Corrigendum: Primary Immunodeficiency Diseases: An Update on the Classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency
 Waleed Al-Herz1,2
Waleed Al-Herz1,2 Aziz Bousfiha3
Aziz Bousfiha3 Jean-Laurent Casanova4,5
Jean-Laurent Casanova4,5 Talal Chatila6Mary Ellen Conley4
Talal Chatila6Mary Ellen Conley4 Charlotte Cunningham-Rundles7
Charlotte Cunningham-Rundles7 Amos Etzioni8
Amos Etzioni8 Jose Luis Franco9
Jose Luis Franco9 H. Bobby Gaspar10*Steven M. Holland11Christoph Klein12
H. Bobby Gaspar10*Steven M. Holland11Christoph Klein12 Shigeaki Nonoyama13
Shigeaki Nonoyama13 Hans D. Ochs14Erik Oksenhendler15,16
Hans D. Ochs14Erik Oksenhendler15,16 Capucine Picard5,17
Capucine Picard5,17 Jennifer M. Puck18
Jennifer M. Puck18 Kate Sullivan19Mimi L. K. Tang20,21,22
Kate Sullivan19Mimi L. K. Tang20,21,22
- 1Department of Pediatrics, Kuwait University, Kuwait City, Kuwait
- 2Allergy and Clinical Immunology Unit, Department of Pediatrics, Al-Sabah Hospital, Kuwait City, Kuwait
- 3Clinical Immunology Unit, Casablanca Children’s Hospital, Ibn Rochd Medical School, King Hassan II University, Casablanca, Morocco
- 4St. Giles Laboratory of Human Genetics of Infectious Diseases, Rockefeller Branch, The Rockefeller University, New York, NY, USA
- 5Laboratory of Human Genetics of Infectious Diseases, Necker Branch, INSERM UMR1163, Imagine Institut, Necker Medical School, University Paris Descartes, Paris, France
- 6Division of Immunology, Children’s Hospital Boston, Boston, MA, USA
- 7Department of Medicine and Pediatrics, Mount Sinai School of Medicine, New York, NY, USA
- 8Meyer Children’s Hospital-Technion, Haifa, Israel
- 9Group of Primary Immunodeficiencies, University of Antioquia, Medellin, Colombia
- 10UCL Institute of Child Health, London, UK
- 11Laboratory of Clinical Infectious Diseases, National Institute of Allergy and Infectious Diseases, Bethesda, MD, USA
- 12Dr. von Hauner Children’s Hospital, Ludwig-Maximilians-University Munich, Munich, Germany
- 13Department of Pediatrics, National Defense Medical College, Saitama, Japan
- 14Department of Pediatrics, Seattle Children’s Research Institute, University of Washington, Seattle, WA, USA
- 15Department of Clinical Immunology, Hôpital Saint-Louis, Assistance Publique-Hôpitaux de Paris, Paris, France
- 16Sorbonne Paris Cité, Université Paris Diderot, Paris, France
- 17Centre d’Étude des Déficits Immunitaires (CEDI), Hôpital Necker-Enfants Malades, AP-HP, Paris, France
- 18Department of Pediatrics, UCSF Benioff Children’s Hospital, University of California San Francisco, San Francisco, CA, USA
- 19Department of Pediatrics, Division of Allergy Immunology, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA
- 20Murdoch Childrens Research Institute, Melbourne, VIC, Australia
- 21Department of Paediatrics, University of Melbourne, Melbourne, VIC, Australia
- 22Department of Allergy and Immunology, Royal Children’s Hospital, Melbourne, VIC, Australia
We report the updated classification of primary immunodeficiencies (PIDs) compiled by the Expert Committee of the International Union of Immunological Societies. In comparison to the previous version, more than 30 new gene defects are reported in this updated version. In addition, we have added a table of acquired defects that are phenocopies of PIDs. For each disorder, the key clinical and laboratory features are provided. This classification is the most up-to-date catalog of all known PIDs and acts as a current reference of the knowledge of these conditions and is an important aid for the molecular diagnosis of patients with these rare diseases.
Background
The International Union of Immunological Societies (IUIS) Expert Committee on Primary Immunodeficiency met in New York on 19th–21st April 2013 to update the classification of human primary immunodeficiencies (PIDs). This report represents the most current and complete catalog of known PIDs. It serves as a reference for these conditions and provides a framework to help in the diagnostic approach to patients suspected to have PID.
As in previous reports, we have classified the conditions into major groups of PIDs and these are now represented in nine different tables. In each table, we list the condition, its genetic defect if known, and the major immunological and in some conditions the non-immunological abnormalities associated with the disease. The classification this year differs slightly from the previous edition in that Table 1 lists combined immunodeficiencies without non-immunologic phenotypes, whereas Table 2 refers to combined immunodeficiencies with syndromic features, as increasing numbers of these are being identified. The title and classification of Tables 3–8 present the same major PID groups as in the previous report.
TABLE 1
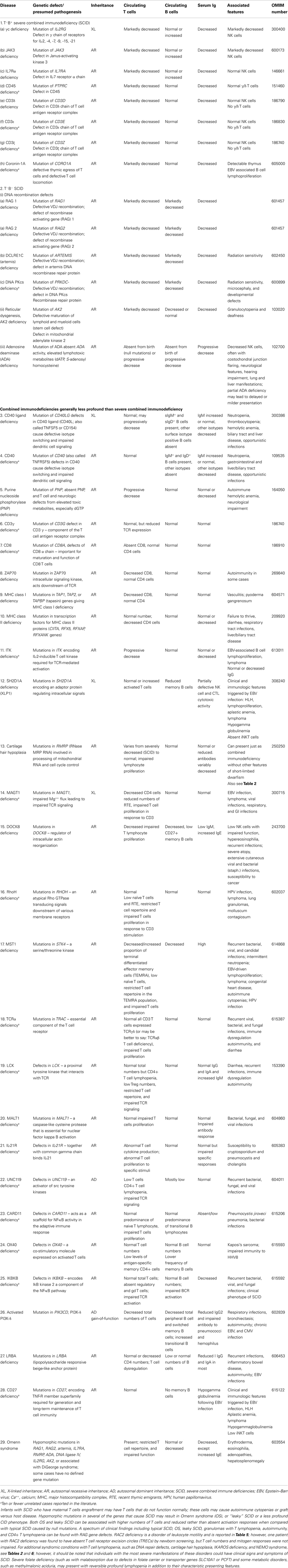
Table 1. Combined immunodeficiencies.
TABLE 2
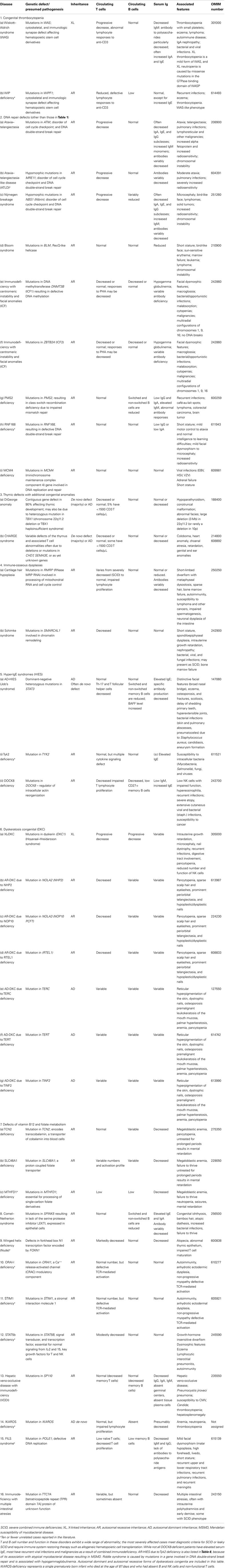
Table 2. Combined immunodeficiencies with associated or syndromic features.
TABLE 3
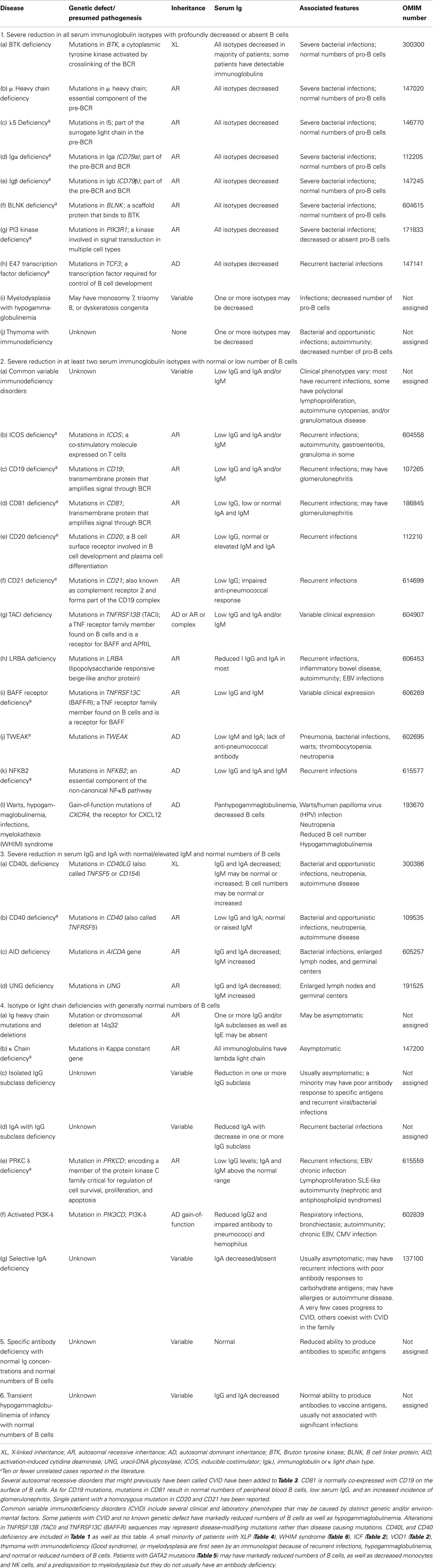
Table 3. Predominantly antibody deficiencies.
TABLE 4
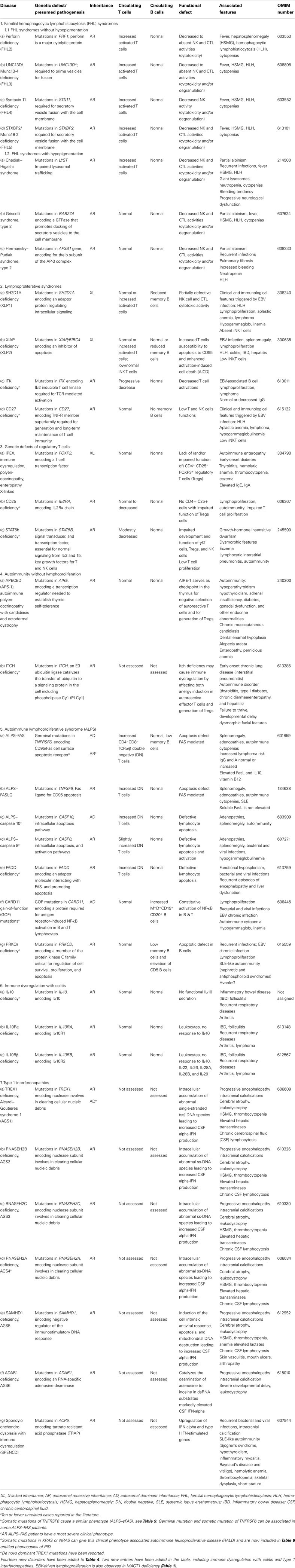
Table 4. Diseases of immune dysregulation.
TABLE 5
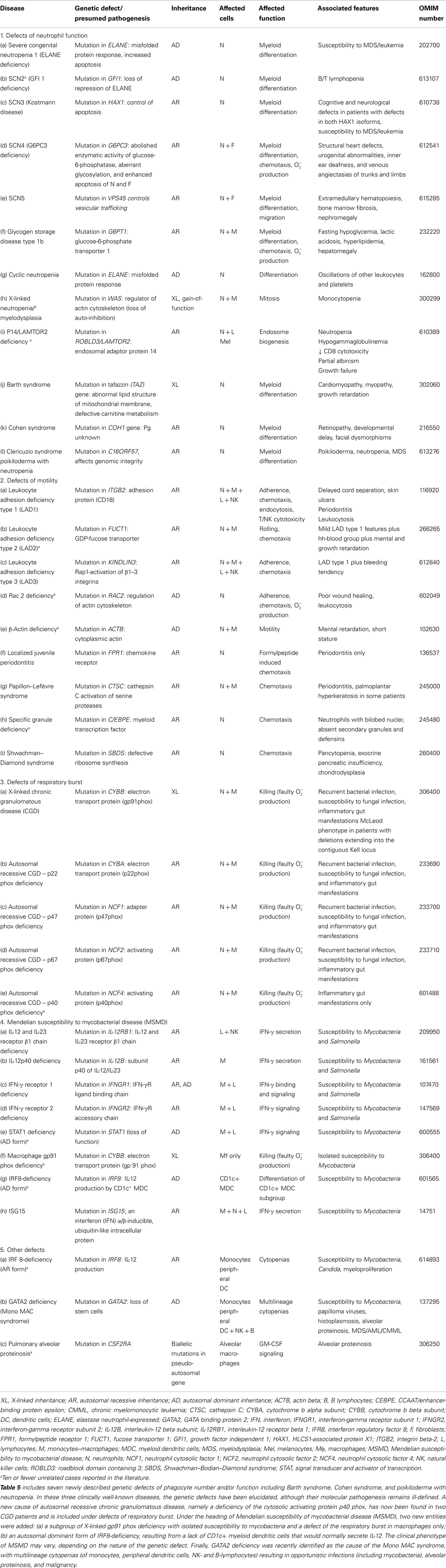
Table 5. Congenital defects of phagocyte number, function, or both.
TABLE 6
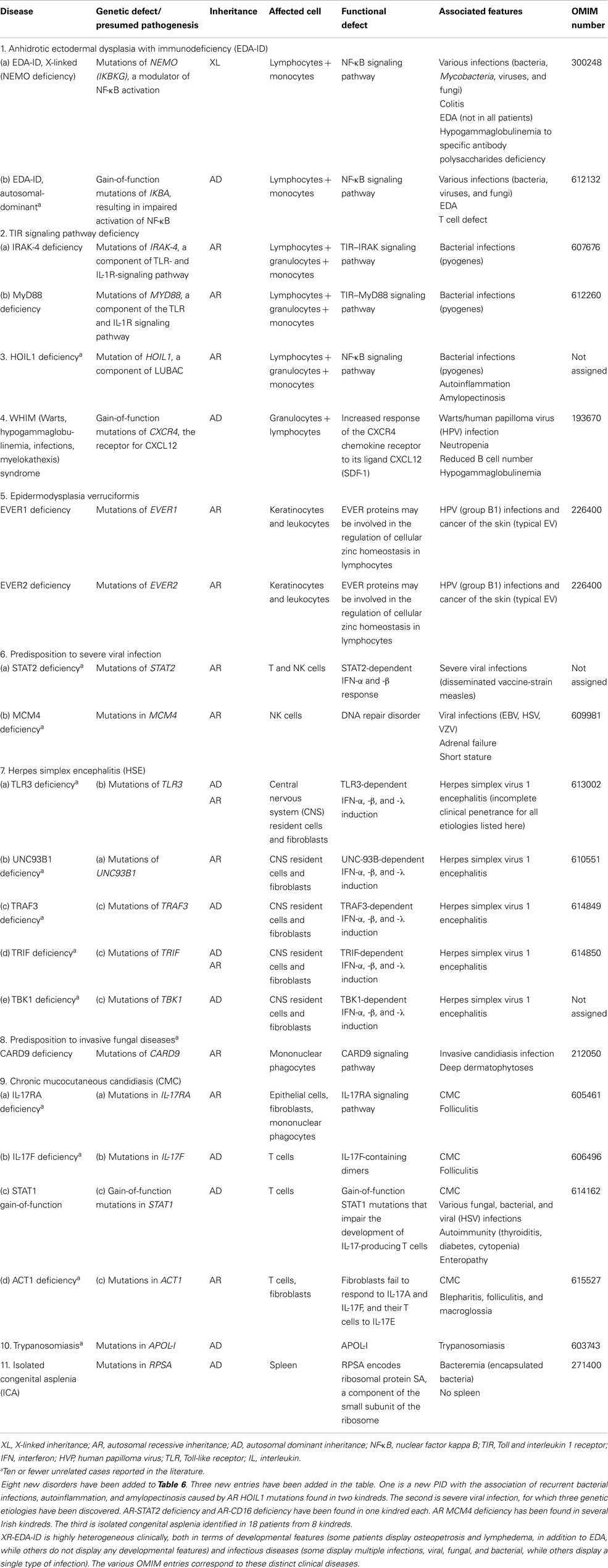
Table 6. Defects in innate immunity.
TABLE 7
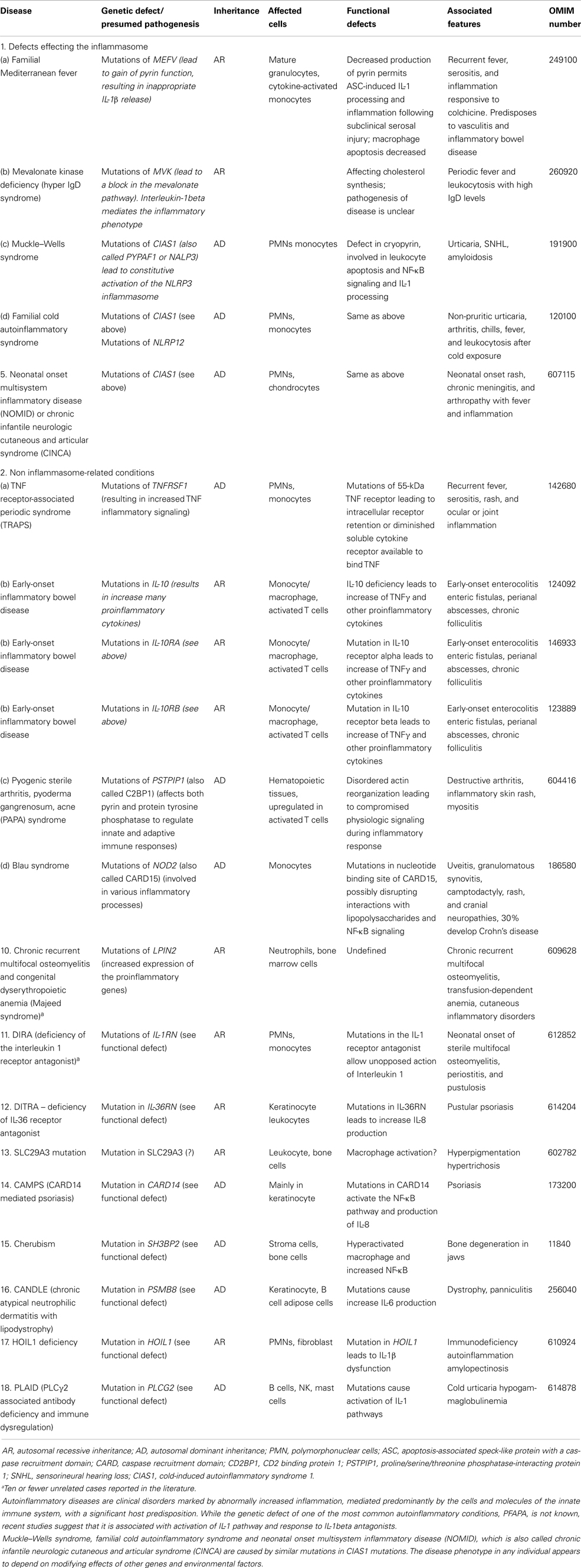
Table 7. Autoinflammatory disorders.
TABLE 8
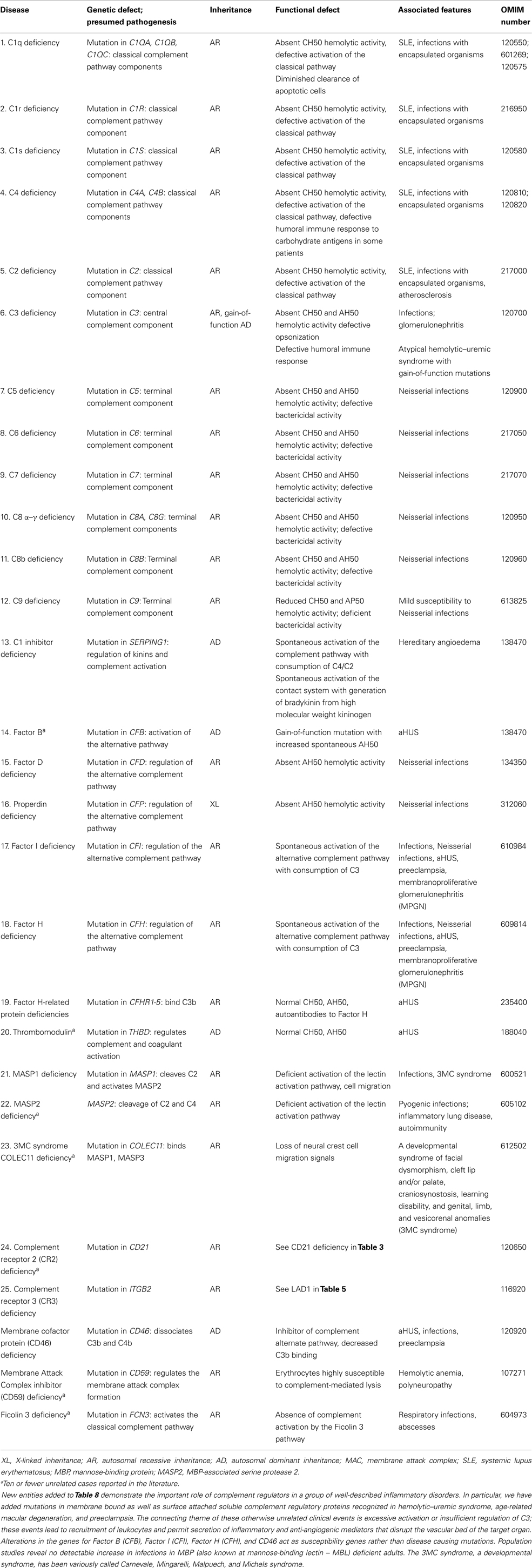
Table 8. Complement deficiencies.
In this updated version, we have added a new category in Table 9 in which “Phenocopies of PID” are listed. This has resulted from our understanding and study of conditions that present as inherited immunodeficiencies, but which are not due to germline mutations and instead arise from acquired mechanisms. Examples include somatic mutations in specific immune cell populations that give rise to the phenotype of autoimmune lymphoproliferative syndrome (ALPS), and also autoantibodies against specific cytokines or immunological factors, with depletion of these factors leading to immunodeficiency. It is likely that increasing numbers of PID phenocopies will be identified in the future, and this may be the start of a much longer table.
TABLE 9
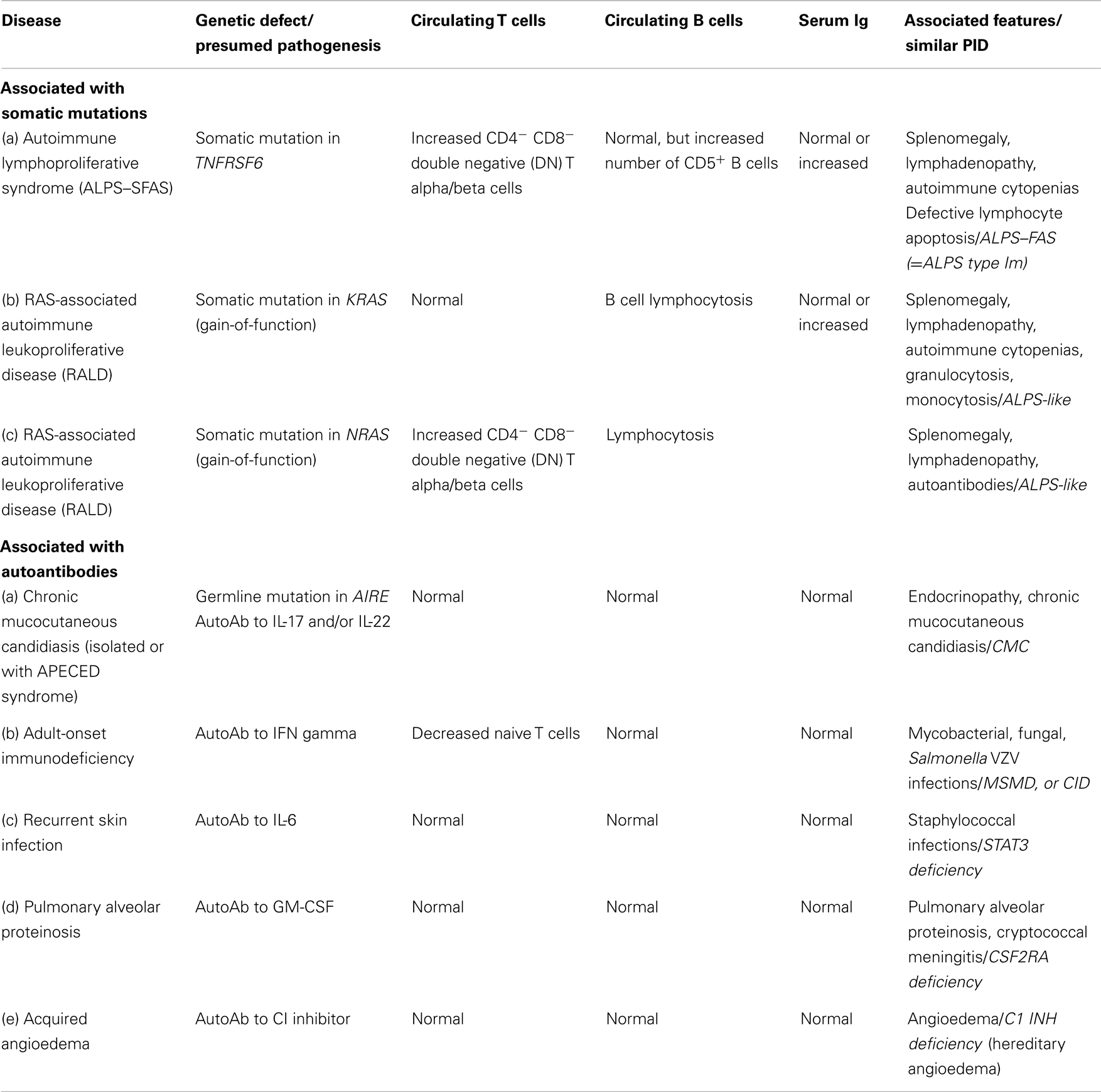
Table 9. Phenocopies of PID.
As with all complex diseases, any classification cannot be strictly adhered to. Certain conditions fall into more than one category and so appear in more than one table. For example, CD40L ligand deficiency is reported in both Tables 1 and 3 as it was initially identified as a defect of B cell isotype switching but is now known to be a defect of co-stimulatory T cell help and function. Similarly, XLP1 due to defects in SH2D1A is listed in Table 1 – combined immunodeficiencies, due to defects of T cell cytotoxicity, T cell help, and B cell maturation, but also in Table 4 – diseases of immune dysregulation, due to the susceptibility to hemophagocytosis. There is a growing appreciation that there can be wide phenotypic viability within a specific genotype that is a product of varied specific mutations between different patients as well as other host and/or environmental factors. The complexities of these conditions in terms of clinical and immunological presentation and heterogeneity cannot be easily captured in the limited space of a table format. For this reason, the furthest left column contains the Online Mendelian Inheritance in Man (OMIM) reference for each condition to allow access to greater detail and updated information.
The rapid advances in gene identification technology, including the widespread use of whole exome and whole genome sequencing, has meant that the ability to identify gene defects in affected families and even single individuals with inherited diseases has grown enormously. In this report, over 30 new gene defects have been added that were identified since the previous classification in November, 2011. These defects can be found in all major groups of PIDs included in this report. In many cases, the mutations are not necessarily in genes formally implicated in immune cell function but are genes involved in essential cell processes. The more detailed analysis and functional consequences of such defects as illustrated by these PIDs will increase our understanding of the interplay between different cellular processes in the development and function of the immune system.
Among the newly identified, gene defects are many that are to date particular to a single pedigree or individual; such defects may prove exceedingly rare, or indeed may not necessarily be found to recur in other individuals. We have marked conditions for which there are 10 or fewer reported individuals with an asterisk, although historically, following the description of the first few cases, additional individuals with a similar PID phenotype and genotype have often been recognized. It is likely that we will uncover many more “personal” or very rare gene defects over time and that the spectrum of PIDs will become increasingly diverse and complex, due to contributions of both environmental exposures and genetic modifiers to each affected individual. The value of this report therefore to capture and catalog the full spectrum at any one time point becomes increasingly important.
The goal of the IUIS Expert Committee on PIDs is to increase awareness, facilitate recognition, and promote optimal treatment for patients with PIDs. In addition to the current report and previous “classification table” publications, the committee has also produced a “Phenotypic Approach for IUIS PID Classification and Diagnosis: Guidelines for Clinicians at the Bedside,” which aims to lead physicians to particular groups of PIDs starting from clinical features and combining routine immunological investigations. Together, these contributions will hopefully allow a practical clinical framework for PID diagnosis. The committee also aims to establish a classification of PIDs based on other aspects and will work on publishing further guidelines in due course.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Keywords: primary immunodeficiencies, IUIS, classification, genetic defects, genotype
Citation: Al-Herz W, Bousfiha A, Casanova J-L, Chatila T, Conley ME, Cunningham-Rundles C, Etzioni A, Franco JL, Gaspar HB, Holland SM, Klein C, Nonoyama S, Ochs HD, Oksenhendler E, Picard C, Puck JM, Sullivan K and Tang MLK (2014) Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front. Immunol. 5:162. doi: 10.3389/fimmu.2014.00162
Received: 16 December 2013; Accepted: 27 March 2014;
Published online: 22 April 2014.
Edited by:
Jordan Orange, Baylor College of Medicine, USAReviewed by:
Jordan Orange, Baylor College of Medicine, USAFrancisco A. Bonilla, Boston Children’s Hospital, USA
Thomas Arthur Fleisher, National Institutes of Health, USA
Fischer Alain, INSERM, France
Copyright: © 2014 Al-Herz, Bousfiha, Casanova, Chatila, Conley, Cunningham-Rundles, Etzioni, Franco, Gaspar, Holland, Klein, Nonoyama, Ochs, Oksenhendler, Picard, Puck, Sullivan and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: H. Bobby Gaspar, Molecular Immunology Unit, UCL Institute of Child Health, 30 Guilford Street, London WC1N 1EH, UK e-mail: h.gaspar@ucl.ac.uk