 Paul N. Goldwater
Paul N. Goldwater- Discipline of Paediatrics, School of Paediatrics and Reproductive Health, University of Adelaide, North Adelaide, SA, Australia
The gut microbiome influences the development of the immune system of young mammals; the establishment of a normal gut microbiome is thought to be important for the health of the infant during its early development. As the role of bacteria in the causation of sudden infant death syndrome (SIDS) is backed by strong evidence, the balance between host immunity and potential bacterial pathogens is likely to be pivotal. Bacterial colonization of the infant colon is influenced by age, mode of delivery, diet, environment, and antibiotic exposure. The gut microbiome influences several systems including gut integrity and development of the immune system; therefore, gut microflora could be important in protection against bacteria and/or their toxins identified in SIDS infants. The aims of the review are to explore (1) the role of the gut microbiome in relation to the developmentally critical period in which most SIDS cases occur; (2) the mechanisms by which the gut microbiome might induce inflammation resulting in transit of bacteria from the lumen into the bloodstream; and (3) assessment of the clinical, physiological, pathological, and microbiological evidence for bacteremia leading to the final events in SIDS pathogenesis.
Introduction
The common bacterial hypothesis (1, 2) and the role of bacteria in the causation of sudden infant death syndrome (SIDS) have not been considered in depth by mainstream researchers nor has there been broad interest in the potential contribution of infection and inflammation to these deaths. Studies on the microbiology of SIDS (3–7) provided explanations for SIDS risk factors and potential mechanisms in which inflammatory responses could affect abnormal arousal, respiration (8, 9), and/or brain stem compromise (10); these are areas that have preoccupied the mainstream of SIDS research for decades without successfully providing hypotheses congruent with epidemiological and pathological features of SIDS.
The infection model of SIDS stands on substantial evidence identifying: Staphylococcus aureus and its enterotoxins (11–14); toxigenic Escherichia coli (15, 16); Clostridium perfringens (3, 4); and recent findings (based on a restricted number of bacterial species identified by culture and PCR) of significant differences in the gut microbiome between SIDS and healthy babies (17). A review of the gut microbiome of babies in the context of immunity and immune/inflammatory responses to bacterial infection was considered timely: to help clarify the role of gut/mucosal immunity in relation to SIDS; to explain the apparent mucosal dysregulation reported for one SIDS infant (18). The basis for a link between the gut microbiome and SIDS is founded upon published evidence that provides a logical explanation of underlying pathomechanisms involved in SIDS. The evidence is summarized in Table 1 and addressed later in the review.
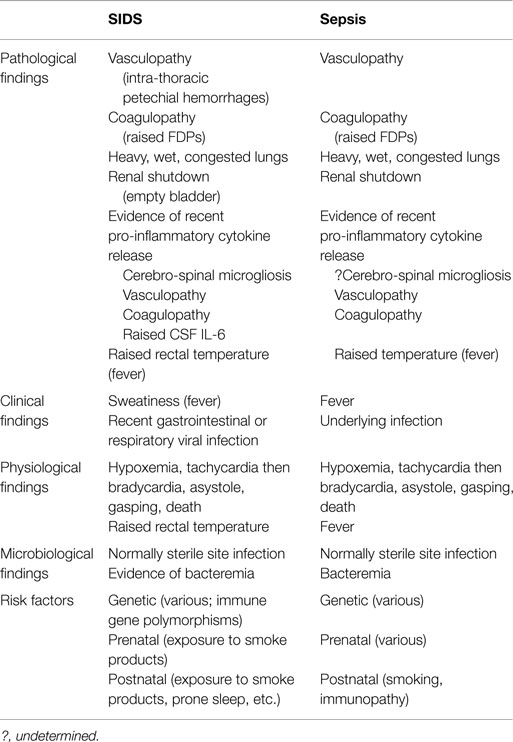
Table 1. Comparison between findings in SIDS compared to sepsis.
Background
It has been known since 1905 that the microbiota of neonates undergo change with growth and development (19). The type of early microbiota is important in terms of development of the immune system, and this could influence susceptibility to infection, induction of gut inflammation, and adverse outcomes of infection. Improved understanding of how the gut microbiota and type of feeding affect immune and neurodevelopment has arisen since the introduction of molecular techniques to identify and quantify bacterial genera and species involved (17, 20–22).
A literature search reviewed recent publications covering development of the infant gut microbiota in conjunction with development of the infant immune system and related functions. Articles pertaining to the gut microbiota and SIDS/sudden unexplained infant deaths (SUDI) were also reviewed. Search engines used included PubMed, MedlineRanker, PubCrawler, Google Scholar, and Open Access Library (OALib).
Role of Gut Microbiome during the Critical Developmental Period Associated with SIDS
Colonization
Bacterial colonization of the human infant colon is influenced by many factors – age, mode of delivery (23), diet, environment, and antibiotic exposure (20, 21, 24–26).
There is growing evidence that cesarean section (CS) (not regarded as a SIDS risk factor when controlled for gestational age) (27) is linked to an impoverishment of natural development of the immune system. Exposure of the neonatal gut to bacterial priming (as occurs with vaginal delivery) appears to be missed in babies born via CS (28). One of the main associations with CS is low gestational age. Prematurity remains a significant and largely unexplained risk factor for SIDS; the vulnerable, immature host notwithstanding (29). It is presumed that the abovementioned mechanisms (27) over and above the vulnerable host would go some way to explain this increased risk.
Diet influences the gut microbiota. The effect of breast milk on the infant immune system is considered to derive benefit via its effect on gut bacterial colonization (30). Breast feeding has a protective effect against SIDS and given the effects on the gut microbiota, suggests that these bacteria might play a role in addition to maternally transferred cellular and humoral immunity. CS remains the most common mode of delivery of preterm infants. The possibility that these babies will receive breast milk or enteral feeds remains remote while the opposite is true for receiving treatment with antibiotics. Animal studies show that a lack of enteral nutrition may be associated with an increased risk of septic shock due to bacterial translocation caused by intestinal epithelial cell apoptosis (31). These factors thus adversely affect preterm infants in terms of their susceptibility to infection and to inflammatory gut disease such as necrotizing enterocolitis (NEC) (30). Because SIDS infants are more likely to have had symptoms of infectious diseases in the last week or the last day before death, they are more often examined by a physician and given antibiotics than control babies (32). It remains unknown if antibiotics specifically contribute to the risk of SIDS.
Gut Immunology and Homeostasis
The intestinal immune system is modulated in response to environmental factors shortly after birth (33). Battersby and Gibbons (34) summarized the emerging knowledge of how the gut maintains immune homeostasis with non-pathogenic bacteria (34). A better understanding of the molecular and cellular mechanisms sustaining homeostasis is emerging (35–37).
Mechanisms by Which the Gut Microbiome Could Induce Inflammation and Transit of Bacteria to the Blood Stream
Microbial Recognition and Inflammation
The nuclear factor kappa B (NF-κB) pathway is responsible for microbial recognition and inflammatory responses in the adult gut. NF-κB, the so-called “master switch” of the immune system, has numerous roles in innate and adaptive immune responses and inflammation. In the mature gut, pathogen-activated molecular patterns (PAMP)-specific region of a gut pathogen binds to its corresponding Toll-like receptor (TLR) on an enterocyte (EC) and causes release of NF-κB (38). The reaction enters the nucleus where genes mediating inflammation are turned on (38–40). In response to bacterial lipopolysaccharide (LPS), fetal ECs upregulate the NF-κB pathway and produce more of the chemokines, CXCL2 and CXCL8 (41).
Tolerance and Commensal Bacteria
First exposure of the neonatal gut to LPS and antigens from non-pathogenic commensals may result in significant inflammatory responses. The mechanisms by which tolerance to non-pathogenic commensals is established at this most critical period require further understanding. Part of the process includes inhibition of the NF-κB pathway by commensal bacteria (40, 42).
As ECs mature, expression of surface TLRs reduces, as do downstream signaling complexes, while inhibitory factor kappa (iκB), a negative regulator, increases. This appears to provide “protection” without adverse inflammation during colonization (43). Alterations in innate immune response genes in fetal ECs contribute to NEC (44) and suggest that an inappropriate inflammatory response takes place in premature babies. Commensal bacteria exert a variety of effects on cytokine responses. These include production of pro-inflammatory cytokines via the IL-25–IL-23–IL-17 axis (45), possible downregulation of IL-17 in Th-17 cells resulting in diminished inflammation, and upregulation of IL-25. IL-25 is thought to suppress IL-23 by gut dendritic cells (DCs) with subsequent diminished IL-17 and an anti-inflammatory effect (45). Commensal bacteria also affect responses of CD103+ DCs to products of ECs (such as retinoic acid, TDF-β, and cytokine thymic stromal lymphopoietin (TSLP) that effectively dampen the adaptive immune response in those (46). In addition, commensal bacteria (such as Bifidobacterium sp.) may upregulate IL-10 production by DCs (47).
Pathogen Recognition
Pathogen recognition by innate immune cells initiates an immune response to infection. Key PAMPs instigate effector responses through activation of pathogen pattern recognition receptors (PRRs), which include well-characterized TLRs. Adults and children carry an increased infection risk if they carry TLR polymorphisms. The same is true for SIDS (48). Neonatal infection with Gram-positive and Gram-negative bacterial infections is associated with enhanced expression of TLR2 and TLR4 (38). Dysregulation of TLR4 expression is associated with development of NEC (49), commonly associated with neonatal sepsis. In addition to activation by exogenous PAMPs such as LPS or viral ssDNA, TLRs occurs through endogenous damage- or danger-associated molecular patterns (DAMPs), intracellular proteins, and inflammatory mediators released by damaged or apoptotic cells. These include endogenous Alarmin High-Mobility Group Box 1 (HMGB1), heat shock proteins, and uric acid; each contributes to the pathophysiology of septic shock. Dysregulated HMGB1 expression associated with progression of sepsis to septic shock (43, 50) perpetuates the inflammatory response. Disruption of EC tight junctions also occurs and leads to increased bacterial translocation (51) and bacteremia.
Recognition of PAMPS or DAMPS by PRRs results in activation of NFkB resulting in pro-inflammatory cytokine and chemokine production and induction of the IRF transcription factors that mediate production of type I interferon. The NOD-like receptor (NLR) family of proteins shares a number of common domains and many are involved in PAMP and DAMP sensing. The process results in NFkB activation and inflammatory gene expression (52, 53). NLRP proteins oligomerize directly or indirectly with caspase1 through the caspase recruitment domain (CARD) to form an inflammasome. Inflammasomes are essentially caspase-activating complexes.
The anti-inflammatory nature of IL-10 is well known, except, perhaps in SIDS where the cytokine might contribute to a fatally defective pathogen recognition (48). In the neonatal gut, IL-10 is reported to reduce inflammation as evidenced by inhibition of key parts of the unfolded protein response (UPR) (38, 54). Paneth and goblet cells respond to abnormal protein handling with the UPR, which results in local tissue inflammation through activation of immune cells including neutrophils (55).
Role of Innate Lymphoid Cells
These cells do not possess a specific antigen receptor; however, innate lymphoid cells (ILCs) are able to secrete a number of cytokines equivalent to those produced by the subsets (subsets TH2, TH17, and TH22.) of T helper cells. Subsets of ILCs have been demonstrated. ILC1 cells differ from natural killer (NK) cells in that they lack CD56, CD16, and CD94 NK cell markers as well as perforin, and granzyme B. ILCs function in lymphoid organogenesis, tissue remodeling, antimicrobial immunity, and inflammation, particularly at barrier surfaces (56). Their ability to respond promptly to insults inflicted by stress-causing microbes strongly suggests that ILCs are critical in first-line immunological defenses. A number of families of ILCs have been described. These include Rorγt-expressing cells involved in lymphoid tissue formation, mucosal immunity, and inflammation. Type 2 ILCs are important for helminth immunity, type 3 for mucosal integrity and healing.
Gut homeostasis depends on minimizing responses to commensal bacteria, which can lead to inflammatory bowel disease (IBD) in genetically predisposed individuals; but there is a need to retain the ability to recognize and control the growth of infectious pathogens (56). Group 3 innate lymphoid cells (ILC3) help maintain intestinal homeostasis by producing the cytokine IL-22, which promotes mucosal healing and maintains barrier integrity. Microbial signals trigger production of IL-23 and IL-1β; these stimulate ILC3s to produce IL-22, leading to induction of antibacterial peptides and epithelial cell regeneration (57). The cell type producing IL-23 in response to microbial signals is unclear and much debated; resident mononuclear phagocytes (MNPs), inflammatory monocytes, and conventional DCs have all been implicated. Longman et al. (57) provide a clue. In mice with Clostridium rodentium infection and in patients with colitis, CX3CR1+ MNPs are superior producers of IL-23 and IL-1β, and they are very efficient in inducing IL-22 production by ILC3 (56, 58).
The development of Peyer’s patches and ILC2 and ILC3 subsets depends on Nfil3 (nuclear factor, interleukin 3-regulated). Loss of Nfil3 selectively decreases Peyer’s patch formation and is associated with defective recruitment and distribution of ILCs within the patches. ILC subsets strongly express Nfil3. Deletion of Nfil3 genes adversely affects development of all subsets, so that Nfil3-/- knockout mice show increased susceptibility to infection or pro-inflammatory agents confirming the importance of the role of Nfil3 in development of ILC subsets upon which the gut depends for protective immunity (59), especially against intestinal pathogens (60). Additionally, development of all innate lymphoid cell subsets depends on Nfil3.
Gut Microbiome, Inflammation, and SIDS
The microbiome contributes to development and sustenance of the immune system. This includes protective immune effector function in the healthy host as well as in cases of disease. It is noted that SIDS cases frequently have been unwell in the days leading up to the death. Diarrheal symptoms are often reported (61). Bacteremia associated with viral gastroenteritis often with an accompanying fever is a known complication (62). Bacteremia without localizing source is also well known (63), and occult bacteremia is not uncommon in infancy (64). Such episodes of bacteremia are usually benign, indicating that most babies’ immune responses are able to cope without lethal consequences. By contrast, SIDS babies might have dysregulated responses (48). In these circumstances, it could be suggested that bacteremia could result in an overwhelming cytokine storm with consequent sepsis/toxemia resulting in the baby’s demise.
In addition to the pathogenic E. coli groups (Bettelheim and Goldwater, this issue), two groups of the gut microbiome have been investigated in relation to SIDS – Bacteroides thetaiotaomicron and Clostridium species.
Gram-Negative Bacteria and Inflammation
It is known that the NLRC4 inflammasome is activated through caspase1 by Gram-negative bacteria containing type III or type IV secretion systems, e.g., Salmonella, Shigella, Legionella, Pseudomonas, Yersinia, and some E. coli. NLRC4 specifically recognizes flagellin; consequently, non-motile strains are unable to activate caspase1. Flagellin protein alone has been shown to activate caspase1. Caspase1 activation appears to be dependent on a rod protein in the type III secretion system that contains a flagellin-like motif (52, 53).
Translocation of bacteria through an inflamed gut wall leading to bacteremia sets the stage for further induction of pro-inflammatory cytokines and perturbation of the clotting cascades. As IL-6 is elevated in the CSF of SIDS cases (65), it is reasonable to surmise that pro-inflammatory cytokines might be responsible for observed organ changes: brain microglial response (10); increased brain weight; myocardial acute inflammatory reaction (66). Pro-inflammatory cytokines are responsible for perturbation of the clotting cascade and loss of endothelial integrity (67) resulting in raised fibrin degradation products and the vasculopathy evidenced as intrathoracic petechial hemorrhages. SIDS babies frequently carry a low-producer polymorphism for the anti-inflammatory cytokine IL-10 (68), which could contribute to reduced control of pro-inflammatory cytokine release.
B. thetaiotaomicron
B. thetaiotaomicron, a Gram-negative obligate anaerobe, colonizing the human gut, is a major endosymbiont of the human gut. It can hydrolyze and utilize as an energy source non-digestible polysaccharides and maltooligosaccharides (69). It contributes to bacteria–host symbiosis, postnatal intestinal development, physiology and metabolism of the host (70), and development of the immune system (71).
Bacteroides thetaiotaomicron is recognized as a major player in the adult intestinal microbiome and is useful as a model for the investigation of human–bacterial interactions. It degrades plant polysaccharides. In the infant, it is important during transition from breast milk to a high plant starch diet. It has been shown to stimulate intestinal angiogenesis in response to microbial products reaching the Paneth cells (72). Postnatally, B. thetaiotaomicron mediates formation of the mucosal gut barrier assisting in protection against pathogenic invasion seemingly through its effect on expression of species-specific protein antibiotics (73). B. thetaiotaomicron allows for adaptive carbohydrate foraging through its environmental sensing system (74). This results in stabilized food webs, and sustenance of bacterial communities (73). This could be important in protection against bacteria and/or toxins purportedly involved in SIDS pathogenesis. The species has been identified among a higher proportion of SIDS infants (30%) compared with control healthy babies (8.8%) (17).
Clostridium spp
Lecithinase-positive clostridia and other clostridia are found significantly more often in formula-fed babies than breast-fed babies (75). Our study found SIDS babies had significantly higher rates of colonization with these anaerobes than live comparison babies (17). Formula feeding seemed to show a trend toward higher colonization rates with clostridia than breastfeeding (17). The role of clostridia in gut inflammation is discussed below. In the context of inflammation, the review by Schuijt et al. (76) considered the hypothesis that the gut has an important detrimental role in promoting systemic inflammation and infection in the critically ill. They demonstrated that during stress and mucosal hypoxia, the mucosa is damaged and host defenses break down allowing bacteria and toxin translocation thought to produce overwhelming inflammation, sepsis, and multiorgan failure (77, 78).
Evidence of Infection in SIDS
Babies dying of SIDS are reported to have sweat-soaked clothing and bedding indicating a febrile episode in the last sleep (61). Rectal temperatures at autopsy of SIDS babies are elevated providing further evidence of fever during the last sleep (79). The basis for a link between the gut microbiome and SIDS is founded upon a substantial body of published evidence and congruence with factors associated with sepsis (Table 1). This provides an explanatory scheme for underlying pathogenic mechanisms involved in events leading to SIDS.
Pathological Findings
Consistent pathological findings include intrathoracic petechiae; liquid heart blood; heavy wet lungs; large heavy brain with microglial response (10); acute myocardial inflammatory reaction (66); elevated fibrin degradation products (80); and recent viral infection (61). These suggest a single, common patho-mechanism (5, 6). The finding of S. aureus and E. coli and other coliforms in normally sterile sites (66, 81, 82) supports the idea of bacteremia occurring as a plausible near-terminal event. Morris’ Common Bacterial Toxin Hypothesis (1, 2, 83) first indicated a key role of bacteremia in the final pathway. How the purported bacteremia arises seems to implicate gut integrity/permeability/immunity, which depend on the gut microbiome (84). Potentially, pathogenic Clostridia species are over-represented in SIDS babies (17); these could influence the integrity of the gut wall through either induction of an inflammatory response or mechanism involving disruption of EC tight junctions leading to increased bacterial translocation (51).
Inflammation and Pro-Inflammatory Cytokines
Inflammasomes are thought to be key in induction of inflammation through production of pro-inflammatory cytokines. Secretion of IL-1β mediates recruitment of cells to the site of proposed clostridia-mediated insult and could play a role in SIDS (52, 53). While gut inflammation is considered to be primary in the events leading to SIDS, the other side of the equation is defective responses to infection, e.g., a defective pathogen recognition pathway.
Clinico-Physiological Events in SIDS
The clinical/physiological events observed in SIDS cases captured on memory monitors (85) can be explained by development of septic/toxic shock resulting from the release of pro-inflammatory cytokines secondary to a bacteremic episode or toxemia. These include fever, tachycardia followed by profound bradycardia, hypoxemia, and gasping (after asystole) (85).
Evidence of Bacteremia/Toxemia in SIDS Infants
The relatively low rate of sterile site infection (16–19% of SIDS cases) could argue against the bacteremia hypothesis. The finding of viable bacteria in a normally sterile site depends on the bacteria’s cultivability. Most of the gut microbiota are unlikely to grow on standard diagnostic artificial culture media (86), and this could contribute to low-positive culture rates. It remains highly probable that non-cultivable bacteria entering the blood stream could induce the cytokine storm thought to underlie the final events in SIDS.
Brain and Heart Abnormalities in SIDS
The findings of brain abnormality (87) and of smaller than normal heart and kidneys (88) seem to relate to abnormal prenatal development. This was refuted by Guntheroth and Spiers (89) and needs to be revisited. The smaller than normal heart (with accompanying possible predisposition to arrythmia) could make the infant more vulnerable to profound immuno-inflammatory events associated with bacteremia/sepsis.
SIDS Risk Factors
Risk factors for SIDS can be categorized into genetic, prenatal, and postnatal. These have been described previously (5, 6). Why prone sleep position is a risk factor has been explained by the acquisition of bacteria from contaminated surfaces (sofas, parental bed, being especially contaminated and high risk) (17). It is important to note that data purportedly showing a relationship between bedsharing and SIDS and inferring overlaying/asphyxia as cause (90) needs re-examination because the reason for bedsharing remains poorly described in published studies. It could be interpreted that bedsharing on the night of the last sleep came about because the baby was unsettled or unwell, indicating subtle symptoms of infection. This is supported in a systematic review of bed sharing in which illness and crying were frequently reported (91). The role of alcohol and drugs by co-sleeping parent(s) has also been interpreted as leading to overlaying. The alternative suggestion of lessened awareness in the co-sleeping parent might lead to failure to notice subtle symptoms of illness in the baby. Using overlaying with consequent asphyxiation as the major explanation of the risk factor of bedsharing requires re-evaluation. Mainstream researchers attribute a “respiratory” cause for intrathoracic petechiae. Differentiation between the intrathoracic petechiae observed in asphyxia and SIDS have been well described (92, 93) and show clear differences; yet, these findings are largely ignored by mainstream researchers.
Future Research
Delineation of the gut microbiome in SIDS and healthy babies might provide additional clues to the processes underlying the role of the gut in SUDI. Extended immunohistological examination of the gut wall for evidence of ILCs and subtle gut inflammatory changes in SIDS babies might provide evidence of inflammation contributing to a functional failure of the gut wall to prevent bacterial translocation from the gut lumen into the bloodstream. Nfil3 gene polymorphisms should be sought as these could contribute to gut vulnerability. In the context of bacteremia, efforts using PCR technology should be made to exclude/demonstrate the presence of non-cultivable bacteria in normally sterile sites. The gut microbiome observed in SIDS might correlate with the presence of ILCs and a “pro-inflammatory gut microbiome.” Such findings might contribute to a new definition of SIDS. The corollary of establishing a possible “anti-inflammatory” gut microbiome in healthy babies via diet or other means (pre- or probiotic) could be explored as a “natural” mechanism of protecting babies in the future but not forgetting the messages advocating a safe sleeping environment and parental avoidance of cigarette smoke and other harmful drugs.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Professor Goldwater has been supported by a grant from the Foundation for the Study of Infant Death (UK).
References
1. Morris JA, Haran D, Smith A. Hypothesis: common bacterial toxins are a possible cause of the sudden infant death syndrome. Med Hypotheses (1987) 22(2):211–22. doi:10.1016/0306-9877(87)90145-9
2. Morris JA. The common bacterial toxins hypothesis of sudden infant death syndrome. FEMS Immunol Med Microbiol (1999) 25:11–7. doi:10.1111/j.1574-695X.1999.tb01322.x
3. Lindsay JA, Mach A, Wilkinson MA, Martin LM, Wallace FM, Keller AM, et al. Clostridium perfringens type-A cytotoxic-enterotoxin(s) as triggers for death in the sudden-infant-death-syndrome development of a toxico-infection hypothesis. Curr Microbiol (1993) 27:51–9. doi:10.1007/BF01576834
4. Murrell WG, Stewart BJ, O’Neill C, Siarakas S, Kariks S. Enterotoxigenic bacteria in the sudden infant death syndrome. J Med Microbiol (1993) 39:114–27. doi:10.1099/00222615-39-2-114
5. Goldwater PN, Bettelheim KA. SIDS risk factors: time for new interpretations the role of bacteria. Pediatrics Res Intern J (2013). Available from: http://www.ibimapublishing.com/journals/PRIJ/prij.html. doi:10.5171/2013.867520
6. Goldwater PN. A perspective on SIDS pathogenesis. The hypotheses: plausibility and evidence. BMC Med (2011) 9:64. doi:10.1186/1741-7015-9-64
7. Blackwell CC, Moscovis SM, Gordon AE, Al Madani OM, Hall ST, Gleeson M, et al. Cytokine responses and sudden infant death syndrome: genetic, developmental, and environmental risk factors. J Leukoc Biol (2005) 78(6):1242–54. doi:10.1189/jlb.0505253
8. Krous HF. Sudden infant death syndrome: pathology and pathophysiology. Pathol Annu (1984) 19:1–14.
9. Tonkin SL, Gunn TR, Bennet L, Vogel SA, Gunn AJ. A review of the anatomy of the upper airway in early infancy and its possible relevance to SIDS. Early Hum Dev (2002) 66:107–21. doi:10.1016/S0378-3782(01)00242-0
10. Kinney HC, Richerson GB, Dymecki SM, Darnall RA, Nattie EE. The brainstem and serotonin in the sudden infant death syndrome. Annu Rev Pathol (2009) 4:517–50. doi:10.1146/annurev.pathol.4.110807.092322
11. Malam JE, Carrick GF, Telford DR, Morris JA. Staphylococcal toxins and sudden infant death syndrome. J Clin Pathol (1992) 45:716–21. doi:10.1136/jcp.45.8.716
12. Zorgani A, Essery SD, Madani OA, Bentley AJ, James VS, MacKenzie DA, et al. Detection of pyrogenic toxins of Staphylococcus aureus in sudden infant death syndrome. FEMS Immunol Med Microbiol (1999) 25(1–2):103–8. doi:10.1111/j.1574-695X.1999.tb01332.x
13. Blackwell CC, Gordon AE, James VS, MacKenzie DA, Mogensen-Buchanan M, El Ahmer OR, et al. The role of bacterial toxins in sudden infant death syndrome (SIDS). Int J Med Microbiol (2002) 291(6–7):561–70. doi:10.1078/1438-4221-00168
14. Highet AR, Goldwater PN. Staphylococcal enterotoxin genes are common in Staphylococcus aureus intestinal flora in sudden infant death syndrome (SIDS) and live comparison infants. FEMS Immunol Med Microbiol (2009) 57(2):151–5. doi:10.1111/j.1574-695X.2009.00592.x
15. Bettelheim KA, Goldwater PN, Dwyer BW, Bourne AJ, Smith DL. Toxigenic Escherichia coli associated with sudden infant death syndrome. Scand J Infect Dis (1990) 22:467–76. doi:10.3109/00365549009027079
16. Bettiol SS, Radcliff FJ, Hunt ALC, Goldsmid JM. Bacterial flora of Tasmanian SIDS infants with special reference to pathogenic strains of Escherichia coli. Epidemiol Infect (1994) 112:275–84. doi:10.1017/S095026880005768X
17. Highet AR, Berry AM, Bettelheim KA, Goldwater PN. Gut microbiome in sudden infant death syndrome (SIDS) differs from that in healthy comparison babies and offers an explanation for the risk factor of prone position. Int J Med Microbiol (2014) 304:735–41. doi:10.1016/j.ijmm.2014.05.007
18. Gleeson M, Clancy RL, Cripps AW. Mucosal immune response in a case of sudden infant death syndrome. Pediatr Res (1993) 33(6):554–6. doi:10.1203/00006450-199306000-00003
19. Tissier H. Repartition des microbes dans l’intestin du nourisson. (Distribution of microorganisms in the newborn intestinal tract). Ann Inst Pasteur (Paris) (1905) 19:109–23.
20. Harmsen HJ, Wildeboer-Veloo AC, Raangs GC, Wagendorp AA, Klijn N, Bindels JG, et al. Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J Pediatr Gastroenterol Nutr (2000) 30:61–7. doi:10.1097/00005176-200001000-00019
21. Stark PL, Lee A. The microbial ecology of the large bowel of breast-fed and formula-fed infants during the first year of life. J Med Microbiol (1982) 15:189–203. doi:10.1099/00222615-15-2-189
22. Forsythe P, Kunze WA. Voices from within: gut microbes and the CNS. Cell Mol Life Sci (2013) 70:55–69. doi:10.1007/s00018-012-1028-z
23. Grölund M-M, Lehtonen O-P, Eerola E, Kero P. Fecal microflora in healthy infants born by different methods of delivery: permanent changes in intestinal flora after cesarean delivery. J Pediatr Gastroenterol Nutr (1999) 28:19–25. doi:10.1097/00005176-199901000-00007
24. Macfarlane GT, McBain AJ. The human colonic microbiota. In: Gibson GR, Roberfroid MB, editors. Colonic Microbiota, Nutrition and Health. Dordecht: Kluwer Academic Publishers (1999). 125 p.
25. Cooperstock MS, Zedd AJ. Intestinal Flora of Infants D.J. Hentges Human Intestinal Microflora in Health and Disease. New York, NY: Academic Press (1983). p. 78–93.
26. Dore J, Sghir A, Hannequart-Gramet G, Corthier G, Pochart P. Design and evaluation of a 16S rRNA-targeted oligonucleotide probe for specific detection and quantitation of human faecal Bacteroides populations. Syst Appl Microbiol (1998) 21:65–71. doi:10.1016/S0723-2020(98)80009-X
27. Highet AR, Goldwater PN. Maternal and perinatal risk factors for SIDS: a novel analysis utilizing pregnancy outcome data. Eur J Pediatr (2013) 172(3):369–72. doi:10.1007/s00431-012-1896-0
28. Biasucci G, Benenati B, Morelli L, Bessi E, Boehm G. Cesarean delivery may affect the early biodiversity of intestinal bacteria. J Nutr (2008) 138:1796S–800S.
29. Malloy MH. Prematurity and sudden infant death syndrome: United States 2005–2007. J Perinatol (2013) 33:470–5. doi:10.1038/jp.2012.158
30. Claud EC, Walker WA. Hypothesis: inappropriate colonization of the premature intestine can cause neonatal necrotizing enterocolitis. FASEB J (2001) 15:1398–403. doi:10.1096/fj.00-0833hyp
31. Ikeda T, Hiromatsu K, Hotokezaka M, Chijiiwa K. Up-regulation of intestinal toll-like receptors and cytokines expressions change after TPN administration and a lack of enteral feeding. J Surg Res (2010) 160:244–52. doi:10.1016/j.jss.2009.01.022
32. Helweg-Larsen K, Lundemose JB, Øyen N, Skjærven R, Alm B, Wennergren G, et al. Interactions of infectious symptoms and modifiable risk factors in sudden infant death syndrome. The Nordic Epidemiological SIDS study. Acta Paediatr (1999) 88:521–7. doi:10.1111/j.1651-2227.1999.tb00168.x
33. Rognum TO, Thrane PS, Stoltenberg L, Vege A, Brandtzaeg P. Development of intestinal mucosal immunity in fetal life and the first postnatal months. Pediatr Res (1992) 32(2):145–9.
34. Battersby AJ, Gibbons DL. The gut mucosal immune system in the neonatal period. Pediatr Allergy Immunol (2013) 24(5):414–21. doi:10.1111/pai.12079
35. Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature (2012) 489:231–41. doi:10.1038/nature11551
36. Newburg DS, Walker WA. Protection of the neonate by the innate immune system of developing gut and of human milk. Pediatr Res (2007) 61:2–8. doi:10.1203/01.pdr.0000250274.68571.18
37. Yang H, Finaly R, Teitelbaum DH. Alteration in epithelial permeability and ion transport in a mouse model of total parenteral nutrition. Crit Care Med (2003) 31:1118–25. doi:10.1097/01.CCM.0000053523.73064.8A
38. Zhang JP, Chen C, Yang Y. Changes and clinical significance of toll-like receptor 2 and 4 expression in neonatal infections. Zhonghua Er Ke Za Zhi Chinese J Pediatr (2007) 45:130–3.
39. Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol (2008) 8:411–20. doi:10.1038/nri2316
40. Walker WA. Development of the intestinal mucosal barrier. J Pediatr Gastroenterol Nutr (2002) 34(Suppl 1):S33–9. doi:10.1097/00005176-200205001-00009
41. Lotz M, Gutle D, Walther S, Menard S, Bogdan C, Hornef MW. Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J Exp Med (2006) 203:973–84. doi:10.1084/jem.20050625
42. Neish AS, Gewirtz AT, Zeng H, Young AN, Hobert ME, Karmali V, et al. Prokaryotic regulation of epithelial responses by inhibition of IkappaB-alpha ubiquitination. Science (2000) 289:1560–3. doi:10.1126/science.289.5484.1560
43. Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol (2005) 5:331–42. doi:10.1038/nri1594
44. Nanthakumar N, Meng D, Goldstein AM, Zhu W, Lu L, Uauy R, et al. The mechanism of excessive intestinal inflammation in necrotizing enterocolitis: an immature innate immune response. PLoS One (2011) 6:e17776. doi:10.1371/journal.pone.0017776
45. Zaph C, Du Y, Saenz SA, Nair MG, Perrigoue JG, Taylor BC, et al. Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J Exp Med (2008) 205:2191–8. doi:10.1084/jem.20080720
46. Barbosa T, Rescigno M. Host-bacteria interactions in the intestine: homeostasis to chronic inflammation. Wiley Interdiscip Rev Syst Biol Med (2010) 2:80–97. doi:10.1002/wsbm.48
47. Hart AL, Lammers K, Brigidi P, Vitali B, Rizzello F, Gionchetti P, et al. Modulation of human dendritic cell phenotype and function by probiotic bacteria. Gut (2004) 53:1602–9. doi:10.1136/gut.2003.037325
48. Highet AR, Berry AM, Goldwater PN. Novel hypothesis for unexplained sudden unexpected death in infancy (SUDI). Arch Dis Child (2009) 94:841–3. doi:10.1136/adc.2009.158352
49. Leaphart CL, Cavallo J, Gribar SC, Cetin S, LI J, Branca MF, et al. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J Immunol (2007) 179:4808–20. doi:10.4049/jimmunol.179.7.4808
50. Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol (2008) 8:776–87. doi:10.1038/nri2402
51. Sappington PL, Yang R, Yang H, Tracey KJ, Delude RL, Fink MP. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology (2002) 123:790–802. doi:10.1053/gast.2002.35391
52. Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity (2011) 34:637–50. doi:10.1016/j.immuni.2011.05.006
53. Nish S, Medzhitov R. Host defense pathways: role of redundancy and compensation in infectious disease phenotypes. Immunity (2011) 34:629–36. doi:10.1016/j.immuni.2011.05.009
54. Shkoda A, Ruiz PA, Daniel H, Kim SC, Rogler G, Sartor RB, et al. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology (2007) 132:190–207. doi:10.1053/j.gastro.2006.10.030
55. Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 linksERstress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell (2008) 134:743–56. doi:10.1016/j.cell.2008.07.021
56. Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol (2012) 30:647–75. doi:10.1146/annurev-immunol-020711-075053)
57. Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, et al. CX3CR1+ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med (2014) 211(8):1571–83. doi:10.1084/jem.20140678
58. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol (2013) 14:221–9. doi:10.1038/NI.2534
59. Seillet C, Rankin LC, Groom JR, Mielke LA, Tellier J, Chopin M, et al. Nfil3 is required for the development of all innate lymphoid cell subsets. J Exp Med (2014) 211(9):1733–40. doi:10.1084/jem.20140145
60. Geiger TL, Abt MC, Gasteiger G, Firth MA, O’Connor MH, Geary CD, et al. Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J Exp Med (2014) 211(9):1723–31. doi:10.1084/jem.20140212
61. Hoffman HJ, Damus K, Hillman L, Krongrad E. Risk factors for SIDS: results of the national institute of child health and human development SIDS cooperative epidemiological study. Ann N Y Acad Sci (1988) 533:13–30. doi:10.1111/j.1749-6632.1988.tb37230.x
62. Gözmen S, Sükran Gözmen K, Apa H, Aktür H, Sorguç Y, Bayra N, et al. Secondary bacteremia in rotavirus gastroenteritis. Pediatr Infect Dis J (2014) 33(7):775–7. doi:10.1097/INF.0000000000000324
63. Ligon J, Kaplan SL, Hulten KG, Mason EO, McNeil C. Staphylococcus aureus Bacteremia without a localizing source in pediatric patients. Pediatr Infect Dis J (2014) 33(5):e132–4. doi:10.1097/INF.0000000000000195
64. Kuppermann N. Occult bacteremia in young febrile children. Pediatr Clin North Am (1999) 46:1073–109. doi:10.1016/S0031-3955(05)70176-0
65. Vege Å, Rognum TO, Scott H, Aasen AO, Saugstad OD. SIDS cases have increased levels of interleukin-6 in cerebrospinal fluid. Acta Paediatr (1995) 84(2):193–6. doi:10.1111/j.1651-2227.1995.tb13608.x
66. Rambaud C, Guibert M, Briand E, Grangeot-Keros L, Coulomb-L’Herminé A, Dehan M. Microbiology in sudden infant death syndrome (SIDS) and other childhood deaths. FEMS Immunology & Medical Microbiology (1999) 25(1–2):59–66.
67. Schouten M, Joost Wiersinga W, Levi M, van der Poll T. Inflammation, endothelium, and coagulation in sepsis. J Leukoc Biol (2008) 83(3):536–45. doi:10.1189/jlb.0607373
68. Summers AM, Summers CW, Drucker DB, Hajeer AH, Barson A, Hutchinson IV. Association of IL-10 genotype with sudden infant death syndrome. Hum Immunol (2000) 61:1270–3. doi:10.1016/S0198-8859(00)00183-X
69. D’Elia JN, Salyers AA. Contribution of a neopullulanase, a pullulanase, and an alpha-glucosidase to growth of Bacteroides thetaiotaomicron on starch. J Bacteriol (1996) 178:7173–9.
70. Xu J, Bjursell MK, Himrod J, Deng S, Carmichael L, Chiang HC, et al. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science (2003) 299:2074–6. doi:10.1126/science.1080029
71. Bauer E, Williams BA, Smidt H, Verstegen MWA, Mosenthin R. Influence of the gastrointestinal microbiota on development of the immune system in young animals. Curr Issues Intestinal Microbiol (2006) 7:35–52.
72. Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci U S A (2002) 99(24):15451–5. doi:10.1073/pnas.202604299
73. Xu J, Gordon JI. Honor thy symbionts. Proc Natl Acad Sci U S A (2003) 100:10452–9. doi:10.1073/pnas.1734063100
74. Sonnenburg ED, Sonnenburg JL, Manchester JK, Hansen EE, Chiang HC, Gordon JI. A hybrid two-component system protein of a prominent human gut symbiont couples glycan sensing in vivo to carbohydrate metabolism. Proc Natl Acad Sci U S A (2006) 103:8834–9. doi:10.1073/pnas.0603249103
75. Benno Y, Sawada K, Mitsuoka T. The intestinal microflora of infants: composition of fecal flora in breast-fed and bottle-fed infants. Microbiol Immunol (1984) 28:975–86. doi:10.1111/j.1348-0421.1984.tb00754.x
76. Schuijt TJ, van der Poll T, Wiersinga WJ. Gut Microbiome and host defense interaction during critical illness. In: Vincent JL, editor. Annual Update in Intensive Care and Emergency Medicine 2012. (Vol. 2012). Berlin, Heidelberg: Springer-Verlag (2012). p. 29–42.
77. Taylor DE. Revving the motor of multiple organ dysfunction syndrome. Gut dysfunction in ARDS and multiorgan failure. Respir Care Clin N Am (1998) 4:611–31.
78. Alverdy JC, Chang EB. The re-emerging role of the intestinal microflora in critical illness and inflammation: why the gut hypothesis of sepsis syndrome will not go away. J Leukoc Biol (2008) 83:461–6. doi:10.1189/jlb.0607372
79. Sunderland R, Emery JL. Febrile convulsions and cot death. Lancet (1981) 2(8239):176–8. doi:10.1016/S0140-6736(81)90359-7
80. Goldwater PN, Williams V, Bourne AJ, Byard RW. Sudden infant death syndrome: a possible clue to causation. Med J Aust (1990) 153:59–60.
81. Weber MA, Klein NJ, Hartley JC, Lock PE, Malone M, Sebire NJ. Infection and sudden unexpected death in infancy: a systematic retrospective case review. Lancet (2008) 371:1848–53. doi:10.1016/S0140-6736(08)60798-9
82. Goldwater PN. Sterile site infection at autopsy in sudden unexpected deaths in infancy. Arch Dis Child (2009) 94:303–7. doi:10.1136/adc.2007.135939
83. Morris JA, Harrison LM, Biswas J, Telford DR. Transient bacteraemia: a possible cause of sudden life threatening events. Med Hypotheses (2007) 69:1032–9. doi:10.1016/j.mehy.2007.02.039
84. Ferreira C, Veldhoen M. Host and microbes date exclusively. Cell (2012) 49:1428–33. doi:10.1016/j.cell.2012.06.005
85. Poets CF, Meny RG, Chobanian MR, Bonofiglo RE. Gasping and other cardiorespiratory patterns during sudden infant deaths. Pediatr Res (1999) 45:350–4. doi:10.1203/00006450-199903000-00010
86. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science (2005) 38(5728):1635–8. doi:10.1126/science.1110591
87. Filiano JJ, Kinney HC. A perspective on neuropathologic findings in victims of the sudden infant death syndrome: the triple-risk model. Biol Neonate (1994) 65:194–7. doi:10.1159/000244052
88. Kelmanson IA. Differences in somatic and organ growth rates in infants who died of sudden infant death syndrome. J Perinat Med (1992) 20(3):183–8. doi:10.1515/jpme.1992.20.3.183
89. Guntheroth WG, Spiers PS. The triple risk hypotheses in sudden infant death syndrome. Pediatrics (2002) 110(5):e64. doi:10.1542/peds.110.5.e64
90. Byard RW. To sleep or not to sleep? Should infants and adults sleep in the same bed together? Med J Aust (2012) 196:10–11. doi:10.5694/mja11.11358
91. Salm Ward TC. Reasons for mother-infant bed-sharing: a systematic narrative synthesis of the literature for future research. Matern Child Health J (2014) 19:675–90. doi:10.1007/s10995-014-1557-1
92. Becroft DM, Thompson JM, Mitchell EA. Epidemiology of intrathoracic petechial hemorrhages in sudden infant death syndrome. Pediatr Dev Pathol (1998) 1(3):200–9. doi:10.1007/s100249900027
Keywords: sudden infant death syndrome, gut, microbiome, immunity
Citation: Goldwater PN (2015) Gut microbiota and immunity: possible role in sudden infant death syndrome. Front. Immunol. 6:269. doi: 10.3389/fimmu.2015.00269
Received: 05 May 2015; Accepted: 15 May 2015;
Published: 03 June 2015
Edited by:
Amanda Cox, Griffith University, AustraliaReviewed by:
Caroline Blackwell, University of Newcastle, AustraliaRichard Luke, La Trobe University, Australia
Copyright: © 2015 Goldwater. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul N. Goldwater, Microbiology and Infectious Diseases Department, SA Pathology at the Women’s and Children’s Hospital, School of Paediatrics and Reproductive Health, University of Adelaide, 72 King William Road, North Adelaide, SA 5006, Australia, paul.goldwater@health.sa.gov.au