated, as the quiescent metabolic state of resting T cells appears to be important for their homeostatic proliferation (23). As detailed later, the energy sensing protein kinase AMPK acts as a master regulator of the metabolic status in resting T cells. Induced by high levels of AMP, AMPK influences mTOR activity through the tumor suppressor tuberous sclerosis complex (24). Comprised of TSC1 and TSC2, the tuberous sclerosis complex itself negatively regulates mTOR activation (25) and is crucial to maintaining homeostatic proliferation of naïve T cells. Supporting this, Yang and colleagues observed that TSC1 deficient naïve T cells possess hyperactive mTOR activity, a resulting loss in quiescence, and a predisposition to undergo apoptosis (26). Though naïve T cells do not initially require mTOR for TCR induced activation, its absence by genetic deletion in mouse CD4 T cells yields a skewed differentiation toward induced T regulatory cells over other effector T cell subsets (27). In addition, mTOR has a central role in the regulation of both activated and long-lived memory T cells as its genetic deletion or pharmacologic inactivation leads to diminished memory T cell activation and function (22, 28, 29).
Metabolic States in Distinct T Cell Subsets
Naïve T cells utilize efficient oxidative metabolism to maintain their quiescent state. Conversely, once T cells are stimulated by antigen, they must quickly expand their numbers to eliminate an antigenic challenge. This view is reminiscent of the Warburg hypothesis, in which heightened glycolysis observed in cancer cells is thought to allow for rapid tumor cell proliferation, particularly under limiting oxygen tension (30). Alternatively, it has been recently proposed that the switch to aerobic glycolysis is instead necessary to support effector T cell differentiation. Pearce and colleagues demonstrated that blockade of glycolysis prevented the expression of interferon gamma in activated T cells, but did not impair clonal expansion (31). Moreover, the rapid recall response observed in memory T cells, cells that often must respond quite rapidly to antigenic rechallenge, is supported by enhanced respiratory and glycolytic capacity (32, 33). It remains to be determined if this may reflect differential survival of unique subpopulations during clonal expansion. Regardless, distinct metabolic processes are clearly involved in providing for the energetic demands of unique T cell subpopulations, with fatty acid oxidation (FAO) and aerobic glycolysis playing significant roles.
Fatty Acid Oxidation
In resting T cells that circulate in the periphery, FAO is the default metabolic state, and the metabolism of these quiescent cells is characterized by a need for basal energy utilization over macromolecular biosynthesis (34). These naïve T cells utilize high energy yielding OXPHOS processes, involving β-oxidation of fatty acids and oxidation of glutamine and pyruvate via the TCA cycle (34). Both naïve CD4 and CD8 T cells also rely on intrinsic IL-7 to maintain homeostasis and quiescent survival (35). Loss of IL-7 receptor (IL-7R) signaling results in defective T cell physiology, characterized by decreased cell size/growth and markedly impaired cell survival (36). The involvement of IL-7R signaling in the maintenance of peripheral T cell homeostasis is complex; it has been recently found that IL-7R signaling must be intermittent and not continuous, as the latter results in sensitization of naïve T cells to cytokine induced cell death (37). While IL-7R signaling promotes survival of quiescent peripheral T cells, and is required for homeostatic proliferation under lymphopenic conditions, it alone is not sufficient to induce naïve T cell activation and the metabolic changes associated with this (38). Instead, with minimal mTOR activity, resting T cells utilize other signaling factors to regulate metabolic pathways. These factors include the nuclear receptors PPARα and PPARγ, both of which regulate fatty acid metabolism and inhibit activation upon TCR engagement (12, 39).
While an increase in glycolysis generally is observed in activated T cells, this is not always the case. Induced regulatory T cells (iTreg), differentiated from peripheral CD4+ T cells, are a unique subset of CD4+ T cells that suppress effector T cells and are vital to immune peripheral tolerance (40). Following a lag phase heavily dependent on glycolysis and glutaminolysis (28), T cells activated in the context of extracellular signals that favor iTreg differentiation (e.g., IL-2 and TGF-β) acquire a metabolic phenotype similar to naïve T cells (41). Relying on lipid oxidation as their primary source of energy, peripherally differentiated iTregs and their thymically derived nTreg counterparts have intermediate to low mTOR activity (18). The distinct metabolic profile of Treg can be replicated through in vitro culture with addition of glycolytic or mTOR inhibitors, such as 2-deoxyglucose or rapamycin, respectively (12). As with naïve T cells, PPARα and PPARγ are important for Tregs, serving as fatty acid sensors, and promoting Foxp3 expression in CD4+ T cells activated in the presence of TGF-β (42).
Fatty acid oxidation also plays a vital role in the maintenance of memory T cell pools. Following clearance of an acute viral infection, the antiviral CD8+ effector T cell pool is radically depleted, with a loss of 90–95% of virus specific CD8+ T cells (43). The surviving cells in turn become long-lived memory T cells (44), possessing unique metabolic characteristics when compared with effector cells (45). Memory CD8+ T cells must be able to withstand periods of both antigenic neglect and rapid antigen specific recall through the acquisition of increased spare respiratory capacity (SRC) through biogenesis of mitochondria and increased glycolytic flux (32). Thus, in contrast to their effector counterparts, these long-lived CD8+ T cells have significantly enhanced SRC. Memory CD8+ T cells share an analogous metabolic profile with resting T cells and Tregs, primarily engaging in FAO to maintain their survival and homeostasis (46). These metabolic processes are maintained by IL-15 signaling, which facilitates the biogenesis of mitochondria and expression of CPT1A, an enzyme responsible for the rate-limiting step of FAO (32).
Glycolysis
As noted above, activated T cells switch their metabolic programing to aerobic glycolysis upon antigenic stimulation (15, 47). This may seem counterproductive, as the effective ATP output per glucose molecule taken into the cell is roughly one fifteenth of the units generated via OXPHOS (12). Instead, it has been proposed that this switch is necessary to facilitate the rapid clonal expansion required to eliminate a microbial infection (45). Growth factor stimulation results in enhanced uptake of glucose through the upregulation of the glucose transporter Glut1 on the surface of cells, along with increased expression of the glycolytic enzymes hexokinase and phosphofructokinase (14), processes activated in T cells by TCR ligation (48). Costimulation through CD28 leads to the induction of Akt, thereby enhancing glycolytic activity in T cells (15), and the prevention of growth factor withdrawal induced cell death (17). Supporting a crucial role for Akt in promoting metabolic changes and the survival of activated T cells, ectopic expression of an active form of Akt leads to increased rates of glycolysis and T cell survival, even in the absence of CD28 signaling (49).
The AMP-dependent protein kinase AMPK serves a critical regulator of cellular metabolism, both in naïve and newly activated T cells (Figure 1A). In resting cells, a high ratio of AMP to ATP leads to elevated AMPK activity and diminished mTOR function. TCR engagement activates LKB1 and in parallel, increases intracellular Ca2+ stores, both leading to an increase in the expression of AMPK (50). LKB1 positively regulates AMPK (51, 52), the latter of which serves as an upstream regulator of TSC1 (52). As TSC1 inhibits mTOR activity in naïve T cells through the tuberous sclerosis complex, AMPK restricts the engagement of metabolic programs associated with clonal expansion. Deletion of the Tsc1 gene leads to metabolic alterations in T cells, most notably, increases in glucose uptake and glycolytic flux (53). AMPK activity is positively influenced by calcium mobilization via its impact on Ca2+ – calmodulin-dependent protein kinase kinase (CaMKK) activity (50, 54). Thus, while AMPK may be associated with a quiescent T cell state, the TCR induced increase of cytosolic Ca2+ enhances AMPK activity (Figure 1B). This effectively conserves ATP by inhibiting mTOR associated anabolic processes, and by promoting OXPHOS (50, 55). With time, the resulting increase in the ATP to AMP ratio leads to diminished AMPK activity, and downstream restriction of mTOR is overcome, allowing the T cell to shift its metabolism from FAO to OXPHOS (Figure 1C).
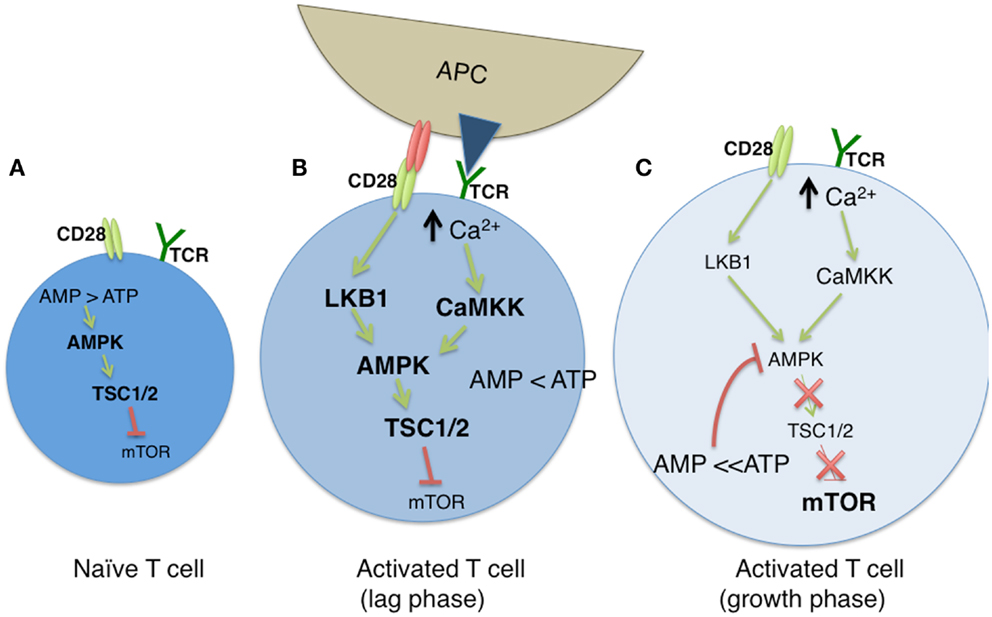
Figure 1. The modulation of cellular metabolism via T cell receptor (TCR) signaling. (A) In naïve T cells, fatty acid oxidation (FAO) is used to maintain basal cellular metabolism. In this quiescent state, the absence of TCR signaling leads to an elevated AMP to ATP ratio, resulting in sustained AMPK and diminished mTOR activities. (B) Upon TCR stimulation by antigen presenting cells (APC), along with CD28-mediated costimulation, glycolysis is greatly enhanced, leading to a diminished AMP:ATP ratio. During a lag phase that precedes T cell clonal expansion, LKB1 and CaMKK (itself induced by high cytosolic Ca2+ levels) promote AMPK function despite reduced AMP levels, and mTOR activity remains low via TSC1/2 mediated inhibition. (C) As T cells exit the lag phase, heightened levels of ATP block AMPK activity, which results in the loss of TSC1/2 activity. This allows for sustained mTOR function, and subsequent mTOR driven clonal expansion and cell growth.
Upon successful engagement of their TCR and the costimulation molecule CD28, the T cell begins to switch toward an activated metabolic program, utilizing aerobic glycolysis, the phosphate pentose pathway (PPP), and glutaminolysis (18). Two transcriptions factors that coordinate metabolic status following T cell activation are Myc and HIF1α (28, 56). Induction of Myc expression in activated T cells is largely responsible for metabolic reprograming at a global gene transcriptome level, as its acute deletion inhibited glycolysis, glutaminolysis, and provoked an overall failure in cell growth and proliferation (28). Myc is also found to intersect with the mTOR pathway, as its loss in activated T cells led to reduced expression and phosphorylation of the downstream mTOR targets S6 and 4E-BP (28). mTOR itself, specifically the mTOR complex 1 (mTORC1), is responsible for activating two key metabolic transcriptional targets, HIF1α and SREBP1/2. Specifically, HIF1α activates downstream targets involved in glycolysis and glucose uptake, while SREBP1/2 activates the PPP and lipid biosynthesis (57). However, while SREBP activity is essential for metabolic reprograming in activated T cells (58), HIF1α appears to play a more selective role in T cell subsets such as Th17 cells (56, 59). It is likely that HIF1β serves a compensatory role in HIF1α-deficient T cells, as loss of HIF1β leads to defective glycolysis, survival, trafficking, and function of CD8+ T cells (60).
Calcium Mobilization Following TCR Ligation
Having considered the modulation of cellular metabolism via TCR proximal signaling pathways, we now turn our attention to calcium mobilization, and how this signaling platform shapes the metabolic response in T cells. As described in detail in this compendium of reviews, TCR stimulation engages numerous signaling cascades, resulting in cellular activation and proliferation. One such molecule that is activated is phospholipase Cγ1 (PLCγ1), which mediates the hydrolysis of phosphatidylinositol-3,4-bisphospahte (PIP2) into inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). Ca2+ release from ER stores is triggered upon the binding of IP3 to inositol trisphosphate receptor (IP3R) found on the ER membrane. The two major ion channels that are known to participate in Ca2+ release from the ER in response to agonist stimulation are IP3Rs (61) and ryanodine receptors (RyRs) (62). Mobilization of intracellular Ca2+ triggers store-operated Ca2+ entry (SOCE), primarily through calcium release activated Ca2+ (CRAC) channels (63). The binding of antigen/MHC to the TCR complex results in the phosphorylation and activation of PLCγ1, which then hydrolyzes phospholipids at the plasma membrane to produce DAG and IP3. IP3 then binds to its respective receptors in the ER, triggering calcium release into the cytosol. This secondarily activates CRAC channels within the plasma membrane, causing a rapid influx of extracellular calcium that sustains high calcium levels required for T cell activation (64). Upon accumulation, cytosolic calcium binds calmodulin (CaM), inducing a conformational change in CaM that promotes its ability to interact with and activate the protein phosphatase calcineurin (65). Calcineurin dephosphorylates the cytoplasmic subunits (NFATc) of nuclear factor of activated T cells (NFAT) transcription complexes, exposing a nuclear localization sequence that results in their import into the nucleus (66).
Cytosolic calcium is a universal second messenger affecting a variety of cellular processes extending from short- to long-term responses in immune cells (67). Unlike other intracellular messengers, Ca2+ is neither synthesized nor metabolized. Rather, Ca2+ storage and release are carefully regulated by a series of channels and pumps that maintain precise Ca2+ concentrations within distinct cellular compartments. In lymphocytes, activation of immune receptors initiates a signaling cascade culminating in the depletion of intracellular Ca2+ stores. Upon depletion, store-operated calcium channels are activated, allowing Ca2+ to enter into the cell, primarily through CRAC channels. The increase in intracellular free calcium is essential for lymphocyte activation, since the transcription factor families NFAT, NF-κB, and AP-1 are all targets of Ca2+ mediated signaling (68). Remarkably, 75% of all activation-regulated genes in T cells show a dependence on Ca2+ influx (68). Supporting this, loss of Ca2+ mobilization dramatically reduces, and often prevents, T cell activation and proliferation (68–70). Below, we detail the regulation of intracellular calcium oscillation by CRAC channels and mitochondria, and describe how Ca2+ influences the generation of and response to cellular oxidants.
Store-Operated Ca2+ Channels
Calcium release activated calcium channels comprise a widespread and highly conserved Ca2+ entry pathway in cells such as lymphocytes, and are activated following the depletion of Ca2+ within the ER. This phenomenon is referred to as SOCE and CRAC activity is proportional to the amount of Ca2+ depletion within the ER. Over the last decade, the key components of the CRAC channel machinery were identified as STIM1, the ER Ca2+ sensor and Orai1/2, pore forming subunits of the CRAC channel (71). Depletion of Ca2+ within the ER is detected by the Ca2+ sensors STIM1 and STIM2, resulting in the activation of store-operated calcium channels. STIM1 and STIM2 are EF-hand containing single-pass transmembrane proteins with both EF hands capable of binding Ca2+. STIM2 is thought to have a lower affinity for Ca2+ since STIM1-deficient mouse T cells and fibroblasts bear a severe impairment in store-operated Ca2+ influx whereas a deficiency in STIM2 has a less dramatic effect (72).
The luminal EF-hand of STIM1 senses Ca2+ depletion within the ER and subsequently causes its oligomerization and translocation toward the plasma membrane (73). STIM1 binds through specialized ER-PM junctions, located within 25 nm of the plasma membrane, to a component of the CRAC channel known as Orai1 (74). Once activated, CRAC channels have a remarkably selective single channel conductance for Ca2+, and sensitive Ca2+ dependent feedback regulation (75). Association of STIM1 and Orai1 depends on the involvement of STIM1 with the phosphoinositides (PIs) of Orai1. Within STIM1, the cytosolic SOAR/CAD domain contains an alpha-helical domain necessary for Orai1 binding (76, 77). Deletion of the SOAR/CAD domain abolishes store depletion-induced clustering of Orai1 monomers and dramatically reduces SOCE, suggesting this domain is required for the assembly of the SOCE complex at ER-PM junctions (76, 77).
Mitochondrial Calcium Buffering and Homeostasis
Mitochondria are recognized as essential calcium signaling organelles. Through calcium buffering, mitochondria influence CRAC channel activity (78). With the ability to sense Ca2+ microdomains, mitochondria are able to translocate to the immunological synapse upon Ca2+ influx, leading to maximal Ca2+ uptake. The positioning of mitochondria correlates with the magnitude of local calcium entry via the reduction of Ca2+ dependent channel inactivation (79). By co-localizing near ER, mitochondria are better suited to buffer cytosolic Ca2+.
Isolated mitochondria take up Ca2+ when supported by exogenous electron transport chain (ETC) substrates (80). Although it has been well known that mitochondria are endowed with a complex array of Ca2+ transporters, the function of these transporters was not well appreciated until the early 1990s when Pozzan, Rizzuto and colleagues examined aequorin, a Ca2+ sensitive bioluminescent protein. They found that agonist-stimulated elevation of cytosolic free Ca2+ results in a rapid and transient increase in mitochondrial Ca2+, an increase blocked by pretreatment with a mitochondrial uncoupler (81). Prior to this discovery, it was believed that mitochondria failed to release much Ca2+ into the cytosol and had little effect on InsP3-mediated Ca2+ release. It has since become appreciated that mitochondrial matrix Ca2+ concentrations are crucial regulators of the Ca2+ dependent enzymes of the TCA cycle (82). Mitochondria also play a vital role in the gating of CRAC channels and do so through Ca2+ buffering (83).
Mitochondria act as Ca2+ buffers through uptake of Ca2+ released by IP3R on proximal ER membranes (Figure 2). Mitochondria accomplish this through the ability to detect changes in Ca2+ microdomains (84); Ca2+ uptake by mitochondria occurs by mitochondrial Ca2+ uniporter (MCU) activity across the inner mitochondrial membrane (85). This Ca2+ buffering is functionally significant as it modifies the Ca2+ dependent inactivation of IP3R, ultimately leading to greater Ca2+ mobilization. Mitochondria also deplete Ca2+ adjacent to the ER, resulting in less available Ca2+ for transport into the ER via SERCA pumps. Together, these events result in robust activation of CRAC channels (86, 87). It has been estimated that between 25 and 50% of the Ca2+ released from ER is taken up by mitochondria, dependent on cell type, and the Ca2+ release channel involved (88). This Ca2+ uptake influences mitochondrial metabolic processes, as three rate-limiting enzymes of the Krebs cycle are activated by a rise in Ca2+ concentration, subsequently causing an increase in mitochondrial ATP generation (89). These enzymes include pyruvate dehydrogenase (PDH), NAD+-isocitrate dehydrogenase (NAD-IDH), and 2-oxoglutarate dehydrogenase (82).
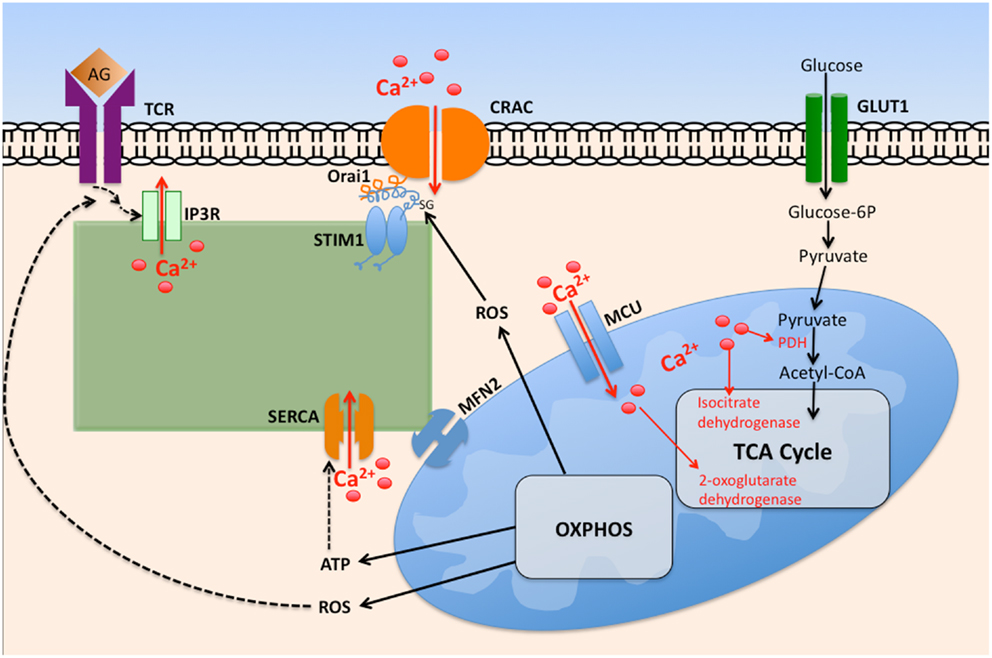
Figure 2. The interplay between intracellular calcium mobilization and cellular metabolism in T cells. Ligation of the T cell receptor (TCR) by antigen (Ag) promotes opening of IP3R and depletion of ER Ca2+ via IP3, itself generated by phospholipase Cγ1. Depletion of ER Ca2+ stores promotes STIM1 interaction with Orai1, promoting oligomerization of Orai monomers, thus forming CRAC channels. The resultant increase in cytosolic Ca2+ promotes its entry into the mitochondrial matrix via the mitochondrial calcium uniporter (MCU). Elevated matrix Ca2+ enhances the activity of key tricarboxylic acid cycle (TCA) enzymes, leading to elevated oxidative phosphorylation (OXPHOS), and ATP generation. Reactive oxygen species (ROS) byproducts of OXPHOS enhance TCR proximal signaling by inhibiting tyrosine phosphatase activity. Elevated cytosolic ROS governs CRAC activity by ROS mediated modification of cysteine residues exposed on Orai1.
In addition to influencing CRAC channel gating, mitochondria also influence STIM1 trafficking (90). Formation of STIM1 puncta below the plasma membrane is diminished, independently of STIM1 oligomerization, upon strong mitochondrial depolarization. This is accomplished by the mitochondrial protein mitofusin 2 (MFN2), an outer mitochondrial membrane GTPase that couples mitochondrial energetic status to STIM1 migration (90). In MFN2-deficient cells, mitochondrial depolarization fails to influence STIM1 trafficking and Ca2+ entry, suggesting that MFN2 confers sensitivity to mitochondrial depolarization. MFN2 assists in tethering mitochondria to the ER and is abundant in the contact sites between the two organelles, though the exact mechanism through which these two organelles interact remains unclear (91). It is speculated that the most likely function of the tethering is to allow mitochondria to localize proximal to Ca2+ released from ER. However, the question as to why STIM1 overexpression partially rescues Ca2+ entry in MFN2 overexpressing cells has yet to be answered.
While both ER and mitochondria serve as important intracellular reservoirs of Ca2+, mitochondria utilize functionally distinct Ca2+ transport mechanisms. Mitochondria possess a large electrochemical gradient across their inner membrane (ΔΨm), which acts as a driving force for the uptake of Ca2+ (92). Unlike with ER, mitochondrial Ca2+ release occurs via ion exchangers, and Ca2+ uptake is mediated via a selective ion channel (93). High calcium levels, in combination with other signals, have been known to trigger the opening of channels within the inner mitochondrial membrane, ultimately resulting in cell death (94). The combined action of these mechanisms allows for the rapid cycling of Ca2+ across the inner mitochondrial membrane (95).
Regulation of STIM1/Orai Signaling by Oxidative Stress
Numerous physiological processes, including proliferation, and cell death, are influenced by ROS (96, 97). To prevent oxidative stress, the cell must maintain a balance between superoxide generation, a byproduct of oxidative energy production, and rapid clearance of ROS. The latter is achieved through circulating antioxidants that act as ROS scavengers. Out of the ∼20 types of ROS, hydrogen peroxide (H2O2) appears to be the major contributor to oxidative damage (96). Of interest here, it has been found that oxidative stress regulates STIM1/Orai1 signaling (98–100). CRAC channels, under the action of STIM1, are stimulated by micromolar concentrations of H2O2 (98–100). Altered calcium signaling during oxidant stress is attributed to a reduction in the Ca2+ binding affinity of STIM1 due to a modification involving its cysteine 56, ultimately resulting in constitutive CRAC activation (100). Upon exposure to ROS, STIM1 becomes S-glutathionylated, thereby triggering its oligomerization and translocation to the plasma membrane (100). Alternatively, Grupe and colleagues demonstrated that H2O2 enhanced the CRAC current, ICRAC-mediated Ca2+ influx by activating IP3R independent of Orai1 (99). Regardless of the route of entry, both groups demonstrate that an increase an ROS leads to enhanced Ca2+ entry into the cell. While several studies support the hypothesis that ROS positively stimulate CRAC channel activity by triggering the oligomerization of STIM1, Bogeski and colleagues report that oxidation via H2O2 specifically blocks the activation of Orai1 channels in human T helper lymphocytes (98). It is speculated that the oxidation of the cysteine, found within the extracellular loop of Orai1, may lock the channel in the closed conformation. Nevertheless, cells with an SOCE deficiency are more susceptible to oxidative stress. Despite having an up-regulated basal antioxidant response, STIM1-deficient mouse embryonic fibroblasts experience an imbalance of ROS production and detoxification upon addition of exogenous oxidants (101). Henke and colleagues conclude that functional SOCE machinery is required to balance ROS production and the cellular antioxidant defense system. Collectively, these studies support the notion that the cellular oxidative stress response is influenced through a dynamic interplay between ROS balance and Ca2+ influx.
Oxidant Dependence of T Cell Activation
Oxidative stress is provoked by an imbalance between the production of mitochondrial superoxide and insufficient scavenging of ROS, ultimately resulting in a wide range of pathological conditions (102). The dynamics of ROS production and scavenging can be detected using several fluorescent dyes, including reduced forms of ethidium bromide (DHE) (103), fluorescein (DCFDA) (104, 105), rhodamine (DHR) (106), and hydroethidine (HE) (107). These dyes remain relatively non-fluorescent until they are oxidized by ROS, and their unquenched fluorescence correlates with increased oxidation. These intracellular dyes provide sensitive probes for multiple reactive species and allow for the detection of fluorescent products using fluorimetry and flow cytometry. In T cells, DCFDA, DHR, and DHE have been utilized to examine ROS production from a variety of stimuli including mitogens (108), viral and bacterial superantigen (109, 110), and TCR peptide agonists (111, 112). ROS generation has been observed following stimulation of T cells by other ligands including TGF-β, insulin, angiotensin II, and EGF (113–116). Collectively, these studies suggest that ROS function as secondary messengers necessary for complete T cell activation. Superoxide is generated from the mitochondrial ETC through complexes I, II, and III (117). Complexes I and II emit superoxide into the mitochondrial matrix, upon which superoxide dismutase 2 (SOD2) converts it into hydrogen peroxide (H2O2). In contrast, complex III emits superoxide into both the matrix and intermembrane spaces. Intermembrane-space superoxide is capable of reaching the cytosol through voltage-dependent anion channels (VDAC) without conversion into H2O2 (118, 119).
Recently, Sena et al. reported that blocking mitochondrial ROS, specifically generated at complex III, results in the inhibition of CD3/CD28 induced IL-2 expression. Additionally, extracellular calcium influx and subsequent Ca2+ uptake by mitochondria were both required for mitochondrial ROS production (120). These findings support the hypothesis that complex III produced mitochondrial ROS, a byproduct of mitochondrial metabolism, facilitate T cell activation and functionality. However, the mechanisms through which oxidants may regulate T cell responses upon stimulation remain poorly defined. Importantly, the timing and subcellular localization of ROS generation are likely of greater influence in T cell responses than overall redox balance (102, 121, 122). ROS clearly play key roles in modulating T cell activation and differentiation, but vital details regarding the influence of these oxidants on specific signaling molecules remain to be clarified.
DRAK2 as a an Intermediary in Calcium and Metabolic Signaling in T Cells
One molecule expressed in T cells that may potentially link calcium and ROS signaling to cellular metabolic regulation is DRAK2/STK17B. DRAK2 is a serine/threonine kinase of the death associated protein kinase (DAPK) family (123), which is comprised of five known members (DAPK1, DAPK2, DAPK3, DRAK1, and DRAK2). All of these kinases are capable of inducing cell death when ectopically expressed in cells, and the prototype DAPK1 possesses a calmodulin regulatory domain that, through binding to calmodulin/Ca2+, regulates its catalytic activity (124). While DRAK2 lacks a calmodulin binding domain, like other DAPK family members, DRAK2 has been shown to induce apoptosis upon overexpression in cell lines (125), and its phosphorylation and nuclear translocation participates in ultraviolet light induced cell death (126).
Although broadly expressed at low levels (127), DRAK2 expression is enriched in cells of hematopoietic origin (128). Loss of expression by virtue of a germline deletion of the Drak2 gene leads to hyperactive Ca2+ mobilization in T cells, especially under suboptimal TCR stimulus conditions (128, 129), supporting the hypothesis that DRAK2 acts as a negative regulator of TCR signaling (130). The notion that DRAK2 serves as a rheostat in calcium signaling following TCR signaling is supported by the finding that it is itself activated by calcium mobilization, and that its ectopic expression in double positive thymocytes raises the threshold for both negative and positive selection (131, 132). Enigmatically, loss of the Drak2 gene leads to diminished autoimmune susceptibility, with reduced clinical severity observed in animal autoimmune models for multiple sclerosis (experimental autoimmune encephalomyelitis) and type I diabetes-prone non-obese diabetic (NOD) mice (129, 133–135). The reduced EAE susceptibility is due, at least in part, to diminished survival of activated effector T cells, perhaps a consequence of impaired calcium homeostasis (133–135).
Using a mass spectrometry based approach, we determined that DRAK2 possesses several autophosphorylation sites, most notably Ser10, Ser12, and Ser351. Using antibodies specific for phosphorylation at Ser12, it was found that TCR stimulation induces DRAK2 Ser12 autophosphorylation (131), and this requires entry of extracellular Ca2+ into T cells. The SERCA inhibitor thapsigargin was found to potently induce DRAK2 autophosphorylation on Ser12, supporting the hypothesis that ER store-operated calcium entry modulates DRAK2 catalytic activity. Recent studies have revealed that TCR induced DRAK2 activation is highly dependent on CRAC function, as DRAK2 was poorly activated following antigenic stimulation in Orai2-deficient T cells (136). Using a panel of small molecule inhibitors and RNA interference approaches, it was found that DRAK2 activation by antigenic stimulation requires protein kinase D (PKD).
Curiously, DRAK2 catalytic activity in activated T cells also depends on mitochondrial ROS, as antioxidants blocked Ser12 autophosphorylation (136). Since the electron transport complex III inhibitor FCCP led to Ser12 phosphorylation independent of antigenic stimulation of T cells, these findings demonstrate that ROS are both necessary and sufficient to promote DRAK2 catalytic activity. PKD activity is itself subject to activation by mitochondrial ROS (137, 138), and thus ROS induced by enhanced mitochondrial respiration that results from TCR stimulation likely participates in PKD-mediated DRAK2 activation (Figure 3). The specific nature of the interaction between mitochondrial ROS and DRAK2’s substrates is poorly understood. However, as DRAK2-deficient T cells bear enhanced CRAC activity (136), and DRAK2-transgenic thymocytes have diminished Ca2+ mobilization following TCR stimulation (132), it is likely that DRAK2 may phosphorylate an intermediate in CRAC signaling. DRAK2 has been shown to phosphorylate S6K1 (139), an important target of mTOR and of TCR signaling that impacts cellular metabolism (22, 57, 140). Thus, DRAK2 may itself serve as an important link between calcium and mTOR signaling, impacting the differentiation and survival of T cell subsets selectively required for immune responsiveness.
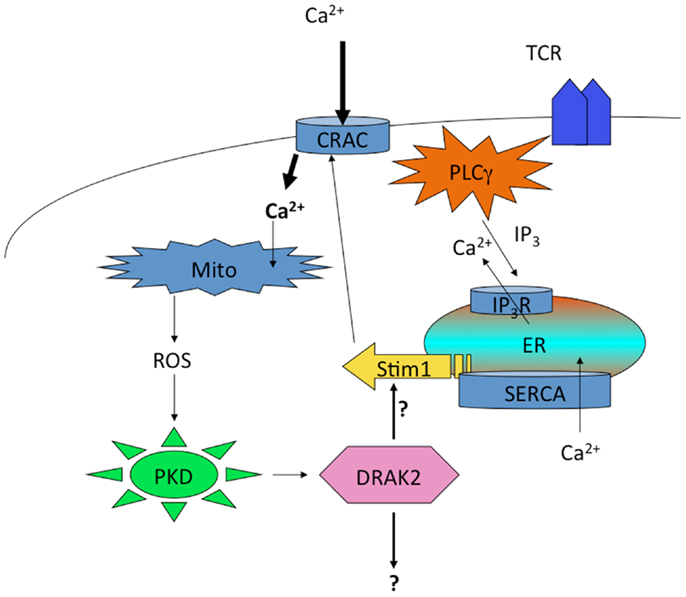
Figure 3. Regulation of DRAK2 activity in T cells via Ca2+-induced respiratory burst. TCR induced Ca2+ depletion from ER stores results in opening of CRAC channels. This high cytosolic [Ca2+] promotes the induction of TCA cycle and OXPHOS in mitochondria, the latter of which release ROS as an OXPHOS byproduct. In addition to inhibiting phosphatases, ROS activate PKD, which itself binds to and activates DRAK2 (likely through transphosphorylation on DRAK2). DRAK2 then impacts Ca2+ signaling by altering SERCA activity (hypothetical), or other unknown substrates.
Conclusion
Research into the regulation of CRAC activity, and the unique involvement of metabolic signaling in T cells, has been intense over the last several years. It is becoming quite evident that both signaling paradigms are highly regulated and vital to T cell activation, differentiation, and homeostasis. These pathways also appear to play selective roles in distinct T cell subsets, with the potential that manipulation of such pathways could influence the balance between immunity vs. self-tolerance in different disease states. A significant challenge to immunologists is to develop selective therapies to influence this balance. In the case of autoimmune disorders, the difficulty is to block autoreactive T cell function without promoting general immunosuppression. In the case of chronic microbial pathogen infections and cancer, therapeutic approaches to reinvigorate antimicrobial or anti-tumor T cell responsiveness are highly sought after. It is thus imperative that novel methods are considered to influence the appropriate immune response. Given the unique crosstalk between calcium mobilization and bioenergetic metabolism, there may be an opportunity to uniquely affect the outcome of immune therapies by targeting the molecules that mediate this crosstalk.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been supported by grants from the Gleis Family Foundation, the University of California, Irvine Multiple Sclerosis Research Center (MSRC) and the National Multiple Sclerosis Foundation.
References
1. Michel F, Attal-Bonnefoy G, Mangino G, Mise-Omata S, Acuto O. CD28 as a molecular amplifier extending TCR ligation and signaling capabilities. Immunity (2001) 15:935–45. doi: 10.1016/S1074-7613(01)00244-8
2. Riley JL, June CH. The CD28 family: a T-cell rheostat for therapeutic control of T-cell activation. Blood (2005) 105:13–21. doi:10.1182/blood-2004-04-1596
3. Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol (2012) 33:168–73. doi:10.1016/j.it.2012.01.010
4. Schreiber S, Crabtree G. The mechanism of action of cyclosporin A and FK506. Immunol Today (1992) 13(4):136–42. doi:10.1016/0167-5699(92)90111-J
5. Lewis RS, Cahalan MD. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regul (1989) 1:99–112.
6. Cahalan MD, Chandy KG. The functional network of ion channels in T lymphocytes. Immunol Rev (2009) 231:59–87. doi:10.1111/j.1600-065X.2009.00816.x
7. Hua X, Thompson CB. Quiescent T cells: actively maintaining inactivity. Nat Immunol (2001) 2:1097–8. doi:10.1038/ni1201-1097
8. Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol (2005) 5:844–52. doi:10.1038/nri1710
9. Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity (2007) 27:173–8. doi:10.1016/j.immuni.2007.07.008
10. Jones RG, Bui T, White C, Madesh M, Krawczyk CM, Lindsten T, et al. The proapoptotic factors Bax and Bak regulate T Cell proliferation through control of endoplasmic reticulum Ca(2+) homeostasis. Immunity (2007) 27:268–80. doi:10.1016/j.immuni.2007.05.023
11. Robey EA, Bluestone JA. Notch signaling in lymphocyte development and function. Curr Opin Immunol (2004) 16:360–6. doi:10.1016/j.coi.2004.03.009
12. Waickman AT, Powell JD. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol Rev (2012) 249:43–58. doi:10.1111/j.1600-065X.2012.01152.x
13. van Stipdonk MJ, Lemmens EE, Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol (2001) 2:423–9.
14. Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol Cell Biol (2001) 21:5899–912. doi:10.1128/MCB.21.17.5899-5912.2001
15. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16:769–77. doi:10.1016/S1074-7613(02)00323-0
16. Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol (2009) 183:6095–101. doi:10.4049/jimmunol.0803510
17. Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol (2003) 23:7315–28. doi:10.1128/MCB.23.20.7315-7328.2003
18. Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol (2012) 13:907–15. doi:10.1038/ni.2386
19. Delgoffe GM, Powell JD. mTOR: taking cues from the immune microenvironment. Immunology (2009) 127:459–65. doi:10.1111/j.1365-2567.2009.03125.x
20. Heikamp EB, Powell JD. Sensing the immune microenvironment to coordinate T cell metabolism, differentiation & function. Semin Immunol (2012) 24:414–20. doi:10.1016/j.smim.2012.12.003
21. von Manteuffel SR, Dennis PB, Pullen N, Gingras AC, Sonenberg N, Thomas G. The insulin-induced signalling pathway leading to S6 and initiation factor 4E binding protein 1 phosphorylation bifurcates at a rapamycin-sensitive point immediately upstream of p70s6k. Mol Cell Biol (1997) 17:5426–36.
22. Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity (2010) 33:301–11. doi:10.1016/j.immuni.2010.09.002
23. Yang K, Chi H. mTOR and metabolic pathways in T cell quiescence and functional activation. Semin Immunol (2012) 24:421–8. doi:10.1016/j.smim.2012.12.004
24. Ojuka EO. Role of calcium and AMP kinase in the regulation of mitochondrial biogenesis and GLUT4 levels in muscle. Proc Nutr Soc (2004) 63:275–8. doi:10.1079/PNS2004339
25. Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene (2004) 23:3151–71. doi:10.1038/sj.onc.1207542
26. Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol (2011) 12:888–97. doi:10.1038/ni.2068
27. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30:832–44. doi:10.1016/j.immuni.2009.04.014
28. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi:10.1016/j.immuni.2011.09.021
29. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460:108–12. doi:10.1038/nature08155
30. Warburg O. The chemical constitution of respiration ferment. Science (1928) 68:437–43. doi:10.1126/science.68.1767.437
31. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153:1239–51. doi:10.1016/j.cell.2013.05.016
32. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36:68–78. doi:10.1016/j.immuni.2011.12.007
33. van der Windt GJ, O’Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci U S A (2013) 110:14336–41. doi:10.1073/pnas.1221740110
34. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259–83. doi:10.1146/annurev-immunol-032712-095956
35. Surh CD, Sprent J. Regulation of mature T cell homeostasis. Semin Immunol (2005) 17:183–91. doi:10.1016/j.smim.2005.02.007
36. Jacobs SR, Michalek RD, Rathmell JC. IL-7 is essential for homeostatic control of T cell metabolism in vivo. J Immunol (2010) 184:3461–9. doi:10.4049/jimmunol.0902593
37. Kimura MY, Pobezinsky LA, Guinter TI, Thomas J, Adams A, Park JH, et al. IL-7 signaling must be intermittent, not continuous, during CD8(+) T cell homeostasis to promote cell survival instead of cell death. Nat Immunol (2013) 14:143–51. doi:10.1038/ni.2494
38. Surh CD, Sprent J. Homeostatic T cell proliferation: how far can T cells be activated to self-ligands? J Exp Med (2000) 192:F9–14. doi:10.1084/jem.192.4.F9
39. Bain G, Cravatt CB, Loomans C, Alberola-Ila J, Hedrick SM, Murre C. Regulation of the helix-loop-helix proteins, E2A and Id3, by the Ras-ERK MAPK cascade. Nat Immunol (2001) 2:165–71. doi:10.1038/84273
40. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi:10.1016/j.cell.2008.05.009
41. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186:3299–303. doi:10.4049/jimmunol.1003613
42. Lei J, Hasegawa H, Matsumoto T, Yasukawa M. Peroxisome proliferator-activated receptor alpha and gamma agonists together with TGF-beta convert human CD4+CD25- T cells into functional Foxp3+ regulatory T cells. J Immunol (2010) 185:7186–98. doi:10.4049/jimmunol.1001437
43. Araki K, Youngblood B, Ahmed R. The role of mTOR in memory CD8 T-cell differentiation. Immunol Rev (2010) 235:234–43. doi:10.1111/j.0105-2896.2010.00898.x
44. Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol (2001) 2:415–22.
45. Michalek RD, Rathmell JC. The metabolic life and times of a T-cell. Immunol Rev (2010) 236:190–202. doi:10.1111/j.1600-065X.2010.00911.x
46. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7. doi:10.1038/nature08097
47. Brand K, Netzker R, Aulwurm U, Hermfisse U, Fabian D, Weigert C, et al. Control of thymocyte proliferation via redox-regulated expression of glycolytic genes. Redox Rep (2000) 5:52–4.
48. Jacobs SR, Rathmell JC. Lymphocyte selection by starvation: glucose metabolism and cell death. Trends Immunol (2006) 27:4–7. doi:10.1016/j.it.2005.11.002
49. Rathmell JC, Elstrom RL, Cinalli RM, Thompson CB. Activated Akt promotes increased resting T cell size, CD28-independent T cell growth, and development of autoimmunity and lymphoma. Eur J Immunol (2003) 33:2223–32. doi:10.1002/eji.200324048
50. Tamas P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, et al. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med (2006) 203:1665–70. doi:10.1084/jem.20052469
51. Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol (2003) 13:2004–8. doi:10.1016/j.cub.2003.10.031
52. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A (2004) 101:3329–35. doi:10.1073/pnas.0308061100
53. MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, et al. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol (2011) 187:4187–98. doi:10.4049/jimmunol.1100367
54. Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res (2007) 100:328–41. doi:10.1161/01.RES.0000256090.42690.05
55. Blagih J, Krawczyk CM, Jones RG. LKB1 and AMPK: central regulators of lymphocyte metabolism and function. Immunol Rev (2012) 249:59–71. doi:10.1111/j.1600-065X.2012.01157.x
56. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208:1367–76. doi:10.1084/jem.20110278
57. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell (2010) 39:171–83. doi:10.1016/j.molcel.2010.06.022
58. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol (2013) 14:489–99. doi:10.1038/ni.2570
59. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146:772–84. doi:10.1016/j.cell.2011.07.033
60. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med (2012) 209:2441–53. doi:10.1084/jem.20112607
61. Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev (2007) 87:593–658. doi:10.1152/physrev.00035.2006
63. Zhou Y, Meraner P, Kwon HT, Machnes D, Oh-hora M, Zimmer J, et al. STIM1 gates the store-operated calcium channel ORAI1 in vitro. Nat Struct Mol Biol (2010) 17:112–6. doi:10.1038/nsmb.1724
64. Lewis RS. Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol (2001) 19:497–521. doi:10.1146/annurev.immunol.19.1.497
65. Vetter SW, Leclerc E. Novel aspects of calmodulin target recognition and activation. Eur J Biochem (2003) 270:404–14. doi:10.1046/j.1432-1033.2003.03414.x
66. Gallo EM, Cante-Barrett K, Crabtree GR. Lymphocyte calcium signaling from membrane to nucleus. Nat Immunol (2006) 7:25–32. doi:10.1038/ni1295
67. Carafoli E. The calcium-signalling saga: tap water and protein crystals. Nat Rev Mol Cell Biol (2003) 4:326–32. doi:10.1038/nrm1073
68. Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A. Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol (2001) 2:316–24. doi:10.1038/86318
69. Fanger CM, Hoth M, Crabtree GR, Lewis RS. Characterization of T cell mutants with defects in capacitative calcium entry: genetic evidence for the physiological roles of CRAC channels. J Cell Biol (1995) 131:655–67. doi:10.1083/jcb.131.3.655
70. Le Deist F, Hivroz C, Partiseti M, Thomas C, Buc HA, Oleastro M, et al. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood (1995) 85:1053–62.
71. Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol (2005) 169:435–45. doi:10.1083/jcb.200502019
72. Oh-Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, et al. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol (2008) 9:432–43. doi:10.1038/ni1574
73. Luik RM, Wu MM, Buchanan J, Lewis RS. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol (2006) 174:815–25. doi:10.1083/jcb.200604015
74. Muik M, Fahrner M, Schindl R, Stathopulos P, Frischauf I, Derler I, et al. STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J (2011) 30:1678–89. doi:10.1038/emboj.2011.79
75. Parekh AB, Putney JW Jr. Store-operated calcium channels. Physiol Rev (2005) 85:757–810. doi:10.1152/physrev.00057.2003
76. Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell (2009) 136:876–90. doi:10.1016/j.cell.2009.02.014
77. Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat Cell Biol (2009) 11:337–43. doi:10.1038/ncb1842
78. Hoth M, Button DC, Lewis RS. Mitochondrial control of calcium-channel gating: a mechanism for sustained signaling and transcriptional activation in T lymphocytes. Proc Natl Acad Sci U S A (2000) 97:10607–12. doi:10.1073/pnas.180143997
79. Schwindling C, Quintana A, Krause E, Hoth M. Mitochondria positioning controls local calcium influx in T cells. J Immunol (2010) 184:184–90. doi:10.4049/jimmunol.0902872
80. Deluca HF, Engstrom GW. Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci U S A (1961) 47:1744–50. doi:10.1073/pnas.47.11.1744
81. Rizzuto R, Simpson AW, Brini M, Pozzan T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature (1992) 358:325–7. doi:10.1038/358325a0
82. Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell (1995) 82:415–24. doi:10.1016/0092-8674(95)90430-1
83. Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol (2000) 529(Pt 1):37–47. doi:10.1111/j.1469-7793.2000.00037.x
84. Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science (1993) 262:744–7. doi:10.1126/science.8235595
85. Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature (2004) 427:360–4. doi:10.1038/nature02246
86. Gilabert JA, Parekh AB. Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca(2+) current I(CRAC). EMBO J (2000) 19:6401–7. doi:10.1093/emboj/19.23.6401
87. Gilabert JA, Bakowski D, Parekh AB. Energized mitochondria increase the dynamic range over which inositol 1,4,5-trisphosphate activates store-operated calcium influx. EMBO J (2001) 20:2672–9. doi:10.1093/emboj/20.11.2672
88. Pacher P, Csordas P, Schneider T, Hajnoczky G. Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J Physiol (2000) 529(Pt 3):553–64. doi:10.1111/j.1469-7793.2000.00553.x
89. Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A (1999) 96:13807–12. doi:10.1073/pnas.96.24.13807
90. Singaravelu K, Nelson C, Bakowski D, de Brito OM, Ng SW, Di Capite J, et al. Mitofusin 2 regulates STIM1 migration from the Ca2+ store to the plasma membrane in cells with depolarized mitochondria. J Biol Chem (2011) 286:12189–201. doi:10.1074/jbc.M110.174029
91. de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature (2008) 456:605–10. doi:10.1038/nature07534
92. Nicholls DG. Mitochondria and calcium signaling. Cell Calcium (2005) 38:311–7. doi:10.1016/j.ceca.2005.06.011
93. Prins D, Michalak M. Organellar calcium buffers. Cold Spring Harb Perspect Biol (2011) 3:1–16. doi:10.1101/cshperspect.a004069
94. Halestrap AP. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans (2006) 34:232–7. doi:10.1042/BST20060232
95. Carafoli E. Mitochondrial uptake of calcium ions and the regulation of cell function. Biochem Soc Symp (1974) 39:89–109.
96. Droge W. Free radicals in the physiological control of cell function. Physiol Rev (2002) 82:47–95.
97. Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science (2006) 312:1882–3. doi:10.1126/science.1130481
98. Bogeski I, Kummerow C, Al-Ansary D, Schwarz EC, Koehler R, Kozai D, et al. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Sci Signal (2010) 3:ra24. doi:10.1126/scisignal.2000672
99. Grupe M, Myers G, Penner R, Fleig A. Activation of store-operated I(CRAC) by hydrogen peroxide. Cell Calcium (2010) 48:1–9. doi:10.1016/j.ceca.2010.05.005
100. Hawkins BJ, Irrinki KM, Mallilankaraman K, Lien YC, Wang Y, Bhanumathy CD, et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J Cell Biol (2010) 190:391–405. doi:10.1083/jcb.201004152
101. Henke N, Albrecht P, Pfeiffer A, Toutzaris D, Zanger K, Methner A. Stromal interaction molecule 1 (STIM1) is involved in the regulation of mitochondrial shape and bioenergetics and plays a role in oxidative stress. J Biol Chem (2012) 287:42042–52. doi:10.1074/jbc.M112.417212
102. Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol (2003) 15:247–54. doi:10.1016/S0955-0674(03)00002-4
103. Rothe G, Valet G. Flow cytometric analysis of respiratory burst activity in phagocytes with hydroethidine and 2’,7’-dichlorofluorescin. J Leukoc Biol (1990) 47:440–8.
104. Szejda P, Parce JW, Seeds MS, Bass DA. Flow cytometric quantitation of oxidative product formation by polymorphonuclear leukocytes during phagocytosis. J Immunol (1984) 133:3303–7.
105. Crow JP. Dichlorodihydrofluorescein and dihydrorhodamine 123 are sensitive indicators of peroxynitrite in vitro: implications for intracellular measurement of reactive nitrogen and oxygen species. Nitric Oxide (1997) 1:145–57. doi:10.1006/niox.1996.0113
106. Rothe G, Oser A, Valet G. Dihydrorhodamine 123: a new flow cytometric indicator for respiratory burst activity in neutrophil granulocytes. Naturwissenschaften (1988) 75:354–5. doi:10.1007/BF00368326
107. Diez B, Cordo Russo R, Teijo MJ, Hajos S, Batlle A, Fukuda H. Ros production by endogenously generated protoporphyrin IX in murine leukemia cells. Cell Mol Biol (2009) 55:15–9.
108. Williams MS, Henkart PA. Role of reactive oxygen intermediates in TCR-induced death of T cell blasts and hybridomas. J Immunol (1996) 157:2395–402.
109. Weber GF, Abromson-Leeman S, Cantor H. A signaling pathway coupled to T cell receptor ligation by MMTV superantigen leading to transient activation and programmed cell death. Immunity (1995) 2:363–72. doi:10.1016/1074-7613(95)90144-2
110. Hildeman DA, Mitchell T, Teague TK, Henson P, Day BJ, Kappler J, et al. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity (1999) 10:735–44. doi:10.1016/S1074-7613(00)80072-2
111. Kwon J, Devadas S, Williams MS. T cell receptor-stimulated generation of hydrogen peroxide inhibits MEK-ERK activation and lck serine phosphorylation. Free Radic Biol Med (2003) 35:406–17. doi:10.1016/S0891-5849(03)00318-6
112. Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med (2002) 195:59–70. doi:10.1084/jem.20010659
113. Thannickal VJ, Day RM, Klinz SG, Bastien MC, Larios JM, Fanburg BL. Ras-dependent and -independent regulation of reactive oxygen species by mitogenic growth factors and TGF-beta1. FASEB J (2000) 14:1741–8. doi:10.1096/fj.99-0878com
114. Mahadev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem (2001) 276:21938–42. doi:10.1074/jbc.C100109200
115. Ushio-Fukai M, Alexander RW, Akers M, Griendling KK. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem (1998) 273:15022–9. doi:10.1074/jbc.273.24.15022
116. Bae YS, Kang SW, Seo MS, Baines IC, Tekle E, Chock PB, et al. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem (1997) 272:217–21. doi:10.1074/jbc.272.1.217
117. Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol (2003) 552:335–44. doi:10.1113/jphysiol.2003.049478
118. Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem (2004) 279:49064–73. doi:10.1074/jbc.M407715200
119. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J (2009) 417:1–13. doi:10.1042/BJ20081386
120. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38:225–36. doi:10.1016/j.immuni.2012.10.020
121. Pani G, Colavitti R, Borrello S, Galeotti T. Redox regulation of lymphocyte signaling. IUBMB Life (2000) 49:381–9. doi:10.1080/152165400410227
122. Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol (2002) 3:1129–34. doi:10.1038/ni1202-1129
123. Bialik S, Kimchi A. The death-associated protein kinases: structure, function, and beyond. Annu Rev Biochem (2006) 75:189–210. doi:10.1146/annurev.biochem.75.103004.142615
124. Cohen O, Feinstein E, Kimchi A. DAP-kinase is a Ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. EMBO J (1997) 16:998–1008. doi:10.1093/emboj/16.5.998
125. Sanjo H, Kawai T, Akira S. DRAKs, novel serine/threonine kinases related to death-associated protein kinase that trigger apoptosis. J Biol Chem (1998) 273:29066–71. doi:10.1074/jbc.273.44.29066
126. Kuwahara H, Nishizaki M, Kanazawa H. Nuclear localization signal and phosphorylation of Serine350 specify intracellular localization of DRAK2. J Biochem (2008) 143:349–58. doi:10.1093/jb/mvm236
127. Mao J, Qiao X, Luo H, Wu J. Transgenic drak2 overexpression in mice leads to increased T cell apoptosis and compromised memory T cell development. J Biol Chem (2006) 281:12587–95. doi:10.1074/jbc.M600497200
128. Friedrich ML, Wen BG, Bain G, Kee BL, Katayama C, Murre C, et al. DRAK2, a lymphoid-enriched DAP kinase, regulates the TCR activation threshold during thymocyte selection. Int Immunol (2005) 17:1379–90. doi:10.1093/intimm/dxh315
129. McGargill MA, Wen BG, Walsh CM, Hedrick SM. A deficiency in Drak2 results in a T cell hypersensitivity and an unexpected resistance to autoimmunity. Immunity (2004) 21:781–91. doi:10.1016/j.immuni.2004.10.008
130. Honey K. DRAK2 puts the brakes on T-cell responses. Nat Rev Immunol (2005) 5:98. doi:10.1038/nri1555
131. Friedrich ML, Cui M, Hernandez JB, Weist BM, Andersen HM, Zhang X, et al. Modulation of DRAK2 autophosphorylation by antigen receptor signaling in primary lymphocytes. J Biol Chem (2007) 282:4573–84. doi:10.1074/jbc.M606675200
132. Gatzka M, Newton RH, Walsh CM. Altered thymic selection and increased autoimmunity caused by ectopic expression of DRAK2 during T cell development. J Immunol (2009) 183:285–97. doi:10.4049/jimmunol.0803530
133. Ramos SJ, Hardison JL, Stiles LN, Lane TE, Walsh CM. Anti-viral effector T cell responses and trafficking are not dependent upon DRAK2 signaling following viral infection of the central nervous system. Autoimmunity (2007) 40:54–65. doi:10.1080/08916930600996700
134. Ramos SJ, Hernandez JB, Gatzka M, Walsh CM. Enhanced T cell apoptosis within Drak2-deficient mice promotes resistance to autoimmunity. J Immunol (2008) 181:7606–16.
135. McGargill MA, Choy C, Wen BG, Hedrick SM. Drak2 regulates the survival of activated T cells and is required for organ-specific autoimmune disease. J Immunol (2008) 181:7593–605.
136. Newton RH, Leverrier S, Srikanth S, Gwack Y, Cahalan MD, Walsh CM. Protein kinase D orchestrates the activation of DRAK2 in response to TCR-induced Ca2+ influx and mitochondrial reactive oxygen generation. J Immunol (2011) 186:940–50. doi:10.4049/jimmunol.1000942
137. Waldron RT, Rozengurt E. Oxidative stress induces protein kinase D activation in intact cells. Involvement of Src and dependence on protein kinase C. J Biol Chem (2000) 275:17114–21. doi:10.1074/jbc.M908959199
138. Storz P, Doppler H, Toker A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol Cell Biol (2005) 25:8520–30. doi:10.1128/MCB.25.19.8520-8530.2005
139. Mao J, Luo H, Han B, Bertrand R, Wu J. Drak2 is upstream of p70S6 kinase: its implication in cytokine-induced islet apoptosis, diabetes, and islet transplantation. J Immunol (2009) 182:4762–70. doi:10.4049/jimmunol.0802255
Keywords: calcium, metabolism, T cells, immune system, CRAC, reactive oxygen, DRAK2
Citation: Fracchia KM, Pai CY and Walsh CM (2013) Modulation of T cell metabolism and function through calcium signaling. Front. Immunol. 4:324. doi: 10.3389/fimmu.2013.00324
Received: 16 July 2013; Accepted: 24 September 2013;
Published online: 11 October 2013.
Edited by:
Gergely Toldi, Semmelweis University, HungaryReviewed by:
Tomasz Zal, University of Texas MD Anderson Cancer Center, USALucette Pelletier, Institute for National and Health and Medical Research, France
Copyright: © 2013 Fracchia, Pai and Walsh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Craig M. Walsh, Department of Molecular Biology and Biochemistry, The Institute for Immunology, University of California Irvine, 3215 McGaugh Hall, Irvine, CA 92697-3900, USA e-mail: cwalsh@uci.edu
†Kelley M. Fracchia and Christine Y. Pai have contributed equally to this work.