 Daniel A. M. Toledo
Daniel A. M. Toledo Heloísa D’Avila
Heloísa D’Avila Rossana C. N. Melo
Rossana C. N. Melo- Laboratory of Cellular Biology, Department of Biology, Institute of Biological Sciences (ICB), Federal University of Juiz de Fora (UFJF), Juiz de Fora, Minas Gerais, Brazil
Pathogens induce several changes in the host cell signaling and trafficking mechanisms in order to evade and manipulate the immune response. One prominent pathogen-mediated change is the formation of lipid-rich organelles, termed lipid bodies (LBs) or lipid droplets, in the host cell cytoplasm. Protozoan parasites, which contribute expressively to the burden of infectious diseases worldwide, are able to induce LB genesis in non-immune and immune cells, mainly macrophages, key players in the initial resistance to the infection. Under host–parasite interaction, LBs not only accumulate in the host cytoplasm but also relocate around and move into parasitophorous vacuoles. There is increasing evidence that protozoan parasites may target host-derived LBs either for gaining nutrients or for escaping the host immune response. Newly formed, parasite-induced LBs may serve as lipid sources for parasite growth and also produce inflammatory mediators that potentially act in the host immune response deactivation. In this mini review, we summarize current knowledge on the formation and role of host LBs as sites exploited by intracellular protozoan parasites as a strategy to maintain their own survival.
Introduction
Protozoan parasitic infections comprise devastating infectious diseases, such as malaria, visceral leishmaniasis, toxoplasmic encephalitis, and trypanosomiasis, which still account for a large proportion of death and disability worldwide (1). Many protozoan parasites have an obligate intracellular existence. The infection is initiated when the parasite enter the host target cell and is internalized within a plasma membrane-derived vacuole, the parasitophorous vacuole (PV) (2). Within the host, protozoan parasites that are mainly intracellular will only cause infectious disease if they are able to survive and multiply within the PV (2, 3).
The events of the PV formation and progression generally occur in parallel with accentuated genesis of lipid-rich organelles, termed lipid bodies (LBs) or lipid droplets, in the host cell cytoplasm [reviewed in Ref. (4, 5)]. It is now well documented that experimental and clinical infections with a range of protozoan parasites trigger LB accumulation (Table 1) and an intriguing interaction with the PV [reviewed in Ref. (5)]. Other pathogens, such as bacteria and viruses, also induce LB formation within different cell types, indicating that LB accumulation in response to infectious diseases is a broad event and may have implications for microbial pathogenesis (5–8).
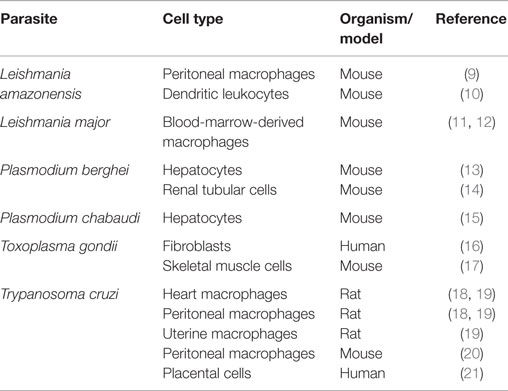
Table 1. Protozoan parasite-induced lipid body formation in host cells.
While the successful replication within the PV is under influence of several factors, there is increasing evidence that LB organelles are important for the rapid intracellular reproduction of protozoan parasites (5). Protozoan parasites require large amounts of lipids necessary for membrane biogenesis of new progenies [reviewed in Ref. (22, 23)] and may take advantage of these organelles as high-energy substrate sources. LBs within infected cells are also able to produce inflammatory mediators that potentially can inhibit the host Th1response, thus, favoring parasite growth [reviewed in Ref. (4)]. In this mini review, we will discuss the role of host LBs as organelles modulated by intracellular protozoan parasites for survival.
LB Structure and Visualization
Lipid bodies are common organelles distributed in the cytoplasm of prokaryotic and eukaryotic cells (5). As a general feature, LBs contain a core rich in neutral lipids, surrounded by a monolayer of phospholipids with structural proteins – the perilipin (PLIN) family proteins (5). Proteins are not restricted to the LB surface. It is documented that many types of proteins are present in the LB internum depending on the cell type (24–26).
In spite of variations in the LB composition, these organelles are seen in the cell cytoplasm as compartmentalized round sites by light microscopy or transmission electron microscopy (TEM). While LB imaging under light microscopy usually requires the use of specific lipid probes, ultrastructural observation does not need any additional labeling because LBs lack a true delimiting unit membrane structure, which enables unambiguous identification by TEM (Figure 1) (4, 8, 26).
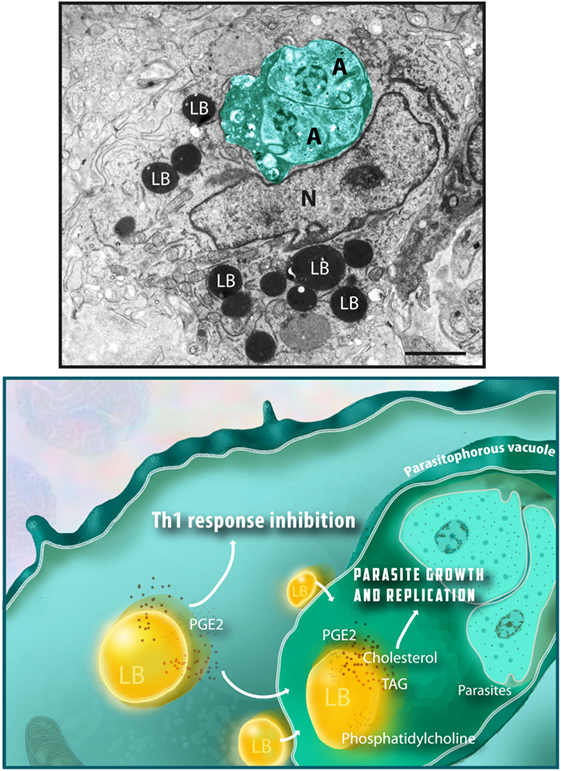
Figure 1. Lipid bodies (LBs) accumulate in the host cell cytoplasm in response to interaction with protozoan parasites and favor parasite survival. Top panel shows an electron micrograph of a heart inflammatory macrophage infected with Trypanosoma cruzi. Note the presence of dividing amastigotes (A) within the parasitophorous vacuole (outlined in green) and the high number of LBs in the cytoplasm. LBs typically appear as round electron dense organelles in the cell cytoplasm. A model to explain how protozoan parasite-induced LB formation favors parasite growth and replication is shown in the bottom panel. Interaction of LBs with the parasitophorous vacuole, an event also triggered by the parasite infection, leads to discharge of LB contents, such as cholesterol, triacylglycerol (TAG), and phosphatidylcholine, which serve as lipid sources for parasite growth. Newly formed LBs are also sites for prostaglandin E2 synthesis (PGE2), a potent inflammation mediator that potentially inhibit the host Th1 immune response, thus decreasing the microbicidal capacity of the parasitophorous vacuole. Nu, nucleus. Scale bar, 600 nm. Top panel was reprinted from Ref. (27) with permission.
Protozoan Parasites Induce LB Accumulation in Host Cells
Several protozoan parasites induce LB formation in a variety of host immune and non-immune cells (Table 1). Under host–parasite interaction, LBs not only accumulate but also increase in size and undergo ultrastructural changes in the host cell cytoplasm. These structural modifications of LBs have been well demonstrated during in vitro and in vivo studies with Trypanosoma cruzi, the causal agent of Chagas’ disease (18, 19). This pathology elicits a strong inflammatory response characterized by elevated infiltration of macrophages in the target organs, mainly the heart (28). Histopathological studies conducted during the acute phase of this disease showed that increased myocardial parasitism is paralleled by increased size of LBs within inflammatory macrophages in which LBs can reach up to 4 μm in diameter (19). Moreover, LBs show varied electron-density in response to the T. cruzi infection, a morphological change likely associated with mobilization and/or in situ synthesis of lipid mediators [reviewed in Ref. (8)]. The capability of host LBs to generate these compounds will be discussed later.
Increase of LB size in parallel with augmented number of these organelles was also found in dendritic leukocytes hosting live Leishmania amazonensis amastigotes (10). Overall, as noted in diverse cell types (Table 1), it is clear that protozoan parasites are able to trigger LB formation in host cells. But how is the mechanism leading to LB genesis? Similar to other studies with mycobacteria (29, 30), it has been demonstrated that parasite uptake potentiates LB formation within host cells but is not essential for triggering this event. After 24 h of infection with T. cruzi, peritoneal macrophages with internalized parasites as well as non-parasitized cells show increased number of LBs compared to control, non-infected cells, suggesting a bystander amplification of the cell response (20). Accordingly, inhibition of parasite phagocytosis did not abolish LB genesis during infection with Leishmania major (11). These authors have recently showed that while phagocytosis of latex beads by macrophages did not trigger LB formation, this phenomenon is equally induced by both live and heat-killed parasites (11). Altogether, these results imply that soluble factors in parasitized cells may act in a paracrine manner to produce LB in non-parasitized bystander cells (11). While LB formation occurs through a toll-like receptor-2 (TLR2)-dependent mechanism as documented during T. cruzi infection in macrophages (20), the identification of downstream signaling pathways involved in this event during parasitic infections awaits further investigation.
Newly Formed LBs are Recruited to the Proximity of Parasites
Interestingly, protozoan parasites trigger a redistribution of the newly formed LBs around parasite-containing phagosomes. As documented by studies with T. cruzi (18, 19), L. major (11, 12), L. amazonensis (10), and Toxoplasma gondii (17, 31), LBs accumulate in close proximity to PVs or even move into these vacuoles, suggesting that these pathogens take advantage of these organelles.
Indeed, TEM analyses have enabled the identification of intimate contact between LBs and the PV membrane (10, 17–20, 31, 32). In 2003, our group was the first to observe that during an in vivo experimental infection with a pathogen (T. cruzi), LBs were internalized into parasite-containing phagosomes (18). Following studies found the same event for infections with different species of bacteria [reviewed in Ref. (5)] and with the protozoan parasites T. gondii (17) and L. major (11).
How LBs translocate across the phagosome membrane? This mechanism has been mainly addressed in studies with bacteria. LB-associated proteins, secreted by bacteria seem to be involved in capturing LB into bacteria-containing vacuoles, while LB translocation seems to involve displacement of the LB structural protein PLIN2/adipose differentiation-related protein (ADRP), which is constitutively associated with the surface of LBs (33). Although studies of membrane sites between pathogen-containing compartments and intracellular host organelles has been gaining more attention in the literature [reviewed in Ref. (34)], the mechanistic details underlying the intriguing LB–phagosome interaction remains to be fully defined.
Host LIPIDS, LBs, and Intracellular Parasite Growth
Lipid metabolic pathways have been demonstrated as major networks modulated by protozoan parasites in host cells. Numerous studies with T. gondii, malaria parasites (Plasmodium berghei, Plasmodium falciparum, and Plasmodium yoelii), L. amazonensis, Cryptosporidium parvum, and Eimeria bovis have documented that these parasites are able to (i) induce accumulation of lipids, such as neutral lipids [triacylglycerol (TAG), diacylglycerol, and cholesterol esters], cholesterol, and/or phospholipids, especially phosphatidylcholine in host cells (10, 12, 35–37); (ii) acquire lipid resources, such as cholesterol and phospholipids from their host environment into the PV (16, 36, 38–40), and/or (iii) use host lipids to synthesize complex own lipids or even own LBs (16, 38, 41).
Thus, although the nutritional requirements of these parasites are intricate, overall it is believed that host lipids are central to support successful parasite replication within the PV (16, 32, 37, 38, 42, 43). Host lipid acquisition is also considered crucial to PV maturation. This vacuole undergoes a pronounced membranous remodeling associated with formation of an internal network of tubules and vesicles as observed during the infection with T. gondii (42, 44). By using different approaches, including fluorescence recovery after photobleaching (FRAP) microscopy, Caffaro and Boothroyd demonstrated that host cells are major lipid contributors to the PV remodeling and that lipids are transferred in a continuous way from the host into the PV (42).
How is the contribution of host LBs as lipid sources for protozoan parasites? Consistent with parasite-induced host LB formation (Table 1), LB genesis in parallel to host lipid reprograming/accumulation (10, 12) and host LB–PV interaction (10–12, 17–19, 31), it is believed that these organelles act as important lipid sources for parasite growth.
Several classes of lipids, including neutral lipids, cholesterol, and phospholipids, make up LBs (45). Because protozoan parasites are not competent or have limited ability to synthesize lipids [reviewed in Ref. (22, 46)], LBs could be an essential source of both TAG and cholesterol for these parasites (10). These molecules are important as precursors for membrane neogenesis for newly formed parasites [reviewed in Ref. (22, 46)]. Moreover, the phospholipid monolayer of LBs consists of numerous phospholipid species of which phosphatidylcholine is the most abundant (47). Thus, it is likely that LB-derived lipids are transferred from host to the PV and taken up by replicating parasites. Indeed, by using a fluorescent probe (BODIPY-phosphatidylcholine) and live imaging microscopy, Charron and Sibley showed that this probe moved from host plasma membrane and/or host LBs, seen as dispersed puncta in the host cytoplasm, to LBs formed within the parasite (16). However, the potential relocation of other lipids from host LBs to the parasite still needs to be better explored in future studies.
Does depletion of host lipids and/or LBs impact protozoan parasites development within host cells? In T. gondii infection, inhibition of cholesterol esterification in the host cell blocks parasite growth (38). Exposure to cholesterol esterification inhibitors led to reduction of cholesteryl ester synthesis, morphological changes in parasite LBs, and deformations of the parasite plasma membrane with discharge of parasite content into the PV (38). Accordingly, the use of inhibitors targeting the host cellular cholesterol de novo synthesis and processing repressed both E. bovis proliferation and LB formation within host endothelial cells (43). On the other hand, depletion of cholesterol content did not impact malaria parasites (P. berghei and P. yoelii) development within hepatocytes (39). In this case, the authors consider that the parasite may exploit alternative sources in these cells to sustain infectivity (39). In fact, protozoan parasites can divert lipids from other sources than the intracellular environment. C. parvum, the causal agent of cryptosporidiosis, a life-threatening diarrheal disease in immunocompromised individuals (48), scavenges cholesterol from plasma low-density protein (LDL) and micelles, and to a lesser extent from the cholesterol pathway within enterocytes (40). Removal of cholesterol from the media, and to lesser extent from host intracellular pools, obstructs parasite reproduction (40).
During the experimental infection with T. cruzi, our group demonstrated that the use of C75, an inhibitor of fatty acid synthase, led to both inhibition of LB formation and parasite division within macrophages (20). While C75 had no direct cytotoxic effect on the parasite, intracellular parasite replication was likely affected by an accentuated reduction of the LB numbers (around 66%) after cell treatment with C75 (20).
Host LBs Produce PGE2 in Response to Infection with Protozoan Parasites
One interesting aspect of LBs is that they are able to change their composition in response to inflammatory events as documented in cells from the immune system [reviewed in Ref. (49)]. LBs contain stores of arachidonic acid (AA), indicating that these organelles are potentially able to initiate cascades that culminate in the formation of inflammatory mediators (eicosanoids) (50, 51). Eicosanoid-generating enzymes (52–55), and in situ synthesis of eicosanoids (prostaglandins and leukotrienes) were indeed documented in these organelles within activated leukocytes and other cells from the immune system during inflammatory conditions [for example, see Ref. (29, 56–58)].
During the infection with T. cruzi (18), T. gondii (17, 31), and L. amazonensis (9), significant correlations between LB formation and enhanced generation of eicosanoids, specifically prostaglandin E2 (PGE2), by host cells have been observed. Moreover, T. gondii infection also elicited a time-dependent increase of cyclooxygenase-2 (COX-2) mRNA levels, indicating that the PGE2 may be a product of an active COX pathway within host cells (17).
By investigating the intracellular specific localization of both COX-2 and PGE2 within T. cruzi-infected macrophages, we found that both molecules were immunolocalized in LBs, indicating that LBs act as sources of PGE2 (20). Interestingly, cell treatment with non-steroidal anti-inflammatory drugs (NSAIDs) inhibited both LB formation and LB-derived PGE2 synthesis in a mechanism independent of COX inhibition (20), as previously documented (59). Therefore, accumulation of LBs in infected host cells may modulate the production of an innate immune response with production of PGE2, which in turn may contribute to a permissive environment for pathogen proliferation. For example, during the infection with T. gondii, the increased numbers of LBs within macrophages correlated with high PGE2 levels, decreased nitric oxide (NO) production and parasite survival (31). In fact, high concentrations of PGE2 potentially inhibit the Th1 response, tumor necrosis factor alpha (TNF-α) and/or NO production (60–62). This scenario is also detected during infections with mycobacteria (29, 30), demonstrating that pathogen-induced LB formation associated with PGE2 synthesis is a broader event that can potentially support intracellular pathogen survival. Taken together, these data suggest a model by which LBs are acting as potential stations for the survival of protozoan parasites within host cells, as depicted in Figure 1.
Final Remarks and Future Directions
Several key issues remain to be addressed to better understand the link between LBs and parasite survival within host cells during infections with intracellular parasitic protozoans. It is now clear that host LBs interact with PVs and that the parasite has a remarkable ability to sequester host lipids. What are the molecular mechanisms involved in LB translocation into the PV and how the LB content is extracted by the parasite? In patients with chronic mycobacterial infection (Mycobacterium leprae), round classical LBs are observed in contact with pathogen-containing phagosomes and intact bacteria are seen completely enmeshed in accumulated lipid content within the vacuole (63). Are LBs important for pathogen survival in chronic parasitic infections? Considering that LBs are sources for parasite development, could these organelles be target by therapeutic treatment? How significant is the contribution of host LBs and/or parasite LBs to parasite development? Moreover, the mechanisms by which new molecules, including natural products, can affect pathways of the parasite lipid metabolism and both host and parasite LB formation have yet to be fully appreciated.
Author Contributions
RM prepared the manuscript. All authors contributed in part to writing and editing the manuscript and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank present and past members of the Laboratory of Cellular Biology (UFJF, Brazil), Centro de Microscopia (Universidade Federal de Minas Gerais – UFMG) and Centro de Aquisição e Processamento de Imagens-CAPI (ICB-UFMG) for the use of their facilities. We acknowledge Kennedy Bonjour for careful assistance with the illustration (Figure 1) used in this paper. We apologize to investigators whose relevant work has not been cited because of space constraints.
Funding
This work was supported by from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil-477475/2013-2; 469995/2014-9, 311083/2014-5; 308291-2012-3), Brazilian Ministry of Health and Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG, Brazil-CBB-APQ-02239-14; CBB-APQ-02477-14). DT is a PhD student supported by a CNPq fellowship.
References
1. Bar AK, Phukan N, Pinheiro J, Simoes-Barbosa A. The interplay of host microbiota and parasitic protozoans at mucosal interfaces: implications for the outcomes of infections and diseases. PLoS Negl Trop Dis (2015) 9(12):e0004176. doi:10.1371/journal.pntd.0004176
2. Sibley LD. Invasion and intracellular survival by protozoan parasites. Immunol Rev (2011) 240(1):72–91. doi:10.1111/j.1600-065X.2010.00990.x
3. Liévin-Le Moal V, Loiseau PM. Leishmania hijacking of the macrophage intracellular compartments. FEBS J (2015) 282(4):598–607. doi:10.1111/febs.13601
4. Melo RCN, D’Avila H, Wan HC, Bozza PT, Dvorak AM, Weller PF. Lipid bodies in inflammatory cells: structure, function, and current imaging techniques. J Histochem Cytochem (2011) 59(5):540–56. doi:10.1369/0022155411404073
5. Melo RCN, Dvorak AM. Lipid body-phagosome interaction in macrophages during infectious diseases: host defense or pathogen survival strategy? PLoS Pathog (2012) 8(7):e1002729. doi:10.1371/journal.ppat.1002729
6. Saka HA, Valdivia R. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu Rev Cell Dev Biol (2012) 28:411–37. doi:10.1146/annurev-cellbio-092910-153958
7. Nicolaou G, Goodall AH, Erridge C. Diverse bacteria promote macrophage foam cell formation via toll-like receptor-dependent lipid body biosynthesis. J Atheroscler Thromb (2012) 19(2):137–48. doi:10.5551/jat.10249
8. Dias FF, Zarantonello VC, Parreira GG, Chiarini-Garcia H, Melo RCN. The intriguing ultrastructure of lipid body organelles within activated macrophages. Microsc Microanal (2014) 20(3):869–78. doi:10.1017/S143192761400066X
9. Pinheiro RO, Nunes MP, Pinheiro CS, D’Avila H, Bozza PT, Takiya CM, et al. Induction of autophagy correlates with increased parasite load of Leishmania amazonensis in BALB/c but not C57BL/6 macrophages. Microbes Infect (2009) 11(2):181–90. doi:10.1016/j.micinf.2008.11.006
10. Lecoeur H, Giraud E, Prevost MC, Milon G, Lang T. Reprogramming neutral lipid metabolism in mouse dendritic leucocytes hosting live Leishmania amazonensis amastigotes. PLoS Negl Trop Dis (2013) 7(6):e2276. doi:10.1371/journal.pntd.0002276
11. Rabhi S, Rabhi I, Trentin B, Piquemal D, Regnault B, Goyard S, et al. Lipid droplet formation, their localization and dynamics during Leishmania major macrophage infection. PLoS One (2016) 11(2):e0148640. doi:10.1371/journal.pone.0148640
12. Rabhi I, Rabhi S, Ben-Othman R, Rasche A, Daskalaki A, Trentin B, et al. Transcriptomic signature of Leishmania infected mice macrophages: a metabolic point of view. PLoS Negl Trop Dis (2012) 6(8):e1763. doi:10.1371/journal.pntd.0001763
13. Rodríguez-Acosta A, Finol HJ, Pulido-Méndez M, Márquez A, Andrade G, González N, et al. Liver ultrastructural pathology in mice infected with Plasmodium berghei. J Submicrosc Cytol Pathol (1998) 30(2):299–307.
14. Pulido-Méndez M, Finol HJ, Girón ME, Aguilar I. Ultrastructural pathological changes in mice kidney caused by Plasmodium berghei infection. J Submicrosc Cytol Pathol (2006) 38(2–3):143–8.
15. Dumont ME, Dei-Cas E, Maurois P, Slomianny C, Prensier G, Houcke-Lecomte M, et al. [Histopathology of the liver and kidney during malaria: relation to malaria-induced dyslipoproteinemia]. Ann Parasitol Hum Comp (1988) 63(3):171–83.
16. Charron AJ, Sibley LD. Host cells: mobilizable lipid resources for the intracellular parasite Toxoplasma gondii. J Cell Sci (2002) 115(Pt 15):3049–59.
17. Gomes AF, Magalhaes KG, Rodrigues RM, de Carvalho L, Molinaro R, Bozza PT, et al. Toxoplasma gondii-skeletal muscle cells interaction increases lipid droplet biogenesis and positively modulates the production of IL-12, IFN-g and PGE2. Parasit Vectors (2014) 7:47. doi:10.1186/1756-3305-7-47
18. Melo RCN, D’Avila H, Fabrino DL, Almeida PE, Bozza PT. Macrophage lipid body induction by Chagas disease in vivo: putative intracellular domains for eicosanoid formation during infection. Tissue Cell (2003) 35(1):59–67. doi:10.1016/S0040-8166(02)00105-2
19. Melo RCN, Fabrino DL, Dias FF, Parreira GG. Lipid bodies: structural markers of inflammatory macrophages in innate immunity. Inflamm Res (2006) 55(8):342–8. doi:10.1007/s00011-006-5205-0
20. D’Avila H, Freire-de-Lima CG, Roque NR, Teixeira L, Barja-Fidalgo C, Silva AR, et al. Host cell lipid bodies triggered by Trypanosoma cruzi infection and enhanced by the uptake of apoptotic cells are associated with prostaglandin E generation and increased parasite growth. J Infect Dis (2011) 204(6):951–61. doi:10.1093/infdis/jir432
21. Fabro AE, Calzolari G. [Increase of lipids in human chagasic placentas: cytochemical and biochemical study]. Rev Fac Cien Med Univ Nac Cordoba (1990) 48(1–2):25–32.
22. Helms JB, Kaloyanova DV, Strating JR, van Hellemond JJ, van der Schaar HM, Tielens AG, et al. Targeting of the hydrophobic metabolome by pathogens. Traffic (2015) 16(5):439–60. doi:10.1111/tra.12280
23. Coppens I. Contribution of host lipids to Toxoplasma pathogenesis. Cell Microbiol (2006) 8(1):1–9. doi:10.1111/j.1462-5822.2005.00647.x
24. Welte MA. Proteins under new management: lipid droplets deliver. Trends Cell Biol (2007) 17(8):363–9. doi:10.1016/j.tcb.2007.06.004
25. Wan HC, Melo RCN, Jin Z, Dvorak AM, Weller PF. Roles and origins of leukocyte lipid bodies: proteomic and ultrastructural studies. FASEB J (2007) 21(1):167–78. doi:10.1096/fj.06-6711com
26. Melo RCN, Weller PF. Unraveling the complexity of lipid body organelles in human eosinophils. J Leukoc Biol (2014) 96(5):703–12. doi:10.1189/jlb.3RU0214-110R
27. Melo RCN, Machado CRS. Trypanosoma cruzi: peripheral blood monocytes and heart macrophages in the resistance to acute experimental infection in rats. Exp Parasitol (2001) 97(1):15–23. doi:10.1006/expr.2000.4576
28. Melo RCN. Acute heart inflammation: ultrastructural and functional aspects of macrophages elicited by Trypanosoma cruzi infection. J Cell Mol Med (2009) 13(2):279–94. doi:10.1111/j.1582-4934.2008.00388.x
29. D’Avila H, Melo RCN, Parreira GG, Werneck-Barroso E, Castro-Faria-Neto HC, Bozza PT. Mycobacterium bovis bacillus Calmette-Guerin induces TLR2-mediated formation of lipid bodies: intracellular domains for eicosanoid synthesis in vivo. J Immunol (2006) 176(5):3087–97. doi:10.4049/jimmunol.176.5.3087
30. Mattos KA, D’Avila H, Rodrigues LS, Oliveira VG, Sarno EN, Atella GC, et al. Lipid droplet formation in leprosy: toll-like receptor-regulated organelles involved in eicosanoid formation and Mycobacterium leprae pathogenesis. J Leukoc Biol (2010) 87(3):371–84. doi:10.1189/jlb.0609433
31. Mota LA, Roberto Neto J, Monteiro VG, Lobato CS, Oliveira MA, Cunha M, et al. Culture of mouse peritoneal macrophages with mouse serum induces lipid bodies that associate with the parasitophorous vacuole and decrease their microbicidal capacity against Toxoplasma gondii. Mem Inst Oswaldo Cruz (2014) 109(6):767–74.
32. Combs TP, Nagajyothi, Mukherjee S, de Almeida CJ, Jelicks LA, Schubert W, et al. The adipocyte as an important target cell for Trypanosoma cruzi infection. J Biol Chem (2005) 280(25):24085–94. doi:10.1074/jbc.M412802200
33. Cocchiaro JL, Kumar Y, Fischer ER, Hackstadt T, Valdivia RH. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc Natl Acad Sci U S A (2008) 105(27):9379–84. doi:10.1073/pnas.0712241105
34. Dumoux M, Hayward RD. Membrane contact sites between pathogen-containing compartments and host organelles. Biochim Biophys Acta (2016). doi:10.1016/j.bbalip.2016.01.018
35. Simoes AP, Roelofsen B, Op den Kamp JA. Incorporation of free fatty acids can explain alterations in the molecular species composition of phosphatidylcholine and phosphatidylethanolamine in human erythrocytes as induced by Plasmodium falciparum. Cell Biol Int Rep (1992) 16(6):533–45. doi:10.1016/S0309-1651(05)80052-9
36. Itoe MA, Sampaio JL, Cabal GG, Real E, Zuzarte-Luis V, March S, et al. Host cell phosphatidylcholine is a key mediator of malaria parasite survival during liver stage infection. Cell Host Microbe (2014) 16(6):778–86. doi:10.1016/j.chom.2014.11.006
37. Hamid PH, Hirzmann J, Kerner K, Gimpl G, Lochnit G, Hermosilla CR, et al. Eimeria bovis infection modulates endothelial host cell cholesterol metabolism for successful replication. Vet Res (2015) 46:100. doi:10.1186/s13567-015-0230-z
38. Nishikawa Y, Quittnat F, Stedman TT, Voelker DR, Choi JY, Zahn M, et al. Host cell lipids control cholesteryl ester synthesis and storage in intracellular Toxoplasma. Cell Microbiol (2005) 7(6):849–67. doi:10.1111/j.1462-5822.2005.00518.x
39. Labaied M, Jayabalasingham B, Bano N, Cha SJ, Sandoval J, Guan G, et al. Plasmodium salvages cholesterol internalized by LDL and synthesized de novo in the liver. Cell Microbiol (2011) 13(4):569–86. doi:10.1111/j.1462-5822.2010.01555.x
40. Ehrenman K, Wanyiri JW, Bhat N, Ward HD, Coppens I. Cryptosporidium parvum scavenges LDL-derived cholesterol and micellar cholesterol internalized into enterocytes. Cell Microbiol (2013) 15(7):1182–97. doi:10.1111/cmi.12107
41. Jackson KE, Klonis N, Ferguson DJ, Adisa A, Dogovski C, Tilley L. Food vacuole-associated lipid bodies and heterogeneous lipid environments in the malaria parasite, Plasmodium falciparum. Mol Microbiol (2004) 54(1):109–22. doi:10.1111/j.1365-2958.2004.04284
42. Caffaro CE, Boothroyd JC. Evidence for host cells as the major contributor of lipids in the intravacuolar network of Toxoplasma-infected cells. Eukaryot Cell (2011) 10(8):1095–9. doi:10.1128/EC.00002-11
43. Hamid PH, Hirzmann J, Hermosilla C, Taubert A. Differential inhibition of host cell cholesterol de novo biosynthesis and processing abrogates Eimeria bovis intracellular development. Parasitol Res (2014) 113(11):4165–76. doi:10.1007/s00436-014-4092-5
44. Sibley LD, Niesman IR, Parmley SF, Cesbron-Delauw MF. Regulated secretion of multi-lamellar vesicles leads to formation of a tubulo-vesicular network in host-cell vacuoles occupied by Toxoplasma gondii. J Cell Sci (1995) 108(Pt 4):1669–77.
45. Farese RV Jr, Walther TC. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell (2009) 139(5):855–60. doi:10.1016/j.cell.2009.11.005
46. Wenk MR. Lipidomics of host-pathogen interactions. FEBS Lett (2006) 580(23):5541–51. doi:10.1016/j.febslet.2006.07.007
47. Penno A, Hackenbroich G, Thiele C. Phospholipids and lipid droplets. Biochim Biophys Acta (2013) 1831(3):589–94. doi:10.1016/j.bbalip.2012.12.001
48. Tzipori S, Ward H. Cryptosporidiosis: biology, pathogenesis and disease. Microbes Infect (2002) 4(10):1047–58. doi:10.1016/S1286-4579(02)01629-5
49. Melo RCN, Weller PF. Lipid droplets in leukocytes: organelles linked to inflammatory responses. Exp Cell Res (2016) 340(2):193–7. doi:10.1016/j.yexcr.2015.10.028
50. Weller PF, Dvorak AM. Arachidonic acid incorporation by cytoplasmic lipid bodies of human eosinophils. Blood (1985) 65(5):1269–74.
51. Weller PF, Monahan-Earley RA, Dvorak HF, Dvorak AM. Cytoplasmic lipid bodies of human eosinophils. Subcellular isolation and analysis of arachidonate incorporation. Am J Pathol (1991) 138(1):141–8.
52. Dvorak AM, Morgan E, Schleimer RP, Ryeom SW, Lichtenstein LM, Weller PF. Ultrastructural immunogold localization of prostaglandin endoperoxide synthase (cyclooxygenase) to non-membrane-bound cytoplasmic lipid bodies in human lung mast cells, alveolar macrophages, type II pneumocytes, and neutrophils. J Histochem Cytochem (1992) 40(6):759–69. doi:10.1177/40.6.1316915
53. Dvorak AM, Morgan ES, Tzizik DM, Weller PF. Prostaglandin endoperoxide synthase (cyclooxygenase): ultrastructural localization to nonmembrane-bound cytoplasmic lipid bodies in human eosinophils and 3T3 fibroblasts. Int Arch Allergy Immunol (1994) 105(3):245–50. doi:10.1159/000236764
54. Bozza PT, Yu W, Penrose JF, Morgan ES, Dvorak AM, Weller PF. Eosinophil lipid bodies: specific, inducible intracellular sites for enhanced eicosanoid formation. J Exp Med (1997) 186(6):909–20. doi:10.1084/jem.186.6.909
55. Bozza PT, Yu W, Cassara J, Weller PF. Pathways for eosinophil lipid body induction: differing signal transduction in cells from normal and hypereosinophilic subjects. J Leukoc Biol (1998) 64(4):563–9.
56. Bandeira-Melo C, Phoofolo M, Weller PF. Extranuclear lipid bodies, elicited by CCR3-mediated signaling pathways, are the sites of chemokine-enhanced leukotriene C4 production in eosinophils and basophils. J Biol Chem (2001) 276(25):22779–87. doi:10.1074/jbc.M101436200
57. Vieira-de-Abreu A, Assis EF, Gomes GS, Castro-Faria-Neto HC, Weller PF, Bandeira-Melo C, et al. Allergic challenge-elicited lipid bodies compartmentalize in vivo leukotriene C4 synthesis within eosinophils. Am J Respir Cell Mol Biol (2005) 33(3):254–61. doi:10.1165/rcmb.2005-0145OC
58. Pacheco P, Vieira-de-Abreu A, Gomes RN, Barbosa-Lima G, Wermelinger LB, Maya-Monteiro CM, et al. Monocyte chemoattractant protein-1/CC chemokine ligand 2 controls microtubule-driven biogenesis and leukotriene B4-synthesizing function of macrophage lipid bodies elicited by innate immune response. J Immunol (2007) 179(12):8500–8. doi:10.4049/jimmunol.179.12.8500
59. Bozza PT, Payne JL, Morham SG, Langenbach R, Smithies O, Weller PF. Leukocyte lipid body formation and eicosanoid generation: cyclooxygenase-independent inhibition by aspirin. Proc Natl Acad Sci U S A (1996) 93(20):11091–6. doi:10.1073/pnas.93.20.11091
60. Renz H, Gong J-H, Schmidt A, Nain M, Gemsa D. Release of tumor necrosis factor-alpha from macrophages. Enhancement and suppression are dose-dependently regulated by prostaglandin E2 and cyclic nucleotides. J Immunol (1988) 141(7):2388–93.
61. Betz M, Fox BS. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J Immunol (1991) 146(1):108–13.
62. Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol (2012) 188(1):21–8. doi:10.4049/jimmunol.1101029
63. Mattos KA, Oliveira VC, Berredo-Pinho M, Amaral JJ, Antunes LC, Melo RCN, et al. Mycobacterium leprae intracellular survival relies on cholesterol accumulation in infected macrophages: a potential target for new drugs for leprosy treatment. Cell Microbiol (2014) 16(6):797–815. doi:10.1111/cmi.12279
Keywords: infectious diseases, lipid droplets, inflammation, phagocytosis, lipid mediators, parasite survival, parasitophorous vacuole
Citation: Toledo DAM, D’Avila H and Melo RCN (2016) Host Lipid Bodies as Platforms for Intracellular Survival of Protozoan Parasites. Front. Immunol. 7:174. doi: 10.3389/fimmu.2016.00174
Received: 20 March 2016; Accepted: 19 April 2016;
Published: 03 May 2016
Edited by:
Alexandre Morrot, Federal University of Rio de Janeiro, BrazilReviewed by:
M. Victoria Delpino, National Scientific and Technical Research Council (CONICET), ArgentinaPatricia Talamás-Rohana, The Center for Research and Advanced Studies of the National Polytechnic Institute (CINVESTAV-IPN), Mexico
Copyright: © 2016 Toledo, D’Avila and Melo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rossana C. N. Melo, rossana.melo@ufjf.edu.br