Corrigendum: Genome-Wide Analysis of DNA Methylation, Copy Number Variation, and Gene Expression in Monozygotic Twins Discordant for Primary Biliary Cirrhosis
 Carlo Selmi1,2*
Carlo Selmi1,2* Francesca Cavaciocchi1,3
Francesca Cavaciocchi1,3 Ana Lleo4
Ana Lleo4 Cristina Cheroni5
Cristina Cheroni5 Raffaele De Francesco5
Raffaele De Francesco5 Simone A. Lombardi1Maria De Santis1,3Francesca Meda1Maria Gabriella Raimondo1Chiara Crotti1
Simone A. Lombardi1Maria De Santis1,3Francesca Meda1Maria Gabriella Raimondo1Chiara Crotti1 Marco Folci1Luca Zammataro1
Marco Folci1Luca Zammataro1 Marlyn J. Mayo6
Marlyn J. Mayo6 Nancy Bach7
Nancy Bach7 Shinji Shimoda8
Shinji Shimoda8 Stuart C. Gordon9
Stuart C. Gordon9 Monica Miozzo10,11
Monica Miozzo10,11 Pietro Invernizzi4Mauro Podda1Rossana Scavelli5
Pietro Invernizzi4Mauro Podda1Rossana Scavelli5 Michelle R. Martin12
Michelle R. Martin12 Michael F. Seldin13,14
Michael F. Seldin13,14 Janine M. LaSalle12
Janine M. LaSalle12 M. Eric Gershwin2
M. Eric Gershwin2
- 1Division of Rheumatology and Clinical Immunology, Humanitas Clinical and Research Center, Milan, Italy
- 2Division of Rheumatology, Allergy, and Clinical Immunology, University of California at Davis, Davis, CA, USA
- 3BIOMETRA Department, University of Milan, Milan, Italy
- 4Liver Unit and Center for Autoimmune Liver Diseases, Humanitas Clinical and Research Center, Milan, Italy
- 5National Institute of Molecular Genetics (INGM), Milan, Italy
- 6University of Texas Southwestern, Dallas, TX, USA
- 7Mt. Sinai University, New York, NY, USA
- 8Clinical Research Center, National Nagasaki Medical Center, Nagasaki, Japan
- 9Henry Ford Hospital, Detroit, MI, USA
- 10Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy
- 11Division of Pathology, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico, Milan, Italy
- 12Genome Center and M.I.N.D. Institute, University of California at Davis, Davis, CA, USA
- 13Department of Biochemistry and Molecular Medicine, University of California at Davis, Davis, CA, USA
- 14Department of Internal Medicine, University of California at Davis, Davis, CA, USA
Primary biliary cirrhosis (PBC) is an uncommon autoimmune disease with a homogeneous clinical phenotype that reflects incomplete disease concordance in monozygotic (MZ) twins. We have taken advantage of a unique collection consisting of genomic DNA and mRNA from peripheral blood cells of female MZ twins (n = 3 sets) and sisters of similar age (n = 8 pairs) discordant for disease. We performed a genome-wide study to investigate differences in (i) DNA methylation (using a custom tiled four-plex array containing tiled 50-mers 19,084 randomly chosen methylation sites), (ii) copy number variation (CNV) (with a chip including markers derived from the 1000 Genomes Project, all three HapMap phases, and recently published studies), and/or (iii) gene expression (by whole-genome expression arrays). Based on the results obtained from these three approaches we utilized quantitative PCR to compare the expression of candidate genes. Importantly, our data support consistent differences in discordant twins and siblings for the (i) methylation profiles of 60 gene regions, (ii) CNV of 10 genes, and (iii) the expression of 2 interferon-dependent genes. Quantitative PCR analysis showed that 17 of these genes are differentially expressed in discordant sibling pairs. In conclusion, we report that MZ twins and sisters discordant for PBC manifest particular epigenetic differences and highlight the value of the epigenetic study of twins.
Introduction
Primary biliary cirrhosis (PBC) is a female-predominant autoimmune liver disease affecting the small interlobular bile ducts, ultimately leading to periportal fibrosis and cirrhosis (1). Similar to most autoimmune diseases, PBC onset results from the interplay of genomic predisposition and environmental factors (2–5) with a possible role for sex factors (6). Recent genome-wide association studies (GWAS) have reported consistent associations with polymorphisms of genes such as IL12RA and HLA class II in subgroups of patients with PBC (7–13) and a pathway analysis was recently performed (13). PBC concordance rates in dizygotic (DZ) and monozygotic (MZ) twins are significantly different being 0 and 63%, respectively, thus supporting the role of both genetic and environmental factors (14) with the latter supported also by epidemiology (15, 16).
Promoter methylation influences gene expression (GEX) and our group recently reported differences in the DNA methylation and expression of two X-linked genes (PIN4 and CLIC2) in MZ twins discordant for PBC (17). On the other hand, copy number variations (CNV) are the result of duplications and other rearrangements (18) occur de novo at much higher rates than single nucleotide variants, and may regulate GEX (19). While sharing their genomic sequence, MZ twins may develop different phenotypes over the years because of increasing differences in DNA methylation (20) and CNV (21, 22).
We have taken advantage of a unique DNA collection of identical and non-identical twins with PBC and performed a genome-wide investigation to determine differences in DNA methylation, CNV, and GEX. Our data identify 17 candidate genes that are significantly under- or up-regulated in affected individuals and we suggest that these might constitute new candidates as disease markers of genetic determinants. The value of this approach is highlighted and suggests the need for the study of a large number of patients and cell subpopulations (23) to support this thesis.
Materials and Methods
Subjects
Blood samples from three MZ twins pairs discordant for PBC whose zygosity had been determined using microsatellite analysis (Ballestar) and eight sister pairs of similar age (within 5 years) discordant for PBC studied (Table 1). Serum antimitochondrial antibodies (AMA) were positive at indirect immunofluorescence in all patients with PBC and none of the healthy twins and sisters. In these subjects, PBC was excluded when serum AMA was negative and serum alkaline phosphatase was within normal range on two different occasions. Genomic DNA was isolated from peripheral blood mononuclear cells (PBMCs) using the QIAamp Blood Midi Kit (Qiagen, Valencia, CA, USA) and stored at −20°C until used. Additional blood samples were obtained using Tempus™ Blood RNA Tubes (Applied Biosystems, Foster City, CA, USA) that were stored at −20°C until mRNA was extracted using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and then stored at −80°C. This study was performed in compliance with the ethical standards of medicine and, following approval by the local IRB, informed consents were obtained from all patients and controls in accordance with the Declaration of Helsinki.
TABLE 1
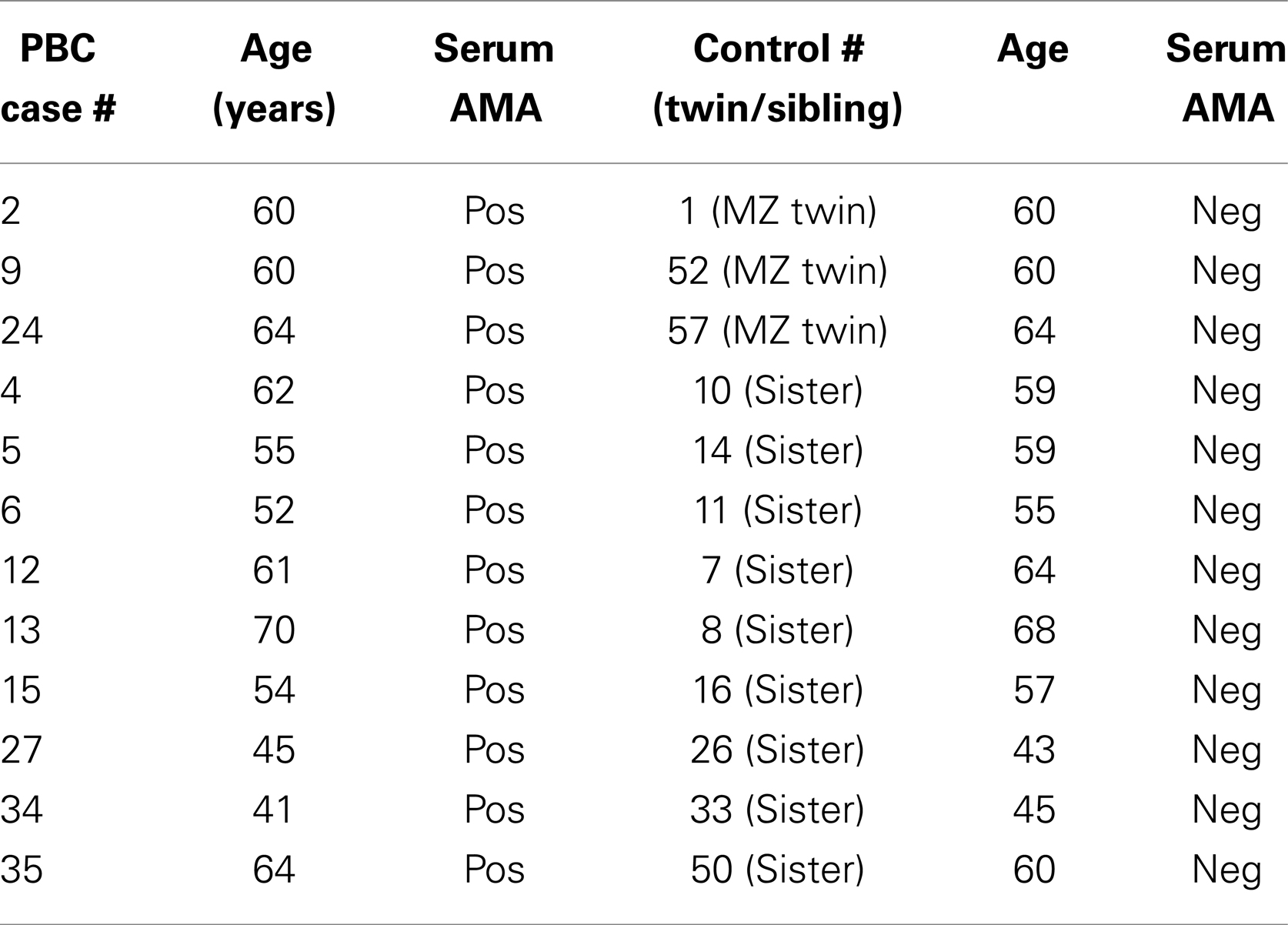
Table 1. Summary of the patients with PBC and the corresponding healthy sibling and twin sisters utilized in the study.
Methylated DNA Immunoprecipitation and Methylation Analysis
DNA samples of three MZ twin sets (#1/2, 9/52, and 24/57; see Table 1) were sonicated and then immunoprecipitated with a monoclonal antibody that specifically recognizes 5-methylcytidine (Roche NimbleGen, Madison, WI, USA). DNA fragments were converted into PCR-amplifiable OmniPlex™ Library molecules flanked by universal primer sites and the library amplified by PCR using universal primers and a limited number of cycles. Immunoprecipitated and reference DNA were tagged, respectively, with cyanine-5 (Cy5) and cyanine-3 (Cy3)-labeled random 9-mers and then hybridized by the NimbleGen Array Hybridization Kit (Roche NimbleGen, Madison, WI, USA).
A four-plex array was custom-designed to include 998 X chromosome and 18,086 randomly selected autosomal chromosome promoter sites (Roche NimbleGen, Madison, WI, USA) and samples analyzed following the manufacturers protocols. First, Nimblescan software (Roche NimbleGen, Madison, WI, USA) was utilized for DNA methylation data analysis using a threshold p-value of 0.05 equivalent to 1.31 based on the Gaussian distribution of data. Second, exclusive elements corresponding to specific microarray probes were identified in affected and healthy subjects and peaks found only in either group were selected for further analysis. Third, elements of interest were inserted into the UCSC Genome Browser (GRCh36/hg19) to identify corresponding genes.
Copy Number Variation Analysis
Copy number variation analysis was performed on genomic DNA from one MZ twin set (#1/2; see Table 1) using the Infinium R HD Assay Super platform (Illumina, San Diego, CA, USA): in particular, we utilized the HumanOmni1-Quad BeadChip that includes markers derived from the 1000 Genomes Project, all three HapMap phases, and recently published studies (7, 9, 24, 25) as well as adequate tools for quality control, CNV calling, and validation. The protocol included the initial DNA preamplification, fragmentation, and precipitation. Data obtained from four-plex chips were analyzed using iScan and Illumina BeadArray system (Illumina, San Diego, CA, USA) followed by the GenomeStudio software (Illumina, San Diego, CA, USA). The position of each probe and the number of copies for each probe were determined using the PennCNV platform based on a hidden Markov model algorithm (26). The UCSC Genome Browser was then used to determine the genes involved and the number of CNV.
Microarray Gene Expression Analysis
We utilized RNA samples from eight pairs of sisters of similar age (Table 1) discordant for PBC. In the first part, we performed a whole-genome microarray comparison of transcripts to detect consistently up- or down-regulated genes in affected subjects. We obtained biotin-labeled cRNA using the Illumina R TotalPrep RNA Amplification Kit (Illumina, San Diego, CA, USA) and used the whole-genome Gene Expression Direct Hybridization Assay (Illumina, San Diego, CA, USA) including 24,500 transcripts. Microarrays were scanned using the BeadArray Reader (Illumina Inc., San Diego, CA, USA) and data were processed using BeadStudio software (Illumina Inc.). Expression data were quantified using a cut-off for significant gene differences of p < 0.05 with a twofold difference in expression as described elsewhere (27).
RT-PCR Expression Analysis
Real-time PCR was utilized to analyze samples prepared from 1 μg total RNA according to high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA) in seven pairs of sisters of similar age (#15/16, 5/14, 6/11, 7/12, 8/13, 26/27, 33/34; see Table 1). Micro-fluidic real-time quantitative PCR cards were customized to include single-plex assays for all candidate genes obtained with DNA methylation, CNV, and GEX analyses. Genes reported by GWAS studies were also included among the candidates (7, 9, 24, 25). All samples were analyzed in duplicate, and included 94 candidate genes and the 18S and β-actin housekeeping genes. Analyses were performed using the ABI Prism 7900HT Sequence Detection System (SDS 2.2.2 software, Applied Biosystems, Foster City, CA, USA). PCR cycle conditions included 50°C for 2 min, 94.5°C for 10 min, and 40 cycles of 97°C for 30 s followed by 59.7°C for 1 min. The preliminary study of all 10 samples defined the maximum allowable cycle threshold (CT) that was set at 38 while outliers exceeding this threshold were excluded from the statistical analysis and no adjustment of p-value was performed. Internal controls for calculating expression levels of candidate genes were 18S and ACTB (beta-actin). The analysis has been performed with Data Assist version 3 statistical software (Applied Biosystems). The software exports data from real-time PCR and performs relative quantification analysis. The data assist analysis contains: Ct data, sample design, assay design, average of Ct values of replicates, ΔCt, normalized versus endogenous controls Ct values ± SD and fold change (RQ) files, which displays RQ min and RQ max for each sample. p-Value is calculated from ΔCt files.
A heat map is used to visualize the data and illustrates, for all case/control sibling pairs, GEX in red/green color based on ΔCt values using Pearson’s correlation. The neutral/middle expression was set as the median of all the ΔCt values from all samples, the red indicated an increase with a ΔCt value below the middle level and the green indicated a decrease with ΔCt above the middle level.
Pathway Analysis
Gene networks were generated through the use of Ingenuity Pathways Analysis software 8.0. Edition (Ingenuity Systems, http://www.ingenuity.com). Each gene identified was mapped to its corresponding gene object in the Ingenuity Pathways Knowledge Base and overlaid onto a global molecular network. The SDS 2.2.2 software (Applied Biosystems, Foster City, CA, USA) was used to determine changes in expression of a target in an experimental sample relative to the same target in a reference sample with the Student’s t-test and p-value <0.05 were considered statistically significant. We utilized Data Assist Software version 3 statistical software (Applied Biosystems) and Stata 8.0 for MacIntosh (Stata Corp, College Station, TX, USA) for statistical analyses.
Results
DNA Methylation
DNA methylation comparison showed 60 differentially methylated regions (DMR) in affected compared to the non-affected twin (p < 0.05 for each of the three discordant twin pairs). These DMR corresponded to 51 genes on the X chromosome and 9 genes on autosomal chromosomes, listed in Table 2. For each DMR, the PBC proband was hypermethylated compared with the non-affected twin.
TABLE 2

Table 2. Differentially methylated genes in PBC-discordant MZ twins.
Copy Number Variations
Ten CNV were discordant between affected and the non-affected twin in one twin set. The healthy twin had four CNVs that were missing in the affected twin and six CNVs were present only in the affected twin. The CNVs were found in the following genes: RYBP ring 1, YY1 binding protein, HERV-V2 envelope glycoprotein ENVV2, POTEK P ankirin domain family member K pseudogene, THSD7A thrombospondin type 1 domain containing 7A = KIAA0960, GOLGA8A golgin A8 family member A, BPTF bromodomain PHD finger transcription factor, and C17orf58 open reading frame. Two additional CNV did not correspond to known genes.
Microarray Gene Expression
Gene expression analysis using the genome-wide microarray showed two genes significantly down-regulated in PBC compared to the healthy sister in each of the eight discordant sister pairs. These genes were IFIT1 (interferon-induced protein with tetratricopeptide repeats; chromosome 10q23.31) and IFI44L (interferon-induced protein 44-like; chromosome 1p31.1) and both are interferon-induced (28).
RT-PCR Analysis
To provide additional support for our initial findings, we used RT-PCR to evaluate expression of each of the candidates that emerged from the DNA methylation (60), CNV (10), and expression studies (2), as well as previously reported GWAS in seven pairs of discordant sisters of similar age (Table 1) (7–9, 12, 13, 24, 25). Our data assist analysis contained: Ct data, sample design, assay design, average of Ct values of replicates, ΔCt, normalized versus endogenous controls Ct values ± SD and fold change (RQ) files, which displays RQ min and RQ max for each sample. p-Value was calculated from ΔCt files. Data assist v3.0 software was used with results exported from real-time PCR and for relative quantification analysis. Graphic result in heat map visualized analyzed data (Figure 1). Heat map showed, for all case/control sibling pairs, genes expression in red/green color based on ΔCt values using Pearson’s correlation. The neutral/middle expression was set as the median of all the ΔCt values from all samples, the red indicated an increase with a ΔCt value below the middle level and the green indicated a decrease with ΔCt value above the middle level. The heat map from all samples is represented in Figure 1. Among the entire set of candidate genes, we found five genes that were underexpressed in at least three of seven sibling pairs with FC < 0.5 (CXCR5, HLA-B, IFI44L, IFIT1, SMARCA1) and one overexpressed gene in at least three of seven pairs with an FC > 2 (IL6). Additional 11 genes showed a widely variable expression profile in each sibling pair (CD80, FAM104B, HLA-DQB1, HLA-DRB1, HLA-G, MTCP1, NHS, PIN4, PRPF38A, THSD7A, and TNFAIP2) (Table 3; Figure 2).
FIGURE 1
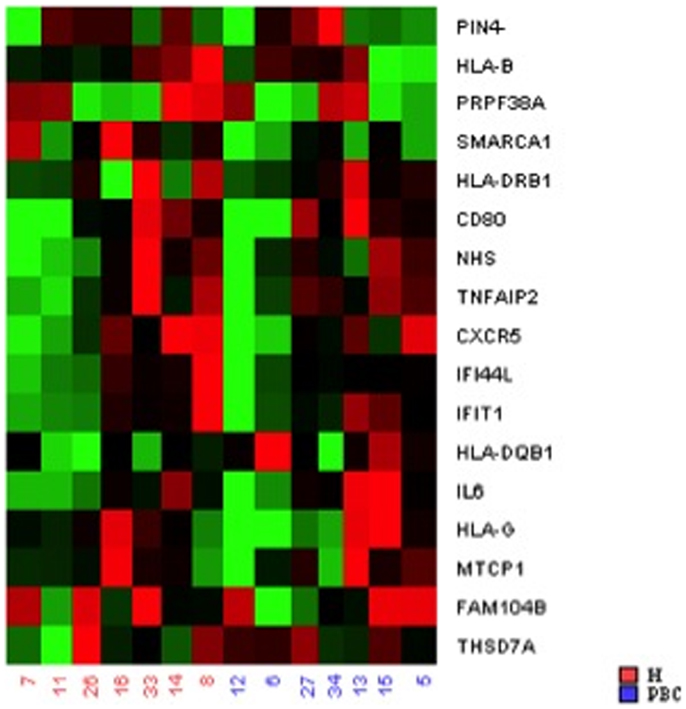
Figure 1. Heat map showed, for all case/control sibling pairs, genes expression in red/green color based on ΔCt values using Pearson’s correlation. The red indicated an increased expression with a ΔCt value below the middle level and the green indicated a decreased expression with ΔCt value above the middle level.
TABLE 3

Table 3. Genes showing consistent differences in DNA methylation, CNV, or expressiona.
FIGURE 2
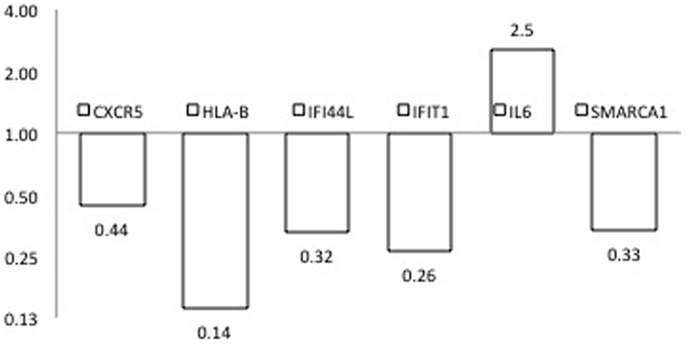
Figure 2. Expression fold changes at RT-PCR of the six genes found differences in PBC versus healthy sibling pairs analysis subjects.
Pathway Analysis
Pathway analyses were performed using the 17 resulting genes from our study and demonstrated that the most representative functions and diseases were inflammatory, immunological, and connective tissue disorders. Furthermore the top canonical pathways involved were: T helper cell differentiation (p = 3.98E−19), dendritic cell maturation (p = 1.39E−13), altered T and B cell signaling in rheumatoid arthritis (p = 1.02E−12), type I diabetes mellitus signaling (p = 1.04E−11), and the crosstalk between dendritic cells and natural killer cells (p = 5.98E−11) (Table 4).
TABLE 4

Table 4. List of PBC-associated genes analyzed with ingenuity pathways analysis software 8.0 IPA.
Discussion
Primary biliary cirrhosis is considered a prototypic autoimmune disease because of the clinical homogeneity between patients and the relative consistency in natural history and pathology. Although relatively uncommon, several independent GWAS (7–13) have identified associations with transcription factors that further suggest a potential role for epigenetic shifts and thus our approach using this unique collection of DNA is a particularly important resource. We are aware of the numerous limitations of our study and that the observed changes in GEX may be stochastic rather than secondary to disease progression or involved in pathways involved in PBC pathogenesis, as suggested for other autoimmune diseases (29–32). The latter includes the possibility of portal hypertension and resulting leukopenia.
We identified 60 DMR and 10 CNV between discordant MZ twins with 14 (20%) also differently expressed between PBC cases and control sisters, thus being stronger candidates as PBC biomarkers or determinants. One of the strengths of our study is the confirmation of identified genes by quantitative PCR and that this approach was extended also to genes identified in recent GWAS allowing identification of six genes differently expressed in PBC mononuclear cells. First, these genes support a down-modulation of Th2-cytokines such as IFIT1, an interferon type I signature represented by IFI44L, in favor of a fibrogenic phenotype as represented by the IL6 up-regulation (33). Regarding this last observation, we note the apparent discrepancy between DNA methylation and GEX of IL6 but we recognize that methylation does not fully correlate with GEX, and the difference could be explained by different mechanisms such as allele-specific methylation (34, 35) (Table 4). Second, a single DMR-associated gene, i.e., hypermethylated SMARCA1, manifested a reduced GEX confirmed in our RT-PCR study of sibling pairs. SMARCA1 is a transcription regulator that modulates the chromatin structure and is involved in apoptosis, DNA damage, and differentiation. Moreover, the gene encodes for a member of the SWI/SNF family of proteins, which are master regulators of GEX, regulating expression among others FOS, CSF-1, CRYAB, MIM-1, p21 (also known as CDKN1A), HSP70, VIM, and CCNA2; SWI/SNF has also been reported to modulate alternative splicing (36). Third, 5/7 sibling pairs had consistent dysregulation of CXCR5 being down-regulated in PBC lymphocytes, which may reflect a compartmentalization of CXCR5+ cells within the liver or may reflect the chronic activation of B cells, as reported in rheumatoid arthritis (37). In fact, the chemokine receptor CXCR5 is expressed by B and T cells and controls their migration within lymph nodes while its ligand BCA-1/CXCL13 is present in lymph nodes and spleen and also in the liver. A downregulation of CXRC5 is correlated with an increased production of IL-2, which may cause the production of immunoglobulins by B cells; IL-2 is normally produced by T cells during an immune response. IL-2 is also necessary during T cell differentiation in Treg, which are involved in self antigens recognition, which could result in autoimmunity (38). Of note, following B cell activation and differentiation into plasma cells and memory cells, CXCR5 becomes down-regulated while the same effect is induced in vitro following anti-CD40 stimulation (39) and CD40L methylation appears to be altered in PBC (40). Fourth, HLA-B is also down-regulated in PBC, similar to several types of cancer (41–43).
The majority of the identified genes map on the X chromosome, in agreement with the female predominance of the disease, and are involved in many cellular pathways. Our group in a previous work assessed the expression of 125 genes with variable X inactivation status and found that two genes (CLIC2 and PIN4) were consistently down-regulated in PBC affected twin of discordant pairs (17). Three genes are differentially methylated in lymphocytes of patients with PBC and systemic sclerosis (32) and may thus be representative of general autoimmunity or fibrosis development; these genes include MTM1 hypermethylated in PBC and in systemic sclerosis while SSR4 and IGH3G are hypomethylated in both diseases. Of note, a recent study reported the up-regulation of the X-linked costimulatory molecule CD40L (40) but our data failed to confirm such hypomethylation in our cohort. The CNV differences observed in our MZ twin set warrant some further observations as the de novo post-twinning CNV frequency was estimated to be as high as 5% on a per-individual basis or 10% per twinning event (21). While the impact of CNV on GEX can vary (44), it would be of great interest to obtain parental information to determine the origin and timing of CNV in the offspring. On the other hand, there are several limitations to our data. PBC is relatively uncommon and our DNA collection reflects a several-year worldwide search; it is nonetheless a limited dataset. In addition, there is only limited information available using PBMC. PBC is an organ-specific disease affecting small intrahepatic bile ducts and thus studies of the portal infiltrating lymphocytes will provide a more valuable resource as would a detailed and well-defined lymphoid cell populations. These comments notwithstanding, the data obtained are intriguing and consistent with our thesis that one explanation for discordant MZ twins is DNA changes on the critical genomic element involved in disease susceptibility and these observations should be recapitulated also in unrelated pairs of patients and controls. With the increased interest in the balance between genetic susceptibility, it becomes critical for research groups to combine resources and improve access to clinical material and data that permits more extensive studies and the potential for more powerful statistical analysis and interpretation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by the NIH grants 5R21DK075400 and R01DK091823 and by the American Liver Foundation research support.
References
1. Selmi C, Bowlus CL, Gershwin ME, Coppel RL. Primary biliary cirrhosis. Lancet (2011) 377:1600–9. doi: 10.1016/S0140-6736(10)61965-4
2. Germolec D, Kono DH, Pfau JC, Pollard KM. Animal models used to examine the role of the environment in the development of autoimmune disease: findings from an NIEHS Expert Panel Workshop. J Autoimmun (2012) 39(4):285–93. doi:10.1016/j.jaut.2012.05.020
3. Miller FW, Alfredsson L, Costenbader KH, Kamen DL, Nelson LM, Norris JM, et al. Epidemiology of environmental exposures and human autoimmune diseases: findings from a National Institute of Environmental Health Sciences Expert Panel Workshop. J Autoimmun (2012) 39(4):259–71. doi:10.1016/j.jaut.2012.05.002
4. Miller FW, Pollard KM, Parks CG, Germolec DR, Leung PS, Selmi C, et al. Criteria for environmentally associated autoimmune diseases. J Autoimmun (2012) 39(4):253–8. doi:10.1016/j.jaut.2012.05.001
5. Selmi C, Leung PS, Sherr DH, Diaz M, Nyland JF, Monestier M, et al. Mechanisms of environmental influence on human autoimmunity: a national institute of environmental health sciences expert panel workshop. J Autoimmun (2012) 39(4):272–84. doi:10.1016/j.jaut.2012.05.007
6. Pollard KM. Gender differences in autoimmunity associated with exposure to environmental factors. J Autoimmun (2012) 38:J177–86. doi:10.1016/j.jaut.2011.11.007
7. Hirschfield GM, Liu X, Xu C, Lu Y, Xie G, Gu X, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med (2009) 360:2544–55. doi:10.1056/NEJMoa0810440
8. Hirschfield GM, Liu X, Han Y, Gorlov IP, Lu Y, Xu C, et al. Variants at IRF5-TNPO3, 17q12-21 and MMEL1 are associated with primary biliary cirrhosis. Nat Genet (2010) 42:655–7. doi:10.1038/ng.631
9. Liu X, Invernizzi P, Lu Y, Kosoy R, Bianchi I, Podda M, et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat Genet (2010) 42:658–60. doi:10.1038/ng.627
10. Tanaka A, Invernizzi P, Ohira H, Kikuchi K, Nezu S, Kosoy R, et al. Replicated association of 17q12-21 with susceptibility of primary biliary cirrhosis in a Japanese cohort. Tissue Antigens (2011) 78:65–8. doi:10.1111/j.1399-0039.2011.01684.x
11. Hirschfield GM, Xie G, Lu E, Sun Y, Juran BD, Chellappa V, et al. Association of primary biliary cirrhosis with variants in the CLEC16A, SOCS1, SPIB and SIAE immunomodulatory genes. Genes Immun (2012) 13:328–35. doi:10.1038/gene.2011.89
12. Juran BD, Hirschfield GM, Invernizzi P, Atkinson EJ, Li Y, Xie G, et al. Immunochip analyses identify a novel risk locus for primary biliary cirrhosis at 13q14, multiple independent associations at four established risk loci and epistasis between 1p31 and 7q32 risk variants. Hum Mol Genet (2012) 21:5209–21. doi:10.1093/hmg/dds359
13. Kar SP, Seldin MF, Chen W, Lu E, Hirschfield GM, Invernizzi P, et al. Pathway-based analysis of primary biliary cirrhosis genome-wide association studies. Genes Immun (2013) 14:179–86. doi:10.1038/gene.2013.1
14. Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology (2004) 127:485–92. doi:10.1053/j.gastro.2004.05.005
15. Selmi C, Invernizzi P, Keeffe EB, Coppel RL, Podda M, Rossaro L, et al. Epidemiology and pathogenesis of primary biliary cirrhosis. J Clin Gastroenterol (2004) 38:264–71. doi:10.1097/00004836-200403000-00013
16. Selmi C. The worldwide gradient of autoimmune conditions. Autoimmun Rev (2010) 9:A247–50. doi:10.1016/j.autrev.2010.02.004
17. Mitchell MM, Lleo A, Zammataro L, Mayo MJ, Invernizzi P, Bach N, et al. Epigenetic investigation of variably X chromosome inactivated genes in monozygotic female twins discordant for primary biliary cirrhosis. Epigenetics (2011) 6:95–102. doi:10.4161/epi.6.1.13405
18. Wong KK, Deleeuw RJ, Dosanjh NS, Kimm LR, Cheng Z, Horsman DE, et al. A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet (2007) 80:91–104. doi:10.1086/510560
19. Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science (2007) 315:848–53. doi:10.1126/science.1136678
20. Ballestar E. Epigenetic alterations in autoimmune rheumatic diseases. Nat Rev Rheumatol (2011) 7:263–71. doi:10.1038/nrrheum.2011.16
21. Bruder CE, Piotrowski A, Gijsbers AA, Andersson R, Erickson S, Diaz De Stahl T, et al. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am J Hum Genet (2008) 82:763–71. doi:10.1016/j.ajhg.2007.12.011
22. Mamtani M, Anaya JM, He W, Ahuja SK. Association of copy number variation in the FCGR3B gene with risk of autoimmune diseases. Genes Immun (2010) 11:155–60. doi:10.1038/gene.2009.71
23. Hughes T, Webb R, Fei Y, Wren JD, Sawalha AH. DNA methylome in human CD4+ T cells identifies transcriptionally repressive and non-repressive methylation peaks. Genes Immun (2010) 11:554–60. doi:10.1038/gene.2010.24
24. Mells GF, Floyd JA, Morley KI, Cordell HJ, Franklin CS, Shin SY, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet (2011) 43:329–32. doi:10.1038/ng.789
25. Nakamura M, Nishida N, Kawashima M, Aiba Y, Tanaka A, Yasunami M, et al. Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am J Hum Genet (2012) 91:721–8. doi:10.1016/j.ajhg.2012.08.010
26. Dellinger AE, Saw SM, Goh LK, Seielstad M, Young TL, Li YJ. Comparative analyses of seven algorithms for copy number variant identification from single nucleotide polymorphism arrays. Nucleic Acids Res (2010) 38:e105. doi:10.1093/nar/gkq040
27. Wozniak MB, Le Calvez-Kelm F, Abedi-Ardekani B, Byrnes G, Durand G, Carreira C, et al. Integrative genome-wide gene expression profiling of clear cell renal cell carcinoma in Czech Republic and in the United States. PLoS One (2013) 8:e57886. doi:10.1371/journal.pone.0057886
28. Malhotra S, Bustamante MF, Perez-Miralles F, Rio J, Ruiz De Villa MC, Vegas E, et al. Search for specific biomarkers of IFNbeta bioactivity in patients with multiple sclerosis. PLoS One (2011) 6:e23634. doi:10.1371/journal.pone.0023634
29. Baranzini SE, Mudge J, Van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA, et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature (2010) 464:1351–6. doi:10.1038/nature08990
30. Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res (2010) 20:170–9. doi:10.1101/gr.100289.109
31. Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, et al. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet (2011) 7:e1002300. doi:10.1371/journal.pgen.1002300
32. Selmi C, Feghali-Bostwick CA, Lleo A, Lombardi SA, De Santis M, Cavaciocchi F, et al. X chromosome gene methylation in peripheral lymphocytes from monozygotic twins discordant for scleroderma. Clin Exp Immunol (2012) 169:253–62. doi:10.1111/j.1365-2249.2012.04621.x
33. Fenoglio D, Bernuzzi F, Battaglia F, Parodi A, Kalli F, Negrini S, et al. Th17 and regulatory T lymphocytes in primary biliary cirrhosis and systemic sclerosis as models of autoimmune fibrotic diseases. Autoimmun Rev (2012) 12:300–4. doi:10.1016/j.autrev.2012.05.004
34. Zhang D, Cheng L, Badner JA, Chen C, Chen Q, Luo W, et al. Genetic control of individual differences in gene-specific methylation in human brain. Am J Hum Genet (2010) 86:411–9. doi:10.1016/j.ajhg.2010.02.005
35. van Eijk KR, De Jong S, Boks MP, Langeveld T, Colas F, Veldink JH, et al. Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMC Genomics (2012) 13:636. doi:10.1186/1471-2164-13-636
36. Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene (2009) 28:1653–68. doi:10.1038/onc.2009.4
37. Henneken M, Dorner T, Burmester GR, Berek C. Differential expression of chemokine receptors on peripheral blood B cells from patients with rheumatoid arthritis and systemic lupus erythematosus. Arthritis Res Ther (2005) 7:R1001–13. doi:10.1186/ar1776
38. Sallusto F, Kremmer E, Palermo B, Hoy A, Ponath P, Qin S, et al. Switch in chemokine receptor expression upon TCR stimulation reveals novel homing potential for recently activated T cells. Eur J Immunol (1999) 29:2037–45. doi:10.1002/(SICI)1521-4141(199906)29:06<2037::AID-IMMU2037>3.3.CO;2-M
39. Brandes M, Legler DF, Spoerri B, Schaerli P, Moser B. Activation-dependent modulation of B lymphocyte migration to chemokines. Int Immunol (2000) 12:1285–92. doi:10.1093/intimm/12.9.1285
40. Lleo A, Liao J, Invernizzi P, Zhao M, Bernuzzi F, Ma L, et al. Immunoglobulin M levels inversely correlate with CD40 ligand promoter methylation in patients with primary biliary cirrhosis. Hepatology (2012) 55:153–60. doi:10.1002/hep.24630
41. Griffioen M, Ouwerkerk IJ, Harten V, Schrier PI. HLA-B down-regulation in human melanoma is mediated by sequences located downstream of the transcription-initiation site. Int J Cancer (1999) 80:573–80. doi:10.1002/(SICI)1097-0215(19990209)80:4<573::AID-IJC15>3.0.CO;2-S
42. Nie Y, Yang G, Song Y, Zhao X, So C, Liao J, et al. DNA hypermethylation is a mechanism for loss of expression of the HLA class I genes in human esophageal squamous cell carcinomas. Carcinogenesis (2001) 22:1615–23. doi:10.1093/carcin/22.10.1615
43. Doyle A, Martin WJ, Funa K, Gazdar A, Carney D, Martin SE, et al. Markedly decreased expression of class I histocompatibility antigens, protein, and mRNA in human small-cell lung cancer. J Exp Med (1985) 161:1135–51. doi:10.1084/jem.161.5.1135
Keywords: autoimmune cholangitis, epigenetics, environment
Citation: Selmi C, Cavaciocchi F, Lleo A, Cheroni C, De Francesco R, Lombardi SA, De Santis M, Meda F, Raimondo MG, Crotti C, Folci M, Zammataro L, Mayo MJ, Bach N, Shimoda S, Gordon SC, Miozzo M, Invernizzi P, Podda M, Scavelli R, Martin MR, Seldin MF, LaSalle JM and Gershwin ME (2014) Genome-wide analysis of DNA methylation, copy number variation, and gene expression in monozygotic twins discordant for primary biliary cirrhosis. Front. Immunol. 5:128. doi: 10.3389/fimmu.2014.00128
Received: 01 November 2013; Accepted: 13 March 2014;
Published online: 28 March 2014.
Edited by:
Paola Zanovello, University of Padova, ItalyReviewed by:
Giovanni Vazza, University of Padova, ItalyDaniele Calistri, IRCCS Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori, Italy
Copyright: © 2014 Selmi, Cavaciocchi, Lleo, Cheroni, De Francesco, Lombardi, De Santis, Meda, Raimondo, Crotti, Folci, Zammataro, Mayo, Bach, Shimoda, Gordon, Miozzo, Invernizzi, Podda, Scavelli, Martin, Seldin, LaSalle and Gershwin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlo Selmi, Division of Rheumatology and Clinical Immunology, Humanitas Research Hospital, via A. Manzoni 56, 20089 Rozzano, Milan, Italy e-mail: carlo.selmi@unimi.it