 Susan Yung
Susan Yung Tak Mao Chan
Tak Mao Chan- Department of Medicine, Queen Mary Hospital, University of Hong Kong, Hong Kong, China
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by a breakdown of self-tolerance, production of auto-antibodies and immune-mediated injury, resulting in damage accrual in multiple organs. Kidney involvement, termed lupus nephritis, is a major cause of morbidity and mortality that affects over half of the SLE population during the course of disease. The etiology of lupus nephritis is multifactorial and remains to be fully elucidated. Accumulating evidence suggests that in addition to forming immune complexes and triggering complement activation, anti-dsDNA antibodies contribute to the pathogenesis of lupus nephritis through binding, either directly or indirectly, to cross-reactive antigens or chromatin materials, respectively, to resident renal cells and/or extracellular matrix components, thereby triggering downstream cellular activation and proliferation as well as inflammatory and fibrotic processes. Several cross-reactive antigens that mediate anti-dsDNA antibody binding have been identified, such as annexin II and alpha-actinin. This review discusses the mechanisms through which anti-dsDNA antibodies contribute to immunopathogenesis in lupus nephritis. Corticosteroids combined with either mycophenolic acid (MPA) or cyclophosphamide is the current standard of care immunosuppressive therapy for severe lupus nephritis. This review also discusses recent data showing distinct effects of MPA and cyclophosphamide on inflammatory and fibrotic processes in resident renal cells.
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that affects multiple organ systems. It follows a relapsing–remitting disease course, and the risk of flare varies between individual patients. Clinical presentation can range from mild to severe depending on the affected organ. Involvement of the kidney, termed lupus nephritis, affects up to 60% of the SLE population and is more prevalent in Asians, Hispanics, Native Americans, and Blacks, especially in females of child-bearing age. Lupus nephritis is characterized by a loss of self-tolerance, production of auto-antibodies, such as those against nuclear antigens, and immune-mediated injury to the kidney. If left untreated, destruction of the normal kidney parenchyma and their replacement with fibrosis tissue will ensue. Clinical manifestations of active lupus nephritis include proteinuria, active urinary sediment, and acute renal injury.
Anti-dsDNA antibodies are specific to SLE and can be detected in patients at least 2 years before diagnosis of clinical disease (1). Serum anti-dsDNA antibody levels often reflect disease activity in lupus nephritis patients (2–5). Evidence that anti-dsDNA antibodies play an important role in disease pathogenesis originates mainly from animal studies. Intra-peritoneal administration of either human or murine anti-dsDNA antibodies to non-autoimmune mice, or their inoculation with the transgene that encodes the secreted form of an IgG anti-dsDNA antibody can induce many features of lupus nephritis (6, 7). Anti-dsDNA antibody production, their presence in immune complexes and deposition in the kidney precedes immune cell infiltration and development of proteinuria in NZBWF1 mice (8). Anti-dsDNA antibodies have also been isolated from glomeruli of lupus nephritis patients with active disease, and diverse histopathological patterns observed in lupus patients is a result of their deposition in distinct locations in the glomerulus (9).
Nephritogenic anti-dsDNA antibodies have been shown to regulate gene and protein expression of inflammatory and fibrotic mediators in resident renal cells, thereby exerting a direct effect on kidney inflammation and fibrosis (10). The precise mechanism through which anti-dsDNA antibodies are deposited in the kidney parenchyma to exert their detrimental effect remains to be fully defined, but the data to date suggest that they can either bind directly to cross-reactive antigens on the surface of resident renal cells or to components of the extracellular matrix, or indirectly through nucleosomes that are bound to constituents of the glomerular basement membrane.
This review discusses the pathogenic role of anti-dsDNA antibodies in the development of lupus nephritis, with particular focus on how they impact on inflammatory and fibrotic processes in resident kidney cells. Mechanisms through which anti-dsDNA antibodies are deposited in the kidney have been discussed elsewhere (5, 11–14).
Detection of Anti-dsDNA Antibodies in SLE Patients
Anti-dsDNA antibodies can be detected by a variety of tests, such as the Farr radioimmunoassay (RIA), Crithidia luciliae indirect immunofluorescence test (CLIFT), and enzyme-linked immunosorbent assays (ELISAs). The Farr RIA and CLIFT are well-established assays that provide both diagnostic and prognostic values for SLE, whereas ELISAs are becoming more common for the measurement of anti-dsDNA antibody levels in routine clinical laboratories (15, 16).
The Farr RIA is a quantitative assay that measures the precipitation of radiolabeled dsDNA/anti-dsDNA antibody complexes. Since high salt conditions are used for precipitation, this assay preferentially detects anti-dsDNA antibodies with high avidity to dsDNA. The source of dsDNA must be carefully selected to ensure it is double-stranded, monodisperse in size with a MW >105 but smaller than 107 kDa to ensure reliable precipitation (17, 18). Circular double-stranded bacteriophage DNA or plasmid DNA, which can be easily iodinated after isolation are preferred (17). This assay does not distinguish between anti-dsDNA antibody Ig subclass. Disadvantages of this assay include the use of radiolabeled dsDNA, a labor-intensive methodology that cannot be automated, and detection of other proteins or compounds capable of precipitating dsDNA, thereby giving false positive results (16).
The CLIFT is a sensitive and relatively specific assay that detects anti-dsDNA antibodies with moderate to high avidity to dsDNA. It relies on indirect immunofluorescence to detect anti-dsDNA antibody binding to circular dsDNA present in the kinetoplast of Crithidia luciliae (19). It is noteworthy that occasional false positive results have been reported, possible due to the putative presence of histones in the kinetoplast, or lipoprotein/IgG complexes in the sample (15).
Enzyme-linked immunosorbent assays, whether “in-house” or commercial, are easy to perform, relatively inexpensive, can be automated and does not involve the use of radioisotopes. They provide quantitative results that can be readily standardized using dsDNA preparations from the World Health Organization (15). When compared to the Farr RIA and CLIFT, ELISAs have high sensitivity but less specificity as they do not distinguish between antibodies with high and low avidity to dsDNA. Discrepancies of results in independent studies have been reported and this may be due to the source and heterogeneity of the coated dsDNA, and MW and conformation of dsDNA used, the latter possibly limiting anti-dsDNA antibody interaction. False positive results may be observed if dsDNA is contaminated with single-stranded DNA or proteins, or if dsDNA coating linkers are used since they may permit binding of Ig that are not directed to dsDNA. The use of biotinylated dsDNA and coating through streptavidin to microtiter plates can reduce such errors. New ELISAs that have been optimized for the detection of anti-dsDNA antibodies of the IgG subclass with high avidity to dsDNA have been reported, and give comparable results to those obtained with the Farr RIA (20).
Anti-dsDNA antibodies can be detected in up to 80% of lupus patients suggesting that the sensitivity of current assays may not be optimal to detect low levels of anti-dsDNA antibodies, or that anti-dsDNA antibodies may be present as immune complexes in sera that prevent them from binding to dsDNA. When interpreting anti-dsDNA antibody results, clinicians should be mindful of the technique used, determine whether the assay can distinguish between high and low avidity anti-dsDNA antibodies, and note values observed in healthy controls and SLE patients with each assay.
Origin of Pathogenic Anti-dsDNA Antibodies
Anti single-stranded DNA antibodies and anti-dsDNA antibodies constitute part of the normal repertoire of natural antibodies in healthy subjects, and are predominantly of relatively low-affinity, belong to the IgM subclass and react weakly with self-antigens (5). In SLE patients, these naturally occurring antibodies may undergo an IgM to IgG class switch or somatic mutations of the Ig-V regions to generate pathogenic anti-dsDNA antibodies. Both molecular processes are catalyzed by activation-induced deaminase (AID) in B cells within germinal centers. The importance of AID in the generation of high affinity IgG anti-dsDNA antibodies and subsequent development of lupus nephritis has been substantiated by Jiang et al. who demonstrated that lupus-prone mice deficient in AID lacked auto-reactive IgG anti-dsDNA antibodies, but produced high levels of IgM anti-dsDNA antibodies that was associated with a significant improvement in glomerulonephritis and survival compared to wild-type mice (21, 22). Class switch and somatic mutations is antigen driven and alters the specificity of B cell receptors and antibodies that they secrete, which when coupled with positive clonal selection for high antigen binding, results in the generation of high affinity antibodies (5, 23, 24). Somatic mutations where positive charged amino acids, such as arginine, asparagine, and lysine residues, are inserted within the CDRs of anti-dsDNA antibodies are critical for high affinity dsDNA binding (23, 25). Anti-dsDNA antibodies of the IgE subclass have also been implicated in the pathogenesis of lupus nephritis and are associated with active disease and increased basophil activation (26, 27).
Clearance of apoptotic bodies is defective in SLE patients and chromatin material, such as dsDNA, released from apoptotic cells would be the most likely auto-antigen to drives B cell clonal expansion. However, independent researchers have suggested that molecular mimicry may be the basis for immunological cross-reactivity (28).
Anti-dsDNA Antibodies and Disease Pathogenicity
Lupus nephritis is initiated by the deposition of anti-dsDNA antibody-containing immune complexes in the kidney parenchyma. Notably, immune complex deposition alone is not sufficient to induce renal injury but must be accompanied by classical complement activation, infiltration of immune cells, release of chemokines, cytokines, and proteolytic enzymes, and oxidative damage, which together induce kidney inflammation and subsequent organ damage. Activation of the classical complement pathway is initiated by C1q following its binding to Ig in immune complexes. This results in a conformational change in its structure that renders it immunological. Defective clearance mechanisms results in the accumulation of C1q and predisposes SLE patients to anti-C1q antibody production (29). Genetic deficiency of C1q is rare and is associated with SLE susceptibility (30). Recent studies have shown that anti-dsDNA antibodies can cross-react with C1q (31).
Although anti-dsDNA antibody levels often correlate with disease activity, some patients can have positive or even high levels of anti-dsDNA antibodies during apparent clinical quiescence, which could indicate that not all anti-dsDNA antibodies that are detected by current assay methods are pathogenic. Factors that endow anti-dsDNA antibodies with their pathogenic potential include immunoglobulin subclasses of IgG1 and IgG3, ability to activate complement and engage Fc receptors, their charge, antigen avidity, and poly-reactivity (2, 5, 9, 32–34). For example, site-mutagenesis studies showed that substitution of aspartic acid with glycine within the heavy chain complementarity determining region of R4A, a pathogenic murine monoclonal anti-dsDNA antibody that binds to glomeruli, resulted in a loss of dsDNA binding (35). By contrast, substitution of three amino acids in R4A resulted in their deposition in renal tubules instead of glomeruli, and when this antibody was administered to SCID mice it resulted in severe proteinuria (35). It was also reported that the degree of pathogenicity of anti-dsDNA antibodies did not necessarily correlate with their affinity for dsDNA (35). These observations, together with the finding that immunization of non-autoimmune mice with mammalian DNA failed to induce nephritogenic anti-dsDNA antibody production or clinical disease manifestations would suggest that auto-reactivity to native DNA per se may not be an important property of pathogenic anti-dsDNA antibodies. In this context, there are data to suggest that their pathogenic nature is associated with their poly-reactivity and ability to bind directly to non-DNA cross-reactive kidney antigens (36, 37). It has been reported that antibodies directed against dsDNA and related nuclear components accounted for <10% of the total IgG eluted from kidneys from lupus nephritis patients (38).
Binding of Anti-dsDNA Antibodies to Non-DNA Cross-Reactive Antigens in Renal Cells
Binding of anti-dsDNA antibodies to cross-reactive antigens in isolated rat kidneys was first reported by Raz et al. (39), who demonstrated that infusion of either murine monoclonal or human polyclonal anti-dsDNA antibodies alone, but not calf-thymus DNA/anti-dsDNA antibody complexes, bound to components in the glomerulus and renal interstitial blood vessels, and was accompanied by increased albumin excretion. However, the conclusion that anti-dsDNA antibodies could bind directly to cross-reactive antigens present in the kidney was subject to debate since the amount of DNA or chromatin material if any, in the anti-dsDNA antibody preparations or within the isolated kidney was not determined (39).
Many studies have focused on the interaction of anti-dsDNA antibodies with resident glomerular cells, namely mesangial and endothelial cells, and to a lesser extent on podocytes (3, 40–43). All three cell types show tri-directional communication, and thus it is not surprising that injury to one cell type will have an impact on the other cell types. The amount and location of immune deposits in the glomerulus correlate with the histopathology and severity of nephritis (9, 32). Proliferative lupus nephritis is associated with immune deposition in the mesangium and subendothelial space, whereas deposition in the subepithelial space results in membranous lesions.
In order for anti-dsDNA antibodies to mediate their detrimental effect on the kidney parenchyma, anti-dsDNA antibodies must first bind to the cell surface of resident renal cells. We and others have shown that anti-dsDNA antibodies can bind directly to annexin II, α-actinin, and ribosomal P protein in mesangial cells and trigger downstream inflammatory, apoptotic, and fibrogenic processes (28, 37, 44, 45). While anti-dsDNA antibodies have been reported to bind directly to myosin 1 and calreticulin in hepatoma cells, lymphocytes, and Chinese hamster ovary cells (46, 47), similar interactions have not been shown in resident renal cells. The mechanisms through which anti-dsDNA antibodies bind to glomerular endothelial cells and podocytes have yet to be fully characterized.
Annexin II
Annexin II is a calcium dependent, phospholipid binding protein than can exit as a monomer, dimer, or heterotetramer in many organs, including the kidney. It is present within the cytosol of cells and translocates to the plasma membrane when the cell is activated by cytokines or growth factors. Annexin II on the surface of endothelial cells has been shown to interact with β2-glycoprotein I and toll-like receptor 4 to mediate cell activation, tissue inflammation, and thrombosis (48–51). Annexin II is also a ligand for C1q, a member of the classical complement pathway that can opsonize apoptotic cells to facilitate their phagocytic clearance (52). Defective clearance of apoptotic bodies in lupus patients is well-established, and auto-antibodies targeting annexin II have been identified in patients with proliferative lupus nephritis (37, 53) as well as in patients with rheumatoid arthritis (54).
Anti-dsDNA antibodies isolated from biopsy-proven patients with diffuse proliferative lupus nephritis can bind to annexin II on the surface of mesangial cells (37). Following their binding, anti-dsDNA antibodies are rapidly internalized and translocate to the cytosol and/or cell nucleus in a time- and temperature-dependent manner. The pathogenic nature of this interaction is highlighted by the induction of PKC activation, secretion of pro-inflammatory cytokines and hyaluronan, and matrix protein deposition (37, 55, 56). Anti-dsDNA antibodies have been reported to be able to penetrate other living cells, such as human mononuclear cells and rat H35 hepatoma cells, and the active energy-requiring internalization process was dependent on the F(ab) region (46, 57). However, Fc-receptor-mediated internalization of anti-dsDNA antibodies cannot be ruled out (58). Yanase et al. demonstrated that a proportion of anti-dsDNA antibodies can be recycled back to the cell surface of rat H35 hepatoma cells, and postulated that should anti-dsDNA antibodies be altered during the recycling process, they may also become immunogenic which could further exacerbate disease progression (59). Whether a similar mechanism occurs in mesangial cells has not been investigated. It is noteworthy that the process of antibody internalization by cells is not specific to anti-dsDNA antibodies since this process has also been observed with ribonucleoprotein, ribosomal P protein, La and Ro antibodies in non-lymphoid cells (60–62). The observation that intra-glomerular annexin II expression is increased in patients and mice with active nephritis and co-localizes with IgG and C3 deposition suggests that it may be involved in disease pathogenesis (37). Our data showed that human anti-dsDNA antibodies could induce annexin II synthesis in mesangial cells, indicating a potential amplification mechanism of immune-mediated inflammation and fibrogenesis (37). A recent report by Seret et al. showed that auto-antibodies from lupus nephritis patients targeted α-actinin and to a lesser extent laminin on the surface of kidney cells (63). That these investigators did not identify annexin II could be related to different experimental methodology and the fact that human embryonic kidney cells used in these studies have low constitutive annexin II expression (64).
Alpha-Actinin
Alpha-actinin (α-actinin) is an ubiquitous F-actin binding protein that is present in cell–cell and cell–matrix contact sites, areas of dense stress fibers and lamellipodia, which plays important roles in determining cell shape and migration (65). Nephritogenic anti-dsDNA antibodies derived from lupus-prone mice have been reported to cross-react with α-actinin, and high titers of anti-α-actinin antibodies have been detected in the serum and kidney eluates of lupus-prone mice (44). Mesangial cells of lupus-prone mice synthesize more α-actinin that cells from BALB/c mice (66). Immunization of non-autoimmune mice with α-actinin-induced anti-chromatin antibody production, glomerular Ig deposition, and proteinuria (67). These experimental data strongly suggest that α-actinin is involved in disease pathogenesis. In the clinical setting, anti-dsDNA antibodies isolated from lupus nephritis patients with active disease, but not patients with non-renal lupus, has been shown to cross-react with α-actinin (68). The association between serum immunoglobulin binding to α-actinin and disease activity in lupus nephritis patients remains controversial (36, 63, 69, 70). Adding further confusion were the findings that anti-dsDNA antibodies could bind to glomerular structures that contained extracellular nucleosomes instead of α-actinin in kidneys of NZBWF1 mice (71), and that α-actinin could not be detected in kidney eluates from nephritic lupus-prone mice (72). These discrepancies may be related to the stage and type of disease when the investigations were performed.
Although not the focus of this review, anti-dsDNA antibodies have also been shown to bind to components of the glomerular basement membrane through nucleosomes released from apoptotic cells. This interaction has been reviewed elsewhere (73, 74).
Downstream Pathogenic Effects of Anti-dsDNA Antibodies Following Their Binding to Resident Glomerular Cells
How anti-dsDNA antibodies contribute to kidney injury remains to be fully elucidated. Early studies showed that nephritogenic murine anti-dsDNA antibodies could interact directly with distinct glomerular and vascular cell surface antigens to induce mesangial expansion and proteinuria (75). Although there is evidence to show direct binding of anti-dsDNA antibodies to cross-reactive antigens in the glomerulus, it is important to note that anti-dsDNA antibody-secreting hybridomas may contain DNA and/or nucleosomes that are released into the supernatant during culture, and without prior DNase treatment these may not be removed from antibody preparations used in experiments (76). More recently, it was reported that anti-dsDNA antibodies that bound directly to basement membrane antigens showed glomerular and mesangial deposition that was not dependent on chromatin material and could activate complement and induce proteinuria in non-autoimmune mice (77).
Cell Proliferation, Apoptosis, and DNase I Synthesis
Interaction of nephritogenic human polyclonal anti-dsDNA antibodies with cultured human or rat mesangial cells induced cell proliferation, apoptosis, PKC activation, secretion of IL-6, IL-1β, TNF-α, TGF-β1, and hyaluronan, and fibronectin synthesis (37, 55, 56, 78, 79), suggesting that anti-dsDNA antibodies may contribute to mesangial expansion, hypercellularity, increased apoptosis, inflammation, and fibrogenesis observed in lupus nephritis. By contrast, using murine anti-dsDNA antibodies Zhang et al. observed no effect on murine mesangial cell proliferation (80).
Lupus is associated with defective clearance of apoptotic cells (81, 82). Also, anti-dsDNA antibodies have been shown to induce apoptosis in rat mesangial cells independent of changes to p53 Fas or c-myc gene expression (79). Several lines of evidence suggest that defective DNase I function may be involved in disease pathogenesis. Madaio et al. reported that murine anti-dsDNA antibodies could bind DNase I and inhibit its enzymatic activity in HL60 promyelocytic cells (83). Reports from Rekvig’s group showed that renal DNase I synthesis was suppressed by the increased intra-glomerular expression of TNF receptor-associated protein-1 in lupus-prone mice, which in turn inhibited chromatin degradation and the retention of chromatin material was associated with activation of TLR-9 and the adaptive immune system (84–86). DNase I null mice showed features present in human lupus nephritis, such as the production of auto-antibodies to chromatin, glomerular immune complex deposition, and glomerulonephritis (87). Serum DNase I activity has been reported to be decreased in patients with SLE compared to healthy individuals (87, 88).
Glomerular Inflammation
Renal inflammation and fibrosis is associated with the activation of NFκβ, MAPK, and PKC signaling pathways and induction of chemokine, cytokine, and growth factor secretion by both infiltrating and resident renal cells. Chemokine production in the glomerulus is an early event during experimental lupus nephritis and precedes cellular infiltration, proteinuria, and kidney damage (89). MCP-1 is one of the most studied chemokines in human and experimental lupus nephritis (90, 91). Anti-dsDNA antibodies can induce MCP-1 secretion in cultured mesangial cells through PKC activation and increased IL-1β secretion (92, 93). Recent studies also showed that stimulation of mesangial cells with nucleosomes could also induce MCP-1 secretion (94). Also, intra-renal expression of MCP-1 increased with disease progression in lupus mice, while MCP-1-deficient mice showed reduced inflammatory cell infiltration and proteinuria, improved kidney histology, and prolonged survival (91). Although the chemotactic properties of MCP-1 are well-established, cDNA microarray analysis of micro-dissected glomeruli from lupus nephritis patients showed that MCP-1 transcript was highly expressed in fibrosis-related gene clusters, suggesting that MCP-1 may also play an important role in the development of glomerulosclerosis (95).
Anti-dsDNA antibodies can induce IL-1β, IL-6, and TNF-α secretion in cultured mesangial and endothelial cells (37, 43, 55). IL-1β can initiate and propagate both immune and inflammatory responses in SLE through induction of itself and downstream pro-inflammatory cytokines, chemokines, hyaluronan, and adhesion molecules (55, 96–98). Although IL-1β is predominantly secreted by infiltrating macrophages, it is also locally synthesized in the kidney of lupus-prone mice and its level correlates with the severity of nephritis (97). Data from IL-1β deficient BALB/c mice demonstrated the importance of IL-1β in anti-dsDNA antibody-induced pro-inflammatory response, and IL-1β deficiency was associated with less immune complex deposition in the kidney and less proteinuria compared to wild-type mice (99). Correlation between serum IL-1β level and disease activity in SLE patients (100) further highlights the importance of IL-1β in the pathogenesis of lupus nephritis.
IL-6 is a pleotropic cytokine secreted by lymphoid and non-lymphoid cells, including resident renal cells (37, 101–106). It is a multi-functional cytokine that possesses both pro- and anti-inflammatory properties. IL-6 is critical for the differentiation and maturation of B cells and mesangial cell proliferation. In lupus nephritis, IL-6 deposition is localized to mesangial cells and podocytes and is also present alongside immune deposits in the glomerulus. Although the mechanisms through which IL-6 secretion is induced in resident renal cells has yet to be fully elucidated, we have demonstrated that binding of anti-dsDNA antibodies to annexin II in mesangial cells can induce IL-6 secretion (37). Furthermore, bidirectional communication exists between mesangial cells and human proximal renal tubular epithelial cells (PTEC) and inflammatory responses occurring in either kidney compartment induced by anti-dsDNA antibodies can provoke a response in the other compartment (104).
Similar to IL-6, TNF-α possesses both pro- and anti-inflammatory properties. In its capacity as a pro-inflammatory cytokine, TNF-α plays an important role in inflammatory processes that potentially lead to tissue damage (107). Failure to regulate TNF-α synthesis at sites of immunological injury leads to chronic activation of innate immune cells and chronic inflammatory responses (108). Serum TNF-α level and bioactivity are increased in SLE patients during flare. In the kidney, TNF-α is synthesized locally by resident renal cells and by infiltrating immune cells, and it acts synergistically with IL-1β to exacerbate intra-renal inflammatory processes. In lupus-prone mice, TNF-α is detected in glomeruli, vascular smooth muscle cells, perivascular infiltrating cells and tubular epithelial cells, and the circulating level and intra-renal expression of TNF-α correlate with proteinuria and disease activity in animal and clinical studies (109–111). Fc-receptor cross-linking on human peripheral blood monocytes induces TNF-α synthesis (112), suggesting a link between immune complex deposition and TNF-α synthesis. In addition to its role in tissue inflammation, there is evidence that TNF-α can down-regulate the adaptive immune response (108). In NZBWF1 mice, TNF-α plays a potentially protective role during the early stage of lupus nephritis since treatment with recombinant TNF-α appeared to delay the development of nephritis (113). Consistent with these findings, depletion of TNF-α accelerated the development of lupus nephritis, thus highlighting a potentially important role of TNF-α in suppressing systemic autoimmunity (114). However, in NZBWF1 mice with established nephritis, TNF-α replacement showed no beneficial effect and could even accelerate disease progression. The data suggest a biphasic role of TNF-α at different stages of disease. Possible roles of TNF-α in the pathogenesis of lupus nephritis have been reviewed (107, 115).
Using complementary DNA microarray gene profiling, Qing et al. demonstrated that incubation of mesangial cells derived from MRL/lpr mice with anti-dsDNA antibodies induced the pro-inflammatory genes CXCL1/KC, CX3CL1 (fractalkine), inducible nitric oxide synthase, and lipocalin 2, which are involved in neutrophil trafficking, production of nitric oxide, and transport of small lipophilic molecules (10). The induction of these pro-inflammatory mediators was facilitated in part through the binding of high-mobility group box 1 protein and engagement of TLR2 and RAGE (116).
Mesangial Fibrogenesis
If not adequately controlled, persistent inflammation induces fibrosis, a process that begins as normal wound healing response aiming to maintain or restore the normal structural and functional integrity of the kidney. Unabated inflammation causes dysregulation of the reparative processes resulting in activation of mesangial cells, endothelial-to-mesenchymal transition, infiltration of cellular mediators and increased secretion of fibrogenic chemokines, cytokines and growth factors, resulting in the over-expression and deposition of matrix proteins (117). The control of inflammation is essential to inhibit or retard progressive kidney fibrosis. In immune-mediated kidney diseases, accumulation of extracellular matrix proteins in the glomerulus results in the development of glomerulosclerosis. Although tubulointerstitial changes better predict long-term renal prognosis (118), Vleming et al. reported that the extent of intra-glomerular fibronectin staining correlated with structural abnormalities in the glomerulus and severity of renal insufficiency (119).
Fibronectin is a large glycoprotein that is essential for renal homeostasis and resident renal cell proliferation, adhesion, migration, differentiation, and survival. In the normal kidney, fibronectin expression is limited to the Bowman’s capsule, mesangial matrix, and glomerular basement membrane where it interacts with other matrix proteins to maintain the structural integrity of the glomerular capillary. We and others have demonstrated that intra-glomerular fibronectin expression is increased in patients and mice with active lupus nephritis (56, 120, 121), and its co-localization with IgG deposition suggests an association between auto-antibody deposition and matrix protein accumulation (56). The underlying mechanism that initiates increased intra-glomerular fibronectin synthesis in lupus nephritis remains to be fully elucidated. We have shown that anti-dsDNA antibodies induced rapid and sustained phosphorylation of PKC-α, PKC-βI, and PKC-βII in cultured mesangial cells, which resulted in increased TGF-β1 bio-activation and subsequent fibronectin synthesis and deposition in the extracellular milieu (56). In line with our findings, Zhang et al. also reported that anti-dsDNA antibodies could induce mediators of fibrosis in mesangial cells (80). The role of TGF-β1 in kidney fibrosis is well-established (122, 123). The importance of PKC-β in the development of lupus nephritis has been highlighted by Oleksyn et al., who demonstrated that PKC-β deficiency abrogated auto-antibody production, proteinuria, and histological features of kidney disease in lupus-prone mice (124).
Binding of Anti-dsDNA Antibodies to Proximal Renal Tubular Epithelial Cells and Their Induction of Inflammatory and Fibrotic Processes
Although lupus nephritis is likely to be initiated in the glomerulus, injury is rarely contained in this compartment and often extends into the tubulo-interstitium. The degree of tubular injury and interstitial fibrosis rather than glomerulosclerosis correlates with progression of renal insufficiency (118). We and others have reported that immune deposits along the tubular basement membrane occurred in up to 70% of patients with diffuse proliferative lupus nephritis. The amount of immune complexes deposited in the tubulointerstitium correlates with circulating anti-dsDNA antibodies, tubulointerstitial inflammatory cell infiltration, IL-6 expression, tubular atrophy, and interstitial fibrosis (104). While tubulointerstitial injury heralds poor long-term kidney prognosis in lupus nephritis patients, the mechanisms leading to tubulointerstitial damage is poorly understood. Anti-dsDNA antibodies can induce epithelial-to-mesenchymal transition (EMT) in cultured PTEC, a process that precedes kidney fibrosis and is characterized by a loss of epithelial markers and replacement of their cobblestone, epithelial morphology by an elongated, fibroblastic appearance, with concomitant de novo synthesis of mesenchymal markers, such as α-smooth muscle actin, β-catenin, SNAIL, fibronectin, and collagen I. EMT markers are increased in renal biopsies from lupus nephritis patients and their expression is associated with renal impairment, interstitial leukocyte infiltration, and tubulointerstitial fibrosis (125, 126).
Anti-dsDNA antibodies can also induce cell-associated and soluble fibronectin synthesis in PTEC through activation of ERK, JNK, p38, PKC-α, and PKC-βII, and subsequent induction of TGF-β1, IL-6, IL-8, MCP-1, and TNF-α secretion (127). It is noteworthy that human anti-dsDNA antibodies can induce secretion of pro-inflammatory cytokines in PTEC, suggesting that even during apparent clinical quiescence, ongoing subclinical inflammation may occur in the kidney. Consistent with our findings, Ronda et al. demonstrated that serum IgG from SLE patients-induced IL-6 secretion in PTEC through ERK activation (128). The ability of anti-dsDNA antibodies to increase both cell-associated and soluble fibronectin suggests that these auto-antibodies can induce tubulointerstitial fibrosis through two distinct mechanisms. While cell-associated fibronectin will deposit in the extracellular matrix and contribute to matrix protein accumulation, soluble fibronectin may amplify fibrotic processes through increased TGF-β1 secretion and induction of collagen I synthesis (127). The observation that the aforementioned pro-inflammatory cytokines can also induce fibronectin synthesis (127) suggests that these peptides not only contribute to kidney inflammation but also to tubulointerstitial fibrosis (127). It is thus possible that these chemokines and cytokines act synergistically with each other or with fibrotic growth factors to facilitate the recruitment of myofibroblasts and other effector cells to sites of injury with mutual signaling pathways involved in both kidney inflammation and fibrosis during lupus nephritis.
How anti-dsDNA antibodies bind to PTEC is currently unknown although our preliminary studies suggest that it is possibly through cross-reactive antigens. Monoclonal murine anti-dsDNA antibodies have been shown to bind PK15 cells, a porcine proximal tubular epithelial cell line, through A and D SnRNP proteins, which mediate their internalization and compartmentalization either in the cytosol or nucleus resulting in modest cell lysis (129). Another sub-set of murine anti-dsDNA antibodies bind PK15 cells, are not internalized and induce significant cell lysis, especially in the presence of complement (129). Whether these observations are relevant to human PTEC remains to be studied, but with regard to cytotoxicity, human polyclonal anti-dsDNA antibodies can induce cell detachment and decrease human PTEC viability, thereby confirming their pathogenic nature (104).
Effects of Mycophenolic Acid and Cyclophosphamide on Inflammatory and Fibrotic Processes Induced by Anti-dsDNA Antibodies
Prevention of fibrosis and preservation of normal kidney histology are essential to ensure long-term renal and patient survival (130). Standard treatment for active lupus nephritis entails the use of corticosteroids combined with an immunosuppressive agent, such as mycophenolate mofetil (MMF) or cyclophosphamide. Although MMF and cyclophosphamide show comparable treatment efficacy, MMF is associated with less side effects (131). Mycophenolic acid (MPA), the active metabolite of MMF, exerts its therapeutic effect through inhibition of lymphocyte proliferation, especially those that are activated (132). We and others have demonstrated that MPA can exert anti-proliferative, anti-inflammatory, and anti-fibrotic effects on non-immune cells that are independent of its immunosuppressive actions (56, 103, 121, 127, 133–135). At clinically relevant doses, MPA significantly reduced anti-dsDNA antibody-induced cell proliferation and fibrogenic processes through the inhibition of PKC activation in human mesangial cells (56). Similarly, MPA inhibited anti-dsDNA antibody-mediated activation of the MAPK and PKC signaling pathways in PTEC, with a concomitant decrease in synthesis of cell-associated and soluble fibronectin, and secretion of TGF-β1, IL-6, IL-8, and TNF-α (127). When compared to cyclophosphamide, MPA was more effective in suppressing anti-dsDNA antibody-induced PKC-α activation, TGF-β1 secretion and fibronectin production (121). Furthermore, MPA and cyclophosphamide could both decrease TGF-β1 and TNF-α-induced fibronectin synthesis in human mesangial cells, whereas IL-6-induced fibronectin was suppressed only by MPA (121). In experimental studies, MMF together with methylprednisolone was more effective than cyclophosphamide and methylprednisolone in suppressing glomerulosclerosis in lupus-prone mice (121). The overall results suggest that anti-dsDNA antibodies contribute to matrix deposition and glomerulosclerosis, and that MPA treatment is associated with more anti-fibrotic effect when compared with cyclophosphamide, thereby may be more beneficial in preventing progression to chronic kidney failure.
Conclusion
Lupus nephritis is characterized by glomerular and tubulointerstitial inflammation, which could lead to progressive glomerulosclerosis and tubulointerstitial fibrosis if not interrupted. How anti-dsDNA antibodies bind to resident renal cells and components of the glomerular basement membrane to trigger downstream kidney inflammation and fibrosis is a topic of much interest and debate, and it is likely that we have only uncovered the tip of the iceberg. Many chemokines, cytokines, and growth factors have multi-faceted roles during lupus nephritis, which could complicate how we perceive the role of each molecule. Inflammatory and fibrotic processes are often intercalated and bidirectional signaling between different compartments of the kidney appears a critical factor to amplify injurious responses. Deciphering how these peptides contribute not only in the initial pathogenic events but also in the effector phase to mediate kidney damage is essential before suitable therapeutic strategies can be devised.
Figure 1 summarizes the putative pathogenic changes that may occur in the kidney following anti-dsDNA antibody binding to resident renal cells.
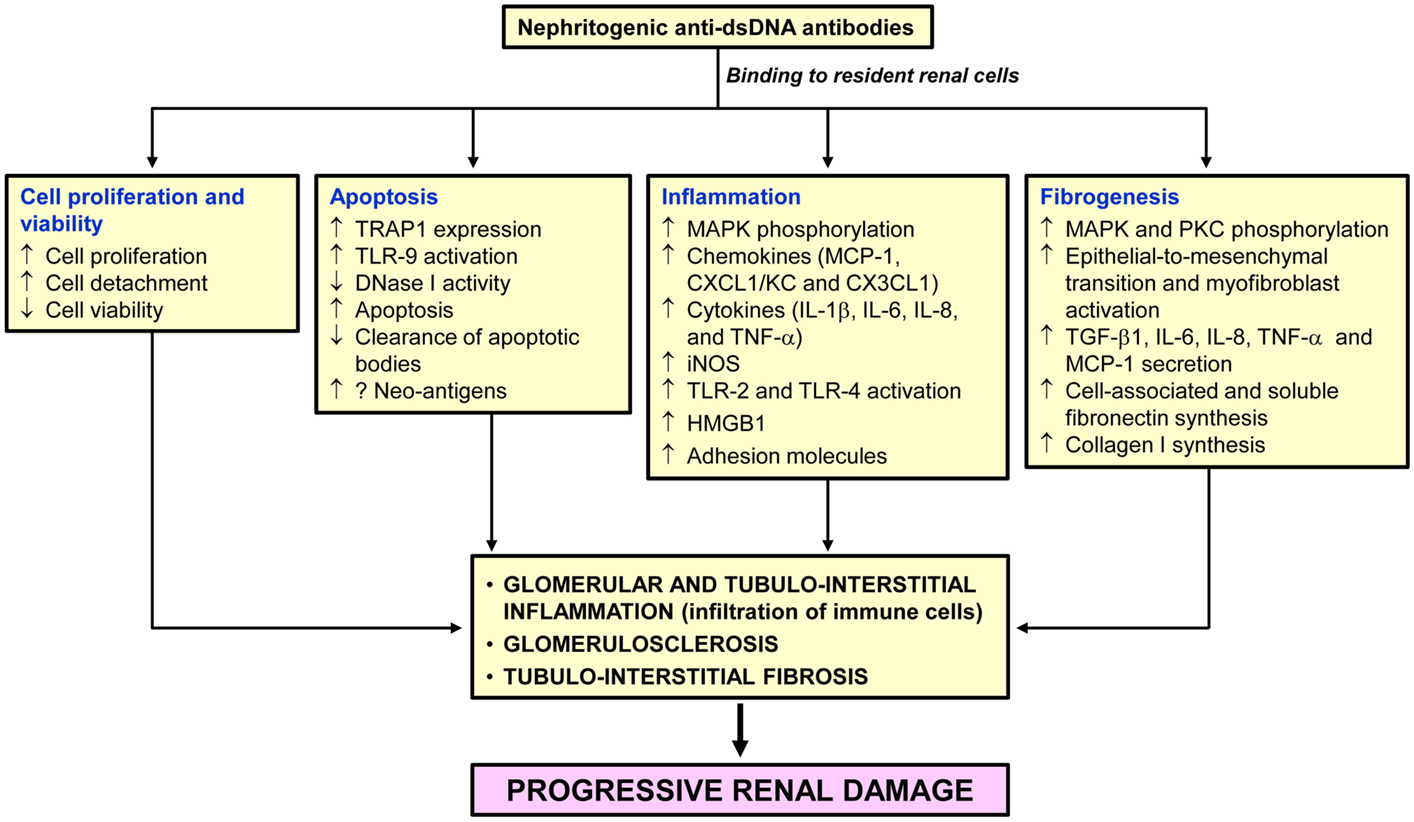
Figure 1. Pathogenic changes in the kidney following anti-dsDNA antibody binding to resident renal cells. Binding of pathogenic anti-dsDNA antibodies to resident glomerular and tubulointerstitial renal cells contributes to cell proliferation, and inflammatory, apoptotic and fibrogenic processes. If not adequately controlled, destruction of the normal kidney parenchyma and their replacement with fibrosis tissue will follow leading to end-stage renal disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was funded by the RGC General Research Fund (HKU 7374/03M, HKU 7366/04M, HKU 7603/09M, HKU 7604/10M, HKU 7607/12M, and 17126814) and Merit Award, the University of Hong Kong Small Project Funding (200807176076 and 201007176285), UGC Matching Grant Schemes (Phases IV, V, and VI), and kind donations from Mr. G. King and Mr. C. S. Yung. SY is supported by the Wai Hung Charitable Foundation Limited and the “Yu Chiu Kwong Professorship in Medicine” Endowment Fund awarded to TMC.
Abbreviations
AID, activation-induced deaminase; C1q, complement 1q; CLIFT, Crithidia luciliae indirect immunofluorescence test; CXCL1, (C–X–C motif) ligand 1; dsDNA, double-stranded DNA; ELISA, enzyme-link immunosorbent assay; EMT, epithelial-to-mesenchymal transition; ERK, extracellular signal-regulated kinase; HMGB1; high-mobility group box 1; IgG, immunoglobulin G; IL-1β, interleukin-1β; IL-6, interleukin-6; IL-8, interleukin-8; iNOS, inducible nitric oxide synthase; JNK, jun N-terminal kinase; MAPK, mitogen-activated protein kinase; MCP-1, monocyte chemoattractant protein-1; MMF, mycophenolate mofetil; MPA, mycophenolic acid; MW, molecular weight; NZBWF1, New Zealand Black and White F1 generation; PKC, protein kinase C; RAGE, receptor for advanced glycation end-products; SLE, systemic lupus erythematosus; TLR, toll-like receptor, TGF-β1, transforming growth factor-β1; TNF-α, tumor necrosis factor-α; TRAP1, TNF receptor-associated protein-1.
References
1. Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med (2003) 349:1526–33. doi: 10.1056/NEJMoa021933
2. Isenberg DA, Manson JJ, Ehrenstein MR, Rahman A. Fifty years of anti-ds DNA antibodies: are we approaching journey’s end? Rheumatology (Oxford) (2007) 46:1052–6. doi:10.1093/rheumatology/kem112
3. Chan TM, Leung JK, Ho SK, Yung S. Mesangial cell-binding anti-DNA antibodies in patients with systemic lupus erythematosus. J Am Soc Nephrol (2002) 13:1219–29. doi:10.1097/01.ASN.0000014223.71109.13
4. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med (2008) 358:929–39. doi:10.1056/NEJMra071297
6. Tsao BP, Ohnishi K, Cheroutre H, Mitchell B, Teitell M, Mixter P, et al. Failed self-tolerance and autoimmunity in IgG anti-DNA transgenic mice. J Immunol (1992) 149:350–8.
7. Vlahakos DV, Foster MH, Adams S, Katz M, Ucci AA, Barrett KJ, et al. Anti-DNA antibodies form immune deposits at distinct glomerular and vascular sites. Kidney Int (1992) 41:1690–700. doi:10.1038/ki.1992.242
8. Fenton K, Fismen S, Hedberg A, Seredkina N, Fenton C, Mortensen ES, et al. Anti-dsDNA antibodies promote initiation, and acquired loss of renal Dnase1 promotes progression of lupus nephritis in autoimmune (NZBxNZW)F1 mice. PLoS One (2009) 4:e8474. doi:10.1371/journal.pone.0008474
9. Waldman M, Madaio MP. Pathogenic autoantibodies in lupus nephritis. Lupus (2005) 14:19–24. doi:10.1191/0961203305lu2054oa
10. Qing X, Zavadil J, Crosby MB, Hogarth MP, Hahn BH, Mohan C, et al. Nephritogenic anti-DNA antibodies regulate gene expression in MRL/lpr mouse glomerular mesangial cells. Arthritis Rheum (2006) 54:2198–210. doi:10.1002/art.21934
11. Yung S, Chan TM. Autoantibodies and resident renal cells in the pathogenesis of lupus nephritis: getting to know the unknown. Clin Dev Immunol (2012) 2012:139365. doi:10.1155/2012/139365
12. Kalaaji M, Mortensen E, Jorgensen L, Olsen R, Rekvig OP. Nephritogenic lupus antibodies recognize glomerular basement membrane-associated chromatin fragments released from apoptotic intraglomerular cells. Am J Pathol (2006) 168:1779–92. doi:10.2353/ajpath.2006.051329
13. Mortensen ES, Rekvig OP. Nephritogenic potential of anti-DNA antibodies against necrotic nucleosomes. J Am Soc Nephrol (2009) 20:696–704. doi:10.1681/asn.2008010112
14. Putterman C. New approaches to the renal pathogenicity of anti-DNA antibodies in systemic lupus erythematosus. Autoimmun Rev (2004) 3:7–11. doi:10.1016/S1568-9972(03)00082-X
15. Rouquette AM, Desgruelles C. Detection of antibodies to dsDNA: an overview of laboratory assays. Lupus (2006) 15:403–7. doi:10.1191/0961203306lu2324oa
16. Riboldi P, Gerosa M, Moroni G, Radice A, Allegri F, Sinico A, et al. Anti-DNA antibodies: a diagnostic and prognostic tool for systemic lupus erythematosus? Autoimmunity (2005) 38:39–45. doi:10.1080/08916930400022616
17. Smeenk RJ. Methodological update detection of antibodies to dsDNA: current insights into its relevance. Clin Exp Rheumatol (2002) 20:294–300.
18. Geisert M, Heicke B, Metzmann E, Zahn RK. Influence of molecular weight of DNA on the determination of anti-DNA antibodies in systemic lupus erythematosus (SLE) sera by radioimmunoassay. Nucleic Acids Res (1975) 2:521–35. doi:10.1093/nar/2.4.521
19. Aarden LA, de Groot ER, Feltkamp TE. Immunology of DNA. III. Crithidia luciliae, a simple substrate for the determination of anti-dsDNA with the immunofluorescence technique. Ann N Y Acad Sci (1975) 254:505–15. doi:10.1111/j.1749-6632.1975.tb29197.x
20. Venner AA, Ibanez D, Gladman DD, Urowitz MB, MacKinnon A, Blasutig IM, et al. Comparison of three anti-dsDNA assays: performance and correlation with systemic lupus erythematosus disease activity. Clin Biochem (2013) 46:317–20. doi:10.1016/j.clinbiochem.2012.12.004
21. Jiang C, Foley J, Clayton N, Kissling G, Jokinen M, Herbert R, et al. Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. J Immunol (2007) 178:7422–31. doi:10.4049/jimmunol.178.11.7422
22. Jiang C, Zhao ML, Scearce RM, Diaz M. Activation-induced deaminase-deficient MRL/lpr mice secrete high levels of protective antibodies against lupus nephritis. Arthritis Rheum (2011) 63:1086–96. doi:10.1002/art.30230
23. Rahman A. Autoantibodies, lupus and the science of sabotage. Rheumatology (Oxford) (2004) 43:1326–36. doi:10.1093/rheumatology/keh354
24. Rahman A, Giles I, Haley J, Isenberg D. Systematic analysis of sequences of anti-DNA antibodies – relevance to theories of origin and pathogenicity. Lupus (2002) 11:807–23. doi:10.1191/0961203302lu302rr
25. Rahman A, Haley J, Radway-Bright E, Nagl S, Low DG, Latchman DS, et al. The importance of somatic mutations in the V(lambda) gene 2a2 in human monoclonal anti-DNA antibodies. J Mol Biol (2001) 307:149–60. doi:10.1006/jmbi.2000.4491
26. Dema B, Pellefigues C, Hasni S, Gault N, Jiang C, Ricks TK, et al. Autoreactive IgE is prevalent in systemic lupus erythematosus and is associated with increased disease activity and nephritis. PLoS One (2014) 9:e90424. doi:10.1371/journal.pone.0090424
27. Dema B, Charles N, Pellefigues C, Ricks TK, Suzuki R, Jiang C, et al. Immunoglobulin E plays an immunoregulatory role in lupus. J Exp Med (2014) 211:2159–68. doi:10.1084/jem.20140066
28. Mostoslavsky G, Fischel R, Yachimovich N, Yarkoni Y, Rosenmann E, Monestier M, et al. Lupus anti-DNA autoantibodies cross-react with a glomerular structural protein: a case for tissue injury by molecular mimicry. Eur J Immunol (2001) 31:1221–7. doi:10.1002/1521-4141(200104)31:4<1221::AID-IMMU1221>3.0.CO;2-P
29. Pickering MC, Botto M. Are anti-C1q antibodies different from other SLE autoantibodies? Nat Rev Rheumatol (2010) 6:490–3. doi:10.1038/nrrheum.2010.56
30. Leffler J, Bengtsson AA, Blom AM. The complement system in systemic lupus erythematosus: an update. Ann Rheum Dis (2014) 73:1601–6. doi:10.1136/annrheumdis-2014-205287
31. Franchin G, Son M, Kim SJ, Ben-Zvi I, Zhang J, Diamond B. Anti-DNA antibodies cross-react with C1q. J Autoimmun (2013) 44:34–9. doi:10.1016/j.jaut.2013.06.002
32. Foster MH, Cizman B, Madaio MP. Nephritogenic autoantibodies in systemic lupus erythematosus: immunochemical properties, mechanisms of immune deposition, and genetic origins. Lab Invest (1993) 69:494–507.
33. Yung S, Chan TM. Anti-DNA antibodies in the pathogenesis of lupus nephritis – the emerging mechanisms. Autoimmun Rev (2008) 7:317–21. doi:10.1016/j.autrev.2007.12.001
34. Radic MZ, Weigert M. Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annu Rev Immunol (1994) 12:487–520. doi:10.1146/annurev.iy.12.040194.002415
35. Katz JB, Limpanasithikul W, Diamond B. Mutational analysis of an autoantibody: differential binding and pathogenicity. J Exp Med (1994) 180:925–32. doi:10.1084/jem.180.3.925
36. Renaudineau Y, Croquefer S, Jousse S, Renaudineau E, Devauchelle V, Gueguen P, et al. Association of alpha-actinin-binding anti-double-stranded DNA antibodies with lupus nephritis. Arthritis Rheum (2006) 54:2523–32. doi:10.1002/art.22015
37. Yung S, Cheung KF, Zhang Q, Chan TM. Anti-dsDNA antibodies bind to mesangial annexin II in lupus nephritis. J Am Soc Nephrol (2010) 21:1912–27. doi:10.1681/ASN.2009080805
38. Mannik M, Merrill CE, Stamps LD, Wener MH. Multiple autoantibodies form the glomerular immune deposits in patients with systemic lupus erythematosus. J Rheumatol (2003) 30:1495–504.
39. Raz E, Brezis M, Rosenmann E, Eilat D. Anti-DNA antibodies bind directly to renal antigens and induce kidney dysfunction in the isolated perfused rat kidney. J Immunol (1989) 142:3076–82.
40. Chan TM, Frampton G, Staines NA, Hobby P, Perry GJ, Cameron JS. Different mechanisms by which anti-DNA MoAbs bind to human endothelial cells and glomerular mesangial cells. Clin Exp Immunol (1992) 88:68–74. doi:10.1111/j.1365-2249.1992.tb03041.x
41. Chan TM, Cheng IK. Identification of endothelial cell membrane proteins that bind anti-DNA antibodies from patients with systemic lupus erythematosus by direct or indirect mechanisms. J Autoimmun (1997) 10:433–9. doi:10.1006/jaut.1997.9998
42. Chan TM, Cheng IK. A prospective study on anti-endothelial cell antibodies in patients with systemic lupus erythematosus. Clin Immunol Immunopathol (1996) 78:41–6. doi:10.1006/clin.1996.0006
43. Lai KN, Leung JC, Bik Lai K, Li PK, Lai CK. Anti-DNA autoantibodies stimulate the release of interleukin-1 and interleukin-6 from endothelial cells. J Pathol (1996) 178:451–7. doi:10.1002/(SICI)1096-9896(199604)178:4<451::AID-PATH499>3.3.CO;2-G
44. Deocharan B, Qing X, Lichauco J, Putterman C. Alpha-actinin is a cross-reactive renal target for pathogenic anti-DNA antibodies. J Immunol (2002) 168:3072–8. doi:10.4049/jimmunol.168.6.3072
45. Sun KH, Liu WT, Tsai CY, Tang SJ, Han SH, Yu CL. Anti-dsDNA antibodies cross-react with ribosomal P proteins expressed on the surface of glomerular mesangial cells to exert a cytostatic effect. Immunology (1995) 85:262–9.
46. Yanase K, Smith RM, Puccetti A, Jarett L, Madaio MP. Receptor-mediated cellular entry of nuclear localizing anti-DNA antibodies via myosin 1. J Clin Invest (1997) 100:25–31. doi:10.1172/JCI119517
47. Seddiki N, Nato F, Lafaye P, Amoura Z, Piette JC, Mazie JC. Calreticulin, a potential cell surface receptor involved in cell penetration of anti-DNA antibodies. J Immunol (2001) 166:6423–9. doi:10.4049/jimmunol.166.10.6423
48. Romay-Penabad Z, Montiel-Manzano MG, Shilagard T, Papalardo E, Vargas G, Deora AB, et al. Annexin A2 is involved in antiphospholipid antibody-mediated pathogenic effects in vitro and in vivo. Blood (2009) 114:3074–83. doi:10.1182/blood-2008-11-188698
49. Ma K, Simantov R, Zhang JC, Silverstein R, Hajjar KA, McCrae KR. High affinity binding of beta 2-glycoprotein I to human endothelial cells is mediated by annexin II. J Biol Chem (2000) 275:15541–8. doi:10.1074/jbc.275.20.15541
50. Canas F, Simonin L, Couturaud F, Renaudineau Y. Annexin A2 autoantibodies in thrombosis and autoimmune diseases. Thromb Res (2015) 135:226–30. doi:10.1016/j.thromres.2014.11.034
51. Zhou H, Yan Y, Xu G, Zhou B, Wen H, Guo D, et al. Toll-like receptor (TLR)-4 mediates anti-beta2GPI/beta2GPI-induced tissue factor expression in THP-1 cells. Clin Exp Immunol (2011) 163:189–98. doi:10.1111/j.1365-2249.2010.04291.x
52. Martin M, Leffler J, Blom AM. Annexin A2 and A5 serve as new ligands for C1q on apoptotic cells. J Biol Chem (2012) 287:33733–44. doi:10.1074/jbc.M112.341339
53. Caster DJ, Korte EA, Merchant ML, Klein JB, Wilkey DW, Rovin BH, et al. Autoantibodies targeting glomerular annexin A2 identify patients with proliferative lupus nephritis. Proteomics Clin Appl (2015). doi:10.1002/prca.201400175
54. Salle V, Maziere JC, Smail A, Cevallos R, Maziere C, Fuentes V, et al. Anti-annexin II antibodies in systemic autoimmune diseases and antiphospholipid syndrome. J Clin Immunol (2008) 28:291–7. doi:10.1007/s10875-008-9188-1
55. Yung S, Tsang RC, Leung JK, Chan TM. Increased mesangial cell hyaluronan expression in lupus nephritis is mediated by anti-DNA antibody-induced IL-1beta. Kidney Int (2006) 69:272–80. doi:10.1038/sj.ki.5000042
56. Yung S, Zhang Q, Zhang CZ, Chan KW, Lui SL, Chan TM. Anti-DNA antibody induction of protein kinase C phosphorylation and fibronectin synthesis in human and murine lupus and the effect of mycophenolic acid. Arthritis Rheum (2009) 60:2071–82. doi:10.1002/art.24573
57. Alarcon-Segovia D, Llorente L. Antibody penetration into living cells. IV. Different effects of anti-native DNA and anti-ribonucleoprotein IgG on the cell cycle of activated T gamma cells. Clin Exp Immunol (1983) 52:365–71.
58. Alarcon-Segovia D. Antinuclear antibodies: to penetrate or not to penetrate, that was the question. Lupus (2001) 10:315–8. doi:10.1191/096120301669579565
59. Madaio MP, Yanase K. Cellular penetration and nuclear localization of anti-DNA antibodies: mechanisms, consequences, implications and applications. J Autoimmun (1998) 11:535–8. doi:10.1006/jaut.1998.0217
60. Alarcon-Segovia D, Ruiz-Arguelles A, Fishbein E. Antibody to nuclear ribonucleoprotein penetrates live human mononuclear cells through Fc receptors. Nature (1978) 271:67–9. doi:10.1038/271067a0
61. Lisi S, Sisto M, Soleti R, Saponaro C, Scagliusi P, D’Amore M, et al. Fcgamma receptors mediate internalization of anti-Ro and anti-La autoantibodies from Sjogren’s syndrome and apoptosis in human salivary gland cell line A-253. J Oral Pathol Med (2007) 36:511–23. doi:10.1111/j.1600-0714.2007.00563.x
62. Koscec M, Koren E, Wolfson-Reichlin M, Fugate RD, Trieu E, Targoff IN, et al. Autoantibodies to ribosomal P proteins penetrate into live hepatocytes and cause cellular dysfunction in culture. J Immunol (1997) 159:2033–41.
63. Seret G, Canas F, Pougnet-Di Costanzo L, Hanrotel-Saliou C, Jousse-Joulin S, Le Meur Y, et al. Anti-alpha-actinin antibodies are part of the anti-cell membrane antibody spectrum that characterize patients with lupus nephritis. J Autoimmun (2015) 61:54–61. doi:10.1016/j.jaut.2015.05.009
64. Hajjar KA, Jacovina AT, Chacko J. An endothelial cell receptor for plasminogen/tissue plasminogen activator. I. Identity with annexin II. J Biol Chem (1994) 269:21191–7.
65. Otey CA, Carpen O. Alpha-actinin revisited: a fresh look at an old player. Cell Motil Cytoskeleton (2004) 58:104–11. doi:10.1002/cm.20007
66. Zhao Z, Deocharan B, Scherer PE, Ozelius LJ, Putterman C. Differential binding of cross-reactive anti-DNA antibodies to mesangial cells: the role of alpha-actinin. J Immunol (2006) 176:7704–14. doi:10.4049/jimmunol.176.12.7704
67. Deocharan B, Zhou Z, Antar K, Siconolfi-Baez L, Angeletti RH, Hardin J, et al. Alpha-actinin immunization elicits anti-chromatin autoimmunity in nonautoimmune mice. J Immunol (2007) 179:1313–21. doi:10.4049/jimmunol.179.2.1313
68. Mason LJ, Ravirajan CT, Rahman A, Putterman C, Isenberg DA. Is alpha-actinin a target for pathogenic anti-DNA antibodies in lupus nephritis? Arthritis Rheum (2004) 50:866–70. doi:10.1002/art.20103
69. Zhao Z, Weinstein E, Tuzova M, Davidson A, Mundel P, Marambio P, et al. Cross-reactivity of human lupus anti-DNA antibodies with alpha-actinin and nephritogenic potential. Arthritis Rheum (2005) 52:522–30. doi:10.1002/art.20862
70. Manson JJ, Ma A, Rogers P, Mason LJ, Berden JH, van der Vlag J, et al. Relationship between anti-dsDNA, anti-nucleosome and anti-alpha-actinin antibodies and markers of renal disease in patients with lupus nephritis: a prospective longitudinal study. Arthritis Res Ther (2009) 11:R154. doi:10.1186/ar2831
71. Mjelle JE, Rekvig OP, Van Der Vlag J, Fenton KA. Nephritogenic antibodies bind in glomeruli through interaction with exposed chromatin fragments and not with renal cross-reactive antigens. Autoimmunity (2011) 44:373–83. doi:10.3109/08916934.2010.541170
72. Kalaaji M, Sturfelt G, Mjelle JE, Nossent H, Rekvig OP. Critical comparative analyses of anti-alpha-actinin and glomerulus-bound antibodies in human and murine lupus nephritis. Arthritis Rheum (2006) 54:914–26. doi:10.1002/art.21622
73. Berden JH, Licht R, van Bruggen MC, Tax WJ. Role of nucleosomes for induction and glomerular binding of autoantibodies in lupus nephritis. Curr Opin Nephrol Hypertens (1999) 8:299–306. doi:10.1097/00041552-199905000-00005
74. van Bavel CC, Fenton KA, Rekvig OP, van der Vlag J, Berden JH. Glomerular targets of nephritogenic autoantibodies in systemic lupus erythematosus. Arthritis Rheum (2008) 58:1892–9. doi:10.1002/art.23626
75. D’Andrea DM, Coupaye-Gerard B, Kleyman TR, Foster MH, Madaio MP. Lupus autoantibodies interact directly with distinct glomerular and vascular cell surface antigens. Kidney Int (1996) 49:1214–21. doi:10.1038/ki.1996.175
76. Blatt NB, Glick GD. Anti-DNA autoantibodies and systemic lupus erythematosus. Pharmacol Ther (1999) 83:125–39. doi:10.1016/S0163-7258(99)00022-4
77. Krishnan MR, Wang C, Marion TN. Anti-DNA autoantibodies initiate experimental lupus nephritis by binding directly to the glomerular basement membrane in mice. Kidney Int (2012) 82:184–92. doi:10.1038/ki.2011.484
78. Yu CL, Sun KH, Tsai CY, Hsieh SC, Yu HS. Anti-dsDNA antibody up-regulates interleukin 6, but not cyclo-oxygenase, gene expression in glomerular mesangial cells: a marker of immune-mediated renal damage? Inflamm Res (2001) 50:12–8. doi:10.1007/s000110050718
79. Yu CL, Huang MH, Tsai CY, Tsai YY, Tsai ST, Sun KH, et al. The effect of human polyclonal anti-dsDNA autoantibodies on apoptotic gene expression in cultured rat glomerular mesangial cells. Scand J Rheumatol (1998) 27:54–60. doi:10.1080/030097498441182
80. Zhang Y, Yang J, Jiang S, Fang C, Xiong L, Cheng H, et al. The lupus-derived anti-double-stranded DNA IgG contributes to myofibroblast-like phenotype in mesangial cells. J Clin Immunol (2012) 32:1270–8. doi:10.1007/s10875-012-9724-x
81. Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol (2010) 6:280–9. doi:10.1038/nrrheum.2010.46
82. Shao WH, Cohen PL. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res Ther (2011) 13:202. doi:10.1186/ar3206
83. Madaio MP, Fabbi M, Tiso M, Daga A, Puccetti A. Spontaneously produced anti-DNA/DNase I autoantibodies modulate nuclear apoptosis in living cells. Eur J Immunol (1996) 26:3035–41. doi:10.1002/eji.1830261232
84. Fismen S, Thiyagarajan D, Seredkina N, Nielsen H, Jacobsen S, Elung-Jensen T, et al. Impact of the tumor necrosis factor receptor-associated protein 1 (Trap1) on renal DNaseI shutdown and on progression of murine and human lupus nephritis. Am J Pathol (2013) 182:688–700. doi:10.1016/j.ajpath.2012.11.013
85. Thiyagarajan D, Fismen S, Seredkina N, Jacobsen S, Elung-Jensen T, Kamper AL, et al. Silencing of renal DNaseI in murine lupus nephritis imposes exposure of large chromatin fragments and activation of toll like receptors and the Clec4e. PLoS One (2012) 7:e34080. doi:10.1371/journal.pone.0034080
86. Zykova SN, Tveita AA, Rekvig OP. Renal Dnase1 enzyme activity and protein expression is selectively shut down in murine and human membranoproliferative lupus nephritis. PLoS One (2010) 5:e12096. doi:10.1371/journal.pone.0012096
87. Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet (2000) 25:177–81. doi:10.1038/76032
88. Chitrabamrung S, Rubin RL, Tan EM. Serum deoxyribonuclease I and clinical activity in systemic lupus erythematosus. Rheumatol Int (1981) 1:55–60. doi:10.1007/BF00541153
89. Perez de Lema G, Maier H, Nieto E, Vielhauer V, Luckow B, Mampaso F, et al. Chemokine expression precedes inflammatory cell infiltration and chemokine receptor and cytokine expression during the initiation of murine lupus nephritis. J Am Soc Nephrol (2001) 12:1369–82.
90. Rovin BH, Doe N, Tan LC. Monocyte chemoattractant protein-1 levels in patients with glomerular disease. Am J Kidney Dis (1996) 27:640–6. doi:10.1016/S0272-6386(96)90097-9
91. Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR. Monocyte chemoattractant protein 1-dependent leukocytic infiltrates are responsible for autoimmune disease in MRL-Fas(lpr) mice. J Exp Med (1999) 190:1813–24. doi:10.1084/jem.190.12.1813
92. Zhang H, Zhao C, Wang S, Huang Y, Wang H, Zhao J, et al. Anti-dsDNA antibodies induce inflammation via endoplasmic reticulum stress in human mesangial cells. J Transl Med (2015) 13:178. doi:10.1186/s12967-015-0536-7
93. Rovin BH, Tan LC. Role of protein kinase pathways in IL-1-induced chemoattractant expression by human mesangial cells. Kidney Int (1994) 46:1059–68. doi:10.1038/ki.1994.367
94. Kanapathippillai P, Hedberg A, Fenton CG, Fenton KA. Nucleosomes contribute to increase mesangial cell chemokine expression during the development of lupus nephritis. Cytokine (2013) 62:244–52. doi:10.1016/j.cyto.2013.03.016
95. Peterson KS, Huang JF, Zhu J, D’Agati V, Liu X, Miller N, et al. Characterization of heterogeneity in the molecular pathogenesis of lupus nephritis from transcriptional profiles of laser-captured glomeruli. J Clin Invest (2004) 113:1722–33. doi:10.1172/JCI200419139
96. McHale JF, Harari OA, Marshall D, Haskard DO. TNF-alpha and IL-1 sequentially induce endothelial ICAM-1 and VCAM-1 expression in MRL/lpr lupus-prone mice. J Immunol (1999) 163:3993–4000.
97. Boswell JM, Yui MA, Burt DW, Kelley VE. Increased tumor necrosis factor and IL-1 beta gene expression in the kidneys of mice with lupus nephritis. J Immunol (1988) 141:3050–4.
98. Brennan DC, Yui MA, Wuthrich RP, Kelley VE. Tumor necrosis factor and IL-1 in New Zealand Black/White mice. Enhanced gene expression and acceleration of renal injury. J Immunol (1989) 143:3470–5.
99. Voronov E, Dayan M, Zinger H, Gayvoronsky L, Lin JP, Iwakura Y, et al. IL-1 beta-deficient mice are resistant to induction of experimental SLE. Eur Cytokine Netw (2006) 17:109–16.
100. McCarthy EM, Smith S, Lee RZ, Cunnane G, Doran MF, Donnelly S, et al. The association of cytokines with disease activity and damage scores in systemic lupus erythematosus patients. Rheumatology (Oxford) (2014) 53:1586–94. doi:10.1093/rheumatology/ket428
101. Chen J, Hartono JR, John R, Bennett M, Zhou XJ, Wang Y, et al. Early interleukin 6 production by leukocytes during ischemic acute kidney injury is regulated by TLR4. Kidney Int (2011) 80:504–15. doi:10.1038/ki.2011.140
102. Sun KH, Yu CL, Tang SJ, Sun GH. Monoclonal anti-double-stranded DNA autoantibody stimulates the expression and release of IL-1beta, IL-6, IL-8, IL-10 and TNF-alpha from normal human mononuclear cells involving in the lupus pathogenesis. Immunology (2000) 99:352–60. doi:10.1046/j.1365-2567.2000.00970.x
103. Baer PC, Wegner B, Geiger H. Effects of mycophenolic acid on IL-6 expression of human renal proximal and distal tubular cells in vitro. Nephrol Dial Transplant (2004) 19:47–52. doi:10.1093/ndt/gfg429
104. Yung S, Tsang RC, Sun Y, Leung JK, Chan TM. Effect of human anti-DNA antibodies on proximal renal tubular epithelial cell cytokine expression: implications on tubulointerstitial inflammation in lupus nephritis. J Am Soc Nephrol (2005) 16:3281–94. doi:10.1681/ASN.2004110917
105. Taflin C, Favier B, Baudhuin J, Savenay A, Hemon P, Bensussan A, et al. Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions. Proc Natl Acad Sci U S A (2011) 108:2891–6. doi:10.1073/pnas.1011811108
106. Chiu YC, Lin CY, Chen CP, Huang KC, Tong KM, Tzeng CY, et al. Peptidoglycan enhances IL-6 production in human synovial fibroblasts via TLR2 receptor, focal adhesion kinase, Akt, and AP-1-dependent pathway. J Immunol (2009) 183:2785–92. doi:10.4049/jimmunol.0802826
107. Aringer M, Smolen JS. The role of tumor necrosis factor-alpha in systemic lupus erythematosus. Arthritis Res Ther (2008) 10:202. doi:10.1186/ar2341
108. Kollias G, Douni E, Kassiotis G, Kontoyiannis D. The function of tumour necrosis factor and receptors in models of multi-organ inflammation, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Ann Rheum Dis (1999) 58(Suppl 1):I32–9. doi:10.1136/ard.58.2008.i32
109. Yokoyama H, Kreft B, Kelley VR. Biphasic increase in circulating and renal TNF-alpha in MRL-lpr mice with differing regulatory mechanisms. Kidney Int (1995) 47:122–30. doi:10.1038/ki.1995.14
110. Herrera-Esparza R, Barbosa-Cisneros O, Villalobos-Hurtado R, Avalos-Diaz E. Renal expression of IL-6 and TNFalpha genes in lupus nephritis. Lupus (1998) 7:154–8. doi:10.1191/096120398678919949
111. Malide D, Russo P, Bendayan M. Presence of tumor necrosis factor alpha and interleukin-6 in renal mesangial cells of lupus nephritis patients. Hum Pathol (1995) 26:558–64. doi:10.1016/0046-8177(95)90253-8
112. Debets JM, Van der Linden CJ, Dieteren IE, Leeuwenberg JF, Buurman WA. Fc-receptor cross-linking induces rapid secretion of tumor necrosis factor (cachectin) by human peripheral blood monocytes. J Immunol (1988) 141:1197–201.
113. Jacob CO, McDevitt HO. Tumour necrosis factor-alpha in murine autoimmune ‘lupus’ nephritis. Nature (1988) 331:356–8. doi:10.1038/331356a0
114. Kontoyiannis D, Kollias G. Accelerated autoimmunity and lupus nephritis in NZB mice with an engineered heterozygous deficiency in tumor necrosis factor. Eur J Immunol (2000) 30:2038–47. doi:10.1002/1521-4141(200007)30:7<2038::AID-IMMU2038>3.0.CO;2-K
115. Yung S, Cheung KF, Zhang Q, Chan TM. Mediators of inflammation and their effect on resident renal cells: implications in lupus nephritis. Clin Dev Immunol (2013) 2013:317682. doi:10.1155/2013/317682
116. Qing X, Pitashny M, Thomas DB, Barrat FJ, Hogarth MP, Putterman C. Pathogenic anti-DNA antibodies modulate gene expression in mesangial cells: involvement of HMGB1 in anti-DNA antibody-induced renal injury. Immunol Lett (2008) 121:61–73. doi:10.1016/j.imlet.2008.08.007
117. Lee SB, Kalluri R. Mechanistic connection between inflammation and fibrosis. Kidney Int Suppl (2010) 78(Suppl 119):S22–6. doi:10.1038/ki.2010.418
118. Nath KA. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am J Kidney Dis (1992) 20:1–17. doi:10.1016/S0272-6386(12)80312-X
119. Vleming LJ, Baelde JJ, Westendorp RG, Daha MR, van Es LA, Bruijn JA. The glomerular deposition of PAS positive material correlates with renal function in human kidney diseases. Clin Nephrol (1997) 47:158–67.
120. Nakapoulou I, Stefanaki K, Zeis PM, Papadakis J, Boletis J, Vosnidis G, et al. The glomerular distribution of laminin and fibronectin in glomerulonephritis. Histol Histopathol (1993) 8:521–6.
121. Yung S, Zhang Q, Chau MK, Chan TM. Distinct effects of mycophenolate mofetil and cyclophosphamide on renal fibrosis in NZBWF1/J mice. Autoimmunity (2015):1–17. doi:10.3109/08916934.2015.1054027
122. Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med (1994) 331:1286–92. doi:10.1056/NEJM199411103311907
123. Liu Y. Renal fibrosis: new insights into the pathogenesis and therapeutics. Kidney Int (2006) 69:213–7. doi:10.1038/sj.ki.5000054
124. Oleksyn D, Pulvino M, Zhao J, Misra R, Vosoughi A, Jenks S, et al. Protein kinase Cbeta is required for lupus development in Sle mice. Arthritis Rheum (2013) 65:1022–31. doi:10.1002/art.37825
125. Rastaldi MP, Ferrario F, Giardino L, Dell’Antonio G, Grillo C, Grillo P, et al. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int (2002) 62:137–46. doi:10.1046/j.1523-1755.2002.00430.x
126. Rastaldi MP. Epithelial-mesenchymal transition and its implications for the development of renal tubulointerstitial fibrosis. J Nephrol (2006) 19:407–12.
127. Yung S, Ng CY, Ho SK, Cheung KF, Chan KW, Zhang Q, et al. Anti-dsDNA antibody induces soluble fibronectin secretion by proximal renal tubular epithelial cells and downstream increase of TGF-beta1 and collagen synthesis. J Autoimmun (2015) 58:111–22. doi:10.1016/j.jaut.2015.01.008
128. Ronda N, Cravedi P, Benozzi L, Lunghi P, Bonati A, Allegri L, et al. Early proinflammatory activation of renal tubular cells by normal and pathologic IgG. Nephron Exp Nephrol (2005) 100:e77–84. doi:10.1159/000084573
129. Koren E, Koscec M, Wolfson-Reichlin M, Ebling FM, Tsao B, Hahn BH, et al. Murine and human antibodies to native DNA that cross-react with the A and D SnRNP polypeptides cause direct injury of cultured kidney cells. J Immunol (1995) 154:4857–64.
130. Yap DY, Tang CS, Ma MK, Lam MF, Chan TM. Survival analysis and causes of mortality in patients with lupus nephritis. Nephrol Dial Transplant (2012) 27:3248–54. doi:10.1093/ndt/gfs073
131. Chan TM, Li FK, Tang CS, Wong RW, Fang GX, Ji YL, et al. Efficacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. Hong Kong-Guangzhou Nephrology Study Group. N Engl J Med (2000) 343:1156–62.
132. Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology (2000) 47:85–118. doi:10.1016/S0162-3109(00)00188-0
133. Copeland JW, Beaumont BW, Merrilees MJ, Pilmore HL. Epithelial-to-mesenchymal transition of human proximal tubular epithelial cells: effects of rapamycin, mycophenolate, cyclosporin, azathioprine, and methylprednisolone. Transplantation (2007) 83:809–14. doi:10.1097/01.tp.0000255680.71816.aa
134. Dubus I, Vendrely B, Christophe I, Labouyrie JP, Delmas Y, Bonnet J, et al. Mycophenolic acid antagonizes the activation of cultured human mesangial cells. Kidney Int (2002) 62:857–67. doi:10.1046/j.1523-1755.2002.00514.x
Keywords: lupus nephritis, anti-dsDNA antibodies, mesangial cells, proximal renal tubular epithelial cells, inflammation, fibrosis, mycophenolic acid, cyclophosphamide
Citation: Yung S and Chan TM (2015) Mechanisms of kidney injury in lupus nephritis – the role of anti-dsDNA antibodies. Front. Immunol. 6:475. doi: 10.3389/fimmu.2015.00475
Received: 25 July 2015; Accepted: 01 September 2015;
Published: 15 September 2015
Edited by:
James Harris, Monash University, AustraliaReviewed by:
Dragana Odobasic, Monash University, AustraliaTri Giang Phan, Garvan Institute, Australia
Copyright: © 2015 Yung and Chan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan Yung and Tak Mao Chan, Department of Medicine, University of Hong Kong, Queen Mary Hospital, 102 Pokfulam Road, Pokfulam, Hong Kong, china, ssyyung@hku.hk; dtmchan@hku.hk