 Andre Kunert1,2*
Andre Kunert1,2* Trudy Straetemans1,2
Trudy Straetemans1,2 Coen Govers1,2
Coen Govers1,2 Cor Lamers1,2
Cor Lamers1,2 Ron Mathijssen2Stefan Sleijfer2
Ron Mathijssen2Stefan Sleijfer2 Reno Debets1,2*
Reno Debets1,2*- 1Laboratory of Experimental Tumor Immunology, Erasmus MC Cancer Institute, Rotterdam, Netherlands
- 2Department of Medical Oncology, Erasmus MC Cancer Institute, Rotterdam, Netherlands
Adoptive transfer of T cells gene-engineered with antigen-specific T cell receptors (TCRs) has proven its feasibility and therapeutic potential in the treatment of malignant tumors. To ensure further clinical development of TCR gene therapy, it is necessary to target immunogenic epitopes that are related to oncogenesis and selectively expressed by tumor tissue, and implement strategies that result in optimal T cell fitness. In addition, in particular for the treatment of solid tumors, it is equally necessary to include strategies that counteract the immune-suppressive nature of the tumor micro-environment. Here, we will provide an overview of the current status of TCR gene therapy, and redefine the following three challenges of improvement: “choice of target antigen”; “fitness of T cells”; and “sensitization of tumor milieu.” We will categorize and discuss potential strategies to address each of these challenges, and argue that advancement of clinical TCR gene therapy critically depends on developments toward each of the three challenges.
TCR Gene Therapy: Clinical Potency and Toxicities
T cells possess distinct properties such as the ability to specifically recognize tumor antigens, serially kill tumor cells, self-replicate, form memory and induce a complete tumor response. It is because of these properties that the therapeutic use of T cells in certain types of cancer may be advantageous when compared to drugs, antibodies, or small molecule inhibitors.
T cell therapy intends to treat cancer by transferring autologous and ex vivo expanded T cells to patients. Therapy with tumor-infiltrating T lymphocytes (TILs) preceded by non-myeloablative lymphodepletion resulted in objective responses in about 50% of metastatic melanoma patients in two different medical centers (1, 2). Equally notable were the durable complete responses observed in these trials that ranged between 10 and 22% (ongoing for more than 3 years) (1, 2). Likewise, adoptive transfer of tumor-specific T cell clones generated from autologous peripheral T cells resulted in regression of individual metastases, and responses in 8 out of 10 melanoma patients (3). In addition, co-culture of peripheral T cells with artificial antigen-presenting cells (APC) loaded with tumor antigens resulted in T cells that were clinically effective in four out of seven evaluable melanoma patients (4). Response rates observed with T cell therapy are generally higher than those observed for other treatments of melanoma, such as chemotherapeutic drugs, high-dose cytokines, inhibitors of kinases, or antibodies against T cell co-inhibitory molecules. See Table 1 for an overview of clinical outcomes of T cell therapies and other treatments of melanoma.
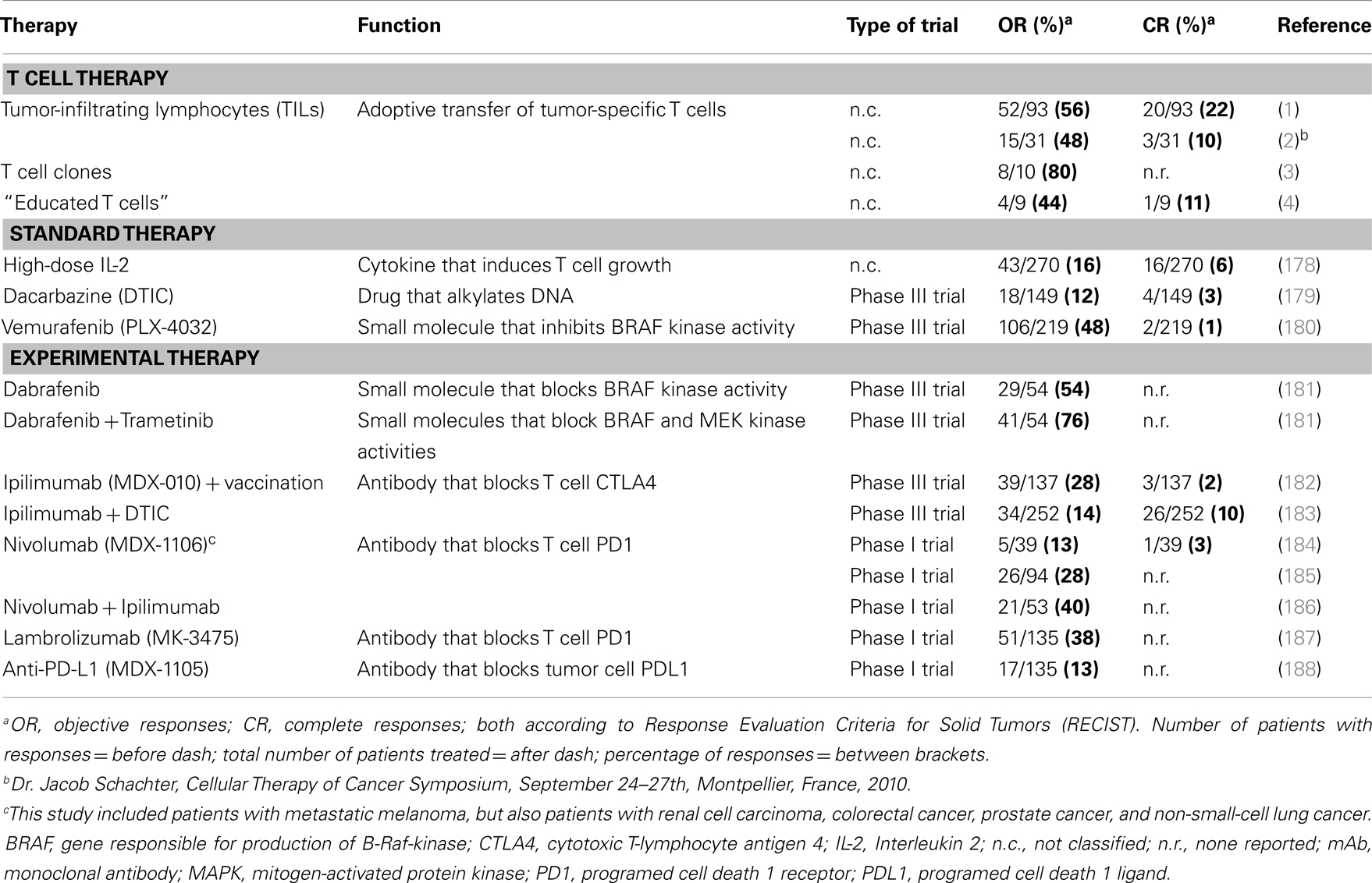
Table 1. Overview of standard and experimental none-gene-based therapies for metastatic melanoma.
Despite its clinical successes, T cell therapy has its limitations in availability and generation of therapeutic T cells for a larger group of patients. Genetic introduction of T cell receptors (TCRs) or chimeric antigen receptors (CARs) into autologous T cells, termed gene-engineering of T cells, can provide an alternative that is more widely applicable and can potentially be extended to multiple types of cancer (5). Key preclinical achievements and clinical tests with TCR-engineered T cells, the focus of the current review, are depicted in Figures 1A,B, respectively. Therapeutic advances with CAR-engineered T cells is reviewed elsewhere (6). The principle of clinical TCR gene therapy is straightforward: transferral of TCRαβ genes into T cells; ex vivo expansion of T cells; and infusion of T cells into the patient. In this way, TCRα and β genes are used as “off the shelf” reagents to confer tumor reactivity to patients whose tumor expresses the appropriate antigen and HLA restriction element. At the moment of writing this review, eight clinical trials using TCR-engineered T cells have reported their results (see Figure 1B and Table 2 for details), and at least another 10 trials using TCR-engineered T cells are open and actively recruiting patients or will recruit patients soon1.
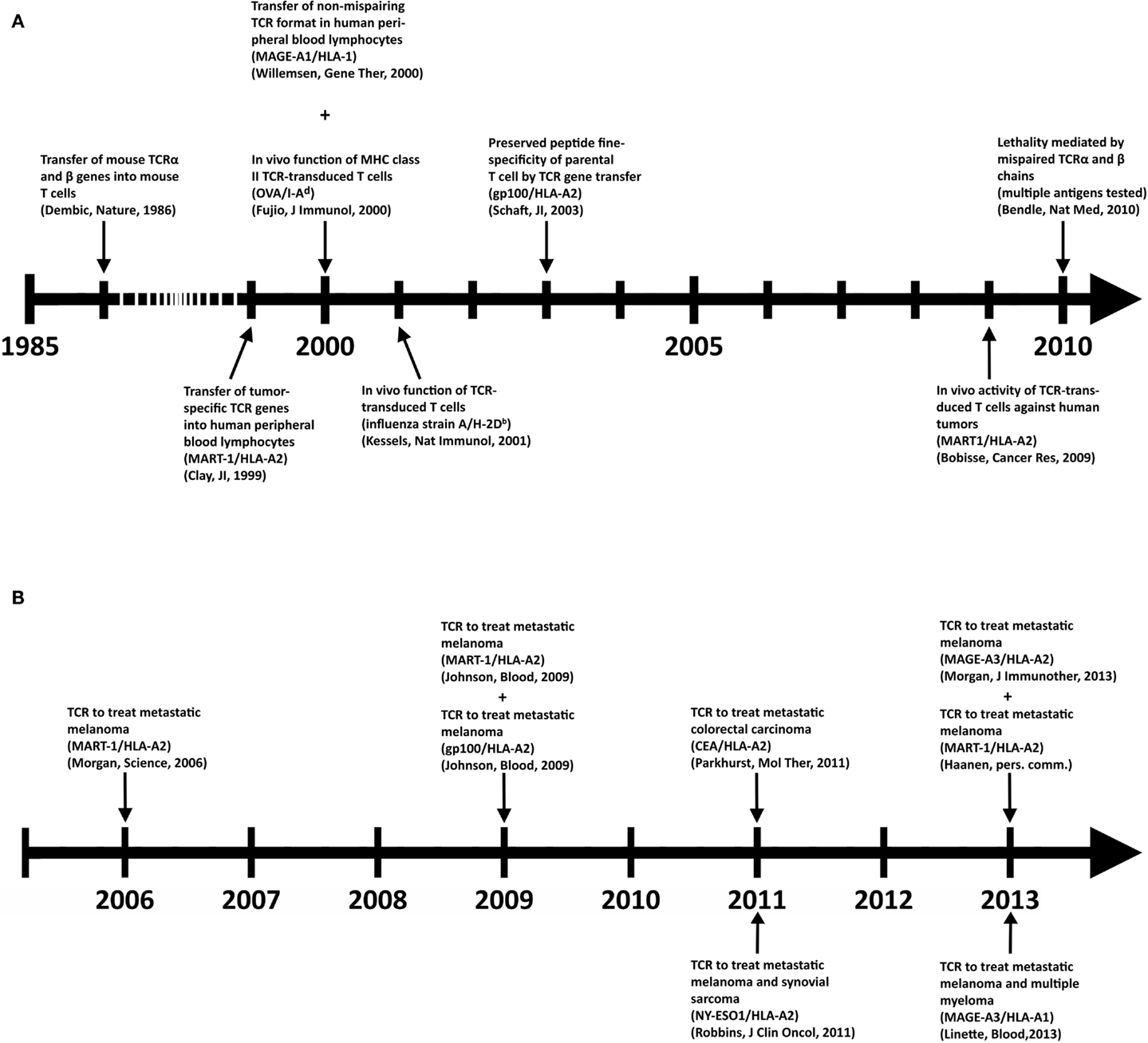
Figure 1. Key achievements in the field of TCR gene therapy directed against solid tumors. (A) Timeline of selected preclinical findings that have contributed to the development of TCR gene therapy. (B) Timeline of clinical findings with TCR gene-engineered T cells. Details with respect to clinically used TCRs can be found in Table 2.
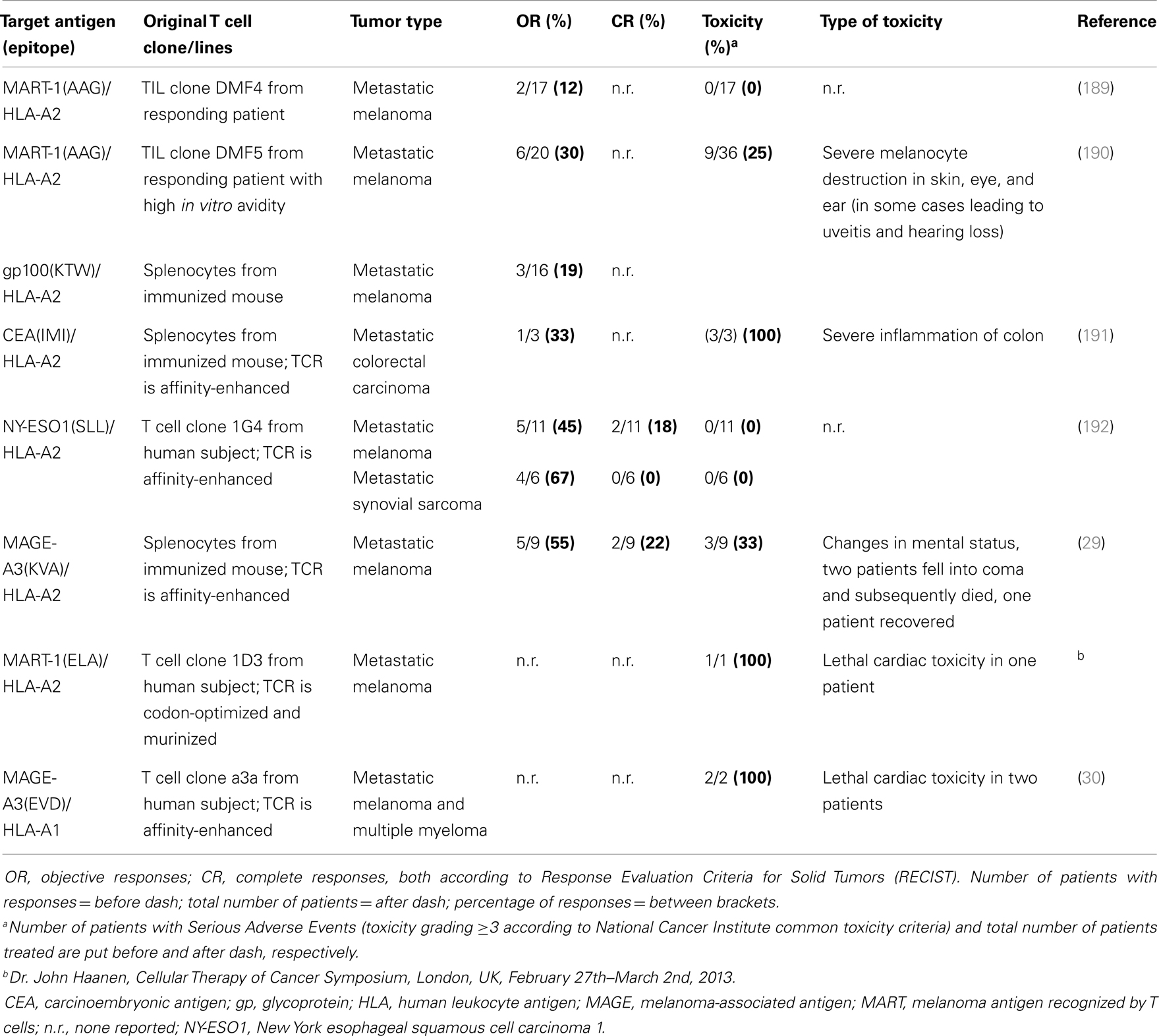
Table 2. T cell receptor gene therapy trials – an update on efficacy and safety.
Most clinical TCRs tested so far were HLA-A2-restricted and directed against either melanoma-associated antigen recognized by T cells 1 (MART-1), glycoprotein (gp) 100, carcinoembryonic antigen (CEA), p53, melanoma-associated antigen (MAGE-)A3, or New York esophageal squamous cell carcinoma antigen (NY-ESO)1. Another TCR tested clinically was HLA-A1-restricted and directed against MAGE-A3. Collectively, these trials have not only demonstrated feasibility but also demonstrated significant clinical responses in patients with metastatic melanoma, colorectal carcinoma, and synovial sarcoma (Table 2). Responses, although variable and tested in a cumulative number of about 80 patients (based on trials listed in Table 2), ranged from 12 to 67%. Notably, the finding that TCR gene-engineered T cells were able to traffic to the central nervous system and cause complete responses of brain metastasis in patients with melanoma was not only encouraging but also underscored the strength of T cell therapy toward metastasized and poorly accessible tumors (7). Clinical testing, however, also clearly demonstrated that therapy is currently hampered by treatment-related toxicity and a transient nature of tumor regression. Treatment-related toxicity became evident from studies with TCRs, in particular those of high-affinity, directed against antigens that are over-expressed on tumors but also expressed on healthy cells. Toxicities included severe but treatable inflammation of skin, eyes, ears (MART-1/HLA-A2; gp100/HLA-A2), and colon (CEA/HLA-A2). In addition, lethal neurological toxicities were observed in two patients when targeting MAGE-A3/HLA-A2, and lethal cardiac toxicities were observed in three patients when targeting MART-1/HLA-A2 (another epitope as above) or MAGE-A3/HLA-A1. The transient nature of tumor regression became evident from observations that anti-tumor responses are initially significant but not sustainable and ultimately incomplete in 80–90% of patients. Table 2 offers an up-to-date and detailed overview of toxicities as well as clinical responses reported for TCR gene therapy trials.
Strategies that aim at preventing or limiting toxicities as well as tumor recurrences have already been developed, some of which need further preclinical testing and some of which have already been implemented in clinical trials. In this review, we have categorized these strategies along three renewed challenges, i.e., “choice of target antigen”; “fitness of T cells,” and “sensitization of micro-milieu for T cell therapy,” as illustrated in Figure 2. We propose and will argue that optimizations along each or combinations of these challenges will contribute most significantly to the advancement of clinical TCR gene therapy.
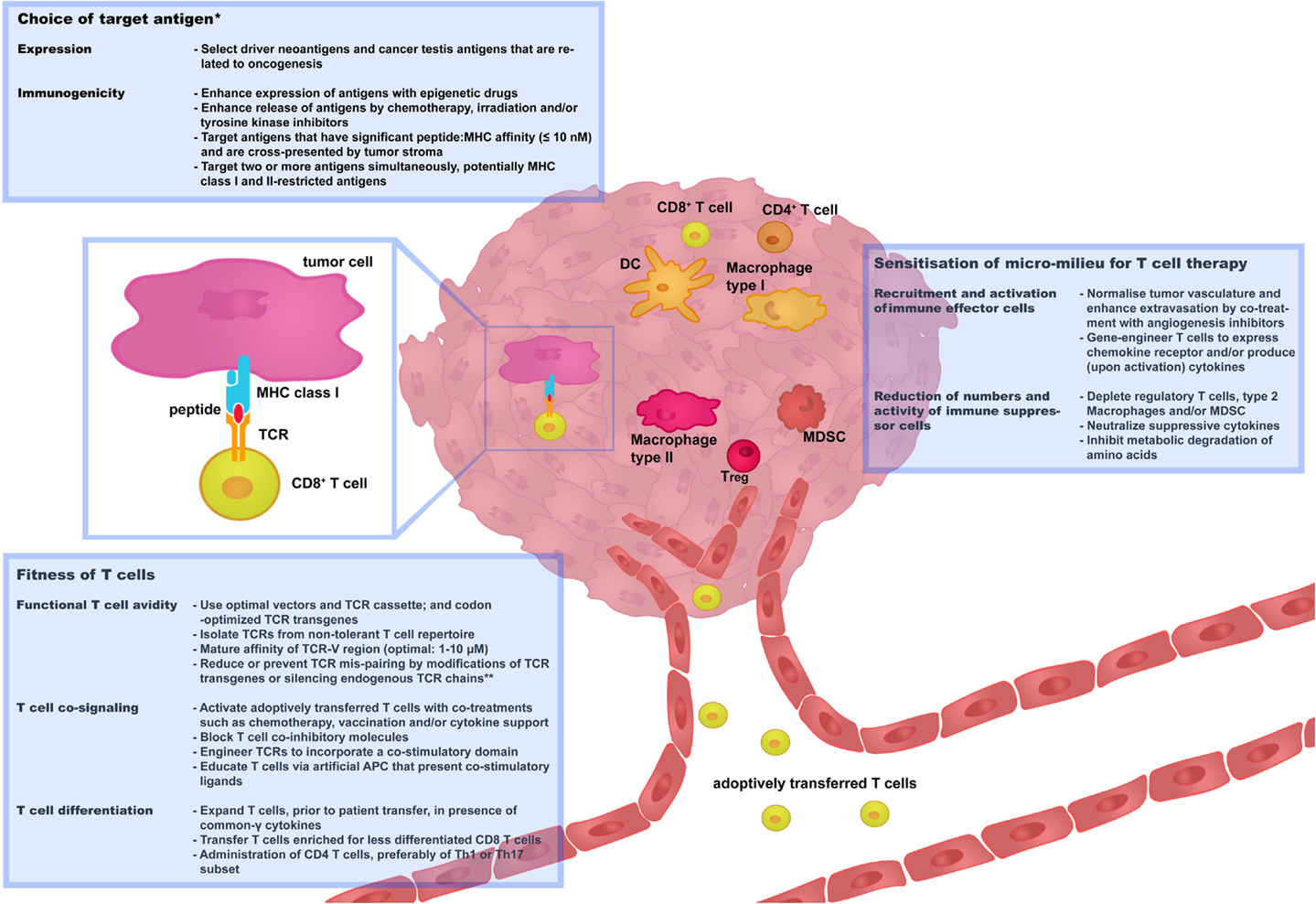
Figure 2. Three challenges that determine the success rate of TCR gene therapy. In this figure, recent and successful strategies to improve TCR gene therapy have been categorized along three renewed challenges: “choice of target antigen”; “fitness of T cells”; and “sensitization of micro-milieu for T cell therapy.” Boxes provide selected strategies that are discussed in more detail in Sections “Choice of Target Antigen,” “Fitness of T cells,” and “Sensitization of Micro-Milieu for T Cell Therapy.” We propose that advancement of clinical TCR gene therapy is guided by the principles of these challenges. *Independent of choice of target antigen, it is recommended to perform stringent in silico analysis and preclinical tests to confirm that healthy cells do not express the target antigen prior to proceeding with the clinical testing of TCR-engineered T cells. **Strategies to reduce or prevent TCR mis-pairing do not only enhance T cell avidity but also reduce the potential risk of off-target toxicity. APC, antigen-presenting cells; DC, Dendritic cells; MDSC, myeloid-derived suppressor cells; Th, T helper cells; Treg, T regulatory cells.
Choice of Target Antigen
Ideally, target antigens are selectively expressed by tumor tissue and not healthy tissue, and hence not expected to evoke a response against self. At the same time, target antigens should have proficient immunogenicity to initiate an effective anti-tumor response.
Selective Expression
Tumor-associated antigens (TAAs) can generally be divided into four groups (8).
• Differentiation antigens: cell surface proteins that are expressed at different stages of tissue development or cell activation. Expression of these antigens may discriminate tumor cells from surrounding healthy cells, but expression by healthy cells is not absent. Examples include MART-1, gp100, CEA, and tyrosinase related protein (TRP)1 and 2.
• Over-expressed antigens: cell surface proteins that are highly, but not selectively, expressed by tumor cells when compared to healthy cells. Examples include the epidermal growth factor receptor (HER)2 or survivin.
• Cancer Testis Antigens (CTAs): proteins that are expressed by tumors and a limited number of healthy and adult cell types. A defined number of CTAs may not be expressed by healthy adult cell types. Examples include MAGE-A1, MAGE-C2, and NY-ESO1.
• Neo-antigens: proteins that result from gene mutations or aberrations in tumor cells. These proteins are uniquely expressed by tumor cells but not healthy cells. Examples include mutated protein (p)53, B-Raf kinase, and cyclin-dependent kinase 4 (CDK4).
Looking at these four groups of TAAs, CTAs, and neo-antigens may represent the best available choices for therapy with TCR-engineered T cells. With respect to CTAs, over several hundreds of genes have been identified (see for a full description of CTAs2). Approximately 40 of these genes belong to multigene families that are located on the X-chromosome. A selected number of mostly X-chromosome-located CTAs may be of interest for T cell therapy. First, these antigens are not expressed by healthy tissues except testes and placentas (determined using RT-PCR), and these latter tissues do not express Major Histocompatibility (MHC) molecules and cannot be targeted by T cells (9). Second, CTAs are expressed by tumor tissues of various histological origins as a result of aberrant epigenetic regulation (9), and expression of CTAs has been associated with advanced stages of disease and unfavorable patient prognosis (10). Along these lines, there is evidence that MAGE proteins are related to oncogenesis as they suppress p53-dependent apoptosis and cause fibronectin-controlled increase in tumor cell proliferation and metastasis (11–15). Third, CTAs are immunogenic proteins that have been reported to induce both humoral and cell-mediated immune responses in patients without the concomitant induction of toxicities (10, 16, 17). Undeniably, current patient studies emphasize the need for careful identification of target CTAs. In one study, Robbins and colleagues demonstrated that a TCR directed against NY-ESO1/HLA-A2 showed significant anti-tumor responses in patients with metastatic melanoma and synovial sarcoma without detectable toxicities (Table 2). Unexpectedly, in another study using a TCR directed against MAGE-A3/HLA-A2, two patients with metastatic melanoma lapsed into coma and died. These adverse events were most likely caused by T cell recognition of rare neurons that were positive for MAGE-A12 and possibly MAGE-A9 antigens, which contain shared or highly similar epitopes compared to MAGE-A3 antigen (Table 2). In a third study, in which a TCR was used directed against MAGE-A3/HLA-A1, one patient with melanoma and one patient with myeloma suffered from cardiovascular toxicity and died. This toxicity was possibly caused by T cell recognition of a similar but not identical peptide from the muscle protein titin (so-called “off-target” toxicity, Table 2).
With respect to neo-antigens, the expression of these antigens may vary significantly among different patients, but their expression is unique to tumor tissues. In case a neo-antigen is the result of “driver mutations,” the antigen may constitute an ideal target for T cell therapy. Driver mutations are related to oncogenesis, may be linked to known genes (∼400), and may provide tumors with a selective growth advantage (18, 19). Nevertheless, it is important to realize that only 15% of up to 100,000 mutations that are encountered in tumor genomes are considered “driver” mutations (18, 20). Moreover, not all driver mutations may result in new immunogenic antigens. A quest for neo-antigen targets does not only require next-generation sequencing techniques to identify tumor-specific mutations (21), but also techniques to determine whether a neo-epitope can be presented by MHC and recognized by T cells (22, 23).
In short, we consider epitopes from selected (non-shared) CTA and neo-antigens as potentially safe T cell target antigens. However, no matter what the antigen, it is recommended to perform stringent in silico analysis and preclinical testing to confirm the antigen’s absence from vital organs. Strategies used to identify titin as a cross-recognized peptide, such as amino acid scanning, gene database searches, and use of three-dimensional cell cultures, are potentially helpful in this respect (24). In addition, one could consider using suicide systems to deplete self-reactive T cells prior to proceeding with clinical testing (25–28). Although suicide genes provide the option to delete TCR-transduced T cells, it is questionable whether such a switch could counteract the fast kinetics of toxicity reported in the above-mentioned trials (29, 30).
Immunogenicity
The immunogenicity of an antigen, i.e., its ability to initiate immune responses, is determined by the level of its expression, how it is processed and presented, and how well it is recognized by T cells.
Level of expression and processing of antigens
Ideally, target antigens should be expressed at high levels by most if not all tumor cells. Such a property is generally restricted to those antigens that are related to oncogenesis and that tumors cannot easily do without (see Selective Expression). It is noteworthy that the production of antigens, such as those of MAGE-A family members and NY-ESO1, is enhanced and becomes more homogeneous within tumors by treatment with demethylation agents and/or histone deacetylases (31–34). In a phase II clinical study, in which hematological malignancies were targeted and which included treatments with epigenetic drugs, it was observed that T cell responses directed against CTA were enhanced with no evidence of adverse events (35). In addition, the production of antigens may depend on immune or intermediate proteasomes, rather than standard proteasomes, and on unconventional post-translational events such as reverse splicing and deamidation of proteins (36–38). Such processing of antigens, in particular when mediated by immune proteasomes, may benefit from local production of interferon (IFN)γ. Finally, the release and hence the availability of antigens may be enhanced via treatment-induced cell death following (co-treatments with) chemotherapy, irradiation, and/or therapy with tyrosine kinase inhibitors (39, 40).
Cross-presentation of antigens
Antigen cross-presentation may take part in the infiltration of antigen-specific CD8 T cells (41) and cause activation of T cells and subsequent stroma destruction, thereby preventing outgrowth of antigen-negative tumor cells. Recently, Engels and colleagues revealed that peptide:MHC affinities of 10 nM or less allowed for cross-presentation of antigens by stromal cells (42). Notably, using an experimental model in which mice transgenic for TCRs with different antigen specificities were used either as donors or recipients of T cells, they showed that the use of peptide targets that can be cross-presented result in complete anti-tumor responses. Destruction of tumor stroma, a bystander response that may put an advantage to T cells over drugs (43, 44), may require optimal T cell fitness (as measured by production of IFNγ) and IFNγ-mediated preservation of Fas expression by stromal cells (45).
Robustness of antigenicity
Loss of tumor antigen expression after infusion of T cells, and its impact on the recurrence of tumors, is an important yet controversial aspect. Decreased antigen expression has been proposed to be a consequence of molecular alterations in tumor cells, such as genetic and epigenetic changes in antigen genes, MHC genes, and genes related to antigen processing and presentation (46–48). Indeed, selective loss of antigen or HLA-A2 expression has been reported in primary and metastatic melanoma lesions in non-treated patients (49, 50) as well as patients treated with T cells (51, 52). Also, Landsberg and colleagues, using a gene-engineered model of melanoma, have eloquently demonstrated that a therapy-resistant phenotype may be directed by an inflammatory milieu and tumor necrosis factor (TNF)α’s ability to lead to epithelial dedifferentiation and decreased expression of melanoma antigens (53). In contrast to these findings, there is increasing evidence to support the view that tumors progress without loss of T cell antigens. In various preclinical models, in which either skin, lung, or ovarium tumors were studied, it was observed that tumors progressed despite continued antigen expression (54–56). In these models, tumor progression was rather a consequence of reduced T cell infiltration and reduced T cell responsiveness. We postulate that in the setting of T cell therapy, loss of target antigen, whether by T cell-dependent selection or epigenetic silencing (57, 58), is not necessarily a driving mechanism in tumor recurrence (Straetemans et al., manuscript submitted).
Target multiple antigens simultaneously
In current TCR gene therapy trials, single MHC class I-restricted antigens are targeted. Preclinical studies have suggested that the targeting of two or more antigens enhances the therapeutic potential of T cells. For example, adoptive transfer of two CD8 T cell populations to simultaneously target ovalbumin and gp100, rather than either one antigen, resulted in delayed recurrence of tumors (59). Interestingly, treatment with viruses positive for three MHC class II-restricted antigens, i.e., neuroblastoma RAS, TRP1, and cytochrome c1, resulted in complete anti-tumor responses that were accompanied by significant CD4 T helper cell type 17 (Th17) responses (60). Since cooperation of CD4 and CD8 T cells appears important in the effector phase of an anti-tumor response and may contribute to the bystander elimination of tumor stroma (61), it may be worthwhile to simultaneously target MHC class I and II targets. With respect to human antigens, it is interesting to note that X-chromosome linked CTAs are coordinately expressed in tumor tissues (62), which may allow the simultaneous targeting of multiple CTAs.
Fitness of T Cells
The responsiveness of T cells toward tumor antigen is generally tuned down, most likely at various levels. First, reactive T cells may be deleted during T cell development in the thymus; second, peripheral T cells may be susceptibility to anergy; and third, intra-tumoral T cells may require enhanced co-stimulation (63). To overcome such T cell tolerizing mechanisms one can optimize T cell fitness. Here, we define T cell fitness according to the following three T cell properties: functional T cell avidity, T cell co-signaling, and T cell differentiation.
Functional T Cell Avidity
Functional T cell avidity is considered as the ability of T cells to respond to a given concentration of cognate peptide antigen, and can be enhanced via strategies, often involving gene-engineering of TCRαβ transgenes, that either increase the level of cell surface expression of TCR chains or the TCR’s affinity for peptide-MHC.
Expression level of TCR transgenes
One angle to enhance the surface expression of TCR transgenes is through optimization of the TCR gene transfer methodology, including choice of gene delivery method, use of optimal vector elements, and use of transgene cassettes [reviewed in Ref. (6, 64)]. Another angle to enhance the surface expression of TCR transgenes is through limitation or abolishment of TCR mis-pairing. TCR mis-pairing is the formation of TCR heterodimers that comprise one transgenic TCR chain and one endogenous TCR chain, and represents a phenomenon that is inherent to the generation of TCR-engineered T cells. Importantly, TCR mis-pairing dilutes the surface expression of the transgenic TCRαβ chains, and mis-paired TCRs are of unknown specificity and can yield self-reactive T cells. Although in clinical trials performed so far, no formal observations of toxicities mediated by TCR mis-pairing have been made, preclinical studies have clearly demonstrated that TCR mis-pairing has the potential to induce harmful recognition of self-antigens (65, 66). Strategies to promote preferential pairing between transgenic TCRα and TCRβ chains (and consequently prevent or reduce TCR mis-pairing) can be grouped according to those that depend on gene-engineering of TCR transgenes and those that do not. The first group of strategies are reviewed in Ref. (67). In short, these strategies include murinization of TCR (68), addition of cysteine amino acids to TCR (69, 70), mutations in TCR transmembrane and constant domains (71, 72), and equipment of TCR with a signaling cassette that replaces TCR transmembrane and intracellular domains with the CD3ζ accessory molecule (73, 74). More recently, a limited number of murine amino acids have been identified that are responsible for enhanced expression and preferential pairing of murinized TCRs (75, 76). Similar efforts to minimize the number of amino acids in a CD3ζ signaling cassette failed, and it was observed that properties of TCRs equipped with CD3ζ signaling cassettes are best preserved when incorporating a complete CD3ζ molecule (77). The other group of strategies includes technologies that enhance expression levels of CD3 molecules in T cells and those that interrupt expression of endogenous TCR chains. Co-transfer of CD3 and TCR genes into T cells resulted in higher levels of TCR expression and allowed T cells to respond to lower concentrations of antigen, and to infiltrate and eliminate tumors with faster kinetics (78). RNA interference techniques have been shown to specifically down-regulate the expression of endogenous but not transgenic TCR chains (79, 80). An alternative method encompasses the use of zinc finger nucleases and a sequential knock-out of endogenous TCRα and β chains, followed by introduction and sorting of TCRα and β transgenes (81). The latter method is relatively new and not yet widely or clinically applied, but holds promise to effectively address TCR mis-pairing.
Affinity-enhancement of TCRαβ transgenes
Affinity-enhancement of tumor-specific TCRs, and its exploitation, relies on the existence of a window for optimal TCR affinities. The existence of such a window is based on observations that TCRs specific for HLA-A2-restricted pathogens have KD values that are generally about 10-fold lower when compared to TCRs specific for HLA-A2-restricted tumor-associated self-antigens (82). In support of this notion are the observations that a high-affinity MART-1/HLA-A2 TCR mediated improved objective response rates compared to a lower affinity MART-1/HLA-A2 TCR, and that an affinity-enhanced NY-ESO1 TCR mediated significant clinical responses (Table 2). Affinity-enhanced TCRs can be obtained through various routes. First, allo-reactive settings can be used to circumvent self-tolerance and yield T cells with a higher avidity when compared to T cells derived from autologous settings (=patients). Examples of such settings include in vitro generation of allo-HLA reactive, peptide-specific T cells (83–85), and immunization of mice transgenic for human-MHC or human TCR (86, 87). Second, TCR affinities can be enhanced by rationally designed mutations of the TCR’s complementarity-determining regions (CDRs) (88, 89). Third, high-affinity TCR variants can be selected from a library of CDR mutants by yeast, phage, or T cell display (90–92). Although the affinity of TCRs significantly contributes to the functional avidity of T cells, recent studies warrant caution when therapeutically implementing this strategy. Clinical reports suggest that CDR mutations in TCRs directed against CEA/HLA-A2, MAGE-A3/HLA-A2, and MAGE-A3/HLA-A1, but not NY-ESO/HLA-A2, were possibly related to patient toxicities (Table 2). Investigations whether defined locations and types of mutations are more prone to lead to toxicities than others would most likely benefit further development of CDR-mutated TCRs. Also, preclinical reports suggest the existence of a functional ceiling with respect to TCR affinity (93, 94). In fact, studies with primary human T cells transduced with affinity-enhanced TCRs directed against NY-ESO1/HLA-A2 (93) or gp100/HLA-A2 (Govers et al., manuscript submitted) pointed to the existence of a KD threshold of 1–5 μM, below which T cell function became compromised. The functional impairment of high avidity T cells in the presence of high levels of antigen, as is often the case in tumors, may be related to enhanced expression of the exhaustion marker programed cell death (PD1) and enhanced activity of its downstream sarcoma homology domain 2 phosphatase (SHP)1 (95, 96).
T Cell Co-Signaling
T cell co-signaling is directed by interactions between co-stimulatory or co-inhibitory molecules and their ligands and determines, in addition to interactions between TCR and peptide-MHC, the functional outcome of T cells [reviewed by Chen and Flies (97)]. The best characterized co-stimulatory and co-inhibitory molecules expressed by T cells are CD28 and cytotoxic T-lymphocyte associated protein (CTLA)4, respectively, which both interact with CD80 and CD86 ligands expressed by APCs. More recent examples of co-stimulatory and co-inhibitory molecules include inducible T cell co-stimulation (ICOS), 4-1BB, OX40, CD40, B and T-lymphocyte attenuator (BTLA), and PD1.
Tumors provide continuous stimulation with antigen often in the absence of co-stimulatory ligands, which may result in exhausted T cells with reduced proliferative capacity, reduced effector function (such as IFNγ production) (98), and up-regulated expression of T cell co-inhibitory molecules (99). Immunotherapy with monoclonal antibodies to block the T cell co-inhibitory molecules CTLA4, PD1, PDL1, or the combination of CTLA4 and PD1 showed clear clinical successes in the treatment of advanced melanoma (see Table 1). These clinical activities have provided an impetus for the development of blocking other co-inhibitory molecules and/or stimulation of co-stimulatory molecules (100–104). The beneficial outcome of targeting T cell co-signaling most likely relies on enhancement of infiltration of T effector cells (Teff) into tumor tissue and activation of Teff, as well as depletion of intra-tumoral T regulatory cells (Treg) (103–105). We would advocate explorative studies to test the combination of blocking T cell co-inhibitory molecules and adoptive transfer of Teff. In addition to this combination of immune therapies, two other approaches to implement T cell co-signaling in protocols of T cell therapy have already been clinically tested. First, TCR transgenes can be equipped with a signaling cassette that harbors a co-stimulatory molecule. Such a signaling cassette, designed in analogy to those used in co-stimulatory CARs (6), typically introduces accessory and co-stimulatory molecules to enhance the function of T cells expressing the TCR transgene. It is noteworthy that clinical trials using CARs containing CD28 or CD137 demonstrated significant objective responses in patients with B cell leukemia (106–108), and while CARs may evoke immune responses, these were directed against murine idiotypes, but never against boundaries between genetically introduced human molecules (109). According to this rationale, single and two-chain TCR genes have been coupled to a combination of CD28 and CD3 molecules and were shown to provide T cells with improved function in vitro (110, 111) (Govers et al., manuscript submitted). Second, T cells can be stimulated ex vivo with human artificial APC (aAPCs) that express co-stimulatory ligands (4, 112). In addition to co-stimulatory ligands, these aAPCs are mostly engineered to express HLA-A × 0201 and used to stimulate T cells in the presence of common-γ cytokines other than interleukin (IL)-2. These combined activations allow for the generation of HLA-A2-restricted, antigen-specific T cells with a less differentiated phenotype (CD45RA+ CD62L+) and superior T cell functions in vivo (112). In a clinical study, T cells educated with aAPC presenting CD80, CD83, and a MART-1 peptide, and cultured in the presence of IL-2 and IL-15, resulted in objective responses in patients with metastatic melanoma (Table 1). Notably, inclusion of T cell co-stimulation by either one of the two above-mentioned approaches relieved the requirement for patient preconditioning with chemotherapy and/or in vivo IL-2 administration (4, 106).
T Cell Differentiation
The differentiation of naïve T cells into mature CD8 Teff or CD4 Th1 or Th17 cells is required for T cells to make full use of their functional attributes directed against tumor cells, such as cytotoxicity and production of IFNγ and TNFα. The differentiation of T cells is largely driven by environmental stimuli, with cytokines being well-studied examples of such stimuli (113, 114). Progression of T cells into a differentiated subset is not necessarily permanent, and in particular T helper cell subsets have shown plasticity and may change into another T helper cell subset (114). Differentiation of CD8 and CD4 T cells, although occurring according to similar principles, follow different routes and show different outcomes. Strategies to manipulate T cell differentiation to advance T cell therapy are discussed separately for both T cell subsets.
CD8 T cells
Naïve CD8 T cells can differentiate, depending on the quantity and quality of the initial antigenic and co-stimulatory stimuli, into stem-cell memory T cells, central memory T cells, effector memory T cells, or T effector cells (115). An important observation that came from preclinical studies was the inverse relationship between CD8 T cell differentiation and proliferation, and hence the inverse relationship between CD8 T cell differentiation and in vivo persistence and therapeutic activity (113). Two strategies have been reported to exploit this inverse relationship and improve adoptive T cell therapy. In one such strategy, as shortly mentioned in Section “T Cell Co-Signaling,” T cells are exposed to common-γ cytokines other than IL-2 prior to adoptive T cell transfer. For example, treatments with either IL-7 + IL-15 or IL-15 + IL-21 generated gene-engineered T cells with a less differentiated CD8 T cell phenotype (i.e., central memory phenotype), prolonged peripheral persistence, and potent antigen reactivity (116, 117). In addition to soluble cytokines, Singh and colleagues reported on aAPC that express membrane-bound IL-15 and IL-21 and facilitate the generation of “young” T cells (112). In other reports, the anti-tumor efficacy of T cells was enhanced either via in vivo administration of IL-15 + IL-21 (118) or conjugation of nanoparticles, encapsulating these cytokines, to the surface of therapeutic T cells (119). In a second strategy, T cells are enriched for less differentiated T cell populations, i.e., based on CD62L expression, and subsequently used as recipient cells for gene transfer (120, 121). A recently identified population of “stem-cell memory” CD8 T cells, expressing high levels of CD95, IL2Rβ and demonstrating increased proliferative potential and ability to mediate anti-tumor responses, may represent a promising subset of T cells for gene-engineering and therapeutic application (122). In fact, Cieri and colleagues have set up a protocol to obtain and gene-modify stem-cell memory CD8 T cells, which includes the use of CD3/CD28 mAbs and IL-7 and IL-15 and could potentially be translated to a clinical setting (123).
CD4 T cells
Naïve CD4 T cells can differentiate into multiple subsets, including Th1, 2, 9, 17, 22, follicular helper and various Tregs, often defined by the expression of “signature cytokines” or typical functions, such as B cell activation or the down-modulation of T cell responses (124). With respect to anti-tumor responses, it appears that upon cell transfer Th1 and Th17 are the most potent CD4 T cell subsets (125, 126). Administration of CD4 T cells, and in particular Th1 cells, has been shown to prevent exhaustion of CD8 T cells, enhance tumor infiltration of CD8 T cells and result in effective tumor eradication (125, 127–130). More recently, it was discovered that adoptive transfer of Th17 cells effectively mediate rejection of TRP1-positive tumors in a TCR-transgenic mouse model (126). Furthermore, Th17 cells appear to be long-lived and their molecular signature resembles that of stem-cell memory CD8 T cells (131). Interestingly, the anti-tumor activity of Th17 cells depended on its (incomplete) differentiation and conversion into Th1 cells, resulting in a co-existence of Th17 and Th1 cells, and it may very well be this multi-potent aspect that provides a therapeutic advantage.
Collectively, these data argue in favor of a combined therapeutic use of CD8 T cells and Th1 or Th17 cells. To this end, CD4 T cells can be functionally endowed with MHC I-restricted TCR and/or CD8 via gene transfer (132–135). Alternatively, one could opt for strategies that induce in vivo conversion of CD4 T cells into Th1 cells, such as IL-12, IFNα, IFNγ, or blocking PD1 ligation (136–139). Also, metabolic signals, such as activation of T cell mammalian target of rapamycin (mTOR) and aerobic glycolysis can enhance differentiation toward IFNγ-producing T cells and may be exploited therapeutically (140, 141).
Sensitization of Micro-Milieu for T Cell Therapy
Tumors, following initial regression upon treatment with T cells, most often become resistant to T cell therapy and recur. Recent understanding suggests that, at least in some tumors, therapy resistance may be part of a negative feedback loop that is initiated once an anti-tumor CD8 T cell has occurred (142). Therapy resistance is often characterized by a dis-balance between numbers and activation state of immune effectors cells versus those of suppressor cells. Strategies to manipulate numbers and activation state of immune cells are discussed separately for effector and suppressor cells.
Recruitment and Activation of Immune Effector Cells
Immune effector cells that have been recognized for their contribution to an anti-tumor response are numerous and, in addition to CD4 and CD8 T cells, include natural killer (NK), natural killer T cells (NKT), macrophages, and neutrophils. Here we will focus on Teff and macrophages and how manipulation of the micro-milieu may enhance their recruitment and activation.
Enhance recruitment of T effector cells
Clinical studies have demonstrated an unfavorable prognostic value of a limited CD8 T cell infiltration in melanoma, colorectal and ovarium carcinomas (143–145). Vascular changes have been reported to contribute to arrested T cell infiltration and include insufficient vascular maturation and enhanced expression of endothelin B receptor, regulator of G-protein signaling 5 (Rgs5) and/or extracellular matrix components [reviewed in Ref. (146)]. Such changes may be targeted, as evidenced by angiostatic therapy in which antibodies directed against vascular endothelial growth factor (VEGF) or angiopoietin 2, or in which T cells gene-engineered with a CAR directed against VEGF receptor (VEGFR)2 resulted in enhanced T cell infiltration (147–149). In addition, drugs that inhibit angiogenesis or endothelin receptor B were able to enhance the expression of intercellular adhesion molecule (ICAM)1 on endothelial cells and to normalize T cell infiltration (150, 151). In various solid tumors, T cell infiltration appears to be facilitated by vessels that closely mimic high endothelial venules (HEV) and which may be part of ectopic lymphoid structures in tumor stroma (152, 153). A better understanding of the development of such HEV in tumor stroma may provide novel targets to improve T cell infiltration in tumors.
In addition to vascular changes, spontaneous cutaneous melanoma tumors in mice demonstrated a decreased mRNA expression of chemoattractants that contribute to recruitment of CD8 T cells, such as chemokine (CC motif) ligand (CCL)5 and chemokine (CXC motif) ligands (CXCL)9 and 10 (146). In a subset of patients with melanoma metastases, lack of chemoattractants coincides with limited migration of CD8 T cells and limited presence of lymphoid structures (154). Current findings from our laboratory suggest that a decreased expression of selected chemoattractants and adhesion molecules are related to a decreased infiltration of CD8 T cells and tumor relapse following T cell therapy (Straetemans et. al., manuscript submitted). Interestingly, Hong and colleagues have shown that the chemotherapeutic drugs dacarbazine, temozolomide, and cisplatin enhanced the expression of CCL5, CXCL9, and CXCL10 in patient melanoma, which in turn correlated with improved immune control of tumors (155). Vice versa, T cells when gene-engineered to express chemokine (CXC motif) receptor (CXCR)2 displayed enhanced trafficking toward tumor cells secreting the corresponding chemokine ligand CXCL1 (156). Also, in xenograft tumor models of mesothelioma and neuroblastoma, the genetic introduction of chemokine (CC motif) receptor (CCR)2 in T cells resulted in increased T cell infiltration in tumors secreting CCL2 and was associated with significantly increased anti-tumor activity (157, 158). Other molecules often present in the micro-milieu that, when targeted, resulted in enhanced T cell accumulation at the tumor site are indoleamine 2,3-dioxygenase (IDO) and reactive nitrogen species. Inhibition of IDO by a small molecule blocks tryptophan depletion, enhances T cell infiltration, and delays tumor growth (159). Reactive nitrogen species induce TIL unresponsiveness (160), nitration of the TCR complex (161), and modification of the chemokine CCL2 (162). Drugs affecting the local production of reactive nitrogen species restore TIL function and improve intra-tumoral T cell migration and an anti-tumor T cell response (160, 162). Taken together, the above studies show the drug-ability of molecules that are involved in T cell extravasation and T cell migration into tumor tissues, and advocate studies to combine such drugs with adoptive T cell therapy.
Enhance T cell effector functions
Early protocols of adoptive T cell therapy already demonstrated the beneficial effects of co-treatments such as chemotherapy, vaccination, and/or cytokine support on T cell activation [reviewed in Ref. (64)]. More recently, additional strategies that enhance anti-tumor functions of Teff have been reported. A first strategy became apparent from clinical success with additional T cell co-stimulation or blocking of T cell co-inhibition (see T Cell Co-Signaling and Table 1). A second strategy relates to the inhibition of T cell suppressive cytokines, such as transforming growth factor (TGF)β. For example, genetic introduction of a dominant-negative TGFβ receptor II in TCR-engineered T cells resulted in increased anti-tumor T cell responses in a spontaneous tumor model of prostate cancer (163). Another study tested the safety of mouse T cells engineered with this dominant-negative receptor, and could not detect spontaneous proliferation of these T cells in vivo (164). Genetic knockdown of negative regulators of T cell activation represents yet another strategy to enhance T cell activation. T cells with siRNA-mediated knockdown of casitas B-lineage lymphoma b (Cbl-b) displayed a lower threshold for T cell activation and, when adoptively transferred in mice with disseminated leukemia, resulted in enhanced anti-tumor effects (165). These latter findings warrant further testing of T cells with enhanced T cell activation, including tests that assess the safe use of these T cells.
Enhance recruitment and activation of macrophages
High numbers of macrophages with a tumor-promoting (M2) phenotype, but not those with a tumor-inhibiting (M1) phenotype, correlate with poor prognosis for patients with various cancers (166). When conjugated to a vascular homing peptide and targeted to tumors, TNFα resulted in a switch from M2 to M1 macrophages, which was accompanied by normalization of tumor vasculature and enhanced infiltration of CD8 T cells (167). Interestingly, T cells gene-engineered to release the cytokine IL-12 were shown to improve the therapeutic efficacy of T cells, an effect that is likely mediated by cells of the innate immune system (168, 169). T cells that express IL-12 under the control of the Nuclear Factor of Activated T cell (NFAT) promoter, and deliver IL-12 locally in the tumor environment upon encounter of cognate antigen, induce destruction of antigen-negative cancer cells with a prominent role for monocytes and monocyte-derived TNFα (168). Such findings are not necessarily restricted to IL-12 since IL-15, when provided locally into tumors, also enhanced the responsiveness of adoptively transferred T cells and facilitated the removal of antigen-negative tumor cells (170).
Reduce Numbers and Activity of Immune Suppressor Cells
T regulatory cells, M2 macrophages, and myeloid-derived suppressor cells (MDSC) are among the major immune-suppressive cell types in the tumor micro-milieu. Immune suppressor cells can reduce T cell infiltration into the tumor and suppress local T cell responses by: release of reactive nitrogen and oxygen species (171); expression of IDO and arginase (159, 172); and production of cytokines such as TGFβ, IL-4, and IL-13 (173). Despite initial removal of these cells by administration of chemotherapeutic agents, the populations of MDSCs and Tregs may recover at a faster rate than CD4 and CD8 Teff (174). Furthermore, Jensen and colleagues demonstrated that therapeutic CD4+ T eff can convert into a Foxp3+CD4+ Treg population (175). Various strategies have been reported to deplete or inactivate Tregs. These strategies include administration of anti-CD25 antibodies, combined intra-tumoral injection of anti-CTLA4 and OX40 mAbs, or blocking IDO (104, 176). Interestingly, blocking IDO may induce conversion from Treg to Th17 helper cells, which can further contribute to anti-tumor T cell responses (176). With respect to MDSCs, it is of interest to note that classical chemotherapeutic agents, such as docetaxel, are able to deplete these cells. Docetaxel-mediated depletion of MDSC, when combined with adoptive T cell therapy and dendritic cell vaccination, was shown to enhance anti-tumor responses (174). Alternatively, differentiation of MDSC into mature myeloid cells, which can be established upon administration of β-glucans (glucose monomers from cell walls), may also provide an angle to relieve immune suppression (177).
Future Perspectives
By now, the feasibility of TCR gene therapy studies has been well established by the pioneering trials listed in Figure 1B, and is further enhanced by current optimizations and standardizations of protocols. TCR gene therapy, alike any cell-based therapy, requires specialized good manufacturing practice (GMP) and patient treatment facilities. Such facilities allow the generation and testing of virus batches and the gene processing and expansion of T cells, and are already integrated in multiple academic and private centers. Notably, parameters, such as time-lines and costs to manufacture a therapeutic T cell product, are considered competitive when compared to other clinical-grade products, such as antibodies. An ongoing EU project to treat metastatic esophagus-gastric cancer and melanoma with NY-ESO1 TCR-engineered T cells, in which we participate, shows that time-lines and costs to obtain a T cell product are about 2 weeks and 36 k per patient (13.5 k for production, quality testing, and test runs of virus batch; and 22.5 k for T cell processing), respectively. For comparison: estimated per patients costs of Ipilimumab (3 mg/kg every 3 weeks, 4 times) and Vemurafenib (0.96 g twice daily for 6 months), both registered treatments for metastasized melanoma in The Netherlands since 2012, are 84 and 57 k [Association of Health Insurances (CVZ), The Netherlands]. The next step, and allowing a more valid comparison, would be the testing of T cell therapy versus standard treatment of care in a randomized trial.
Clinical testing of TCR-engineered T cells, when looking at single trials, demonstrated impressive and unprecedented efficacy but at the same time is hampered by treatment-related toxicity and a transient nature of tumor regression (Table 2). There exists a multitude of strategies that are developed and tested toward advanced safety and efficacy of TCR gene therapy. Here, we have defined three challenges and have categorized recent and successful strategies along these three challenges, which have been schematically depicted in Figure 2. With respect to the first challenge, i.e., choice for target antigen, an important criterion is minimal or no expression of such an antigen by healthy tissues. In this respect, non-shared and tumor-restricted CTAs as well as neo-antigens should be considered as potentially safe target antigens. Advances in the isolation and characterization of anti-tumor T cells from individual patient samples may increase the number of CTAs and neo-antigens that may qualify as target antigens. T cell-based recognition of similar, but unrelated peptides should be excluded, and to this end it is strongly recommended to perform stringent in silico analysis and preclinical tests to confirm that cross-reactive antigens are absent in healthy tissue. In order to improve patient safety further, measures to allow directed killing of engineered T cells have been tested and should be considered, at least for novel TCRs tested in the near future. In addition to tumor-restricted expression, another criterion to choose target antigens is maximal immunogenicity. Peptide epitopes that are cross-presented or the targeting of a more than a single peptide have been reported to induce complete anti-tumor responses, and may represent examples to consider when selecting target antigens.
With respect to the second and third challenges, i.e., fitness of T cells and sensitization of tumor micro-milieu, we would like to propose a two-step treatment protocol. The first step represents the transfer of fit T cells. T cell fitness involves optimal T cell avidity, additional T cell co-signaling, and using T cells with a preferred differentiation stage. T cell avidity can be optimized by enhancement of TCR affinity, yet reported treatment-related toxicities warrant caution when using affinity-enhanced TCRs (Table 2) and recommend further studies to define rules of TCR binding of cognate versus non-cognate peptides. With respect to T cell co-signaling, antibodies that block T cell co-inhibitory molecules and T cells gene-engineered with co-stimulatory receptors have demonstrated clinical successes. The implementation of such strategies in T cell therapy protocols holds promise for future trials. Also, developments to obtain and gene-modify early differentiation stages of CD8 T cells, including stem-cell memory CD8 T cells, are at the brim of being translated to a clinical setting. Whatever the chosen route, an important measure for T cell fitness in vivo is the ability of these cells, whether it be CD8 T cells or certain subsets of CD4 T cells, to produce IFNγ and TNFα. The production of these cytokines not only determines T cell responsiveness, but also to what extent innate immune cells are recruited into the tumor and become activated to further improve an anti-tumor response and potentially avoid tumor relapse. The second step represents antagonism of an immune-suppressed milieu. Various strategies, such as antibodies or drugs to mediate angiostasis, chemotherapeutic agents to enhance intra-tumoral T cell infiltration, and local (T cell-mediated) delivery of cytokines, have proven beneficial to enhance the local ratio between effector and suppressor immune cells. Development of such a two-step protocol, together with the targeting of a selected antigen, is the way forward and expected to further enhance the success rate of TCR gene therapy to treat solid tumors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
1. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol (2008) 26(32):5233–9. doi:10.1200/JCO.2008.16.5449
2. Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L, et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res (2010) 16(9):2646–55. doi:10.1158/1078-0432.CCR-10-0041
3. Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A (2002) 99(25):16168–73. doi:10.1073/pnas.242600099
4. Butler MO, Friedlander P, Milstein MI, Mooney MM, Metzler G, Murray AP, et al. Establishment of antitumor memory in humans using in vitro-educated CD8+ T cells. Sci Transl Med (2011) 3(80):80ra34. doi:10.1126/scitranslmed.3002207
5. Park TS, Rosenberg SA, Morgan RA. Treating cancer with genetically engineered T cells. Trends Biotechnol (2011) 29(11):550–7. doi:10.1016/j.tibtech.2011.04.009
6. Gilham DE, Debets R, Pule M, Hawkins RE, Abken H. CAR-T cells and solid tumors: tuning T cells to challenge an inveterate foe. Trends Mol Med (2012) 18(7):377–84. doi:10.1016/j.molmed.2012.04.009
7. Hong JJ, Rosenberg SA, Dudley ME, Yang JC, White DE, Butman JA, et al. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res (2010) 16(19):4892–8. doi:10.1158/1078-0432.CCR-10-1507
8. Lucas S, Coulie PG. About human tumor antigens to be used in immunotherapy. Semin Immunol (2008) 20(5):301–7. doi:10.1016/j.smim.2008.02.001
9. Hofmann O, Caballero OL, Stevenson BJ, Chen YT, Cohen T, Chua R, et al. Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci U S A (2008) 105(51):20422–7. doi:10.1073/pnas.0810777105
10. Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci (2009) 100(11):2014–21. doi:10.1111/j.1349-7006.2009.01303.x
11. Monte M, Simonatto M, Peche LY, Bublik DR, Gobessi S, Pierotti MA, et al. MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc Natl Acad Sci U S A (2006) 103(30):11160–5. doi:10.1073/pnas.0510834103
12. Yang B, O’Herrin SM, Wu J, Reagan-Shaw S, Ma Y, Bhat KM, et al. MAGE-A, mMage-b, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in MAGE-positive cell lines. Cancer Res (2007) 67(20):9954–62. doi:10.1158/0008-5472.CAN-07-1478
13. Liu W, Cheng S, Asa SL, Ezzat S. The melanoma-associated antigen A3 mediates fibronectin-controlled cancer progression and metastasis. Cancer Res (2008) 68(19):8104–12. doi:10.1158/0008-5472.CAN-08-2132
14. Doyle JM, Gao J, Wang J, Yang M, Potts PR. MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol Cell (2010) 39(6):963–74. doi:10.1016/j.molcel.2010.08.029
15. Nardiello T, Jungbluth AA, Mei A, Diliberto M, Huang X, Dabrowski A, et al. MAGE-A inhibits apoptosis in proliferating myeloma cells through repression of Bax and maintenance of survivin. Clin Cancer Res (2011) 17(13):4309–19. doi:10.1158/1078-0432.CCR-10-1820
16. Schuler-Thurner B, Schultz ES, Berger TG, Weinlich G, Ebner S, Woerl P, et al. Rapid induction of tumor-specific type 1 T helper cells in metastatic melanoma patients by vaccination with mature, cryopreserved, peptide-loaded monocyte-derived dendritic cells. J Exp Med (2002) 195(10):1279–88. doi:10.1084/jem.20012100
17. Lurquin C, Lethé B, De Plaen E, Corbière V, Théate I, van Baren N, et al. Contrasting frequencies of antitumor and anti-vaccine T cells in metastases of a melanoma patient vaccinated with a MAGE tumor antigen. J Exp Med (2005) 201(2):249–57. doi:10.1084/jem.20041378
18. Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science (2011) 331(6024):1553–8. doi:10.1126/science.1204040
19. Lee W, Jiang Z, Liu J, Haverty PM, Guan Y, Stinson J, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature (2010) 465(7297):473–7. doi:10.1038/nature09004
20. Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science (2007) 318(5853):1108–13. doi:10.1126/science.1145720
21. Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet (2010) 11(10):685–96. doi:10.1038/nrg2841
22. Heemskerk B, Kvistborg P, Schumacher TN. The cancer antigenome. EMBO J (2013) 32(2):194–203. doi:10.1038/emboj.2012.333
23. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19(6):747–52. doi:10.1038/nm.3161
24. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med (2013) 5(197):197ra103. doi:10.1126/scitranslmed.3006034
25. Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science (1997) 276(5319):1719–24. doi:10.1126/science.276.5319.1719
26. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med (2011) 365(18):1673–83. doi:10.1056/NEJMoa1106152
27. Kieback E, Charo J, Sommermeyer D, Blankenstein T, Uckert W. A safeguard eliminates T cell receptor gene-modified autoreactive T cells after adoptive transfer. Proc Natl Acad Sci U S A (2008) 105(2):623–8. doi:10.1073/pnas.0710198105
28. Marin V, Cribioli E, Philip B, Tettamanti S, Pizzitola I, Biondi A, et al. Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum Gene Ther Methods (2012) 23(6):376–86. doi:10.1089/hgtb.2012.050
29. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (2013) 36(2):133–51. doi:10.1097/CJI.0b013e3182829903
30. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood (2013) 122(6):863–71. doi:10.1182/blood-2013-03-490565
31. Coral S, Sigalotti L, Altomonte M, Engelsberg A, Colizzi F, Cattarossi I, et al. 5-aza-2′-deoxycytidine-induced expression of functional cancer testis antigens in human renal cell carcinoma: immunotherapeutic implications. Clin Cancer Res (2002) 8(8):2690–5.
32. Chou J, Voong LN, Mortales CL, Towlerton AM, Pollack SM, Chen X, et al. Epigenetic modulation to enable antigen-specific T-cell therapy of colorectal cancer. J Immunother (2012) 35(2):131–41. doi:10.1097/CJI.0b013e31824300c7
33. Pollack SM, Li Y, Blaisdell MJ, Farrar EA, Chou J, Hoch BL, et al. NYESO-1/LAGE-1s and PRAME are targets for antigen specific T cells in chondrosarcoma following treatment with 5-Aza-2-deoxycitabine. PLoS One (2012) 7(2):e32165. doi:10.1371/journal.pone.0032165
34. Wargo JA, Robbins PF, Li Y, Zhao Y, El-Gamil M, Caragacianu D, et al. Recognition of NY-ESO-1+ tumor cells by engineered lymphocytes is enhanced by improved vector design and epigenetic modulation of tumor antigen expression. Cancer Immunol Immunother (2009) 58(3):383–94. doi:10.1007/s00262-008-0562-x
35. Goodyear O, Agathanggelou A, Novitzky-Basso I, Siddique S, McSkeane T, Ryan G, et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood (2010) 116(11):1908–18. doi:10.1182/blood-2009-11-249474
36. Skipper JC, Hendrickson RC, Gulden PH, Brichard V, Van Pel A, Chen Y, et al. An HLA-A2-restricted tyrosinase antigen on melanoma cells results from posttranslational modification and suggests a novel pathway for processing of membrane proteins. J Exp Med (1996) 183(2):527–34. doi:10.1084/jem.183.2.527
37. Dalet A, Robbins PF, Stroobant V, Vigneron N, Li YF, El-Gamil M, et al. An antigenic peptide produced by reverse splicing and double asparagine deamidation. Proc Natl Acad Sci U S A (2011) 108(29):E323–31. doi:10.1073/pnas.1101892108
38. Guillaume B, Stroobant V, Bousquet-Dubouch MP, Colau D, Chapiro J, Parmentier N, et al. Analysis of the processing of seven human tumor antigens by intermediate proteasomes. J Immunol (2012) 189(7):3538–47. doi:10.4049/jimmunol.1103213
39. Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol (2005) 26(2):111–7. doi:10.1016/j.it.2004.12.003
40. Blank CU, Hooijkaas AI, Haanen JB, Schumacher TN. Combination of targeted therapy and immunotherapy in melanoma. Cancer Immunol Immunother (2011) 60(10):1359–71. doi:10.1007/s00262-011-1079-2
41. Walch JM, Zeng Q, Li Q, Oberbarnscheidt MH, Hoffman RA, Williams AL, et al. Cognate antigen directs CD8+ T cell migration to vascularized transplants. J Clin Invest (2013) 123(6):2663–71. doi:10.1172/JCI66722
42. Engels B, Engelhard VH, Sidney J, Sette A, Binder DC, Liu RB, et al. Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell (2013) 23(4):516–26. doi:10.1016/j.ccr.2013.03.018
43. Anders K, Buschow C, Herrmann A, Milojkovic A, Loddenkemper C, Kammertoens T, et al. Oncogene-targeting T cells reject large tumors while oncogene inactivation selects escape variants in mouse models of cancer. Cancer Cell (2011) 20(6):755–67. doi:10.1016/j.ccr.2011.10.019
44. Briesemeister D, Sommermeyer D, Loddenkemper C, Loew R, Uckert W, Blankenstein T, et al. Tumor rejection by local interferon gamma induction in established tumors is associated with blood vessel destruction and necrosis. Int J Cancer (2011) 128(2):371–8. doi:10.1002/ijc.25350
45. Listopad JJ, Kammertoens T, Anders K, Silkenstedt B, Willimsky G, Schmidt K, et al. Fas expression by tumor stroma is required for cancer eradication. Proc Natl Acad Sci U S A (2013) 110(6):2276–81. doi:10.1073/pnas.1218295110
46. Algarra I, Cabrera T, Garrido F. The HLA crossroad in tumor immunology. Hum Immunol (2000) 61(1):65–73. doi:10.1016/S0198-8859(99)00156-1
47. Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol (2000) 74:181–273. doi:10.1016/S0065-2776(08)60911-6
48. Seliger B, Maeurer MJ, Ferrone S. Antigen-processing machinery breakdown and tumor growth. Immunol Today (2000) 21(9):455–64. doi:10.1016/S0167-5699(00)01692-3
49. Maeurer MJ, Gollin SM, Martin D, Swaney W, Bryant J, Castelli C, et al. Tumor escape from immune recognition: lethal recurrent melanoma in a patient associated with downregulation of the peptide transporter protein TAP-1 and loss of expression of the immunodominant MART-1/Melan-A antigen. J Clin Invest (1996) 98(7):1633–41. doi:10.1172/JCI118958
50. Wang Z, Marincola FM, Rivoltini L, Parmiani G, Ferrone S. Selective histocompatibility leukocyte antigen (HLA)-A2 loss caused by aberrant pre-mRNA splicing in 624MEL28 melanoma cells. J Exp Med (1999) 190(2):205–15. doi:10.1084/jem.190.2.205
51. Khong HT, Wang QJ, Rosenberg SA. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J Immunother (2004) 27(3):184–90. doi:10.1097/00002371-200405000-00002
52. Mackensen A, Meidenbauer N, Vogl S, Laumer M, Berger J, Andreesen R. Phase I study of adoptive T-cell therapy using antigen-specific CD8+ T cells for the treatment of patients with metastatic melanoma. J Clin Oncol (2006) 24(31):5060–9. doi:10.1200/JCO.2006.07.1100
53. Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature (2012) 490(7420):412–6. doi:10.1038/nature11538
54. Scarlett UK, Rutkowski MR, Rauwerdink AM, Fields J, Escovar-Fadul X, Baird J, et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J Exp Med (2012) 209(3):495–506. doi:10.1084/jem.20111413
55. Ward PL, Koeppen HK, Hurteau T, Rowley DA, Schreiber H. Major histocompatibility complex class I and unique antigen expression by murine tumors that escaped from CD8+ T-cell-dependent surveillance. Cancer Res (1990) 50(13):3851–8.
56. DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, et al. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell (2011) 19(1):72–85. doi:10.1016/j.ccr.2010.11.011
57. DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature (2012) 482(7385):405–9. doi:10.1038/nature10803
58. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature (2012) 482(7385):400–4. doi:10.1038/nature10755
59. Kaluza KM, Kottke T, Diaz RM, Rommelfanger D, Thompson J, Vile R. Adoptive transfer of cytotoxic T lymphocytes targeting two different antigens limits antigen loss and tumor escape. Hum Gene Ther (2012) 23(10):1054–64. doi:10.1089/hum.2012.030
60. Pulido J, Kottke T, Thompson J, Galivo F, Wongthida P, Diaz RM, et al. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat Biotechnol (2012) 30(4):337–43. doi:10.1038/nbt.2157
61. Schietinger A, Philip M, Liu RB, Schreiber K, Schreiber H. Bystander killing of cancer requires the cooperation of CD4(+) and CD8(+) T cells during the effector phase. J Exp Med (2010) 207(11):2469–77. doi:10.1084/jem.20092450
62. Gure AO, Chua R, Williamson B, Gonen M, Ferrera CA, Gnjatic S, et al. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin Cancer Res (2005) 11(22):8055–62. doi:10.1158/1078-0432.CCR-05-1203
63. Cheung AF, Dupage MJ, Dong HK, Chen J, Jacks T. Regulated expression of a tumor-associated antigen reveals multiple levels of T-cell tolerance in a mouse model of lung cancer. Cancer Res (2008) 68(22):9459–68. doi:10.1158/0008-5472.CAN-08-2634
64. Coccoris M, Straetemans T, Govers C, Lamers C, Sleijfer S, Debets R. T cell receptor (TCR) gene therapy to treat melanoma: lessons from clinical and preclinical studies. Expert Opin Biol Ther (2010) 10(4):547–62. doi:10.1517/14712591003614756
65. Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med (2010) 16(5):565–70. doi:10.1038/nm.2128
66. van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, et al. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci U S A (2010) 107(24):10972–7. doi:10.1073/pnas.1005802107
67. Govers C, Sebestyén Z, Coccoris M, Willemsen RA, Debets R. T cell receptor gene therapy: strategies for optimizing transgenic TCR pairing. Trends Mol Med (2010) 16(2):77–87. doi:10.1016/j.molmed.2009.12.004
68. Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res (2006) 66(17):8878–86. doi:10.1158/0008-5472.CAN-06-1450
69. Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, et al. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood (2007) 109(6):2331–8. doi:10.1182/blood-2006-05-023069
70. Thomas S, Xue SA, Cesco-Gaspere M, San José E, Hart DP, Wong V, et al. Targeting the Wilms tumor antigen 1 by TCR gene transfer: TCR variants improve tetramer binding but not the function of gene modified human T cells. J Immunol (2007) 179(9):5803–10.
71. Haga-Friedman A, Horovitz-Fried M, Cohen CJ. Incorporation of transmembrane hydrophobic mutations in the TCR enhance its surface expression and T cell functional avidity. J Immunol (2012) 188(11):5538–46. doi:10.4049/jimmunol.1103020
72. Kuball J, Hauptrock B, Malina V, Antunes E, Voss RH, Wolfl M, et al. Increasing functional avidity of TCR-redirected T cells by removing defined N-glycosylation sites in the TCR constant domain. J Exp Med (2009) 206(2):463–75. doi:10.1084/jem.20082487
73. Willemsen RA, Weijtens ME, Ronteltap C, Eshhar Z, Gratama JW, Chames P, et al. Grafting primary human T lymphocytes with cancer-specific chimeric single chain and two chain TCR. Gene Ther (2000) 7(16):1369–77. doi:10.1038/sj.gt.3301253
74. Sebestyén Z, Schooten E, Sals T, Zaldivar I, San José E, Alarcón B, et al. Human TCR that incorporate CD3zeta induce highly preferred pairing between TCRalpha and beta chains following gene transfer. J Immunol (2008) 180(11):7736–46.
75. Sommermeyer D, Uckert W. Minimal amino acid exchange in human TCR constant regions fosters improved function of TCR gene-modified T cells. J Immunol (2010) 184(11):6223–31. doi:10.4049/jimmunol.0902055
76. Bialer G, Horovitz-Fried M, Ya’Acobi S, Morgan RA, Cohen CJ. Selected murine residues endow human TCR with enhanced tumor recognition. J Immunol (2010) 184(11):6232–41. doi:10.4049/jimmunol.0902047
77. Govers C, Sebestyén Z, Berrevoets C, Venselaar H, Debets R. T cell receptor fused to CD3ζ: transmembrane domain of CD3ζ prevents TCR mis-pairing, whereas complete CD3ζ directs functional TCR expression. Open Gene Ther J (2011) 4:11–22. doi:10.2174/1875037001104010011
78. Ahmadi M, King JW, Xue SA, Voisine C, Holler A, Wright GP, et al. CD3 limits the efficacy of TCR gene therapy in vivo. Blood (2011) 118(13):3528–37. doi:10.1182/blood-2011-04-346338
79. Ochi T, Fujiwara H, Okamoto S, An J, Nagai K, Shirakata T, et al. Novel adoptive T-cell immunotherapy using a WT1-specific TCR vector encoding silencers for endogenous TCRs shows marked antileukemia reactivity and safety. Blood (2011) 118(6):1495–503. doi:10.1182/blood-2011-02-337089
80. Okamoto S, Mineno J, Ikeda H, Fujiwara H, Yasukawa M, Shiku H, et al. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res (2009) 69(23):9003–11. doi:10.1158/0008-5472.CAN-09-1450
81. Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, Reik A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med (2012) 18(5):807–15. doi:10.1038/nm.2700
82. Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang KM, et al. Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur J Immunol (2012) 42(12):3174–9. doi:10.1002/eji.201242606
83. Sadovnikova E, Jopling LA, Soo KS, Stauss HJ. Generation of human tumor-reactive cytotoxic T cells against peptides presented by non-self HLA class I molecules. Eur J Immunol (1998) 28(1):193–200. doi:10.1002/(SICI)1521-4141(199801)28:01<193::AID-IMMU193>3.3.CO;2-B
84. Savage P, Gao L, Vento K, Cowburn P, Man S, Steven N, et al. Use of B cell-bound HLA-A2 class I monomers to generate high-avidity, allo-restricted CTLs against the leukemia-associated protein Wilms tumor antigen. Blood (2004) 103(12):4613–5. doi:10.1182/blood-2003-11-3903
85. Wilde S, Geiger C, Milosevic S, Mosetter B, Eichenlaub S, Schendel DJ. Generation of allo-restricted peptide-specific T cells using RNA-pulsed dendritic cells: a three phase experimental procedure. Oncoimmunology (2012) 1(2):129–40. doi:10.4161/onci.1.2.18216
86. Stanislawski T, Voss RH, Lotz C, Sadovnikova E, Willemsen RA, Kuball J, et al. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat Immunol (2001) 2(10):962–70. doi:10.1038/ni1001-962
87. Li LP, Lampert JC, Chen X, Leitao C, Popovic J, Müller W, et al. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nat Med (2010) 16(9):1029–34. doi:10.1038/nm.2197
88. Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol (2008) 180(9):6116–31.
89. Zoete V, Michielin O. Comparison between computational alanine scanning and per-residue binding free energy decomposition for protein-protein association using MM-GBSA: application to the TCR-p-MHC complex. Proteins (2007) 67(4):1026–47. doi:10.1002/prot.21395
90. Holler PD, Chlewicki LK, Kranz DM. TCRs with high affinity for foreign pMHC show self-reactivity. Nat Immunol (2003) 4(1):55–62. doi:10.1038/ni863
91. Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol (2005) 23(3):349–54. doi:10.1038/nbt1070
92. Chervin AS, Aggen DH, Raseman JM, Kranz DM. Engineering higher affinity T cell receptors using a T cell display system. J Immunol Methods (2008) 339(2):175–84. doi:10.1016/j.jim.2008.09.016
93. Schmid DA, Irving MB, Posevitz V, Hebeisen M, Posevitz-Fejfar A, Sarria JC, et al. Evidence for a TCR affinity threshold delimiting maximal CD8 T cell function. J Immunol (2010) 184(9):4936–46. doi:10.4049/jimmunol.1000173
94. Thomas S, Xue SA, Bangham CR, Jakobsen BK, Morris EC, Stauss HJ. Human T cells expressing affinity-matured TCR display accelerated responses but fail to recognize low density of MHC-peptide antigen. Blood (2011) 118(2):319–29. doi:10.1182/blood-2010-12-326736
95. Brentville VA, Metheringham RL, Gunn B, Durrant LG. High avidity cytotoxic T lymphocytes can be selected into the memory pool but they are exquisitely sensitive to functional impairment. PLoS One (2012) 7(7):e41112. doi:10.1371/journal.pone.0041112
96. Hebeisen M, Baitsch L, Presotto D, Baumgaertner P, Romero P, Michielin O, et al. SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J Clin Invest (2013) 123(3):1044–56. doi:10.1172/JCI65325
97. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13(4):227–42. doi:10.1038/nri3405
98. Zippelius A, Batard P, Rubio-Godoy V, Bioley G, Liénard D, Lejeune F, et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res (2004) 64(8):2865–73. doi:10.1158/0008-5472.CAN-03-3066
100. Murphy TL, Murphy KM. Slow down and survive: enigmatic immunoregulation by BTLA and HVEM. Annu Rev Immunol (2010) 28:389–411. doi:10.1146/annurev-immunol-030409-101202
101. Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res (2011) 71(10):3540–51. doi:10.1158/0008-5472.CAN-11-0096
102. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med (2010) 207(10):2175–86. doi:10.1084/jem.20100637
103. Curran MA, Kim M, Montalvo W, Al-Shamkhani A, Allison JP. Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS One (2011) 6(4):e19499. doi:10.1371/journal.pone.0019499
104. Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest (2013) 123(6):2447–63. doi:10.1172/JCI64859
105. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med (2013) 210(9):1695–710. doi:10.1084/jem.20130579
106. Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med (2011) 3(95):95ra73. doi:10.1126/scitranslmed.3002842
107. Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood (2011) 118(18):4817–28. doi:10.1182/blood-2011-04-348540
108. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood (2012) 119(12):2709–20. doi:10.1182/blood-2011-10-384388
109. Lamers CH, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood (2011) 117(1):72–82. doi:10.1182/blood-2010-07-294520
110. Aggen DH, Chervin AS, Schmitt TM, Engels B, Stone JD, Richman SA, et al. Single-chain ValphaVbeta T-cell receptors function without mispairing with endogenous TCR chains. Gene Ther (2012) 19(4):365–74. doi:10.1038/gt.2011.104
111. Schaft N, Lankiewicz B, Drexhage J, Berrevoets C, Moss DJ, Levitsky V, et al. T cell re-targeting to EBV antigens following TCR gene transfer: CD28-containing receptors mediate enhanced antigen-specific IFNgamma production. Int Immunol (2006) 18(4):591–601. doi:10.1093/intimm/dxh401
112. Singh H, Figliola MJ, Dawson MJ, Huls H, Olivares S, Switzer K, et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res (2011) 71(10):3516–27. doi:10.1158/0008-5472.CAN-10-3843
113. Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A (2005) 102(27):9571–6. doi:10.1073/pnas.0503726102
114. Nakayamada S, Takahashi H, Kanno Y, O’Shea JJ. Helper T cell diversity and plasticity. Curr Opin Immunol (2012) 24(3):297–302. doi:10.1016/j.coi.2012.01.014
115. Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer (2012) 12(10):671–84. doi:10.1038/nrc3322
116. Kaneko S, Mastaglio S, Bondanza A, Ponzoni M, Sanvito F, Aldrighetti L, et al. IL-7 and IL-15 allow the generation of suicide gene-modified alloreactive self-renewing central memory human T lymphocytes. Blood (2009) 113(5):1006–15. doi:10.1182/blood-2008-05-156059
117. Pouw N, Treffers-Westerlaken E, Kraan J, Wittink F, ten Hagen T, Verweij J, et al. Combination of IL-21 and IL-15 enhances tumour-specific cytotoxicity and cytokine production of TCR-transduced primary T cells. Cancer Immunol Immunother (2010) 59(6):921–31. doi:10.1007/s00262-010-0818-0
118. Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med (2005) 201(1):139–48. doi:10.1084/jem.20041057
119. Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat Med (2010) 16(9):1035–41. doi:10.1038/nm.2198
120. Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest (2008) 118(1):294–305. doi:10.1172/JCI32103
121. Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci U S A (2009) 106(41):17469–74. doi:10.1073/pnas.0907448106
122. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med (2011) 17(10):1290–7. doi:10.1038/nm.2446
123. Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood (2013) 121(4):573–84. doi:10.1182/blood-2012-05-431718
124. Hirahara K, Poholek A, Vahedi G, Laurence A, Kanno Y, Milner JD, et al. Mechanisms underlying helper T-cell plasticity: implications for immune-mediated disease. J Allergy Clin Immunol (2013) 131(5):1276–87. doi:10.1016/j.jaci.2013.03.015
125. Nishimura T, Iwakabe K, Sekimoto M, Ohmi Y, Yahata T, Nakui M, et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J Exp Med (1999) 190(5):617–27. doi:10.1084/jem.190.5.617
126. Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood (2008) 112(2):362–73. doi:10.1182/blood-2007-11-120998
127. Hunziker L, Klenerman P, Zinkernagel RM, Ehl S. Exhaustion of cytotoxic T cells during adoptive immunotherapy of virus carrier mice can be prevented by B cells or CD4+ T cells. Eur J Immunol (2002) 32(2):374–82. doi:10.1002/1521-4141(200202)32:2<374::AID-IMMU374>3.0.CO;2-9
128. Marzo AL, Kinnear BF, Lake RA, Frelinger JJ, Collins EJ, Robinson BW, et al. Tumor-specific CD4+ T cells have a major “post-licensing” role in CTL mediated anti-tumor immunity. J Immunol (2000) 165(11):6047–55.
129. Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol (2005) 174(5):2591–601.
130. Gyobu H, Tsuji T, Suzuki Y, Ohkuri T, Chamoto K, Kuroki M, et al. Generation and targeting of human tumor-specific Tc1 and Th1 cells transduced with a lentivirus containing a chimeric immunoglobulin T-cell receptor. Cancer Res (2004) 64(4):1490–5. doi:10.1158/0008-5472.CAN-03-2780
131. Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez-Perez L, et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity (2011) 35(6):972–85. doi:10.1016/j.immuni.2011.09.019
132. Kuball J, Schmitz FW, Voss RH, Ferreira EA, Engel R, Guillaume P, et al. Cooperation of human tumor-reactive CD4+ and CD8+ T cells after redirection of their specificity by a high-affinity p53A2.1-specific TCR. Immunity (2005) 22(1):117–29. doi:10.1016/j.immuni.2004.12.005
133. Willemsen R, Ronteltap C, Heuveling M, Debets R, Bolhuis R. Redirecting human CD4+ T lymphocytes to the MHC class I-restricted melanoma antigen MAGE-A1 by TCR alphabeta gene transfer requires CD8alpha. Gene Ther (2005) 12(2):140–6. doi:10.1038/sj.gt.3302388
134. Morris EC, Tsallios A, Bendle GM, Xue SA, Stauss HJ. A critical role of T cell antigen receptor-transduced MHC class I-restricted helper T cells in tumor protection. Proc Natl Acad Sci U S A (2005) 102(22):7934–9. doi:10.1073/pnas.0500357102
135. Willemsen RA, Sebestyén Z, Ronteltap C, Berrevoets C, Drexhage J, Debets R. CD8 alpha coreceptor to improve TCR gene transfer to treat melanoma: down-regulation of tumor-specific production of IL-4, IL-5, and IL-10. J Immunol (2006) 177(2):991–8.
136. Mukasa R, Balasubramani A, Lee YK, Whitley SK, Weaver BT, Shibata Y, et al. Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity (2010) 32(5):616–27. doi:10.1016/j.immuni.2010.04.016
137. Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, et al. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci U S A (2001) 98(26):15137–42. doi:10.1073/pnas.261570598
138. Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol (2002) 3(6):549–57. doi:10.1038/ni794
139. Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med (2011) 3(111):111ra120. doi:10.1126/scitranslmed.3003130
140. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30(6):832–44. doi:10.1016/j.immuni.2009.04.014
141. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi:10.1016/j.cell.2013.05.016
142. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci Transl Med (2013) 5(200):200ra116. doi:10.1126/scitranslmed.3006504
143. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313(5795):1960–4. doi:10.1126/science.1129139
144. Kalialis LV, Drzewiecki KT, Klyver H. Spontaneous regression of metastases from melanoma: review of the literature. Melanoma Res (2009) 19(5):275–82. doi:10.1097/CMR.0b013e32832eabd5
145. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med (2003) 348(3):203–13. doi:10.1056/NEJMoa020177
146. Rahir G, Moser M. Tumor microenvironment and lymphocyte infiltration. Cancer Immunol Immunother (2012) 61(6):751–9. doi:10.1007/s00262-012-1253-1
147. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res (2010) 70(15):6171–80. doi:10.1158/0008-5472.CAN-10-0153
148. Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell (2011) 19(4):512–26. doi:10.1016/j.ccr.2011.02.005
149. Chinnasamy D, Yu Z, Theoret MR, Zhao Y, Shrimali RK, Morgan RA, et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest (2010) 120(11):3953–68. doi:10.1172/JCI43490
150. Dirkx AE, oude Egbrink MG, Castermans K, van derSchaft DW, Thijssen VL, Dings RP, et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J (2006) 20(6):621–30. doi:10.1096/fj.05-4493com
151. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med (2008) 14(1):28–36. doi:10.1038/nm1699
152. Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie JJ, et al. Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res (2011) 71(17):5678–87. doi:10.1158/0008-5472.CAN-11-0431
153. Cipponi A, Mercier M, Seremet T, Baurain JF, Théate I, van den Oord J, et al. Neogenesis of lymphoid structures and antibody responses occur in human melanoma metastases. Cancer Res (2012) 72(16):3997–4007. doi:10.1158/0008-5472.CAN-12-1377
154. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res (2009) 69(7):3077–85. doi:10.1158/0008-5472.CAN-08-2281
155. Hong M, Puaux AL, Huang C, Loumagne L, Tow C, Mackay C, et al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res (2011) 71(22):6997–7009. doi:10.1158/0008-5472.CAN-11-1466
156. Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM, et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther (2002) 13(16):1971–80. doi:10.1089/10430340260355374
157. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother (2010) 33(8):780–8. doi:10.1097/CJI.0b013e3181ee6675
158. Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res (2011) 17(14):4719–30. doi:10.1158/1078-0432.CCR-11-0351
159. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9(10):1269–74. doi:10.1038/nm934
160. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol (2005) 5(8):641–54. doi:10.1038/nri1668
161. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13(7):828–35. doi:10.1038/nm1609
162. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med (2011) 208(10):1949–62. doi:10.1084/jem.20101956
163. Zhang Q, Yang X, Pins M, Javonovic B, Kuzel T, Kim SJ, et al. Adoptive transfer of tumor-reactive transforming growth factor-beta-insensitive CD8+ T cells: eradication of autologous mouse prostate cancer. Cancer Res (2005) 65(5):1761–9. doi:10.1158/0008-5472.CAN-04-3169
164. Lacuesta K, Buza E, Hauser H, Granville L, Pule M, Corboy G, et al. Assessing the safety of cytotoxic T lymphocytes transduced with a dominant negative transforming growth factor-beta receptor. J Immunother (2006) 29(3):250–60. doi:10.1097/01.cji.0000192104.24583.ca
165. Stromnes IM, Blattman JN, Tan X, Jeevanjee S, Gu H, Greenberg PD. Abrogating Cbl-b in effector CD8(+) T cells improves the efficacy of adoptive therapy of leukemia in mice. J Clin Invest (2010) 120(10):3722–34. doi:10.1172/JCI41991
166. Lee HW, Choi HJ, Ha SJ, Lee KT, Kwon YG. Recruitment of monocytes/macrophages in different tumor microenvironments. Biochim Biophys Acta (2013) 1835(2):170–9.
167. Johansson A, Hamzah J, Ganss R. Intratumoral TNFalpha improves immunotherapy. Oncoimmunology (2012) 1(8):1395–7. doi:10.4161/onci.20981
168. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res (2011) 71(17):5697–706. doi:10.1158/0008-5472.CAN-11-0103
169. Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z, et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res (2010) 70(17):6725–34. doi:10.1158/0008-5472.CAN-10-0735
170. Liu RB, Engels B, Schreiber K, Ciszewski C, Schietinger A, Schreiber H, et al. IL-15 in tumor microenvironment causes rejection of large established tumors by T cells in a noncognate T cell receptor-dependent manner. Proc Natl Acad Sci U S A (2013) 110(20):8158–63. doi:10.1073/pnas.1301022110
171. Grohmann U, Bronte V. Control of immune response by amino acid metabolism. Immunol Rev (2010) 236:243–64. doi:10.1111/j.1600-065X.2010.00915.x
172. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med (2005) 202(7):931–9. doi:10.1084/jem.20050715
173. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol (2011) 29:235–71. doi:10.1146/annurev-immunol-031210-101324
174. Kodumudi KN, Weber A, Sarnaik AA, Pilon-Thomas S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J Immunol (2012) 189(11):5147–54. doi:10.4049/jimmunol.1200274
175. Jensen SM, Twitty CG, Maston LD, Antony PA, Lim M, Hu HM, et al. Increased frequency of suppressive regulatory T cells and T cell-mediated antigen loss results in murine melanoma recurrence. J Immunol (2012) 189(2):767–76. doi:10.4049/jimmunol.1103822
176. Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Chandler P, et al. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood (2009) 113(24):6102–11. doi:10.1182/blood-2008-12-195354
177. Tian J, Ma J, Ma K, Guo H, Baidoo SE, Zhang Y, et al. beta-Glucan enhances antitumor immune responses by regulating differentiation and function of monocytic myeloid-derived suppressor cells. Eur J Immunol (2013) 43(5):1220–30. doi:10.1002/eji.201242841
178. Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol (1999) 17(7):2105–16.
179. Middleton MR, Grob JJ, Aaronson N, Fierlbeck G, Tilgen W, Seiter S, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol (2000) 18(1):158–66.
180. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med (2011) 364(26):2507–16. doi:10.1056/NEJMoa1103782
181. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med (2012) 367(18):1694–703. doi:10.1056/NEJMoa1210093
182. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
183. Robert C, Thomas L, Bondarenko I, O’Day S, M D JW, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364(26):2517–26. doi:10.1056/NEJMoa1104621
184. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol (2010) 28(19):3167–75. doi:10.1200/JCO.2009.26.7609
185. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
186. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi:10.1056/NEJMoa1302369
187. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med (2013) 369(2):134–44. doi:10.1056/NEJMoa1305133
188. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi:10.1056/NEJMoa1200694
189. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (2006) 314(5796):126–9. doi:10.1126/science.1129003
190. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood (2009) 114(3):535–46. doi:10.1182/blood-2009-03-211714
191. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther (2011) 19(3):620–6. doi:10.1038/mt.2010.272
Keywords: antigens, inhibitory micro-milieu, solid tumors, T cell avidity, T cell co-stimulation, T cells, TCR affinity, TCR transgenes
Citation: Kunert A, Straetemans T, Govers C, Lamers C, Mathijssen R, Sleijfer S and Debets R (2013) TCR-engineered T cells meet new challenges to treat solid tumors: choice of antigen, T cell fitness, and sensitization of tumor milieu. Front. Immunol. 4:363. doi: 10.3389/fimmu.2013.00363
Received: 13 August 2013; Accepted: 24 October 2013;
Published online: 08 November 2013.
Edited by:
Bruno Laugel, Cardiff University School of Medicine, UKReviewed by:
Gavin Bendle, University of Birmingham, UKAnna Mondino, San Raffaele Scientific Institute, Italy
Copyright: © 2013 Kunert, Straetemans, Govers, Lamers, Mathijssen, Sleijfer and Debets. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andre Kunert and Reno Debets, Department of Medical Oncology, Laboratory of Experimental Tumor Immunology, Erasmus MC Cancer Institute, Dr. Molewaterplein 50, Rotterdam 3015 GE, Netherlands e-mail: a.kunert@erasmusmc.nl; j.debets@erasmusmc.nl