 Faith C. Robertson
Faith C. Robertson Jay A. Berzofsky*
Jay A. Berzofsky* Masaki Terabe
Masaki Terabe- Vaccine Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
CD1d-restricted natural killer T (NKT) cells lie at the interface between the innate and adaptive immune systems and are important mediators of immune responses and tumor immunosurveillance. These NKT cells uniquely recognize lipid antigens, and their rapid yet specific reactions influence both innate and adaptive immunity. In tumor immunity, two NKT subsets (type I and type II) have contrasting roles in which they not only cross-regulate one another, but also impact innate immune cell populations, including natural killer, dendritic, and myeloid lineage cells, as well as adaptive populations, especially CD8+ and CD4+ T cells. The extent to which NKT cells promote or suppress surrounding cells affects the host’s ability to prevent neoplasia and is consequently of great interest for therapeutic development. Data have shown the potential for therapeutic use of NKT cell agonists and synergy with immune response modifiers in both pre-clinical studies and preliminary clinical studies. However, there is room to improve treatment efficacy by further elucidating the biological mechanisms underlying NKT cell networks. Here, we discuss the progress made in understanding NKT cell networks, their consequent role in the regulation of tumor immunity, and the potential to exploit that knowledge in a clinical setting.
Introduction
Effective tumor immunosurveillance is mediated by players from both the innate and adaptive immune systems. Innate immune cells (including macrophages, neutrophils, dendritic cells (DCs), and natural killer cells) rapidly respond to an immunological threat such as cancer. However, it is important that information from this front-line defense be transmitted to the adaptive immune system. Adaptive immune cells, including T and B lymphocytes, mediate tumor immunity in a more precise and lasting manner via antigen-specific and effector memory responses. A few key players in immunosurveillance lie at the interface of innate and adaptive immunity and facilitate this transition, one of which is natural killer T (NKT) cells. In an innate fashion, NKT cells have the ability to react quickly, producing a broad range of cytokines within minutes to hours of antigenic stimulation and also activate antigen-presenting DCs. This rapid effect can trigger powerful innate and acquired immune responses both directly and indirectly. Thereby, NKT cells bridge two major defense systems to modulate immunity and are important in influencing host immune responses to cancer and other diseases. Here, we review the current understanding of NKT cells regarding their role in enhancing or suppressing tumor immunity, as well as ongoing strategies using NKT cells for cancer immunotherapy and discuss future approaches that might optimize NKT cell-based anti-tumor responses.
Natural Killer T Cells
Natural Killer T cells are a heterogeneous lymphoid population that exhibits characteristics from both the innate and adaptive arms of the immune system. Similar to innately functioning NK cells, these lymphocytes react quickly to stimuli and produce an array of cytokines and chemokines to modulate the immune response (1, 2). NKT cells respond in an antigen-specific manner through a true T cell receptor (TCR). However, unlike TCRs from conventional T cells that recognize one epitope (or a few at most), a single TCR from NKT cells can react with numerous antigens, both self and foreign. Thus, even though the absolute frequency of NKT cells is low (e.g., ~1% in mouse spleen), their number is high in the context of T cells specific for one antigen, high enough to initiate a significant immune response (3–5). Additionally, they differ from conventional T cells that recognize peptide antigen presented by class I or II major histocompatibility complex molecules (MHCI or II) on antigen-presenting cells (APCs). Instead, NKT cells interact with lipid antigens presented by an MHC-like molecule known as CD1d. In fact, because many NKT cells do not express NK cell markers, the defining characteristic of NKT cells is now CD1d restriction, and NK-like markers are primarily used to characterize subsets (6).
Natural killer T cells may have both immune enhancing and immunosuppressive roles. Upon antigenic stimulation, NKT cells are able to produce a range of cytokines including interferon-gamma (IFN-γ), interleukins (IL)-2, -4, -10, -13, -17, -21, and 22, granulocyte-macrophage colony-stimulating factor (GM-CSF), and tumor necrosis factor-alpha (TNF-α) (7–9). The cytokine profile produced differs slightly among subsets, but interestingly, NKT cells can simultaneously secrete Th1/pro-inflammatory (e.g., IFN-γ, TNF-α) and Th2/anti-inflammatory (IL-4, IL-10, IL-13) cytokines. The cytokines secreted activate other immune cells such as NK cells, T cells, and B cells, potentiating a cascade of cytokine and chemokine production and galvanizing additional cytolytic populations to mediate tumor surveillance (2). Because, NKT cell activation contributes to DC maturation (10), and in turn enhances both priming and boosting of CD8+ T cells, co-administering peptide, or protein antigen with NKT cell agonists has an adjuvant effect. This activity can be channeled toward improving vaccines for cancer, infectious diseases, and other therapeutic settings (10–12).
The NKT cell population is composed of several phenotypically and functionally different subpopulations, and can be characterized further according to tissue location, surface markers, specific TCR usage, and effector functions (Figure 1). Surface markers CD4 and CD8 broadly divide NKT cells, where a majority is CD4+ (approximately 90% in mice) and the remainder is CD4−CD8− double negative (DN). In human beings, additional minute populations of CD8αα and CD8αβ NKT cells exist (13). Activated human DN NKT cells predominantly produce cytokines that instigate a Th1 response, while CD4+ NKT cells generate both Th1 and Th2 cytokines (1, 14, 15). NKT cells can also be subdivided into NK1.1+ or negative, with different functional consequences (16). Aside from CD4 and CD8 expression, functional differences in the nature and magnitude of the NKT cell response can arise from developmental differentiation and tissue location. For instance, NKT cells from human peripheral blood generate cytokines similar to those isolated from spleens but differ significantly from NKT cells in the thymus or cord blood (17). Peripheral blood NKT cells are also distinct from liver NKT cells in terms of surface marker expression (18). Because, human NKT cells from peripheral blood are commonly used as a surrogate for systemic NKT cell analysis, this discrepancy suggests that current protocols analyzing NKT cell number and functionality may be misleading. NKT cell frequency and expression of cell surface proteins has been shown to differ between various sites in the small and large intestine as well (19). Functional differences of NKT cells from different tissues have also been demonstrated in the context of tumor (20). These comparative analyses provide insight that NKT cell tissue location should be considered when extrapolating information from experimental or clinical data, and underscores that a clearer description of tissue-specific characteristics of NKT cells is needed.
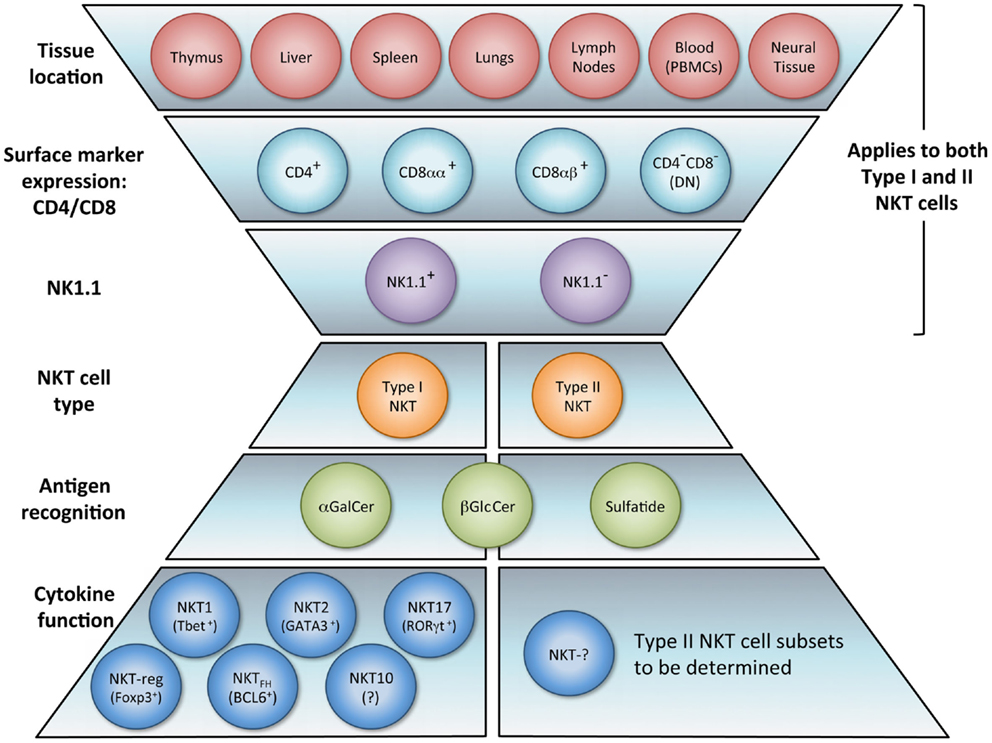
Figure 1. NKT cell subsets. The natural killer T (NKT) cell population encompasses several phenotypically and functionally different subpopulations. Tissue location and surface markers (CD4, CD8, and NK1.1) are defining characteristics that broadly divide NKT cells and contribute to functionality. Differences in TCR rearrangements allow separation into two major subsets, type I and type II. Type I NKT cells express a semi-invariant TCRα chain, while type II NKT cells display a more diverse repertoire. It has been proposed that these NKT cell subsets recognize distinct lipid antigens. The prototypic antigen able to activate all type I NKT cells is α-GalCer. Type II NKT cells recognize a greater variety of antigens, one being sulfatide. Though these two subsets have been reported to recognize some common antigens, e.g., β-GlcCer, the biochemical structure is slightly different between the antigen recognized by type I versus type II NKT cells. Lastly, type I NKT cells are functionally heterogeneous. NKT1, NKT2, NKT17, NKTreg, NKTFH, and NKT10 subsets have been described. Overall, an in vivo NKT cell response likely depends on which subsets are activated.
Heterogeneity of TCR rearrangements has allowed NKT cells to be separated into two categories, type I and type II (as described below). In the context of tumor immunity, these subsets have been shown to differentially impact innate and adaptive immune cell populations. Type I NKT cells are usually associated with the promotion of tumor immunity whereas type II NKT cells seem to suppress it (21–27).
Type I NKT Cells
Type I NKT cells express a semi-invariant TCRα chain (Vα14-Jα18 TCR in mice, Vα24-Jα18 in human beings) paired with a limited repertoire of Vβ chains (primarily Vβ8, 7 and 2 in mice, Vβ11 in human beings) and are consequently referred to as invariant or iNKT cells. In type I NKT cells, it appears that a combination of activation variables dictates NKT cell function: the affinity of the antigen presented to the NKT TCR; the presence of costimulatory molecules; and the tissue environment in which the interaction takes place (7, 28). The prototypic antigen for type I NKT cells is α-galactosylceramide (α-GalCer or KRN7000), a synthetic form of a glycolipid isolated from a marine sponge (29, 30). Type I NKT cells also recognize microbial glycolipids and self-antigens, e.g., Sphingomonas and Ehrlichia lipids, lyso-phosphatidylcholine (lyso-PC), and isoglobotrihexosylceramide (iGb3) (31–35). α-GalCer is a potent activator of all type I NKT cells, causing them to produce copious amounts of IFN-γ, which helps activate both CD8+ T cells and APCs (36). NKT cells specifically stimulate DCs through the CD1d-TCR complex and CD40–CD40L interaction, which induces DC maturation and IL-12 secretion (37, 38). IL-12 stimulates both NK, NKT, and other T cells to produce more IFN-γ, and the two cytokines together significantly impact the activation of downstream effector populations such as NK cells, CD8+ T cells, and γδ T cells (39). NKT cell activation also causes DCs to upregulate costimulatory receptors (e.g., CD70, CD80, and CD86). CD70 expression by DCs is essential for cross-priming CD8+ T cells to promote adaptive immunity (40–42). IL-2 produced by activated NKT cells induces the proliferation of memory CD4+ T helper 1 (Th1) and Th2 cells (43). Additionally, because differentiation of CD4+ T cells into T helper cell subsets depends on the cytokine milieu, cytokines from NKT cells may facilitate polarization into Th1, Th2, and/or Th17 subsets. Having these innate and acquired immune reactions occur simultaneously is integral for a potent immunological response, especially for eradication of tumor masses, which frequently contain both MHC-negative cells (targeted by NK cells) and MHC-positive cells (targeted by CD8+ T cells) (44).
Of recent interest are unique cytokine producing subsets of type I NKT cells, particularly those making IL-17. A study analyzing subsets according to tissue origin and CD4 and NK1.1 marker expression found significant diversity of cytokine production by distinct subsets, especially CD4−NK1.1− NKT cells that produce high levels of IL-17 (16, 45). IL-17 has potent pro-inflammatory functions including the induction of IL-6 and TNF-α, as well as the recruitment and enhancement of neutrophils. Analogous to CD4+ Th17, primary producers of IL-17, this NKT cell lineage constitutively expresses the RORγ-t transcription factor, as well as IL-23R (46). However, the NKT17 population was isolated from naïve animals without priming, and was able to secrete IL-17 as soon as 2–3 h following antigen stimulation, whereas naïve CD4+ T cells must undergo a differentiation period of a few days before antigen can polarize the cell into Th17 phenotype and elicit such a response. Other reports have further described this NKT cell subset by IL-17R expression and lack of NK1.1 expression, or added that mature differentiation of CD44+CD4−NK1.1−cells manifests in stage 2 of thymic development (46, 47). Additional type I NKT cells have been characterized by their transcription factor expression and effector functions in a manner comparable to other CD4+ T helper subsets [Th1, Th2, Tregs, T follicular helper cells (TFH)] (48–50). Lee et al. report that NKT1, NKT2, and NKT17 cells are programed intrathymically to elicit a particular cytokine profile (49). Others showed that Foxp3+ type I NKT cells behave similarly to Tregs and suppress conventional CD4+ T cell proliferation in a contact-dependent and antigen-independent manner (48). A subset of IL-10-producing NKT cells was recently reported by Sag et al. (51). This demonstrates diverse lineages of molecularly distinct type I NKT cell subsets that differ functionally in their production of particular cytokines. Our incomplete knowledge of the heterogeneity within type I NKT cells has likely hindered progress in harnessing the true potential of NKT cells; NKT cell responses in vivo likely depend on which subsets are activated. A better understanding of NKT cell subcategories could inform more selective and focused mechanisms for immunological intervention.
Type II NKT Cells
In contrast, CD1d-restricted NKT cells not expressing the semi-invariant TCR are known as type II. This subset recognizes glycolipid antigens distinct from those recognized by type I NKT cells and is less well characterized. For those reactive to sulfatide, a myelin-derived glycolipid found abundantly in the nervous system, the type II NKT TCR repertoire shows preferential use of alpha gene segments from Vα1 and Vα3, paired with Vβ8.1/Vβ8.3 (52). Because, type II NKT cells do not utilize the Vα14-Jα18 gene segment, they can be studied by comparing wild type (WT) mice that have both type I and II NKT cells, to Jα18 knockout (KO) mice that lack type I NKT cells but retain type II, and to CD1d KO mice, which are deficient in all NKT cells. Type II NKT cells have also been studied with TCR transgenic mice expressing the 24αβ-TCR from the CD4+ type II NKT cell hybridoma VIII24 that expresses a Vα3.2 and Vβ9 rearrangement and is not reactive to sulfatide (53). Additionally, because NKT cells constitutively express IL-4 mRNA transcripts, the use of IL-4 GFP enhanced transcript (4get) mice has introduced another type II NKT subset (54, 55). Liver NKT cells from 4get Jα18-deficient mice were sorted to obtain TCRβ+GFP+ α-GalCer/CD1d tetramer-negative cells. This population produced IFN-γ when CD1d-expressing bone marrow-derived DCs presented several types of lipids, including β-d-glucopyranosyl ceramide (β-GlcCer), β-GalCer, and Lyso-PE. The pool of β-GlcCer reactive type II NKT cells did not respond to sulfatide and favored TCR gene segments from Vα8 and Vβ8.1/8.2 (55). It is apparent that there are many distinct populations within the type II NKT cell division; however, even with the recent methodological advances that have enhanced our ability to study this non-invariant subset, much less is known about type II than type I NKT cells.
NKT Cell Enhancement of Tumor Immunity
In tumor immunosurveillance, NKT cells can directly kill malignant cells, or combat cancer indirectly via activation of additional immune cells (Figure 2). Type I NKT cells employ various mechanisms of cytolytic activity. For instance, both mouse and human NKT cells can directly lyse tumor cells by a perforin-dependent mechanism (56), and intracellular granzyme B expression may also potentiate cell killing (57). Tumor cells expressing CD1d may be especially susceptible to direct NKT cell lysis, as shown in vitro (58–60). This pattern is consistent with in vivo observations (61, 62) and there is evidence that high CD1d expression levels correlate with lower metastasis rates (60).
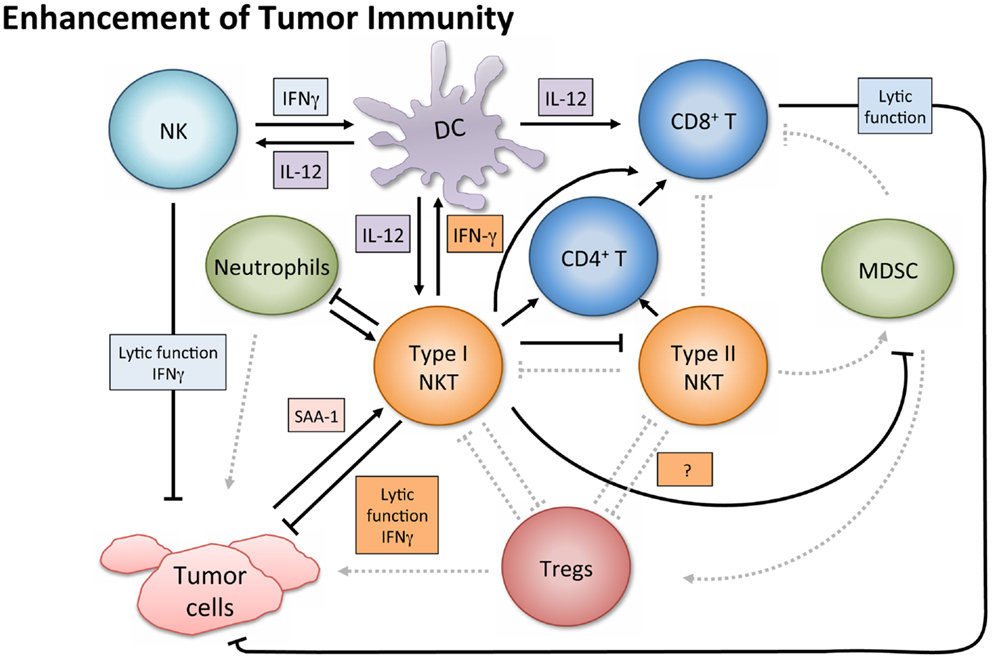
Figure 2. Enhancement of tumor immunity. Upon antigenic stimulation, type I NKT cells produce copious amounts of IFN-γ, which helps activate both CD8+ T cells and DCs. NKT cells specifically induce DC maturation by engaging the CD1d-TCR complex and CD40–CD40L interaction. DCs then upregulate costimulatory receptors essential for the cross-priming of CD8+ T cells to promote adaptive immunity. Additionally, IL-12 production by DCs stimulates NK, NKT, and other T cells to produce more IFN-γ and the two cytokines together significantly impact the activation of downstream effector populations. Both type I and type II NKT cells have been shown to enhance proliferation of memory CD4+ T cells, which can help CD8+ T cells as well. NK and type I NKT cells are able to directly lyse tumor cells utilizing various mechanisms which include perforin, granzyme, and FasL. Type I NKT cell stimulation can also enhance tumor immunity by hindering immunosuppressive populations. Type I NKT cells regulate effects of type II and expansion of MDSCs. Tumor-induced inflammatory proteins like serum amyloid A 1 (SAA-1) have been shown to increase neutrophilic CD1d-dependent stimulation of type I NKT cells, which then mitigates the detrimental activity of suppressive neutrophils by hindering production of IL-10 and enhancing IL-12.
While type I NKT cells are undoubtedly capable of mediating tumor lysis directly, several studies in both human beings and mice demonstrate NKT cell-mediated anti-tumor effects that are independent of lytic capacity and largely dependent on activating other innate and adaptive immune cells (20, 51, 63, 64). This recruitment of anti-tumor cytolytic cell populations primarily involves initiating Th1 cytokine cascades. As mentioned above, α-GalCer was the first NKT cell ligand identified and is a potent activator of type I NKT cells. It showed clinical potential when application of its synthetic form (KRN7000) increased survival in mice bearing the B16 melanoma (29, 58). The copious amount of IFN-γ released upon α-GalCer stimulation appears necessary for tumor protection (12, 65). In lung and liver metastasis models, production of IFN-γ by NKT cells and NK cells was absolutely required to obtain anti-metastatic activity of α-GalCer, and IL-12 and IL-18 were necessary to achieve optimal serum IFN-γ induction and tumor immunity (63). Type I NKT cells can also mediate tumor immunosurveillance in the absence of exogenous antigen. In a model of MCA-induced fibrosarcoma, Jα18 KO mice lacking type I NKT cells had a greater susceptibility to the disease. Adoptive transfer of liver DN type I NKT cells from WT into Jα18 KO mice restored the NKT cell population and rescued tumor immunity. However, CD1d KO mice were not protected upon NKT cell transfer, demonstrating that the protection was CD1d-dependent (20, 22). Additionally, only the DN liver subset conferred protection in this model; the CD4+ subset of liver NKT cells were not protective, and when type I NKT cells from the thymus were adoptively transferred, only slight protection was observed. Notably, the thymic NKT cells produced more IL-4 than those from liver or spleen. Thus, even in the absence of exogenous antigenic stimulation, it is becoming clearer that different subsets of NKT cells play distinct roles in immunosurveillance (66).
While surface marker expression and anatomical origin may influence NKT cell function, different antigens can also impact the immunological capacity of NKT cells. Examining both the cytokine profile and tumor protection capacity within a panel of type I NKT agonists, there was a strong correlation between the Th1 profile (in terms of IFN-γ:IL-4 ratio or IFN-γ production) and magnitude of protection in the mice from tumor growth in CT26 lung metastasis model (67). Least effective in the panel was OCH, which induced some IFN-γ but was the most Th2 skewed (IL-4 and IL-13) cytokine response in this set. OCH still suppressed tumor growth, but less than antigens eliciting greater Th1 skewing. Because, IFN-γ production appears to be the best correlate for tumor protection, and because, simultaneous NKT cell production of both Th1 and Th2 type cytokines may lessen the beneficial Th1 profile effects, multiple groups have tried enhancing α-GalCer to heighten IFN-γ production (68–72). A synthetic C-glycoside analog, α-C-galactosylceramide (α-C-GalCer), stimulated prolonged production of IFN-γ and IL-12 in vivo, and decreased production of the Th2 cytokine IL-4 compared with α-GalCer (69), as confirmed in our hands as well (O’Konek unpublished). This enhanced protection against B16 melanoma pulmonary metastases (69). An induced fit antigen with enhanced binding to CD1d, naphthylurea (NU)-α-GalCer, heightened Th1 biased cytokine skewing, and conferred better tumor protection than α-GalCer (72). In the same study, a type I NKT cell agonist that stimulated a weak Th2 biased cytokine production (xylo-α-GalCer) had no protective effect. Analog 7DW8-5, which possesses a fluorinated benzene ring, improved NKT cell stimulation, and exhibited an adjuvant effect superior to that of α-GalCer on HIV and malaria vaccines (73). Analogs containing phenyl group(s) on the lipid tail of α-GalCer also seem to steer NKT cells toward a Th1-skewed response (70), and exhibited more potent anticancer activities than α-GalCer in mice by inducing neither NKT cell anergy nor expansion of myeloid-derived suppressor cells (MDSCs) (70, 74). Overall, superior Th1-biasing antigens have shown enhanced tumor protection and may warrant translation to the clinic (72).
Aside from augmenting protective cell responses in both arms of the immune system, NKT cells can enhance tumor immunity by altering the effects of immunosuppressive cells. For instance, in a study on influenza A virus infection, the absence of type I NKT cells resulted in the expansion of MDSCs, which suppressed CD8+ T cell immune responses (75). Adoptively transferred liver NKT cells restored the WT phenotype, and the result was shown to be CD1d-dependent. Although the protection did not require stimulation of transferred type I NKT cells by an exogenous antigen, activating NKT cells with α-GalCer reversed MDSC-mediated suppression in a CD40–CD40L manner. Because some MDSCs (e.g., those activated by tumor-derived IL-1β) impair NK cell development and function (76), having NKT cells present to prevent MDSC expansion may enhance NK cell activity as well. NKT cells can also counter tumor-driven immunosuppressive neutrophils (77). Tumor-induced inflammation causing secretion of serum amyloid A1 (SAA-1) induced differentiation of suppressive IL-10-producing neutrophils. However, SAA-1 also increased interaction between neutrophils and type I NKT cells. The suppressive neutrophils expressed both CD40 and CD1d and the presence of SAA-1 triggered a neutrophilic CD1d-dependent activation of type I NKT cells comparable to stimulation with α-GalCer-pulsed neutrophils. Type I NKT cells not only mitigated the detrimental effect of neutrophils by hindering production of IL-10, and enhancing IL-12, but also restored proliferation of antigen-specific CD8+ T cells. Because this effect was proportional to the numbers of type I NKT cells added in vitro, it would be interesting to see if greater quantities of NKT cells in vivo would shift the equilibrium in immune reaction to SAA-1 further, decreasing IL-10 mediated suppression, and restoring tumor immunity. This balance and crosstalk underscores the importance of type I NKT cells in the regulation of tumor immunity and introduces an opportunity to exploit NKT agonists in tumor settings where inflammatory proteins like SAA-1 are released.
NKT Cells in Suppression of Tumor Immunity
In contrast to their immune enhancing role in cancer, NKT cells, especially type II, have displayed immunosuppressive activity in tumor immunology (Figure 3). A series of studies using multiple tumor models demonstrated that type II NKT cells were sufficient for down-regulation of tumor immunosurveillance, within the context of type I and type II NKT cells (26, 27, 61, 78). CD4+ type II NKT cells produced more of IL-13 and IL-4 than type I cells, and the NKT cell-dependent IL-13 was necessary for tumor recurrence in a growth-regression-recurrence pattern 15-12RM fibrosarcoma tumor model (24). The immunosuppressive effect seemed mediated by the sulfatide-reactive subset of type II NKT cells, as sulfatide treatment enhanced tumor growth in a CD1d-dependent manner (27). IL-13 initiated signaling through the IL-4R-STAT6 pathway, which together with TNF-α led to an increase in production of transforming growth factor beta (TGF-β) by a CD11b+Gr1+ population, sometimes called MDSCs (25, 79). TGF-β is a pleiotropic cytokine with three isoforms and TGF-β1 has been shown to down-regulate cytotoxic T cell lymphocyte (CTL) activity and in turn repress tumor immunosurveillance in several mouse tumor models. Blocking TGF-β or depleting Gr-1+ cells in vivo prevented tumor recurrence (25). Type II NKT cells were also shown to suppress immunity against CD1d-transfected B cell lymphoma; amounts of IL-13, TGF-β, and MDSCs correlated with immune suppression (61).
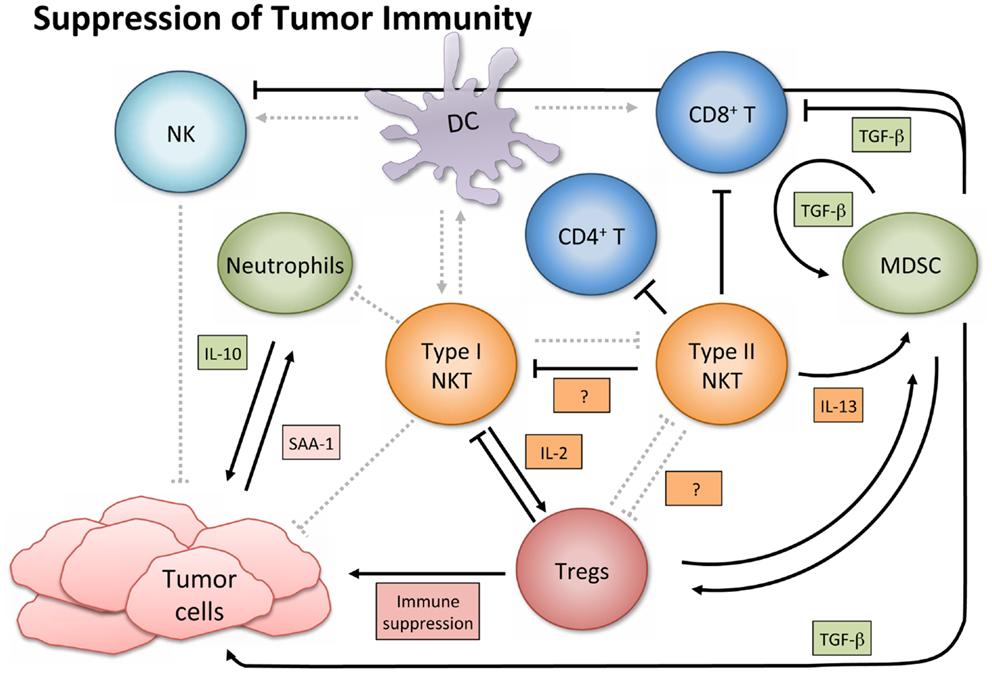
Figure 3. Suppression of tumor immunity. Activated type I NKT cells have been shown to support immunosuppressive Tregs through IL-2 production, and are then suppressed by Tregs in a cell-contact-dependent manner. Treg cells can then suppress CD8+ and CD4+ T cells and NK cells (not shown). Sulfatide-reactive type II NKT cells suppress CD8+ T cells and inhibit proliferation of naïve, but not memory, CD4+ T cells. Type II NKT cells also suppress Type I, and while the exact mechanism in cancer settings remains unknown, the mechanism in a con-A-induced hepatitis model is believed to involve pDCs. Type II NKT cell production of IL-13 functions with TNF-α from other cells to increase production of TGF-β by a CD11b+Gr1+ population known as myeloid-derived suppressor cells (MDSCs). MDSCs not only directly support tumor growth with TGF-β production but also suppress other immune cells (e.g., CD8+ T and NK cells), feed into an autocrine loop to enhance development of additional tumor-associated MDSCs, and aid in expansion of Tregs. When tumor-induced inflammatory proteins like SAA-1 stimulate suppressive IL-10 producing neutrophils, and type I NKT cells are not activated to alter that reaction, increased levels of IL-10 can induce Treg cells and potentiate further tumor growth.
Myeloid-derived suppressor cells accumulate in the bone marrow and lymphoid organs of cancer patients and can counteract T cell responses. Because type II NKT cells appear to enhance MDSC activity, manipulating the NKT-MDSC immune axis has therapeutic implications. However, what works in one tumor model does not always translate to others. We investigated the anti-tumor effects of neutralizing TGF-β in conjunction with a peptide vaccine (80). In a pre-clinical study using a TC1 tumor in C57BL/6 mice, a pan-inhibitor of all three TGF-β isoforms had no impact as a single agent, suggesting that the mechanism of immunosuppression was different from that in 15-12RM. However, TGF-β neutralization significantly and synergistically improved cancer vaccine efficacy by reducing growth of primary tumors, and increasing tumor antigen-specific CTLs (80). Similar synergy between a vaccine and a TGF-β antagonist has been reported in multiple tumor models with multiple vaccine platforms (81–83). Currently, because the third isoform (TGF-β3) is hypothesized to have beneficial physiological effects in cancer (84), we are investigating the role of different TGF-β isoforms in the tumor setting and in conjunction with a cancer vaccine that contains a tumor antigen and a type I NKT agonist. It may not be necessary to block all three isoforms of TGF-β to promote tumor immunity or enhance cancer vaccine efficacy (Terabe et al., manuscript in preparation). This could be utilized in a push-pull approach (85) to maximize vaccine efficacy. TGF-β1 production by MDSCs not only directly suppresses other immune cells (e.g., CD8+ T cells) but also feeds into an autocrine loop to enhance development of additional tumor-associated MDSCs (86). MDSCs have also been shown to induce expansion of immunosuppressive, tumor-specific Tregs (87). Consequently, whether or not the MDSC activation is mediated by IL-13 from type II NKT cells, neutralizing TGF-β could interrupt autocrine and paracrine TGF-β loops driving suppression of tumor immunity (86, 88). Simultaneously, a type I NKT cell agonist could aid in the maturation of DCs, the suppression of MDSCs, and the enhancement of NK and CD8+ T cells to restore the anti-tumor immune response, essentially killing several birds with one stone.
There are additional mechanisms of NKT cell-mediated tumor suppression as well. In a K7M2 mouse osteosarcoma model, it was found that 88% of NKT cell-deficient mice allowed for the rejection of osteosarcoma primary tumors, compared to a 24% rejection rate in WT mice containing both type I and type II NKT cells (89). Tumor rejection was shown to be CD8+ T cell-dependent, and CD1d KO mice that rejected the tumor had significantly higher numbers of tumor infiltrating lymphocytes. While previously TGF-β and IL-13 mediated such suppression, TGF-β and IL-13 were not the drivers of immunosuppression in this model, and even though the exact mechanism was not revealed, this study highlighted that alternative pathways exist for CD1d-dependent NKT cell-mediated immunosuppression. For instance, we recently found that sulfatide-reactive type II NKT cells inhibit proliferation of naïve, CD4+ T cells (90), whereas they may not suppress memory CD4+ T cells (43). Because CD4+ T cells aid CD8+ T cells, it is possible that in situations where NKT cells interrupt CD8+ T cell-immunity and tumor rejection, NKT cell suppression of naïve CD4+ T cells may contribute (90).
Although in the majority of tumor settings the immunosuppressive role of NKT cells has been ascribed to type II NKT cells, while type I are associated with an improved anti-tumor immune response, there are exceptions (91–93). Paradoxically, activated type I NKT cells have been shown to support immunosuppressive Tregs through IL-2 production, though the NKT cells are subsequently suppressed by Tregs in a cell-contact-dependent manner (94). Two studies of note have reported that type I NKT cells directly suppress tumor immunity in hematological malignancies. In a model of Burkett’s-like B cell lymphoma, Jα18 KO mice deficient in type I NKT cells had significantly fewer splenic tumors than WT or CD1d KO mice. While stimulation of type I NKT cells with α-GalCer did not increase tumor burden, it decreased tumor-specific CD8+ T cells (91). Given a T cell lymphoma cell line transfected with CD1d, NKT deficient mice had augmented CTL activity and greater survival rates than WT mice (92). Accordingly, the authors suggested that tumor expression of CD1d might shift the NKT cell response toward a Th2 skewed anti-inflammatory reaction, which produces more IL-13, TGF-β, and inhibition of CTL and NK cell activity.
Cross Regulation
Type I and type II NKT subsets have been shown to not only differentially impact cell populations but also cross-regulate one another. This immunoregulatory axis was realized when Jα18 KO mice, which lacked type I NKT cells showed higher susceptibility to tumor growth than WT mice in a CT26 tumor pulmonary metastasis model, whereas CD1d KO mice deficient in both types showed strong resistance against the tumor. This led us to believe that type I NKT cells may be acting to reduce the suppressive effect of type II NKT cells (27). Sulfatide activation of type II NKT cells in vivo enhanced tumorigenesis, and abolished or reduced the beneficial clinical effects of α-GalCer when the two were administered together. Thus, it appeared that type II NKT cells may also suppress type I NKT cell activation (26, 27). The latter effect likely results from decreased pro-inflammatory cytokine secretion by type I cells. In vitro, α-GalCer-induced IFN-γ, IL-2, and IL-4 production was inhibited upon addition of the type II antigen sulfatide (27, 95). Whereas this effect may not always be entirely due to type II mediated suppression, but occur partially or in some cases because sulfatide competes with α-GalCer in binding CD1d (95), this competitive binding mechanism was excluded in the settings described by Ambrosino et al. (27) In a model of concanavalin A-induced hepatitis, it was demonstrated that activation of sulfatide-reactive type II NKT cells and plasmacytoid DCs in the liver contributed to the anergy, or hyporesponsiveness of type I NKT cells (96). It is unclear if this mechanism is the same in the tumor setting. Nonetheless, the discovery that protective type I and suppressive type II NKT cells cross-regulate one another defines an NKT regulatory axis, provides a better understanding of the tumor immunity equilibrium, and opens a window of opportunity to exploit that knowledge in a clinical setting.
Clinical Applications
In addition to the positive results of harnessing NKT cell potential in pre-clinical studies, clinical observations suggested diminished NKT cell function in cancer. For example, reports have found patients battling multiple myeloma and solid tumors to have significantly fewer NKT cells than healthy donors (74, 97). Several studies showed that the number of type I NKT cells in tumors or in peripheral blood correlated with lower rates of lymph node metastases, highly significant rates of overall and disease-free survival and served as an independent predictor for patient prognosis (98–100). Taken together with pre-clinical studies in mice, data imply that stimulation of type I NKT cells might be a useful strategy for cancer therapy in human beings. However, no clinical response was observed in the original clinical trial using α-GalCer/KRN7000 to treat solid tumors (97). The less-than-expected efficacy in human trials was believed to have multiple contributing factors. Potential problems include insufficient drug delivery, too low NKT cell numbers at the time of treatment, induction of NKT cell anergy, existence of natural anti-α-gal-antibodies, or other confounding factors like cross regulation of beneficial NKT cell activity (Table 1).
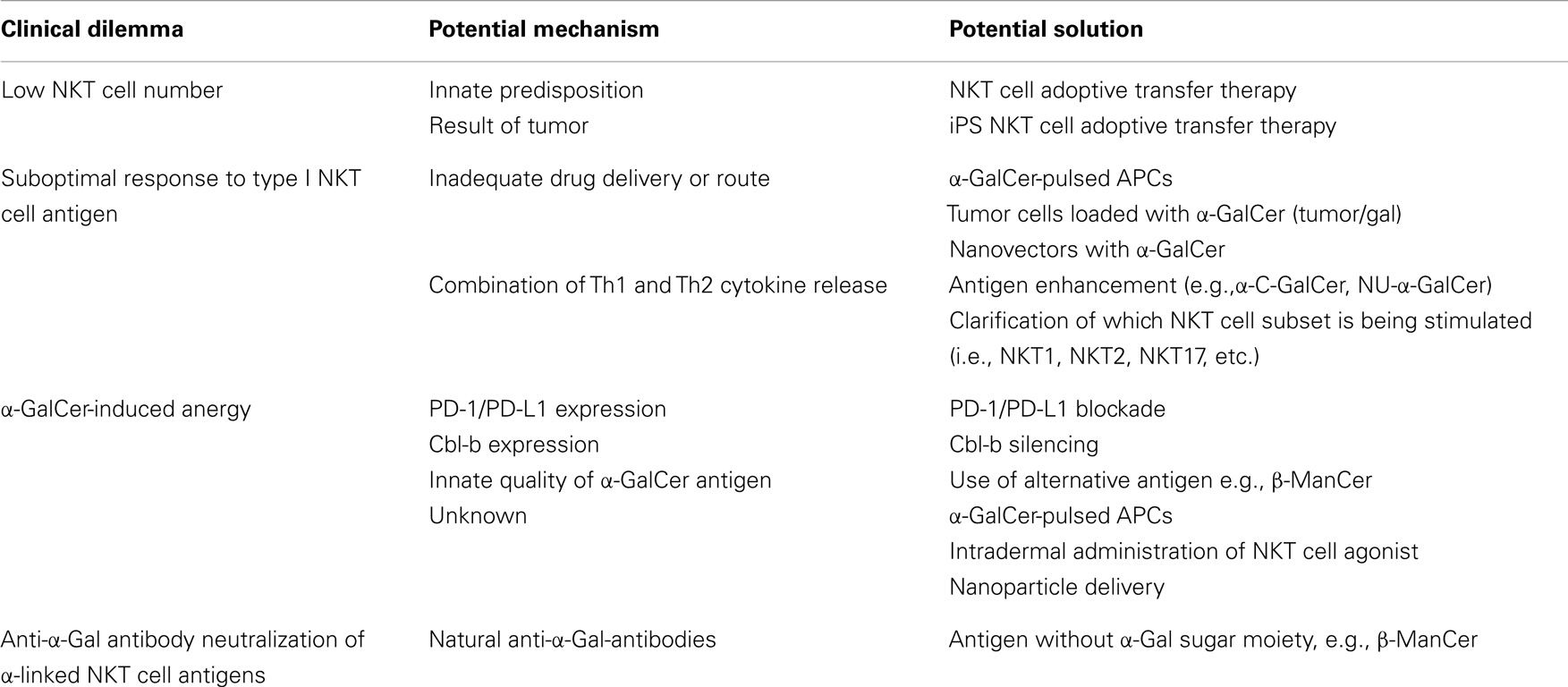
Table 1. Clinical dilemmas, possible mechanisms, and potential solutions.
Adequate Stimulation
Because pre-clinical data showed APCs preloaded with antigen better augmented NKT cell-based anti-tumor responses than free α-GalCer, α-GalCer-pulsed APCs were administered intravenously to patients (101), reviewed in Ref. (102). These α-GalCer-pulsed APCs are delivered to the lung parenchyma, where they activate type I NKT cells and initiate both direct and indirect anti-tumor effects. Multiple phase I and II clinical studies using monotherapy of α-GalCer-pulsed APCs on non-small cell lung cancer (NSCLC) and head and neck squamous cell carcinoma (HNSCC) reported objective anti-tumor responses, albeit at low frequency. The combination of this therapy with infusion of pre-activated type I NKT cells in HNSCC resulted in objective responses in about 40% of patients, and a majority of those treated displayed enhanced numbers of IFN-γ-expressing type I NKT cells in peripheral blood (103). Alternative routes of administration for α-GalCer-pulsed APCs are under investigation (104, 105), and it will be interesting to see whether these NKT cell-specific immune responses can be optimized for other cancer types as well.
The immense variability in NKT cell number from patient to patient may also contribute to variability in clinical response. One potential solution to overcome this issue is NKT cell adoptive transfer therapy. A recent study by Fujii et al. (106) establishing induced pluripotent stem (iPS) cells that can generate unlimited numbers of NKT cells with adjuvant activity may realize this type of therapy.
Anergy
Anergy may also hinder the beneficial and/or lasting effects of NKT cell-mediated treatment (107). NKT cell anergy refers to a tolerance mechanism following repeated antigen stimulation in which the lymphocyte remains in a hyporesponsive state for an extended period of time (108). New protocols, which prevent anergy induction are being employed, such as intradermal administration or nanoparticle delivery of the NKT cell agonist (105, 109). The exact mechanism of NKT cell long-term functional anergy induction is under investigation by numerous groups (107, 110–114). Marked increase in programed death-1 marker (PD-1) expression after α-GalCer stimulation prompted multiple studies on the role of PD-1 in type I NKT cell anergy, as PD-1 is an exhaustion marker and negative regulator of T cell antigen receptor signaling. However, whether PD-1 blockade can prevent or reverse anergy remains controversial (110, 111, 115). Also, it was demonstrated that the PD-1/PD-L1 pathway was essential for CD4+ type II NKT cells to suppress CD4+ T cells (116). With the observations that PD-1 is upregulated by the exhausted CD8+ T cells (117), and has been shown to be a mechanism for tumor evasion (118, 119), targeting PD-1 is an attractive therapeutic approach. PD-1/PDL1 blockade may not only prevent or reverse type I NKT cell anergy and decrease suppressive activity of type II NKT cells on CD4+ T cells but it may also augment tumor immunity by preserving CD8+ T cells. Combining anti-PD-1 treatment with an NKT cell agonist may also be beneficial, for that could stimulate NKT cell-mediated DC maturation, NK cell activity, and cross-priming of those “rescued” CD8+ T cells. This is particularly interesting when considering that ongoing clinical trials using anti-PD-1 in treatment for melanoma, NSCLC, and renal cell carcinoma show early evidence for some efficacy, but could be improved (120).
Additionally, PD-1 and the E3 ubiquitin ligase, casitas B-lineage lymphoma-b (Cbl-b) are both regulated by Egr2/3, which is overexpressed during α-GalCer-induced anergy (74). Cbl-b is increased when type I NKT cells become anergic and Cbl-b deficiency permits rescue of IFN-γ but not IL-4 production in anergic NKT cells (112). Therefore, blocking Cbl-b with small molecule or peptide antagonists may prevent anergy induction by α-GalCer.
Natural Anti-α-Gal-Antibodies
Another potential roadblock to α-GalCer treatment involves the fact that human and non-human primates cannot make α-linked sugar moieties and have natural anti-α-gal-antibodies (121). These antibodies may neutralize the α-linked component of the drug. Although it was originally believed that an alpha-linked sugar moiety was necessary for glycosphingolipids to activate type I NKT cells, a recent study showed that a ceramide with a beta-linked sugar moiety could induce a comparable protective effect (67). The β-glycolipid analog β-mannosylceramide (β-ManCer) acts through an independent mechanism from that of the α-linked KRN7000 (nitric oxide synthase/TNF-α pathways, as opposed to primarily IFN-γ). It not only protected mice against tumors, but also synergized with α-GalCer, so lower doses of both drugs could be used (67). Additionally, β-ManCer failed to induce long-term NKT cell anergy (122). In contrast to α-GalCer, β-ManCer may achieve protection with less cytokine production, decreasing toxicity, less likelihood of anergy, and without potential neutralization by anti-α-gal-antibodies. Thus, this new class of type I NKT cell agonists represented by β-ManCer may overcome several of the problems that could be interfering with clinical efficacy of α-GalCer and its close analogs.
DCs and Vaccines
A recurring theme seems to suggest that a single agent α-GalCer may not be sufficient for cancer treatment. However, the NKT cell-mediated activation of NK, B, CD4+, and CD8+ T cells suggests many opportunities to use NKT cell agonists as vaccine adjuvants. As mentioned, type I NKT cells enable DCs to cross-prime CD8+ T cells. Coadministration of α-GalCer with peptide or protein antigens enhances the priming and boosting of CD8+ T cells (10–12). A recent approach with vaccines involving nanovectors, CD1d-containing endosomes that serve as nanoparticle carriers of α-GalCer, is an attractive method to stimulate NKT cells. The carriers are taken up by APCs, and because they release α-GalCer more slowly than current methods (soluble infusion or DC-pulsing); they require lower drug amounts for similar biological effects (123). The microspheres can be decorated with specific antibodies to focus them on particular targets, and particles can be engineered to enhance cross-priming of CD8+ T cells by targeting CD8+DCs (123).
A separate approach to enhance cross-priming of CD8+ T cells that differs from simply administering antigen alongside an NKT cell agonist has been tried in cancer. In a pre-clinical study, tumor cells were loaded with α-GalCer (named tumor/gal), irradiated, and infused intravenously (124). Tumor/gal cells were lysed by NKT and NK cells in an innate fashion but NKT cells also reacted to the α-GalCer/CD1d complex on splenic DCs, which had taken up debris from lysed tumors. This allowed DCs to mature and tumor antigen was cross-presented to CD8+ T cells to induce a tumor-specific CTL response, which protected mice from tumor challenge. More research is needed to determine which types of cancer cells to inject for tumor/Gal therapy. For instance, because cancer stem cells or cancer-initiating cells may drive malignancy and immune-escape, injecting glycolipid-coated, irradiated cancer stem cells could enhance the therapeutic effect (124).
Chemotherapy
Combining an NKT cell agonist with certain types of chemotherapy is another option for harnessing the potential of NKT cell-mediated protection (59, 125). In multiple myeloma, a small cohort of patients had an enhanced immune response when α-GalCer-stimulated NKT cells were administered in conjunction with lenolidomide (an approved treatment for multiple myeloma) (125). The therapy caused an activation of type I NKT cells and consequent induction of activated NK cells and eosinophils.
Conclusion
The role that NKT cells play in tumor immunity is becoming increasingly clear, especially as we develop a more accurate and holistic understanding of how NKT cell subsets differentially impact the innate and adaptive immune cell populations. Initial attempts to harness the immune enhancing effects of type I NKT cells in the context of cancer were met with limited success during translation into the clinic. However, that clinical research has encouraged further investigation and broader understanding of NKT cell subsets in cancer. The immunological potential of type I NKT cells in vivo is being optimized through antigen enhancement, tactical delivery, and even ex vivo expansion. Insight on the suppressive mechanisms of type II NKT cells has informed methods of intervention to restore tumor immunity (e.g., anti-TGF-β). Finally, taking advantage of the adjuvant effect mediated by NKT cells in vaccines and chemotherapy treatment has proven to augment tumor immunity in animal models. While there is optimism that NKT cells could be an effective tool in enhancing the anti-tumor immune response, an incomplete understanding of NKT cell subsets has hampered the progress of translating the basic and pre-clinical NKT cell studies into the clinic. The successful strides noted in this review will be enhanced further with a greater understanding of what drives context-specific interactions between NKT cells and other cell populations, as well as by further clarification of the role of differentiated subsets within type I (NKT1, NKT2, NKT17, NKTreg, NKTFH), as well as the heterogeneous subset of non-invariant or type II NKT cells.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and the Gui Foundation.
References
1. Matsuda JL, Naidenko OV, Gapin L, Nakayama T, Taniguchi M, Wang CR, et al. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J Exp Med (2000) 192(5):741–54. doi: 10.1084/jem.192.5.741
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Godfrey DI, Kronenberg M. Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest (2004) 114(10):1379–88. doi:10.1172/JCI23594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Rhost S, Lofbom L, Rynmark BM, Pei B, Mansson JE, Teneberg S, et al. Identification of novel glycolipid ligands activating a sulfatide-reactive, CD1d-restricted, type II natural killer T lymphocyte. Eur J Immunol (2012) 42(11):2851–60. doi:10.1002/eji.201142350
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Gapin L, Godfrey DI, Rossjohn J. Natural killer T cell obsession with self-antigens. Curr Opin Immunol (2013) 25(2):168–73. doi:10.1016/j.coi.2013.01.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Terabe M, Berzofsky JA. The immunoregulatory role of type I and type II NKT cells in cancer and other diseases. Cancer Immunol Immunother (2014) 63(3):199–213. doi:10.1007/s00262-013-1509-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L. NKT cells: what’s in a name? Nat Rev Immunol (2004) 4(3):231–7. doi:10.1038/nri1309
7. Terabe M, Berzofsky JA. The role of NKT cells in tumor immunity. Adv Cancer Res (2008) 101:277–348. doi:10.1016/S0065-230X(08)00408-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Bai L, Picard D, Anderson B, Chaudhary V, Luoma A, Jabri B, et al. The majority of CD1d-sulfatide-specific T cells in human blood use a semiinvariant Vdelta1 TCR. Eur J Immunol (2012) 42(9):2505–10. doi:10.1002/eji.201242531
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Parekh VV, Wu L, Olivares-Villagomez D, Wilson KT, Van Kaer L. Activated invariant NKT cells control central nervous system autoimmunity in a mechanism that involves myeloid-derived suppressor cells. J Immunol (2013) 190(5):1948–60. doi:10.4049/jimmunol.1201718
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med (2003) 198(2):267–79. doi:10.1084/jem.20030324
11. Silk JD, Hermans IF, Gileadi U, Chong TW, Shepherd D, Salio M, et al. Utilizing the adjuvant properties of CD1d-dependent NK T cells in T cell-mediated immunotherapy. J Clin Invest (2004) 114(12):1800–11. doi:10.1172/JCI22046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Gonzalez-Aseguinolaza G, Van Kaer L, Bergmann CC, Wilson JM, Schmieg J, Kronenberg M, et al. Natural killer T cell ligand alpha-galactosylceramide enhances protective immunity induced by malaria vaccines. J Exp Med (2002) 195(5):617–24. doi:10.1084/jem.20011889
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Gadola SD, Dulphy N, Salio M, Cerundolo V. Valpha24-JalphaQ-independent, CD1d-restricted recognition of alpha-galactosylceramide by human CD4(+) and CD8alphabeta(+) T lymphocytes. J Immunol (2002) 168(11):5514–20. doi:10.4049/jimmunol.168.11.5514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Gumperz JE, Miyake S, Yamamura T, Brenner MB. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J Exp Med (2002) 195(5):625–36. doi:10.1084/jem.20011786
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Lee PT, Benlagha K, Teyton L, Bendelac A. Distinct functional lineages of human Va24 natural killer cells. J Exp Med (2002) 195(5):637–41. doi:10.1084/jem.20011908
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Coquet JM, Chakravarti S, Kyparissoudis K, McNab FW, Pitt LA, McKenzie BS, et al. Diverse cytokine production by NKT cell subsets and identification of an IL-17-producing CD4-NK1.1- NKT cell population. Proc Natl Acad Sci U S A (2008) 105(32):11287–92. doi:10.1073/pnas.0801631105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Chan AC, Leeansyah E, Cochrane A, d’Udekem d’Acoz Y, Mittag D, Harrison LC, et al. Ex-vivo analysis of human natural killer T cells demonstrates heterogeneity between tissues and within established CD4(+) and CD4(-) subsets. Clin Exp Immunol (2013) 172(1):129–37. doi:10.1111/cei.12045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Kenna T, Mason LG, Porcelli SA, Koezuka Y, Hegarty JE, O’Farrelly C, et al. NKT cells from normal and tumor-bearing human livers are phenotypically and functionally distinct from murine NKT cells. J Immunol (2003) 171(4):1775–9. doi:10.4049/jimmunol.171.4.1775
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Wingender G, Stepniak D, Krebs P, Lin L, McBride S, Wei B, et al. Intestinal microbes affect phenotypes and functions of invariant natural killer T cells in mice. Gastroenterology (2012) 143(2):418–28. doi:10.1053/j.gastro.2012.04.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Crowe NY, Smyth MJ, Godfrey DI. A critical role for natural killer T cells in immunosurveillance of methylcholanthrene-induced sarcomas. J Exp Med (2002) 196(1):119–27. doi:10.1084/jem.20020092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, et al. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science (1997) 278(5343):1623–6. doi:10.1126/science.278.5343.1623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Smyth MJ, Thia KY, Street SE, Cretney E, Trapani JA, Taniguchi M, et al. Differential tumor surveillance by natural killer (NK) and NKT cells. J Exp Med (2000) 191(4):661–8. doi:10.1084/jem.191.4.661
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Smyth MJ, Godfrey DI. NKT cells and tumor immunity – a double-edged sword. Nat Immunol (2000) 1(6):459–60. doi:10.1038/82698
24. Terabe M, Matsui S, Noben-Trauth N, Chen H, Watson C, Donaldson DD, et al. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat Immunol (2000) 1(6):515–20. doi:10.1038/82771
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Terabe M, Matsui S, Park J-M, Mamura M, Noben-Trauth N, Donaldson DD, et al. Transforming growth factor-b production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med (2003) 198(11):1741–52. doi:10.1084/jem.20022227
26. Terabe M, Swann J, Ambrosino E, Sinha P, Takaku S, Hayakawa Y, et al. A nonclassical non-Va14Ja18 CD1d-restricted (type II) NKT cell is sufficient for down-regulation of tumor immunosurveillance. J Exp Med (2005) 202(12):1627–33. doi:10.1084/jem.20051381
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Ambrosino E, Terabe M, Halder RC, Peng J, Takaku S, Miyake S, et al. Cross-regulation between type I and type II NKT cells in regulating tumor immunity: a new immunoregulatory axis. J Immunol (2007) 179:5126–36. doi:10.4049/jimmunol.179.8.5126
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Park JJ, Lee JH, Seo KC, Bricard G, Venkataswamy MM, Porcelli SA, et al. Syntheses and biological activities of KRN7000 analogues having aromatic residues in the acyl and backbone chains with varying stereochemistry. Bioorg Med Chem Lett (2010) 20(3):814–8. doi:10.1016/J.Bmcl.2009.12.103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Kobayashi E, Motoki K, Uchida T, Fukushima H, Koezuka Y. KRN7000, a novel immunomodulator, and its antitumor activities. Oncol Res (1995) 7(10–11):529–34.
30. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science (1997) 278(5343):1626–9. doi:10.1126/science.278.5343.1626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Brutkiewicz RR. CD1d ligands: the good, the bad, and the ugly. J Immunol (2006) 177(2):769–75. doi:10.4049/jimmunol.177.2.769
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Tupin E, Kinjo Y, Kronenberg M. The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol (2007) 5(6):405–17. doi:10.1038/nrmicro1657
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol (2007) 25:297–336. doi:10.1146/annurev.immunol.25.022106.141711
34. Chang DH, Deng H, Matthews P, Krasovsky J, Ragupathi G, Spisek R, et al. Inflammation associated lysophospholipids as ligands for CD1d restricted T cells in human cancer. Blood (2008) 112:1308–16. doi:10.1182/blood-2008-04-149831
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Zhou D, Mattner J, Cantu C III, Schrantz N, Yin N, Gao Y, et al. Lysosomal glycosphingolipid recognition by NKT cells. Science (2004) 306(5702):1786–9. doi:10.1126/science.1103440
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. La Cava A, Van Kaer L, Fu Dong S. CD4+CD25+ Tregs and NKT cells: regulators regulating regulators. Trends Immunol (2006) 27(7):322–7. doi:10.1016/j.it.2006.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Kitamura H, Iwakabe K, Yahata T, Nishimura S, Ohta A, Ohmi Y, et al. The natural killer T (NKT) cell ligand alpha-galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL)-12 production by dendritic cells and IL-12 receptor expression on NKT cells. J Exp Med (1999) 189(7):1121–8. doi:10.1084/jem.189.7.1121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Tomura M, Yu WG, Ahn HJ, Yamashita M, Yang YF, Ono S, et al. A novel function of Valpha14+CD4+NKT cells: stimulation of IL-12 production by antigen-presenting cells in the innate immune system. J Immunol (1999) 163(1):93–101.
39. Paget C, Chow MT, Duret H, Mattarollo SR, Smyth MJ. Role of gammadelta T Cells in alpha-galactosylceramide-mediated immunity. J Immunol (2012) 188(8):3928–39. doi:10.4049/jimmunol.1103582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Taraban VY, Martin S, Attfield KE, Glennie MJ, Elliott T, Elewaut D, et al. Invariant NKT cells promote CD8+ cytotoxic T cell responses by inducing CD70 expression on dendritic cells. J Immunol (2008) 180(7):4615–20. doi:10.4049/jimmunol.180.7.4615
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Fujii S, Shimizu K, Hemmi H, Steinman RM. Innate Valpha14(+) natural killer T cells mature dendritic cells, leading to strong adaptive immunity. Immunol Rev (2007) 220:183–98. doi:10.1111/j.1600-065X.2007.00561.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Semmling V, Lukacs-Kornek V, Thaiss CA, Quast T, Hochheiser K, Panzer U, et al. Alternative cross-priming through CCL17-CCR4-mediated attraction of CTLs toward NKT cell-licensed DCs. Nat Immunol (2010) 11(4):313–20. doi:10.1038/ni.1848
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Iwamura C, Shinoda K, Endo Y, Watanabe Y, Tumes DJ, Motohashi S, et al. Regulation of memory CD4 T-cell pool size and function by natural killer T cells in vivo. Proc Natl Acad Sci U S A (2012) 109(42):16992–7. doi:10.1073/pnas.1203494109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol (2002) 3(11):999–1005. doi:10.1038/ni1102-999
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Michel ML, Keller AC, Paget C, Fujio M, Trottein F, Savage PB, et al. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med (2007) 204(5):995–1001. doi:10.1084/jem.20061551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Michel ML, Mendes-da-Cruz D, Keller AC, Lochner M, Schneider E, Dy M, et al. Critical role of ROR-gammat in a new thymic pathway leading to IL-17-producing invariant NKT cell differentiation. Proc Natl Acad Sci U S A (2008) 105(50):19845–50. doi:10.1073/pnas.0806472105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Watarai H, Sekine-Kondo E, Shigeura T, Motomura Y, Yasuda T, Satoh R, et al. Development and function of invariant natural killer T cells producing T(h)2- and T(h)17-cytokines. PLoS Biol (2012) 10(2):e1001255. doi:10.1371/journal.pbio.1001255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Moreira-Teixeira L, Resende M, Devergne O, Herbeuval JP, Hermine O, Schneider E, et al. Rapamycin combined with TGF-beta converts human invariant NKT cells into suppressive Foxp3+ regulatory cells. J Immunol (2012) 188(2):624–31. doi:10.4049/jimmunol.1102281
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Lee YJ, Holzapfel KL, Zhu J, Jameson SC, Hogquist KA. Steady-state production of IL-4 modulates immunity in mouse strains and is determined by lineage diversity of iNKT cells. Nat Immunol (2013) 14(11):1146–54. doi:10.1038/ni.2731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Chang PP, Barral P, Fitch J, Pratama A, Ma CS, Kallies A, et al. Identification of Bcl-6-dependent follicular helper NKT cells that provide cognate help for B cell responses. Nat Immunol (2012) 13(1):35–43. doi:10.1038/ni.2166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Sag D, Krause P, Hedrick CC, Kronenberg M, Wingender G. IL-10-producing NKT10 cells are a distinct regulatory invariant NKT cell subset. J Clin Invest (2014) 124(9):3725–40. doi:10.1172/JCI72308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Arrenberg P, Halder R, Dai Y, Maricic I, Kumar V. Oligoclonality and innate-like features in the TCR repertoire of type II NKT cells reactive to a beta-linked self-glycolipid. Proc Natl Acad Sci U S A (2010) 107(24):10984–9. doi:10.1073/pnas.1000576107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Skold M, Faizunnessa NN, Wang CR, Cardell S. CD1d-specific NK1.1+ T cells with a transgenic variant TCR. J Immunol (2000) 165(1):168–74. doi:10.4049/jimmunol.165.1.168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Zeissig S, Murata K, Sweet L, Publicover J, Hu Z, Kaser A, et al. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat Med (2012) 18(7):1060–8. doi:10.1038/nm.2811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Zhao J, Weng X, Bagchi S, Wang CR. Polyclonal type II natural killer T cells require PLZF and SAP for their development and contribute to CpG-mediated antitumor response. Proc Natl Acad Sci U S A (2014) 111(7):2674–9. doi:10.1073/pnas.1323845111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Kawano T, Nakayama T, Kamada N, Kaneko Y, Harada M, Ogura N, et al. Antitumor cytotoxicity mediated by ligand-activated human V alpha24 NKT cells. Cancer Res (1999) 59(20):5102–5.
57. Coquet JM, Kyparissoudis K, Pellicci DG, Besra G, Berzins SP, Smyth MJ, et al. IL-21 is produced by NKT cells and modulates NKT cell activation and cytokine production. J Immunol (2007) 178(5):2827–34. doi:10.4049/jimmunol.178.5.2827
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Sato H, et al. Natural killer-like nonspecific tumor cell lysis mediated by specific ligand-activated Valpha14 NKT cells. Proc Natl Acad Sci U S A (1998) 95(10):5690–3. doi:10.1073/pnas.95.10.5690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Fallarini S, Paoletti T, Orsi Battaglini N, Lombardi G. Invariant NKT cells increase drug-induced osteosarcoma cell death. Br J Pharmacol (2012) 167(7):1533–49. doi:10.1111/j.1476-5381.2012.02108.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Hix LM, Shi YH, Brutkiewicz RR, Stein PL, Wang CR, Zhang M. CD1d-expressing breast cancer cells modulate NKT cell-mediated antitumor immunity in a murine model of breast cancer metastasis. PLoS One (2011) 6(6):e20702. doi:10.1371/journal.pone.0020702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Renukaradhya GJ, Khan MA, Vieira M, Du W, Gervay-Hague J, Brutkiewicz RR. Type I NKT cells protect (and type II NKT cells suppress) the host’s innate antitumor immune response to a B-cell lymphoma. Blood (2008) 111(12):5637–45. doi:10.1182/blood-2007-05-092866
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Dhodapkar MV, Geller MD, Chang DH, Shimizu K, Fujii S, Dhodapkar KM, et al. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J Exp Med (2003) 197(12):1667–76. doi:10.1084/jem.20021650
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Smyth MJ, Crowe NY, Pellicci DG, Kyparissoudis K, Kelly JM, Takeda K, et al. Sequential production of interferon-gamma by NK1.1(+) T cells and natural killer cells is essential for the antimetastatic effect of alpha-galactosylceramide. Blood (2002) 99(4):1259–66. doi:10.1182/blood.V99.4.1259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Terabe M, Berzofsky JA. Immunoregulatory T cells in tumor immunity. Curr Opin Immunol (2004) 16(2):157–62. doi:10.1016/j.coi.2004.01.010
65. Street SE, Cretney E, Smyth MJ. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood (2001) 97(1):192–7. doi:10.1182/blood.V97.1.192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Crowe NY, Coquet JM, Berzins SP, Kyparissoudis K, Keating R, Pellicci DG, et al. Differential antitumor immunity mediated by NKT cell subsets in vivo. J Exp Med (2005) 202(9):1279–88. doi:10.1084/jem.20050953
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. O’Konek JJ, Illarionov P, Khursigara DS, Ambrosino E, Izhak L, Castillo BF II, et al. Mouse and human iNKT cell agonist beta-mannosylceramide reveals a distinct mechanism of tumor immunity. J Clin Invest (2011) 121:683–94. doi:10.1172/JCI42314
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Sakai T, Ehara H, Koezuka Y. Synthesis of NBD-alpha-galactosylceramide and its immunologic properties. Org Lett (1999) 1(3):359–61. doi:10.1021/ol9900111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Schmieg J, Yang G, Franck RW, Tsuji M. Superior protection against malaria and melanoma metastases by a C-glycoside analogue of the natural killer T cell ligand alpha-galactosylceramide. J Exp Med (2003) 198(11):1631–41. doi:10.1084/jem.20031192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Chang YJ, Huang JR, Tsai YC, Hung JT, Wu D, Fujio M, et al. Potent immune-modulating and anticancer effects of NKT cell stimulatory glycolipids. Proc Natl Acad Sci U S A (2007) 104(25):10299–304. doi:10.1073/pnas.0703824104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Wu TN, Lin KH, Chang YJ, Huang JR, Cheng JY, Yu AL, et al. Avidity of CD1d-ligand-receptor ternary complex contributes to T-helper 1 (Th1) polarization and anticancer efficacy. Proc Natl Acad Sci U S A (2011) 108(42):17275–80. doi:10.1073/pnas.1114255108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Aspeslagh S, Li Y, Yu ED, Pauwels N, Trappeniers M, Girardi E, et al. Galactose-modified iNKT cell agonists stabilized by an induced fit of CD1d prevent tumour metastasis. EMBO J (2011) 30(11):2294–305. doi:10.1038/emboj.2011.145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Li X, Fujio M, Imamura M, Wu D, Vasan S, Wong CH, et al. Design of a potent CD1d-binding NKT cell ligand as a vaccine adjuvant. Proc Natl Acad Sci U S A (2010) 107(29):13010–5. doi:10.1073/pnas.1006662107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Huang JR, Tsai YC, Chang YJ, Wu JC, Hung JT, Lin KH, et al. Alpha-galactosylceramide but not phenyl-glycolipids induced NKT cell anergy and IL-33-mediated myeloid-derived suppressor cell accumulation via upregulation of egr2/3. J Immunol (2014) 192(4):1972–81. doi:10.4049/jimmunol.1302623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. De Santo C, Salio M, Masri SH, Lee LY, Dong T, Speak AO, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest (2008) 118(12):4036–48. doi:10.1172/JCI36264
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Elkabets M, Ribeiro VS, Dinarello CA, Ostrand-Rosenberg S, Di Santo JP, Apte RN, et al. IL-1beta regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur J Immunol (2010) 40(12):3347–57. doi:10.1002/eji.201041037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. De Santo C, Arscott R, Booth S, Karydis I, Jones M, Asher R, et al. Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat Immunol (2010) 11(11):1039–46. doi:10.1038/ni.1942
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Izhak L, Ambrosino E, Kato S, Parish ST, O’Konek JJ, Weber H, et al. Delicate balance among three types of T cells in concurrent regulation of tumor immunity. Cancer Res (2013) 73(5):1514–23. doi:10.1158/0008-5472.CAN-12-2567
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Fichtner-Feigl S, Terabe M, Kitani A, Young CA, Fuss I, Geissler EK, et al. Restoration of tumor immunosurveillance via targeting of interleukin-13 receptor-alpha 2. Cancer Res (2008) 68(9):3467–75. doi:10.1158/0008-5472.CAN-07-5301
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Terabe M, Ambrosino E, Takaku S, O’Konek JJ, Venzon D, Lonning S, et al. Synergistic enhancement of CD8+ T cell-mediated tumor vaccine efficacy by an anti-transforming growth factor-beta monoclonal antibody. Clin Cancer Res (2009) 15(21):6560–9. doi:10.1158/1078-0432.CCR-09-1066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Ueda R, Fujita M, Zhu X, Sasaki K, Kastenhuber ER, Kohanbash G, et al. Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin Cancer Res (2009) 15(21):6551–9. doi:10.1158/1078-0432.CCR-09-1067
82. Gunn GR, Zubair A, Peters C, Pan ZK, Wu TC, Paterson Y. Two Listeria monocytogenes vaccine vectors that express different molecular forms of human papilloma virus-16 (HPV-16) E7 induce qualitatively different T cell immunity that correlates with their ability to induce regression of established tumors immortalized by HPV-16. J Immunol (2001) 167(11):6471–9. doi:10.4049/jimmunol.167.11.6471
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Kim S, Buchlis G, Fridlender ZG, Sun J, Kapoor V, Cheng G, et al. Systemic blockade of transforming growth factor-beta signaling augments the efficacy of immunogene therapy. Cancer Res (2008) 68(24):10247–56. doi:10.1158/0008-5472.CAN-08-1494
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Laverty HG, Wakefield LM, Occleston NL, O’Kane S, Ferguson MW. TGF-beta3 and cancer: a review. Cytokine Growth Factor Rev (2009) 20(4):305–17. doi:10.1016/j.cytogfr.2009.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Ahlers JD, Belyakov IM, Terabe M, Koka R, Donaldson DD, Thomas E, et al. A push-pull approach to maximize vaccine efficacy: abrogating suppression with an IL-13 inhibitor while augmenting help with GM-CSF and CD40L. Proc Natl Acad Sci U S A (2002) 99(20):13020–5. doi:10.1073/pnas.192251199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Pang YL, Gara SK, Achyut BR, Li ZY, Yan HH, Day CP, et al. TGF-beta signaling in myeloid cells is required for tumor metastasis. Cancer Discov (2013) 3(8):936–51. doi:10.1158/2159-8290.Cd-12-0527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res (2008) 68(13):5439–49. doi:10.1158/0008-5472.can-07-6621
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Moses HL, Yang EY, Pietenpol JA. TGF-beta stimulation and inhibition of cell proliferation: new mechanistic insights. Cell (1990) 63(2):245–7. doi:10.1016/0092-8674(90)90155-8
89. Terabe M, Khanna C, Bose S, Melchionda F, Mendoza A, Mackall CL, et al. CD1d-restricted NKT cells can down-regulate tumor immunosurveillance independent of IL-4R-STAT6 or TGF-ß. Cancer Res (2006) 66(7):3869–75. doi:10.1158/0008-5472.CAN-05-3421
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Robertson FC, Lu X, Terabe M, Berzofsky JA. Type II NKT cells play a role in the regulation of CD4 T cell subsets. J Immunol (2014) 192(1 Suppl):194.1.
91. Bjordahl RL, Gapin L, Marrack P, Refaeli Y. iNKT cells suppress the CD8+ T cell response to a murine Burkitt’s-like B cell lymphoma. PLoS One (2012) 7(8):e42635. doi:10.1371/journal.pone.0042635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Renukaradhya GJ, Sriram V, Du W, Gervay-Hague J, Van Kaer L, Brutkiewicz RR. Inhibition of antitumor immunity by invariant natural killer T cells in a T-cell lymphoma model in vivo. Int J Cancer (2006) 118:3045–53. doi:10.1002/ijc.21764
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Yang W, Li H, Mayhew E, Mellon J, Chen PW, Niederkorn JY. NKT cell exacerbation of liver metastases arising from melanomas transplanted into either the eyes or spleens of mice. Invest Ophthalmol Vis Sci (2011) 52(6):3094–102. doi:10.1167/iovs.10-7067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Jiang S, Game DS, Davies D, Lombardi G, Lechler RI. Activated CD1d-restricted natural killer T cells secrete IL-2: innate help for CD4+CD25+ regulatory T cells? Eur J Immunol (2005) 35(4):1193–200. doi:10.1002/eji.200425899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Kanamori M, Tasumi Y, Iyoda T, Ushida M, Inaba K. Sulfatide inhibits alpha-galactosylceramide presentation by dendritic cells. Int Immunol (2012) 24(2):129–36. doi:10.1093/intimm/dxr108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Halder RC, Aguilera C, Maricic I, Kumar V. Type II NK T cell-mediated anergy induction in type I NK T cells prevents inflammatory liver disease. J Clin Invest (2007) 117:2302–12. doi:10.1172/JCI31602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Giaccone G, Punt CJ, Ando Y, Ruijter R, Nishi N, Peters M, et al. A phase I study of the natural killer T-cell ligand alpha-galactosylceramide (KRN7000) in patients with solid tumors. Clin Cancer Res (2002) 8(12):3702–9.
98. Tachibana T, Onodera H, Tsuruyama T, Mori A, Nagayama S, Hiai H, et al. Increased intratumor Valpha24-positive natural killer T cells: a prognostic factor for primary colorectal carcinomas. Clin Cancer Res (2005) 11(20):7322–7. doi:10.1158/1078-0432.CCR-05-0877
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Molling JW, Langius JA, Langendijk JA, Leemans CR, Bontkes HJ, van der Vliet HJ, et al. Low levels of circulating invariant natural killer T cells predict poor clinical outcome in patients with head and neck squamous cell carcinoma. J Clin Oncol (2007) 25(7):862–8. doi:10.1200/JCO.2006.08.5787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Najera Chuc AE, Cervantes LA, Retiguin FP, Ojeda JV, Maldonado ER. Low number of invariant NKT cells is associated with poor survival in acute myeloid leukemia. J Cancer Res Clin Oncol (2012) 138(8):1427–32. doi:10.1007/s00432-012-1251-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Nieda M, Nicol A, Koezuka Y, Kikuchi A, Lapteva N, Tanaka Y, et al. TRAIL expression by activated human CD4(+)V alpha 24NKT cells induces in vitro and in vivo apoptosis of human acute myeloid leukemia cells. Blood (2001) 97(7):2067–74. doi:10.1182/blood.V97.7.2067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Motohashi S, Okamoto Y, Nakayama T. Clinical trials of invariant natural killer T cell-based immunotherapy for cancer. In: Terabe M, Berzofsky JA, editors. Natural Killer T Cells: Balancing the Regulation of Tumor Immunity. New York: Springer Science+Business Media, LLC (2011). pp. 185–98.
103. Kunii N, Horiguchi S, Motohashi S, Yamamoto H, Ueno N, Yamamoto S, et al. Combination therapy of in vitro-expanded natural killer T cells and alpha-galactosylceramide-pulsed antigen-presenting cells in patients with recurrent head and neck carcinoma. Cancer Sci (2009) 100(6):1092–8. doi:10.1111/j.1349-7006.2009.01135.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Nicol AJ, Tazbirkova A, Nieda M. Comparison of clinical and immunological effects of intravenous and intradermal administration of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells. Clin Cancer Res (2011) 17(15):5140–51. doi:10.1158/1078-0432.CCR-10-3105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Bontkes HJ, Moreno M, Hangalapura B, Lindenberg JJ, de Groot J, Lougheed S, et al. Attenuation of invariant natural killer T-cell anergy induction through intradermal delivery of alpha-galactosylceramide. Clin Immunol (2010) 136(3):364–74. doi:10.1016/j.clim.2010.04.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Fujii S, Shimizu K, Okamoto Y, Kunii N, Nakayama T, Motohashi S, et al. NKT cells as an ideal anti-tumor immunotherapeutic. Front Immunol (2013) 4:409. doi:10.3389/fimmu.2013.00409
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Parekh VV, Wilson MT, Olivares-Villagomez D, Singh AK, Wu L, Wang CR, et al. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest (2005) 115(9):2572–83. doi:10.1172/JCI24762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Schwartz RH. T cell anergy. Annu Rev Immunol (2003) 21:305–34. doi:10.1146/annurev.immunol.21.120601.141110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Thapa P, Zhang G, Xia C, Gelbard A, Overwijk WW, Liu C, et al. Nanoparticle formulated alpha-galactosylceramide activates NKT cells without inducing anergy. Vaccine (2009) 27(25–26):3484–8. doi:10.1016/j.vaccine.2009.01.047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Chang WS, Kim JY, Kim YJ, Kim YS, Lee JM, Azuma M, et al. Cutting edge: programmed death-1/programmed death ligand 1 interaction regulates the induction and maintenance of invariant NKT cell anergy. J Immunol (2008) 181(10):6707–10. doi:10.4049/jimmunol.181.10.6707
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Parekh VV, Lalani S, Kim S, Halder R, Azuma M, Yagita H, et al. PD-1/PD-L blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J Immunol (2009) 182(5):2816–26. doi:10.4049/jimmunol.0803648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Kojo S, Elly C, Harada Y, Langdon WY, Kronenberg M, Liu YC. Mechanisms of NKT cell anergy induction involve Cbl-b-promoted monoubiquitination of CARMA1. Proc Natl Acad Sci U S A (2009) 106(42):17847–51. doi:10.1073/pnas.0904078106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Finnefrock AC, Tang A, Li F, Freed DC, Feng M, Cox KS, et al. PD-1 blockade in rhesus macaques: impact on chronic infection and prophylactic vaccination. J Immunol (2009) 182(2):980–7. doi:10.4049/jimmunol.182.2.980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Wu J, Shin J, Xie D, Wang H, Gao J, Zhong XP. Tuberous sclerosis 1 promotes invariant NKT cell anergy and inhibits invariant NKT cell-mediated antitumor immunity. J Immunol (2014) 192(6):2643–50. doi:10.4049/jimmunol.1302076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Iyoda T, Ushida M, Kimura Y, Minamino K, Hayuka A, Yokohata S, et al. Invariant NKT cell anergy is induced by a strong TCR-mediated signal plus co-stimulation. Int Immunol (2010) 22(11):905–13. doi:10.1093/intimm/dxq444
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Kadri N, Korpos E, Gupta S, Briet C, Lofbom L, Yagita H, et al. CD4(+) type II NKT cells mediate ICOS and programmed death-1-dependent regulation of type 1 diabetes. J Immunol (2012) 188(7):3138–49. doi:10.4049/jimmunol.1101390
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443(7109):350–4. doi:10.1038/nature05115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8(8):793–800. doi:10.1038/nm0902-1039c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer (2010) 116(7):1757–66. doi:10.1002/cncr.24899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Taube JM, Klein AP, Brahmer JR, Xu H, Pan X, Kim JH, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res (2014) 20(19):5064–74. doi:10.1158/1078-0432.CCR-13-3271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Galili U, Shohet SB, Kobrin E, Stults CL, Macher BA. Man, apes, and old world monkeys differ from other mammals in the expression of alpha-galactosyl epitopes on nucleated cells. J Biol Chem (1988) 263(33):17755–62.
122. O’Konek JJ, Kato S, Takao S, Izhak L, Xia Z, Illarionov P, et al. Beta-mannosylceramide activates type I natural killer T cells to induce tumor immunity without inducing long-term functional anergy. Clin Cancer Res (2013) 19:4404–11. doi:10.1158/1078-0432.CCR-12-2169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Faveeuw C, Trottein F. Optimization of natural killer T cell-mediated immunotherapy in cancer using cell-based and nanovector vaccines. Cancer Res (2014) 74(6):1632–8. doi:10.1158/0008-5472.Can-13-3504
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Shimizu K, Kurosawa Y, Taniguchi M, Steinman RM, Fujii S. Cross-presentation of glycolipid from tumor cells loaded with alpha-galactosylceramide leads to potent and long-lived T cell mediated immunity via dendritic cells. J Exp Med (2007) 204(11):2641–53. doi:10.1084/jem.20070458
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Richter J, Neparidze N, Zhang L, Nair S, Monesmith T, Sundaram R, et al. Clinical regressions and broad immune activation following combination therapy targeting human NKT cells in myeloma. Blood (2013) 121(3):423–30. doi:10.1182/blood-2012-06-435503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: NKT cell, tumor immunology, immune regulation, immune network, NKT cell subsets
Citation: Robertson FC, Berzofsky JA and Terabe M (2014) NKT cell networks in the regulation of tumor immunity. Front. Immunol. 5:543. doi: 10.3389/fimmu.2014.00543
Received: 30 September 2014; Accepted: 13 October 2014;
Published online: 28 October 2014.
Edited by:
Dennis O. Adeegbe, Osaka University Immunology Frontier Research Center, JapanReviewed by:
William L. Redmond, Earle A. Chiles Research Institute, USAWilliam J. Magner, Roswell Park Cancer Institute, USA
Copyright: © 2014 Robertson, Berzofsky and Terabe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jay A. Berzofsky, Vaccine Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Building 41, Room D702D, Bethesda, MD 20892, USA e-mail: berzofsk@helix.nih.gov