 Megan Woods
Megan Woods Yong-Rui Zou
Yong-Rui Zou Anne Davidson
Anne Davidson- Center for Autoimmunity and Musculoskeletal Diseases, Feinstein Institute for Medical Research, New York, NY, USA
Germinal centers (GCs) are the primary site at which clonal expansion and affinity maturation of B cells occur. B cells encounter antigen and receive T cell help in the GC light zone (LZ) and then migrate to the dark zone where they proliferate and undergo somatic mutation before cycling back to the LZ for further rounds of selection. Tolerance to autoantigens is frequently lost de novo as GC B cells undergo class switching and somatic mutation. This loss of tolerance is regulated by a variety of mechanisms including cell death, failure to compete for T cell help, and failure to differentiate into effector cells. Systemic lupus erythematosus (SLE) is characterized by loss of tolerance to nucleic acid antigens. While defects in tolerance occur in the naïve repertoire of SLE patients, pathogenic autoantibodies also arise in the GC by somatic mutation from non-autoreactive precursors. Several B cell defects contribute to the loss of GC tolerance in SLE, including polymorphisms of genes encoded by the Sle1 locus, excess TLR7 signaling, defects in FcRIIB expression, or defects of B cell apoptosis. Extrinsic soluble factors, such as Type-1 IFN and B cell-activating factor, or an increased number of T follicular helper cells in the GC also alter B cell-negative selection. Finally, defects in clearance of apoptotic debris within the GC result in BCR-mediated internalization of nucleic acid containing material and stimulation of autoantibody production by endosomal TLR-driven mechanisms.
Introduction – Germinal Center Formation and Structure
In quiescent lymphoid follicles, a chemokine gradient restricts CCR7+/PSGL-1+ T cells to a central area and CCR7−/CXCR5+ B cells to a ring surrounding the T cell zone. Major changes in this structure occur upon antigen encounter. Activated T cells downregulate CCR7 and PSGL-1 and upregulate CXCR5, promoting their migration to the interfollicular zone where they contact antigen-activated B cells that have upregulated CCR7. The interaction of ICOSL on B cells with ICOS on T cells initiates the differentiation of T follicular helper cells (TFH), whereas T cell help promotes the expansion of extrafollicular (EF) B cell foci (1–3). B cell fate decision is then regulated by several factors including the affinity of the B cell receptor (BCR) for antigen, costimulatory signals from T cells, and the cytokine milieu (4, 5). Some B cells will remain outside the follicle and upregulate Blimp1, becoming short-lived low-affinity plasmablasts. Other B cells downregulate EBV-induced molecule-2 (EBI2) that interacts with a chemokine-like gradient established by 7α,25-dihydroxycholesterol (6–9); these cells, as well as CXCR5+ TFH, move inside the follicle to form the germinal center (GC) (6). Expression of particular transcription and survival factors including Bcl6 and IRF4 in T cells and Bcl6, Myc, and Mcl1 in B cells is required for the differentiation and survival of GC cells [reviewed in Ref. (10, 11)].
As the GC matures, stromal cells orchestrate the spatial segregation of centrocytes and centroblasts into the light zone (LZ) and dark zone (DZ) regions, respectively (Figure 1). Follicular dendritic cells (FDC), usually located inside quiescent B cell follicles (12), form clusters within the developing LZ; these cells capture and retain immune complexes via the complement receptors, CD21 and CD35 (13, 14). FDCs also produce CXCL13 that recruits CXCR5+ centrocytes and TFH cells into the LZ, thus forming a platform for cognate B–T cell interactions (15). By contrast, CXCR4hi centroblasts are segregated in the DZ by reticular cells expressing high levels of CXCL12 (16). Selection of B cells begins as early as the pre-GC stage and continues in the LZ where centrocytes are exposed to FDC-bound native antigen. High-affinity B cells endocytose and present more antigen to T cells thereby competing most efficiently for help from TFH. Centrocytes then upregulate CXCR4 and migrate into the DZ where they proliferate and accumulate somatic mutations in their variable regions before downregulating CXCR4 and cycling back to the LZ for further rounds of affinity-based selection (17–21). When FDCs are depleted, GC B cells and TFH cells die rapidly, whereas in the absence of CXCR4, GC B cells are confined to the LZ and have fewer somatic mutations (20, 22).
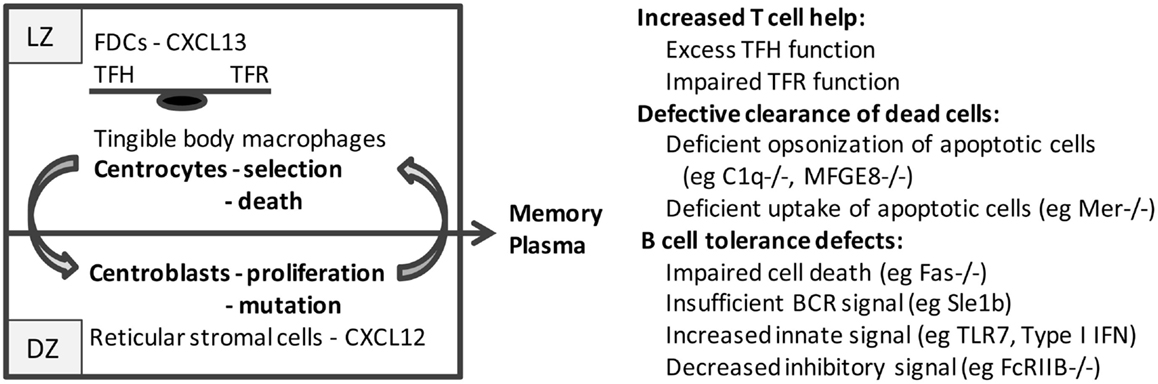
Figure 1. Schematic of the germinal center: germinal centers consist of two major anatomic regions. The major cell types in the light zone (LZ) and dark zone (DZ) are shown. B cell centrocytes undergo selection in the light zone and then migrate to the dark zone where the B cell centroblasts proliferate and undergo somatic mutation before cycling back to the light zone. After several cycles, B cells differentiate into memory cells or plasma cells and exit the germinal center. Defects in germinal center cells or functions that result in spontaneous germinal center formation and loss of tolerance to nucleic acid antigens are shown on the right.
Several types of T cells populate the LZ and secrete a variety of cytokines that drive GC B cell differentiation and effector functions. The characteristics of TFH cells have recently been reviewed and will not be further addressed here (23–25). GC regulatory T cells have also been described (26). Foxp3+CD4+ T follicular regulatory cells (TFR) express CXCR5 and Bcl6 and act by downregulating CD80 and CD86 expression on B cells, thereby diminishing the strength of cognate B–T interactions (27, 28). Qa1-restricted ICOSL+CXCR5+ regulatory CD8 T cells interact directly with Qa1-expressing TFH cells (29). The balance between TFH and TFR sets the threshold at which B cells compete for T cell help; this in turn regulates B cell entry and dwell time in the DZ.
Regulation of B Cell Selection in Germinal Centers
Most high-affinity naïve autoreactive B cells are anergized or deleted before the GC stage (30, 31). However, mechanisms need to be in place to purge those B cells that increase their autoreactivity or acquire autoreactivity de novo as a result of random somatic mutations occurring in the GC (31). How this is achieved is still not fully understood. Elegant studies have been performed in the hen egg lysozyme (HEL)/anti-HEL model using HEL variants with differing affinities and patterns of tissue expression. These studies have led to the paradigm that engagement of the BCR by self-antigen but in the absence of T cell costimulatory signals results in B cell death before the plasma cell stage. Tolerance can be broken if the self-antigen crossreacts with a foreign antigen, and B cells are therefore able to recruit help from anti-foreign T cells, or if the self-antigen is not present in high enough concentrations within the GC to mediate deletion (32).
Regulation of autoreactivity involving nucleic acid autoantigens is, however, more complex because low-affinity IgM anti-nuclear autoantibodies are required to opsonize and promote clearance of nucleic acid antigens that are shed from apoptotic cells; loss of this IgM can induce or accelerate autoimmunity (33). By contrast, IgG autoantibodies directed to nuclear autoantigens can penetrate tissues and initiate inflammatory cascades. There has been much work directed at understanding whether the class-switched autoreactive B cells that arise in systemic lupus erythematosus (SLE) are derived from naïve autoreactive B1, marginal zone, or follicular B cells that undergo clonal expansion either inside or outside the GC or whether they arise de novo by somatic mutation. Mice with site-directed transgenes that encode autoreactive immunoglobulin genes capable of class switching and somatic mutation have been used to address this question.
i. D42 is an anti-dsDNA hybridoma derived from the NZB/W lupus-prone strain. Anti-DNA activity of D42 is conferred by its basic VHCDR3 region as well as by its associated light chain Vκ16–104. In non-autoimmune D42 heavy chain transgenic (D42hTg) mice, autoreactivity is regulated by clonal deletion at the immature stage, clonal anergy, and receptor editing. D42 hybridomas derived from these mice have low-affinity for DNA and use diverse light chains. In lupus-prone D42hTg NZB/W mice, clonal deletion initially appears intact but high-affinity IgG anti-DNA antibodies appear in the serum with age. In this strain, receptor editing of the light chain results in a preference in the naïve repertoire for Vκ4-55*01 that confers low-affinity polyreactivity. Nevertheless, nearly all IgG anti-DNA hybridomas from D42hTg NZB/W mice use Vκ16–104 rearranged to Jk5, a combination generated by receptor editing that confers high affinity for DNA. Thus, receptor editing can protect from autoimmunity but may also generate potentially dangerous antibodies (34–37). Using cell sorting and single cell analyses, we have shown that B cells expressing a restricted repertoire of light chains, including Vκk4–55*01, that confer no or low-affinity autoreactivity are positively selected into the naïve B cell pool of D42hTg NZB/W mice. By contrast, D42/Vκ16-104 expressing B cells are mostly deleted by the late transitional B cell stage, but are then preferentially selected and expanded in the GC as the mice age (38).
ii. The 3H9 heavy chain, also derived from an anti-DNA hybridoma, pairs with a wide variety of light chains to generate DNA and non-DNA binding, as well as low-affinity anti-cardiolipin antibodies (39). In non-autoimmune 3H9 heavy chain transgenic (3H9hTg) mice, autoreactivity is regulated by receptor editing and anergy (40). Those 3H9 B cells that do enter the GC and undergo somatic mutations fail to develop into plasma cells (41). By contrast, 3H9-encoded anti-dsDNA antibodies using pathogenic light chains arise in autoimmune strains (42). We showed in 3H9hTg NZB/W and NZW/BXSB mice that the GC and plasma cell repertoire is dominated by 3H9/Vκ5 pairs and that the Jκ region influences the avidity of such pairs for chromatin and the stringency for GC entry. The acquisition of somatic mutations then increases the affinity for chromatin and confers new binding for DNA or cardiolipin (43).
Taken together, the studies in D42 and 3H9 transgenic NZB/W mice show that failure to regulate GC entry and clonal expansion of rare naïve high-affinity autoreactive B cells (D42) and failure to regulate autoimmunity acquired by somatic mutation in the GC (3H9) can both contribute to loss of tolerance to nuclear autoantigens.
One approach to address the same question in humans is to express immunoglobulin genes from single B cells and test them for autoreactivity. Pioneering studies showed a high frequency of autoreactivity among early immature bone marrow B cells that is subsequently censored during the late immature and early transitional stages. These tolerance checkpoints are compromised in patients with SLE even during periods of remission (44, 45). Normal individuals also regulate autoreactivity within the IgM memory and bone marrow plasma cell compartments (46, 47). Sanz and colleagues showed that autoreactive VH4-34 encoded 9G4 idiotype+ B cells from normal tonsils have an anergic phenotype and fail to mature to a GC phenotype or enter the memory compartment. 9G4 B cells from SLE patients are overrepresented among GC and memory cells, suggesting tolerance failure in the GC (48). By contrast, single cell analyses have demonstrated that autoreactive B cells from the class-switched memory compartment can be derived from non-autoreactive precursors (49). Thus, in humans, as in mice, failure of regulation of autoreactive B cells can occur both at the GC entry checkpoint and at the post-somatic mutation checkpoint.
Many Models of SLE are Characterized by Spontaneous GC Development
The Sle1 mouse that bears a region of chromosome 1 derived from the lupus-prone NZM2410 strain develops spontaneous GCs and high titers of anti-chromatin autoantibodies without systemic inflammation or clinical lupus (50, 51). The Sle1b locus harbors polymorphisms in genes from the signaling lymphocyte activation molecule (SLAM) family of receptors that contribute to loss of GC tolerance in a B cell-intrinsic manner. Paradoxically, Sle1b B cells manifest lower Ca2+ flux and less cell death than their wild-type counterparts, protecting them from negative selection in the GC (52). They also form poorer T–B conjugates, perhaps allowing them to interact more promiscuously with T cells (53).
Germinal center B cells upregulate the inhibitory FcRIIB receptor and this regulates the final differentiation of memory B and plasma cells. Failure to upregulate FcRIIB on GC B cells occurs in the Sle1 model (54) and in human lupus patients (55) resulting in loss of tolerance at the late B cell checkpoint. In addition, several lupus models have a polymorphism that confers lower levels of expression of FcRIIB (56); lupus in these strains is attenuated by restoration of FcRIIB on B cells (57).
Not surprisingly, TFH numbers influence GC reaction and regulate tolerance to autoantigens. One interesting model is deficient in Roquin1, an RNA-binding protein that post-transcriptionally represses expression of ICOS and the costimulatory molecule Ox-40 (58). Roquin1-deficient mice have increased numbers of TFH and develop GC-dependent autoimmunity with a lupus-like phenotype (59). In humans, increased expression of Ox-40L on myeloid antigen-presenting cells is induced by RNA-containing immune complexes and this in turn amplifies TFH responses once tolerance is broken (60). Failure of TFR can also induce lupus. For example, mice deficient in GC CD8 Tregs have large GCs, autoantibodies, and lupus-like glomerulonephritis (29).
Innate immune pathways also contribute to spontaneous GC formation in lupus mice. We showed that administration of a single dose of IFN-expressing adenovirus to young NZB/W and NZW/BXSB mice induces GC formation and autoantibodies in a T cell-dependent manner (61). Using single cell analysis and expression of 3H9 heavy/light chain pairs of interest, we further showed in NZW/BXSB mice that type-I IFN relaxes the stringency for GC entry and clonal expansion resulting in increased autoreactivity of the GC-derived repertoire (43).
Type-1 IFNs are induced through the activation of endosomal TLRs that specifically recognize nucleic acids. TLR7 is needed to generate antibodies against RNA antigens and cardiolipin, whereas TLR9 is needed for spontaneous generation of anti-dsDNA autoantibodies. TLR7-overexpressing mice develop severe inflammatory kidney disease and increased mortality (62), whereas disease is ameliorated in TLR7-deficient lupus mice (63). Surprisingly, TLR9-deficient lupus mice have increased autoimmune disease; this is associated with greater production of anti-RNA autoantibodies (63, 64). Lupus-prone mice deficient in both TLR7 and TLR9 or in their adaptor protein MyD88 do not develop either autoantibodies or lupus nephritis (63, 65).
Mice deficient in TLR7 on B cells have a significantly reduced GC response upon challenge with an RNA virus and have a relative decrease in the size of the DZ where proliferation and somatic mutation occur. This results in decreased production of high-affinity anti-viral antibodies (66). In lupus models, B cell-intrinsic TLR7 expression drives GC formation and autoantibody production (67). In the BWAS−/− model in which B cells are deficient in the Wiskott–Aldrich syndrome protein, GC formation depends on B cell-intrinsic TLR7 (64) but is unaffected by Type 1 IFN (American College of Rheumatology, Abstract 2870, 2014). In the pristane model of SLE, induction of TLR7 is downstream of Type-I IFN (68). These studies both suggest that the mechanism of action of TLR7 in GC formation and autoantibody induction is not mediated via induction of Type-I IFN. Using 3H9 NZW/BXSB male mice that bear two copies of TLR7, we showed that excess TLR7, like excess Type-I IFN, results in the failure of exclusion of high-affinity autoreactive B cells from the GC, with subsequent clonal expansion and somatic mutation of these cells that then can enter the plasma cell compartment (69).
The Role of Baff in the Germinal Center
B cell-activating factor (BAFF) is made by stromal cells in the secondary lymphoid organs including FDCs. The interaction of BAFF with BAFF-R is required for B cell survival and regulates the selection of naïve autoreactive B cells whereas BAFF excess can induce lupus-like autoimmunity (70). The GC is in general a BAFF poor environment, and expression of BAFF receptors is also downregulated, with BAFF-R being the sole receptor on GC B cells (71). BAFF and BAFF-R KO mice form smaller GCs that decline prematurely, indicating that BAFF is needed for GC maintenance (72). The mechanism for this dependence is not yet clear. BAFF influences ICOSL expression on B cells, thus regulating the ability of B cells to promote TFH expansion during GC development (73). TFH, in turn, produce BAFF and this may enhance the survival and selection of high-affinity B cells (74). Acting via a receptor other than BAFF-R, BAFF is required for the development of a mature FDC reticulum (72). Furthermore, activated T cells are also responsive to BAFF, releasing IFNγ that amplifies autoimmune effector responses (75). TFH cells express both BAFF-R and BCMA. In a lupus-prone strain with T cell-specific BCMA deficiency, TFH cells are expanded and signaling through BAFF-R results in enhanced IFNγ release (76). Whether BAFF modulates the selection of autoreactive B cells in the GC either in mice or humans is not yet known.
Extrafollicular Clonal Expansion and Somatic Mutation in SLE
Extrafollicular B cells can undergo clonal expansion and limited somatic mutation. Although T cell help is usually rather limited in EF foci, TFH-like cells have been found in these foci in several lupus-prone mouse strains, raising the possibility that somatic mutations leading to high-affinity autoreactivity can occur in these sites without the usual regulatory mechanisms in place (77, 78). In NZW/BXSB mice, GCs are found early in the disease but then disappear around the time of disease onset and are replaced by a disorganized splenic architecture with many EF TFH cells (79). Induction of class-switched autoreactivity outside GCs also occurs in BAFF transgenic mice; this requires MyD88 but not T cell help suggesting that under some circumstances, autoreactivity can be driven solely by innate mechanisms (80). Whether humans with lupus generate autoantibodies through this mechanism has not yet been determined. Recent studies in the 9G4 system have shown that during SLE flares, activated naïve B cells are the source of oligoclonal expansions of plasma cells with only low frequencies of somatic mutation, suggesting that some autoantibodies may derive from EF sources in humans (81).
Cell Death in Germinal Centers
Germinal center B cell survival is a result of the balance of pro-survival and pro-apoptotic molecules whose expression is regulated by affinity-based BCR signals, extrinsic signals from other cells, including cytokines and contact-dependent signals, and stochastic effects, that are either random or reflect local signals at critical times (11). The death molecule Fas is highly upregulated in GC B cells and its loss increases the size of the GC but decreases affinity maturation as low-affinity antigen-specific B cells fail to die (82). Similarly, transgenic overexpression of the anti-apoptotic Bcl2 protein results in survival of low-affinity antigen-specific B cells and B cells cross-reactive to self-antigens (83). More recent studies have dissected the role of individual Bcl2 family members in GC and post-GC B cell survival and selection (84).
Given the high death rate in the GC, specialized mechanisms are in place to clear dead cells before necrotic material activates innate pathways that could induce autoimmunity. Apoptotic cells are opsonized by soluble molecules, such as IgM, C1q, and MFGE8, and/or are rapidly cleared by highly phagocytic tingible body macrophages that express a large variety of scavenger receptors (85). C1q deficiency is a highly penetrant genetic cause of SLE in humans (86). MFGE8 recognizes phosphatidyl serine on apoptotic cells in the GC. Its absence in mice results in the induction of autoantibodies with age; this loss of tolerance is accelerated by repeated immunizations that induce GC formation (87).
Targeting the GC in SLE
Considering the important role of the GC in the acquisition of autoreactivity in SLE, targeting of the GC has been a major focus of therapeutic endeavor. Early studies using the costimulatory antagonists, anti-CD40L, to abort ongoing GCs or CTLA4Ig to prevent GC formation were highly effective at preventing lupus onset in mouse models in which GCs predominate as a source of autoantibodies. A short-term combination of CTLA4Ig and anti-CD40L was even more effective than either agent alone (88). We showed that in NZB/W mice, this was due to tolerance induction in the GCs, preventing the formation of cross-reactive autoantibodies upon immunization with foreign antigen (89). Similar restoration of tolerance occurs after administration of an agonist antibody to CD137 (4-1BB) (90). Other GC targets include TLR7 and TLR pathways, BAFF, ICOS that is important not only for GC development but also for the function of effector T cells, and IL-21 that is expressed both by GC and EF TFH cells. An alternate strategy is to increase clearance of nucleic acids using RNAse or DNAse.
In mouse models, therapies targeting the GC lose efficacy once long-lived autoreactive memory and plasma cells have already developed (88). Class-switched memory cells do not need to reenter the GC to rapidly differentiate into plasma cells, and long-lived plasma cells require neither antigen nor T cells to survive. GC-directed therapy might therefore be most effective in the early stages of disease or as maintenance therapy during disease quiescence. Another potential use of GC-directed therapy might be to prevent the emergence of autoantibodies that arise in ectopic tissue GCs during inflammation and are directed to tissue antigens, such as vimentin (91). Finally, it is possible that targeting ectopic TFH could prevent ongoing formation of EF foci of short-lived plasma cells.
Conclusion
Defective tolerance induction in the GC is a feature of many SLE models and can be due both to impaired selection of naïve autoreactive B cells into the GC and impaired regulation of B cells that acquire autoreactivity by somatic mutation. Defects in GC T cells and insufficient clearance of the large numbers of dead cells that are generated in the GC can lead to loss of tolerance to nuclear autoantigens. Impaired signaling through the BCR leading to insufficient B cell death and excess innate signals leading to a decrease in stringency of B cell selection both lead to increased B cell autoreactivity. Many strategies are currently being tested for targeting of the GC, but the clinical efficacy and optimal applications of this approach remain to be tested in humans.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH AR064811-01.
References
1. Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity (2011) 34(6):947–60. doi: 10.1016/j.immuni.2011.03.024
2. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity (2011) 34(6):932–46. doi:10.1016/j.immuni.2011.03.023
3. Liu D, Xu H, Shih C, Wan Z, Ma X, Ma W, et al. T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature (2015) 517(7533):214–8. doi:10.1038/nature13803
4. Dal Porto JM, Haberman AM, Shlomchik MJ, Kelsoe G. Antigen drives very low affinity B cells to become plasmacytes and enter germinal centers. J Immunol (1998) 161(10):5373–81.
5. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity (2008) 29(1):138–49. doi:10.1016/j.immuni.2008.05.009
6. Gatto D, Paus D, Basten A, Mackay CR, Brink R. Guidance of B cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses. Immunity (2009) 31(2):259–69. doi:10.1016/j.immuni.2009.06.016
7. Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP, et al. Oxysterols direct immune cell migration via EBI2. Nature (2011) 475(7357):524–7. doi:10.1038/nature10280
8. Liu C, Yang XV, Wu J, Kuei C, Mani NS, Zhang L, et al. Oxysterols direct B-cell migration through EBI2. Nature (2011) 475(7357):519–23. doi:10.1038/nature10226
9. Pereira JP, Kelly LM, Xu Y, Cyster JG. EBI2 mediates B cell segregation between the outer and centre follicle. Nature (2009) 460(7259):1122–6. doi:10.1038/nature08226
10. Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol (2012) 30:429–57. doi:10.1146/annurev-immunol-020711-075032
11. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol (2015) 15(3):137–48. doi:10.1038/nri3804
12. Jarjour M, Jorquera A, Mondor I, Wienert S, Narang P, Coles MC, et al. Fate mapping reveals origin and dynamics of lymph node follicular dendritic cells. J Exp Med (2014) 211(6):1109–22. doi:10.1084/jem.20132409
13. El Shikh ME, El Sayed RM, Sukumar S, Szakal AK, Tew JG. Activation of B cells by antigens on follicular dendritic cells. Trends Immunol (2010) 31(6):205–11. doi:10.1016/j.it.2010.03.002
14. Heesters BA, Myers RC, Carroll MC. Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol (2014) 14(7):495–504. doi:10.1038/nri3689
15. Aguzzi A, Kranich J, Krautler NJ. Follicular dendritic cells: origin, phenotype, and function in health and disease. Trends Immunol (2014) 35(3):105–13. doi:10.1016/j.it.2013.11.001
16. Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol (2004) 5(9):943–52. doi:10.1038/ni1100
17. Allen CD, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science (2007) 315(5811):528–31. doi:10.1126/science.1136736
18. Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, Henrickson SE, et al. Definition of germinal-center B cell migration in vivo reveals predominant intrazonal circulation patterns. Immunity (2007) 26(5):655–67. doi:10.1016/j.immuni.2007.04.008
19. Schwickert TA, Lindquist RL, Shakhar G, Livshits G, Skokos D, Kosco-Vilbois MH, et al. In vivo imaging of germinal centres reveals a dynamic open structure. Nature (2007) 446(7131):83–7. doi:10.1038/nature05573
20. Bannard O, Horton RM, Allen CD, An J, Nagasawa T, Cyster JG. Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection. Immunity (2013) 39(5):912–24. doi:10.1016/j.immuni.2013.08.038
21. Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature (2014) 509(7502):637–40. doi:10.1038/nature13300
22. Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, et al. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. J Exp Med (2011) 208(12):2497–510. doi:10.1084/jem.20111449
23. Cannons JL, Lu KT, Schwartzberg PL. T follicular helper cell diversity and plasticity. Trends Immunol (2013) 34(5):200–7. doi:10.1016/j.it.2013.01.001
24. Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol (2012) 8(6):337–47. doi:10.1038/nrrheum.2012.58
25. Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol (2015) 16(2):142–52. doi:10.1038/ni.3054
26. Sage PT, Sharpe AH. T follicular regulatory cells in the regulation of B cell responses. Trends Immunol (2015) 36(7):410–8. doi:10.1016/j.it.2015.05.005
27. Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol (2010) 11(1):7–13. doi:10.1038/ni.1818
28. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi:10.1126/science.1160062
29. Kim HJ, Cantor H. Regulation of self-tolerance by Qa-1-restricted CD8(+) regulatory T cells. Semin Immunol (2011) 23(6):446–52. doi:10.1016/j.smim.2011.06.001
30. Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell (2007) 130(1):25–35. doi:10.1016/j.cell.2007.06.033
31. Goodnow CC, Vinuesa CG, Randall KL, Mackay F, Brink R. Control systems and decision making for antibody production. Nat Immunol (2010) 11(8):681–8. doi:10.1038/ni.1900
32. Brink R. The imperfect control of self-reactive germinal center B cells. Curr Opin Immunol (2014) 28:97–101. doi:10.1016/j.coi.2014.03.001
33. Manson JJ, Mauri C, Ehrenstein MR. Natural serum IgM maintains immunological homeostasis and prevents autoimmunity. Springer Semin Immunopathol (2005) 26(4):425–32. doi:10.1007/s00281-004-0187-x
34. Yachimovich-Cohen N, Fischel R, Bachar N, Yarkoni Y, Eilat D. Autoimmune NZB/NZW F1 mice utilize B cell receptor editing for generating high-affinity anti-dsDNA autoantibodies from low-affinity precursors. Eur J Immunol (2003) 33(9):2469–78. doi:10.1002/eji.200324025
35. Yachimovich N, Mostoslavsky G, Yarkoni Y, Verbovetski I. The efficiency of B cell receptor (BCR) editing is dependent on BCR light chain rearrangement status. Eur J Immunol (2002) 32(4):1164–74. doi:10.1002/1521-4141(200204)32
36. Friedmann D, Yachimovich N, Mostoslavsky G, Pewzner-Jung Y, Ben-Yehuda A, Rajewsky K, et al. Production of high affinity autoantibodies in autoimmune New Zealand Black/New Zealand white F1 mice targeted with an anti-DNA heavy chain. J Immunol (1999) 162(8):4406–16.
37. Pewzner-Jung Y, Friedmann D, Sonoda E, Jung S, Rajewsky K, Eilat D. B cell deletion, anergy, and receptor editing in “knock in” mice targeted with a germline-encoded or somatically mutated anti-DNA heavy chain. J Immunol (1998) 161(9):4634–45.
38. Huang W, Moisini I, Bethunaickan R, Sahu R, Akerman M, Eilat D, et al. BAFF/APRIL inhibition decreases selection of naive but not antigen-induced autoreactive B cells in murine systemic lupus erythematosus. J Immunol (2011) 187(12):6571–80. doi:10.4049/jimmunol.1101784
39. Radic MZ, Mascelli MA, Erikson J, Shan H, Weigert M. Ig H and L chain contributions to autoimmune specificities. J Immunol (1991) 146(1):176–82.
40. Radic MZ, Erikson J, Litwin S, Weigert M. B lymphocytes may escape tolerance by revising their antigen receptors. J Exp Med (1993) 177(4):1165–73. doi:10.1084/jem.177.4.1165
41. Paul E, Lutz J, Erikson J, Carroll MC. Germinal center checkpoints in B cell tolerance in 3H9 transgenic mice. Int Immunol (2004) 16(2):377–84. doi:10.1093/intimm/dxh035
42. Li Y, Li H, Ni D, Weigert M. Anti-DNA B cells in MRL/lpr mice show altered differentiation and editing pattern. J Exp Med (2002) 196(12):1543–52. doi:10.1084/jem.20021560
43. Moisini I, Huang W, Bethunaickan R, Sahu R, Ricketts PG, Akerman M, et al. The Yaa locus and IFN-alpha fine-tune germinal center B cell selection in murine systemic lupus erythematosus. J Immunol (2012) 189(9):4305–12. doi:10.4049/jimmunol.1200745
44. Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, et al. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med (2005) 201(5):703–11. doi:10.1084/jem.20042251
45. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science (2003) 301(5638):1374–7. doi:10.1126/science.1086907
46. Scheid JF, Mouquet H, Kofer J, Yurasov S, Nussenzweig MC, Wardemann H. Differential regulation of self-reactivity discriminates between IgG+ human circulating memory B cells and bone marrow plasma cells. Proc Natl Acad Sci U S A (2011) 108(44):18044–8. doi:10.1073/pnas.1113395108
47. Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussenzweig MC, Wardemann H. Autoreactivity in human IgG+ memory B cells. Immunity (2007) 26(2):205–13. doi:10.1016/j.immuni.2007.01.009
48. Cappione A III, Anolik JH, Pugh-Bernard A, Barnard J, Dutcher P, Silverman G, et al. Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J Clin Invest (2005) 115(11):3205–16. doi:10.1172/JCI24179
49. Mietzner B, Tsuiji M, Scheid J, Velinzon K, Tiller T, Abraham K, et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc Natl Acad Sci U S A (2008) 105(28):9727–32. doi:10.1073/pnas.0803644105
50. Mohan C, Alas E, Morel L, Yang P, Wakeland EK. Genetic dissection of SLE pathogenesis. Sle1 on murine chromosome 1 leads to a selective loss of tolerance to H2A/H2B/DNA subnucleosomes. J Clin Invest (1998) 101(6):1362–72.
51. Vuyyuru R, Mohan C, Manser T, Rahman ZS. The lupus susceptibility locus Sle1 breaches peripheral B cell tolerance at the antibody-forming cell and germinal center checkpoints. J Immunol (2009) 183(9):5716–27. doi:10.4049/jimmunol.0804215
52. Wong EB, Soni C, Chan AY, Domeier PP, Abraham T, Limaye N, et al. B cell-intrinsic CD84 and Ly108 maintain germinal center B cell tolerance. J Immunol (2015) 194(9):4130–43. doi:10.4049/jimmunol.1403023
53. Sinai P, Dozmorov IM, Song R, Schwartzberg PL, Wakeland EK, Wulfing C. T/B-cell interactions are more transient in response to weak stimuli in SLE-prone mice. Eur J Immunol (2014) 44(12):3522–31. doi:10.1002/eji.201444602
54. Rahman ZS, Manser T. Failed up-regulation of the inhibitory IgG Fc receptor Fc gamma RIIB on germinal center B cells in autoimmune-prone mice is not associated with deletion polymorphisms in the promoter region of the Fc gamma RIIB gene. J Immunol (2005) 175(3):1440–9. doi:10.4049/jimmunol.175.3.1440
55. Mackay M, Stanevsky A, Wang T, Aranow C, Li M, Koenig S, et al. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. J Exp Med (2006) 203(9):2157–64. doi:10.1084/jem.20051503
56. Pritchard NR, Cutler AJ, Uribe S, Chadban SJ, Morley BJ, Smith KG. Autoimmune-prone mice share a promoter haplotype associated with reduced expression and function of the Fc receptor FcgammaRII. Curr Biol (2000) 10(4):227–30. doi:10.1016/S0960-9822(00)00344-4
57. Fukuyama H, Nimmerjahn F, Ravetch JV. The inhibitory Fcgamma receptor modulates autoimmunity by limiting the accumulation of immunoglobulin G+ anti-DNA plasma cells. Nat Immunol (2005) 6(1):99–106. doi:10.1038/ni1151
58. Vogel KU, Edelmann SL, Jeltsch KM, Bertossi A, Heger K, Heinz GA, et al. Roquin paralogs 1 and 2 redundantly repress the Icos and Ox40 costimulator mRNAs and control follicular helper T cell differentiation. Immunity (2013) 38(4):655–68. doi:10.1016/j.immuni.2012.12.004
59. Linterman MA, Rigby RJ, Wong R, Silva D, Withers D, Anderson G, et al. Roquin differentiates the specialized functions of duplicated T cell costimulatory receptor genes CD28 and ICOS. Immunity (2009) 30(2):228–41. doi:10.1016/j.immuni.2008.12.015
60. Jacquemin C, Schmitt N, Contin-Bordes C, Liu Y, Narayanan P, Seneschal J, et al. OX40 ligand contributes to human lupus pathogenesis by promoting T Follicular helper response. Immunity (2015) 42(6):1159–70. doi:10.1016/j.immuni.2015.05.012
61. Liu Z, Bethunaickan R, Huang W, Lodhi U, Solano I, Madaio MP, et al. Interferon-alpha accelerates murine systemic lupus erythematosus in a T cell-dependent manner. Arthritis Rheum (2011) 63(1):219–29. doi:10.1002/art.30087
62. Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A (2006) 103(26):9970–5. doi:10.1073/pnas.0603912103
63. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity (2006) 25(3):417–28. doi:10.1016/j.immuni.2006.07.013
64. Jackson SW, Scharping NE, Kolhatkar NS, Khim S, Schwartz MA, Li QZ, et al. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol (2014) 192(10):4525–32. doi:10.4049/jimmunol.1400098
65. Christensen SR, Shlomchik MJ. Regulation of lupus-related autoantibody production and clinical disease by Toll-like receptors. Semin Immunol (2007) 19(1):11–23. doi:10.1016/j.smim.2006.12.005
66. Clingan JM, Matloubian M. B Cell-intrinsic TLR7 signaling is required for optimal B cell responses during chronic viral infection. J Immunol (2013) 191(2):810–8. doi:10.4049/jimmunol.1300244
67. Hwang SH, Lee H, Yamamoto M, Jones LA, Dayalan J, Hopkins R, et al. B cell TLR7 expression drives anti-RNA autoantibody production and exacerbates disease in systemic lupus erythematosus-prone mice. J Immunol (2012) 189(12):5786–96. doi:10.4049/jimmunol.1202195
68. Thibault DL, Chu AD, Graham KL, Balboni I, Lee LY, Kohlmoos C, et al. IRF9 and STAT1 are required for IgG autoantibody production and B cell expression of TLR7 in mice. J Clin Invest (2008) 118(4):1417–26. doi:10.1172/JCI30065
69. Boneparth A, Huang W, Bethunaickan R, Woods M, Sahu R, Arora S, et al. TLR7 influences germinal center selection in murine SLE. PLoS One (2015) 10(3):e0119925. doi:10.1371/journal.pone.0119925
70. Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol (2009) 9(7):491–502. doi:10.1038/nri2572
71. Goenka R, Scholz JL, Sindhava VJ, Cancro MP. New roles for the BLyS/BAFF family in antigen-experienced B cell niches. Cytokine Growth Factor Rev (2014) 25(2):107–13. doi:10.1016/j.cytogfr.2014.01.001
72. Rahman ZS, Rao SP, Kalled SL, Manser T. Normal induction but attenuated progression of germinal center responses in BAFF and BAFF-R signaling-deficient mice. J Exp Med (2003) 198(8):1157–69. doi:10.1084/jem.20030495
73. Ou X, Xu S, Lam KP. Deficiency in TNFRSF13B (TACI) expands T-follicular helper and germinal center B cells via increased ICOS-ligand expression but impairs plasma cell survival. Proc Natl Acad Sci U S A (2012) 109(38):15401–6. doi:10.1073/pnas.1200386109
74. Goenka R, Matthews AH, Zhang B, O’Neill PJ, Scholz JL, Migone TS, et al. Local BLyS production by T follicular cells mediates retention of high affinity B cells during affinity maturation. J Exp Med (2014) 211(1):45–56. doi:10.1084/jem.20130505
75. Scapini P, Hu Y, Chu CL, Migone TS, Defranco AL, Cassatella MA, et al. Myeloid cells, BAFF, and IFN-{gamma} establish an inflammatory loop that exacerbates autoimmunity in Lyn-deficient mice. J Exp Med (2010) 207(8):1757–73. doi:10.1084/jem.20100086
76. Coquery CM, Loo WM, Wade NS, Bederman AG, Tung KS, Lewis JE, et al. BAFF regulates follicular helper t cells and affects their accumulation and interferon-gamma production in autoimmunity. Arthritis Rheumatol (2015) 67(3):773–84. doi:10.1002/art.38950
77. Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, et al. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med (2008) 205(12):2873–86. doi:10.1084/jem.20080840
78. Bubier JA, Sproule TJ, Foreman O, Spolski R, Shaffer DJ, Morse HC III, et al. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc Natl Acad Sci U S A (2009) 106(5):1518–23. doi:10.1073/pnas.0807309106
79. Ramanujam M, Kahn P, Huang W, Tao H, Madaio MP, Factor SM, et al. Interferon-alpha treatment of female (NZW x BXSB)F(1) mice mimics some but not all features associated with the Yaa mutation. Arthritis Rheum (2009) 60(4):1096–101. doi:10.1002/art.24414
80. Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, et al. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med (2007) 204(8):1959–71. doi:10.1084/jem.20062567
81. Tipton CM, Fucile CF, Darce J, Chida A, Ichikawa T, Gregoretti I, et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol (2015) 16(7):755–65. doi:10.1038/ni.3175
82. Takahashi Y, Ohta H, Takemori T. Fas is required for clonal selection in germinal centers and the subsequent establishment of the memory B cell repertoire. Immunity (2001) 14(2):181–92. doi:10.1016/S1074-7613(01)00100-5
83. Ray SK, Putterman C, Diamond B. Pathogenic autoantibodies are routinely generated during the response to foreign antigen: a paradigm for autoimmune disease. Proc Natl Acad Sci U S A (1996) 93(5):2019–24. doi:10.1073/pnas.93.5.2019
84. Peperzak V, Vikstrom IB, Tarlinton DM. Through a glass less darkly: apoptosis and the germinal center response to antigen. Immunol Rev (2012) 247(1):93–106. doi:10.1111/j.1600-065X.2012.01123.x
85. Rahman ZS. Impaired clearance of apoptotic cells in germinal centers: implications for loss of B cell tolerance and induction of autoimmunity. Immunol Res (2011) 51(2–3):125–33. doi:10.1007/s12026-011-8248-4
86. Leffler J, Bengtsson AA, Blom AM. The complement system in systemic lupus erythematosus: an update. Ann Rheum Dis (2014) 73(9):1601–6. doi:10.1136/annrheumdis-2014-205287
87. Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science (2004) 304(5674):1147–50. doi:10.1126/science.109435
88. Davidson A, Aranow C. Lupus nephritis: lessons from murine models. Nat Rev Rheumatol (2010) 6(1):13–20. doi:10.1038/nrrheum.2009.240
89. Wang X, Huang W, Mihara M, Sinha J, Davidson A. Mechanism of action of combined short-term CTLA4Ig and anti-CD40 ligand in murine systemic lupus erythematosus. J Immunol (2002) 168(4):2046–53. doi:10.4049/jimmunol.168.4.2046
90. Foell J, Strahotin S, O’Neil SP, McCausland MM, Suwyn C, Haber M, et al. CD137 costimulatory T cell receptor engagement reverses acute disease in lupus-prone NZB x NZW F1 mice. J Clin Invest (2003) 111(10):1505–18. doi:10.1172/JCI200317662
Keywords: SLE, B cell, germinal center, autoimmunity, tolerance mechanisms
Citation: Woods M, Zou Y-R and Davidson A (2015) Defects in germinal center selection in SLE. Front. Immunol. 6:425. doi: 10.3389/fimmu.2015.00425
Received: 06 July 2015; Accepted: 03 August 2015;
Published: 14 August 2015
Edited by:
James Harris, Monash University, AustraliaReviewed by:
Laurence Morel, University of Florida, USADavid Tarlinton, Walter and Eliza Hall Institute, Australia
Copyright: © 2015 Woods, Zou and Davidson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne Davidson, Center for Autoimmunity and Musculoskeletal Diseases, Feinstein Institute for Medical Research, 350 Community Drive, Manhasset, New York, NY 11030, USA, adavidson1@nshs.edu