 Agnieszka M. Kabat
Agnieszka M. Kabat Johanna Pott
Johanna Pott Kevin J. Maloy
Kevin J. Maloy- Sir William Dunn School of Pathology, University of Oxford, Oxford, UK
The gastrointestinal tract presents a unique challenge to the mucosal immune system, which has to constantly monitor the vast surface for the presence of pathogens, while at the same time maintaining tolerance to beneficial or innocuous antigens. In the intestinal mucosa, specialized innate and adaptive immune components participate in directing appropriate immune responses toward these diverse challenges. Recent studies provide compelling evidence that the process of autophagy influences several aspects of mucosal immune responses. Initially described as a “self-eating” survival pathway that enables nutrient recycling during starvation, autophagy has now been connected to multiple cellular responses, including several aspects of immunity. Initial links between autophagy and host immunity came from the observations that autophagy can target intracellular bacteria for degradation. However, subsequent studies indicated that autophagy plays a much broader role in immune responses, as it can impact antigen processing, thymic selection, lymphocyte homeostasis, and the regulation of immunoglobulin and cytokine secretion. In this review, we provide a comprehensive overview of mucosal immune cells and discuss how autophagy influences many aspects of their physiology and function. We focus on cell type-specific roles of autophagy in the gut, with a particular emphasis on the effects of autophagy on the intestinal T cell compartment. We also provide a perspective on how manipulation of autophagy may potentially be used to treat mucosal inflammatory disorders.
Introduction
The gastrointestinal tract contains a vast network of non-lymphoid and secondary lymphoid tissues that host numerous populations of leukocytes, many of which are intestine-specific subpopulations (1). The gut-associated lymphoid tissue (GALT) comprises the immune cells residing in the intestinal epithelium and lamina propria (LP) compartments, as well as various secondary lymphoid structures, including the mesenteric lymph nodes (mLNs), the Peyer’s patches (PP) of the small intestine, and isolated lymphoid follicles and cryptopatches that are distributed throughout the intestine (2). The intestinal mucosa, comprising the epithelium, the underlying LP, and the muscularis mucosa, is the site where majority of immunological processes occur (Figure 1).
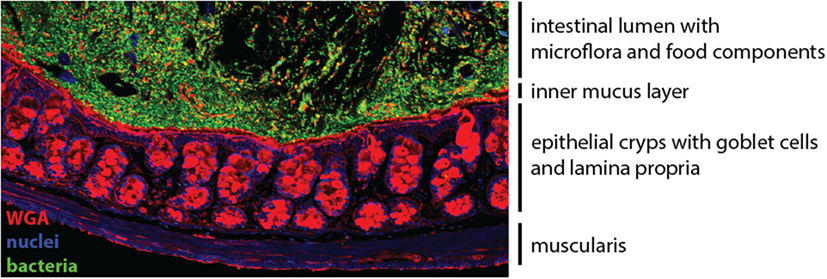
Figure 1. The colonic mucosa. The epithelial cell layer of the intestinal mucosa separates the luminal content harboring the microflora (green) from the underlying lamina propria. Specialized secretory epithelial cells, termed goblet cells, produce and secrete mucus to enforce the barrier (red). Cell nuclei are stained in blue.
The immune system has evolved to prevent invasion of the host by microbial species. However, the mammalian gastrointestinal tract is a preferential site for colonization of the host by commensals, a diverse community consisting of fungal, viral, and bacterial species. The presence of a commensal microbiota is vital for optimal digestion and nutrient acquisition, as well as resistance to pathogenic infection, but commensal microbes also contribute to the development, maturation, and activation of the host immune system by influencing both innate and adaptive immune responses (3). A mutualistic dialog between the microbiota and intestinal immune system maintains peaceful coexistence, through multiple mechanisms that we are just beginning to understand (3, 4). Besides microbial communities, the intestinal immune system constantly encounters a vast dietary antigenic load, and the induction of a state of immune unresponsiveness toward these antigens (oral tolerance) is key function of the mucosal immune system (5). Therefore, the intestinal immune system often employs different rules than the systemic immune system to ensure the right balance between tolerance and immunity is maintained. Disruption of this equilibrium can lead to the chronic immune-mediated pathologies of the gastrointestinal tract, such as inflammatory bowel disease (IBD) or food allergies (5, 6).
The term IBD describes a spectrum of chronic incurable inflammatory disorders affecting the gastrointestinal tract, but often with extra-intestinal manifestations, with the two most common forms being Crohn’s disease (CD) and ulcerative colitis (UC) (7). IBD is a complex multifactorial disease that emerges on a background of many genetic and environmental factors (6). In recent years, there has been a tremendous progress in understanding the genetics of IBD susceptibility, facilitated by technological progress that led to large-scale genome-wide association studies (GWAS), followed by meta-analysis and targeted genotype arrays (Immunochip) (8). Currently, 163 loci associated with IBD have been identified, which are far more than any other complex immunological disease to date (9). A considerable proportion of IBD-associated genes are involved in immune cell function, innate and adaptive immune responses, promotion of epithelial barrier integrity and bacterial handling (9). Among these, a single-nucleotide polymorphism (SNP) in ATG16L1 was identified as a strongly associated risk locus for CD, suggesting for the first time a role for the macroautophagy (herein referred to as autophagy) pathway in IBD (10, 11). Importantly, it was recently shown that homozygous expression of the CD susceptibility variant T300A allele of ATG16L1 results in defective autophagy during stress conditions (12, 13). Additionally, SNPs in several other autophagy-associated genes, including IRGM, LRRK2, SMURF1, and NDP52, have been linked to IBD susceptibility (9), strongly suggesting that it is the classical autophagy pathway that connects these genetic alterations to impaired intestinal homeostasis. However, the identification of susceptibility polymorphisms provides only correlative evidence for the involvement of specific genes or pathways, and an understanding of the functional consequences of the majority of genetic polymorphisms is still lacking. In the case of autophagy, however, we are beginning to understand how modulation of this pathway affects various aspects of mucosal immune cell physiology.
The Autophagy Pathway
Degradation and recycling of cellular components is critical for all eukaryotic cells in order to maintain cellular homeostasis. The autophagy pathway degrades large cytoplasmic components, including organelles, long-lived proteins, and protein aggregates, as well as intracellular pathogens, by sequestering these constituents in double-membrane vesicles and delivering this cargo for lysosomal degradation (14). Autophagy is an evolutionary conserved process occurring throughout the eukaryotic phylogenetic tree, with the core autophagy machinery proteins showing great homology between yeast and mammalian cells (15, 16), and its essential physiological role is indicated by the observation that mice lacking essential autophagy genes are unable to survive the neonatal starvation period and die shortly after birth (17).
During autophagy, sequestration of the cytosolic cargo involves “de novo” formation of an isolation membrane that surrounds the cytosolic material to be degraded, forming an intermediate vesicle called an autophagosome. The autophagosome subsequently fuses with the lysosome leading to the formation of the digestive compartment – the autolysosome. Lysosomal enzymes degrade the content of the vesicle, which facilitates the permease-mediated release of the recycled molecules via the lysosomal membrane (16, 18, 19) (Figure 2). While autophagy is the primary cell response to the stress of nutrient deprivation, in recent years, more complex and cell type-specific functions have emerged, including roles in innate and adaptive immune responses (20, 21).
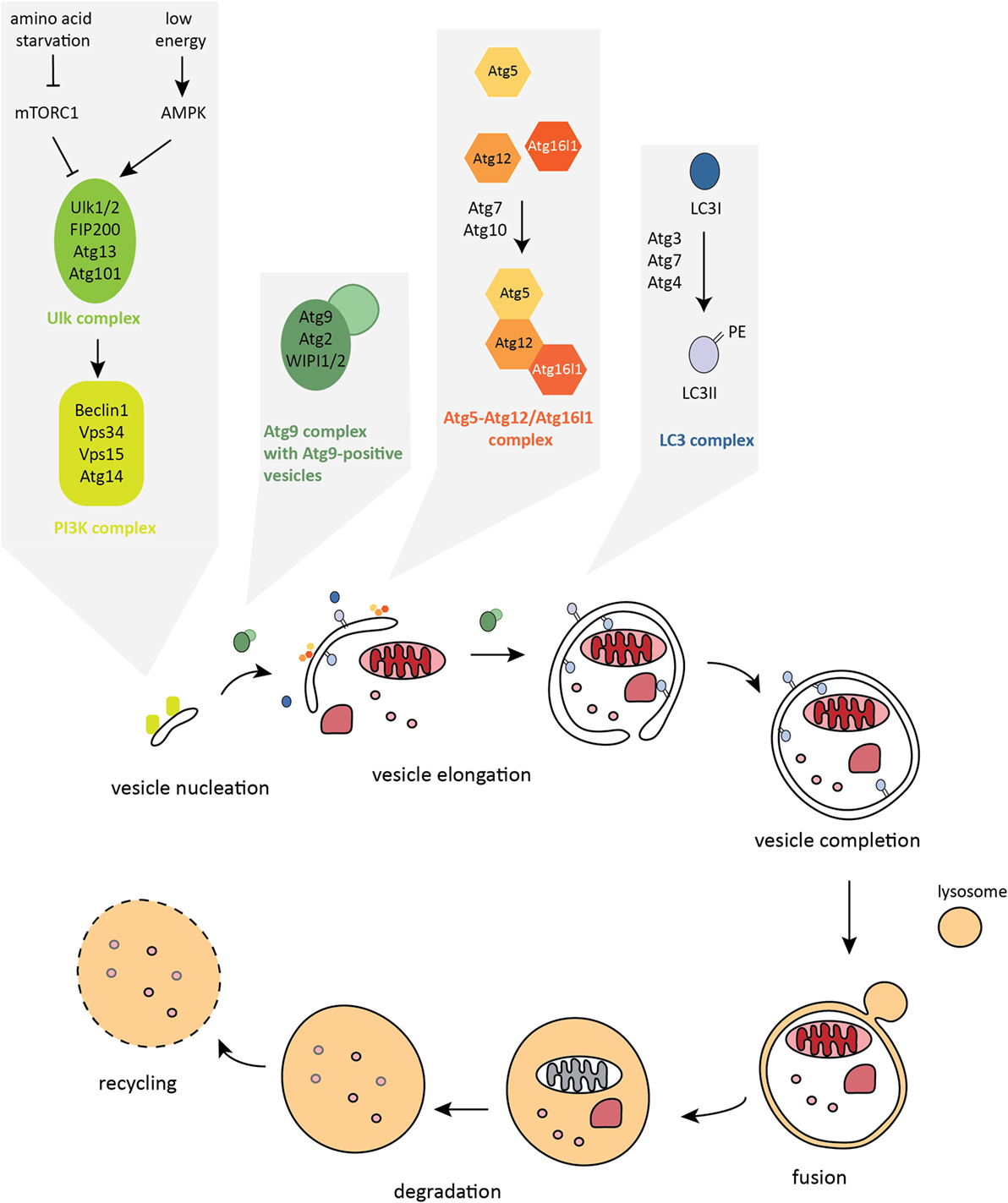
Figure 2. The autophagy pathway. Autophagosome formation is a stepwise process characterized by dynamic remodeling of cytoplasmic membranes. Proteins that control activation, elongation, and completion of an autophagosome are grouped into five functional complexes that are active at different stages of the autophagy pathway.
Although originally described as a non-selective pathway for bulk degradation, autophagy can also act as a highly selective process. While metabolic stress mainly triggers non-selective autophagy where a portion of the cytoplasm is targeted for degradation, intracellular pathogens or damaged organelles are targeted in a selective fashion. This is achieved by the use of cargo-specific autophagy adaptors. These adaptors are able to recognize ubiquitinated substrates and target them to the autophagosome, a process that also requires adaptor binding to the protein LC3 (microtubule-associated protein L chain 3) or γ-aminobutyric acid receptor-associated proteins (GABARAP), through a specific amino acid sequence called the LC3-interacting region (LIR) (22, 23). To date, there is evidence for the targeted sequestration and selective autophagy of a diverse array of cytosolic cargos, including aggregate-prone or misfolded proteins (aggrephagy) (22, 24–26), protein complexes in signaling cascades (27–29), peroxisomes (pexophagy) (30, 31), mitochondria (mitophagy) (32–38), surplus ER (reticulophagy) (39, 40), ribosomes (ribophagy) (40, 41), ferritin (ferritinophagy) (42, 43), bacteria and viruses (xenophagy) (21), lipid droplets (lipophagy) (44), and glycogen (glycophagy) (45, 46). The diversity of autophagy targets highlights the complex role of this pathway in regulating many aspects of cellular physiology during steady-state and in stress responses.
Molecular Mechanisms of Autophagy
Formation of the double-membrane autophagosome structure is the key step in autophagy. There are nearly 40 autophagy-related (Atg) proteins that facilitate crucial steps of autophagosome formation and degradation (47). Autophagy is initiated by the formation of the isolation membrane, also called a phagophore, at the phagophore assembly site (PAS). The core autophagy machinery in mammalian cells can be categorized into five functional groups (Figure 2). The primary initiation complex, comprising unc-51-like kinase-1 or 2 (Ulk1/2) – Atg13 – FIP200 – Atg101, is reciprocally controlled by mechanistic target of rapamycin complex 1 (mTORC1) and AMP-activated protein kinase (AMPK). Activated Ulk1/2 complex translocates to the site of autophagosome formation and activates the second functional complex, the class III phosphatidylinositol 3-kinase (PI3K) complex, whose main components are vacuolar protein sorting 34 (Vps34), Beclin1, autophagy/beclin-1 regulator 1 (AMBRA1), and Atg14 (48–51). The PI3K complex mediates the nucleation step of phagophore formation. Once activated, this complex produces phosphatidylinositol-3-phosphate (PI3P) leading to recruitment of PI3P-binding effector proteins to the phagophore membrane, including WD-repeat domain phosphatidylinositide-interacting-1 or -2 (WIPI1/2) (52–54). The third complex consists of Atg9, the only known transmembrane Atg protein, and its cycling system, involving Atg2 and WIPI1/2, that shuttle among endosomes, autophagosomes, and the Golgi apparatus. Atg9-positive vesicles are thought to provide membrane for the growing autophagosome (55, 56). Once initiated, elongation of the isolation membrane requires the subsequent action of two ubiquitin-like (UBL) conjugation systems: the Atg5–Atg12/Atg16l1 complex, which is assembled through the action of Atg7 (E1-like enzyme) and Atg10 (E2-like enzyme) and then locates to the growing autophagosome membrane (57–61); and the LC3 complex, containing LC3A/B/C, GABARAP, GABARAPL1/2, Atg7, Atg3 (E2-like enzyme), and the cysteine protease Atg4, which cleaves LC3 at the C-terminus to expose glycine, allowing the conjugation of the membrane lipid phosphatidylethanolamine (PE) to the soluble form of LC3, named LC3-I, and subsequent incorporation of LC3-PE (also called LC3-II) into the inner and outer membranes of autophagosomes (57, 62–64). The Atg5–Atg12/Atg16l1 complex acts as an E3-like enzyme during LC3 lipidation (58, 65). LC3-II that is bound to the isolation membrane is thought to play not only a scaffolding role in membrane growth and is needed for autophagosome closure but is also important for the binding of autophagy adaptors and thus in mediating selective types of autophagy (66). The mature autophagosome migrates into close proximity to the lysosome through the action of the dynein motor complex and microtubules (67). Fusion with the lysosome requires the action of the HOPS complex (homotypic fusion and protein sorting) and SNARE proteins (soluble NSF attachment protein receptors) (68–71). Upon formation of the autolysosome compartment, lysosomal acidic hydrolases degrade the inner membrane and the luminal content. Ultimately, this provides building blocks for anabolic processes and fuel for ATP synthesis. Eventually, the autolysosome fissions to release lysosomes and autophagy are terminated (72).
Cytoplasmic and Nuclear Regulation of Autophagy
Autophagy is modulated in response to adverse micronenvironmental conditions, including nutrient depletion, hypoxia, growth factor withdrawal, inflammatory cytokines, and infection. As such, a network of regulatory pathways governs its activity. For a long time, autophagy was believed to be predominantly regulated at the post-transcriptional level by signaling mediators. Recently, however, mechanisms of transcriptional, translational, and epigenetic regulation of the autophagy pathway have emerged. It is now thought that cytosolic regulation generally serves as a more rapid, short-term response, whereas transcriptional modulation provides long-term regulation, although some nuclear events can also have a rapid effect on autophagy (72–74).
The primary role of autophagy is to respond to cellular metabolic perturbations. Many signals that modulate autophagy levels do so by converging on the mechanistic target of rapamycin complex 1 (mTORC1). mTOR is a conserved serine/threonine kinase that integrates signals from various stimuli, including amino acids, growth factors, energy, glucose, and oxygen levels (75). The autophagy pathway is regulated by mTORC1 in several ways. In the presence of nutrients, including amino acids, mTORC1 is active and suppresses autophagy through inhibitory phosphorylation of Ulk1, Atg13, AMBRA, and Atg14 (51, 76–80). During starvation, mTORC1 is inhibited, which activates autophagy. Autophagy activity eventually leads to increased nutrient levels, which in turn reactivates mTORC1 to terminate autophagy. This reactivation of mTORC1 that occurs during prolonged starvation is critical for the restoration of lysosomal homeostasis after prolonged autophagy (81). Another key sensor that coordinates cellular metabolic responses is AMPK, which, next to mTOR, can be considered a signaling hub for autophagy modulation. AMPK is a serine/threonine kinase that senses decreased energy levels by detecting changes in the ATP:ADP:AMP ratio in the cytoplasm (82). In response to decreased intracellular ATP levels, AMPK initiates metabolic reprograming toward catabolic reactions, including stimulation of autophagy through direct phosphorylation of the Ulk1/2 (76, 83, 84). AMPK can also phosphorylate various components of the Vps34 complexes that do not contain proautophagic adaptors, leading to the inhibition of its non-autophagic functions in Golgi–endosome trafficking (85). In addition to direct interactions, AMPK can indirectly activate autophagy by inhibiting mTORC1 (86, 87). Importantly though, AMPK activity is not indispensable for autophagy induction, as starvation still induces autophagy in AMPK-null cells, suggesting a role for this kinase in fine-tuning autophagy modulation (76).
Transcriptional regulation is now appreciated to be one of the main regulatory mechanisms of autophagy. The transcription factor EB (TFEB) is a master regulator of lysosomal and autophagy gene expression (88). TFEB controls the gene network called coordinated lysosomal expression and regulation (CLEAR), which contains the majority of genes encoding lysosomal proteins (89). TFEB also regulates the expression of several genes encoding proteins belonging to the core autophagy machinery, including Lc3, p62, and Atg9 (73, 88). Upon autophagy induction, TFEB is rapidly recruited from the cytosol to the nucleus, and this is at least partially mediated by inhibition of mTORC1. Active mTORC1 mediates the phosphorylation of TFEB, which results in its sequestration in the cytoplasm (90–92). Acting in opposition to TFEB, the DNA-binding protein zinc-finger protein with KRAB and SCAN domains 3 (ZKSCAN3) represses an extensive set of autophagy genes, including Lc3 and WIPI2 (93). During starvation, ZKSCAN3 accumulates in the cytoplasm, and its activity is inhibited. Thus, TFEB and ZKSCAN3 seem to provide a switch mechanism during starvation-induced autophagy (73). In summary, multiple intersecting pathways modulate autophagy on many levels, reflecting the importance of this complex homeostatic pathway in the cellular adaptations to environmental factors.
Autophagy Has Diverse Roles in Cells of the Intestinal Mucosa
Intestinal Epithelial Cells in Barrier Function and Immune Homeostasis
Intestinal epithelial cells (IECs) form a single cell layer separating the intestinal mucosa from the lumen. The primary function of these cells is nutrient absorption from the lumen; however, their interactions with the intestinal microbiota and host leukocytes strongly influence immune responses (94). The IEC monolayer is composed of several specialized cell types: stem cells, Paneth cells, goblet cells, neuroendocrine cells, and enteroabsorptive cells (95). Multipotent Lgr5+ stem cells are located at the bottom of the intestinal crypts and by division these cells give rise to either transient amplifying cells or stem cells. The transient amplifying cells rapidly proliferate and differentiate and thereby ensure the renewal of the epithelial layer every 4–5 days (95). Paneth cells that localize to the base of small intestinal crypts are specialized secretory cells that produce large amounts of antimicrobial molecules, including lysozyme, α-defensins, and Reg3γ (Reg3α in humans) (96). Antimicrobial peptides (AMP) play a crucial role not only in the defense against enteric pathogens but also in shaping the host microbiota, as mice lacking MMP7, an enzyme required for the maturation of α-defensins, exhibited significant changes in the microbiota composition (97). In addition, AMP have modulatory functions in chemotaxis, toll-like receptors (TLR) signaling, and wound healing (98). Paneth cells also participate in maintaining crypt stem cell activity through production of EGF, TGF-α, Wnt3, and the Notch ligand Dll4 (99). Goblet cells are another class of secretory cells that produce heavily glycosylated mucins which, after secretion to the lumen, form a mucus gel layer (94). This serves as a protective physical barrier and as a matrix loaded with secretory IgA (sIgA) and AMP, which fortify the mucosal barrier (4). Recent studies have suggested that mucus also has an additional role in promoting tolerogenic responses toward food and commensal antigens (100, 101). The observation that mice with defects in MUC2 production develop spontaneous colitis emphasizes that mucus is essential for intestinal homeostasis (102, 103). IECs are actively engaged in the dialog between the microbiota and the immune system. Sensing of bacterial metabolites and structural components by IECs fortifies barrier integrity and protects from pathogen invasion (104). For example, recent studies underlined the crucial role of inflammasome signaling in the epithelium in regulating microbiota composition and for protection against infectious colitis (105–107). In addition, the metabolite acetate, produced by commensal bacteria belonging to the genus Bifidobacterium, protected against mortality during enterohemorrhagic Escherichia coli infection by promoting anti-apoptotic responses in IECs (108). IECs also influence the recruitment, activation and differentiation of leukocytes by producing various other modulatory factors in response to commensal microbiota, including thymic stromal lymphopoietin (TSLP), TGF-β1, RA, IL-25, and IL-18 (4, 109).
Autophagy Reinforces Barrier and Secretory Functions of IECs
The IEC monolayer is in close proximity to microbiota communities within the gastrointestinal tract and is an entry site for mucosal pathogens. Recent studies that assessed the impact of autophagy deficiency on bacterial handling by IECs found that autophagy was essential for protection against intracellular bacteria, including Salmonella typhimurium and Shigella flexneri, by acting to limit bacterial replication and subsequent dissemination to other tissues (13, 110–112) (Figure 3). Furthermore, a recent study showed that autophagy induction reduced tight junction permeability in IECs by targeting the pore forming protein claudin-2 for degradation (113), highlighting an additional mechanism through which autophagy may enhance barrier function.
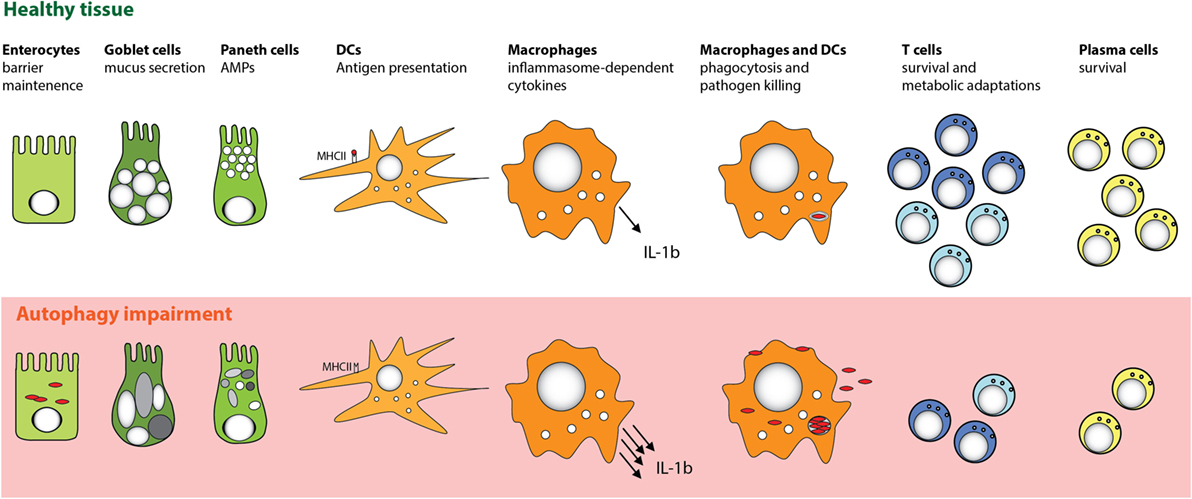
Figure 3. Cell-specific functions of autophagy in the intestinal mucosa. Autophagy pathway is essential for several key functions of distinct cell types that promote intestinal immune homeostasis. Perturbation in the autophagy pathway results in decreased antibacterial defense in IECs and MPs. Autophagy also facilitates secretory functions of Paneth cells and goblet cells, is involved in antigen presentation by DC, and limits proinflammatory cytokine production from MP. Furthermore, defects in autophagy pathway strongly compromise the survival of particular subsets of T cells and B cells.
Many studies of autophagy in IECs have concentrated on the functional role of ATG16L1, and in trying to understand how CD-associated polymorphisms in ATG16L1 may impact on epithelial homeostasis. It has been reported that Paneth cells from patients with CD homozygous for the ATG16L1 T300A variant allele, or from mice with a hypomorphic mutation of Atg16l1, exhibit abnormal granule structure and reduced AMP secretion (114, 115) (Figure 3). However, Atg16l1 hypomorphic mice did not exhibit signs of spontaneous intestinal inflammation, although they showed increased susceptibility to DSS-induced colitis. This increased susceptibility of Atg16l1 hypomorphic mice, as well as their Paneth cell abnormalities, was only observed when the mice harbored a commensal microbiota that contained a persistent enteric norovirus (MNV) (114, 115). These studies suggested that decreased autophagy levels drive Paneth cell abnormalities only when additional triggering factors are present. A recent study showed that ER stress could also be such a trigger (116). Analyses of mice with an IEC-specific deletion of the unfolded protein response (UPR) response element Xbp1 demonstrated that defects in the UPR pathway in Paneth cells were partially compensated by increased autophagy. However, when autophagy was also impaired, through IEC-specific deletion of Atg16l1 or Atg7, ER stress could not be resolved and this double defect led to the development of severe intestinal inflammation (116). Interestingly, Paneth cells from patients with the ATG16L1 T300A variant allele showed increased ER stress markers (117).
Consistent with the notion that manifestation of defective autophagy in Paneth cells could depend on additional environmental or genetic factors, two recent studies reported contradictory results when mice with a “knock-in” of the risk-associated Atg16l1 gene variant (T316A) were analyzed. Murthy et al. reported no changes in the morphology of Paneth cells in the knock-in mice (12), whereas Lassen et al. observed spontaneous Paneth cell abnormalities (13). One possible explanation for these differences could be the distinct microbiota composition of mice housed in different facilities. Beyond Paneth cells, autophagy could also play an important functional role in other secretory IEC types, as Atg5-deficient colonic goblet cells were also reported to show impaired granule formation (118). Taken together, these studies indicate that autophagy plays an important role in fortifying intestinal epithelial barrier function by enhancing resistance to intracellular bacteria and by regulating the functions of secretory IECs (Figure 3).
Mononuclear Phagocytes Regulate Intestinal Immune Homeostasis
Mucosal mononuclear phagocytes (MPs) comprise dendritic cells (DCs) and macrophages. These cells are key players in intestinal homeostasis, as they provide a crucial link between innate and adaptive immunity, and in maintaining functional compartmentalization of the systemic and mucosal immune system. Intestinal MPs are a heterogeneous population; expression of CX3C chemokine receptor 1 (CX3CR1) and CD103 (αE integrin) can be used to identify two major intestinal MP populations, which appear to promote intestinal tolerance in different ways (119, 120). Under homeostatic conditions, CD103+ DCs acquire intestinal antigens and migrate from the intestinal LP to the mLNs, where they initiate T cell responses, promoting intestinal tropism through induction of homing receptors CCR9 and α4β7, and preferentially inducing tolerogenic Treg cells through production of TGF-β1 and RA (121–123). The ability to convert latent TGF-β1 into its active form is important for this tolerogenic CD103+ DC function (124, 125). Additionally, this CD103+ DC subset also acts on B cells, promoting differentiation of naive B cells into IgA+ plasma cells within the intestinal LP (126). CD103+ DCs have also been identified as a crucial subset in promoting oral tolerance against food antigens (5), although CX3CR1+ MPs may augment Treg cell induction by CD103+ DCs through the transfer of soluble food antigens via gap junctions (127). CX3CR1+ MPs (a population comprising both DCs and macrophages) sample luminal contents through extended dendrites but appear to be non-migratory and have poor abilities for naive T cell priming (128–130). However, CX3CR1+ MPs may be involved in the secondary expansion of Tregs in the LP that had been primed initially in gut-draining lymph nodes (131).
During infection or inflammation, intestinal MPs adopt a different phenotype, characterized by the production of proinflammatory cytokines and chemokines that coordinate host protective immune responses, but excessive activation of mucosal myeloid cells has also been associated with chronic inflammatory conditions (132–134). Commensal bacteria are able to directly modulate intestinal MP functions to regulate effector T cell responses in the LP. For instance, Atarashi et al. showed that ATP produced by commensal bacteria activates CX3CR1+ MPs and leads to the induction of Th17 cells (135), and microbiota-derived signals were shown to induce IL-1β production from mucosal MPs that is essential for the induction of Th17 cells in the steady-state gut (136).
Autophagy Regulates Proinflammatory Signaling in Mononuclear Phagocytes
Antigen presentation by intestinal MP is crucial in orchestrating protective and tolerogenic responses in the mucosa. The role of autophagy in major histocompatibility complex class II (MHC II) antigen presentation is well documented; autophagy can enhance MHC II expression on MPs and is directly engaged in delivering cytoplasmic antigens, including bacterial and viral antigens, into MHC II compartments (137–140) (Figure 3). There is also evidence that autophagy may contribute to processing of viral antigens for MHC I presentation (141, 142).
As professional phagocytic cells, DCs and macrophages are particularly well equipped to handle bacteria, and autophagy is now appreciated to play an important role in intracellular bacterial killing (Figure 3). Autophagy can be activated in MPs by pattern recognition receptor (PRR) triggering, including activation of TLR (143–147) or NOD-like receptors (NLR) (112, 148). In particular, activation of DCs and macrophages with the NOD2 ligand muramyl di-peptide (MDP) induces autophagy and thereby enhanced bacterial killing, and this activation is reduced in DCs with the ATG16L1 T300A allele variant (112, 148). Activation of NOD1 and NOD2 in the cytoplasm directs the autophagy machinery by recruiting ATG16L1 to the site of bacterial entry, although this interaction was not affected in cells with homozygous expression of the T300A variant (112). Of note, autophagy induction downstream of PRRs is not limited to myeloid cells, as autophagy is also activated in epithelial cells in response to NOD1 and NOD2 triggering (112), and bacterial outer membrane vesicles (OMV) were shown to selectively activate NOD1-dependent autophagy in an epithelial cell line (149).
Sequestration of cytosolic bacteria by xenophagy requires the coordinated action of specialized autophagy adaptors that recognize ubiquitin-tagged or galectin-tagged pathogens for degradation (150). The importance of autophagy in defense against cytosolic pathogens is highlighted by the fact that several pathogens have developed sophisticated adaptations to inhibit specific stages of autophagy (21, 151) and others even hijack the pathway for their own propagation (152). An essential role for autophagy in intestinal pathogen handling in DCs and macrophages has been observed in Salmonella, Shigella, and Listeria infections (153, 154).
In addition to PRR triggering, inflammatory cytokines can also influence autophagy in MPs. For instance, autophagy is induced by IFN-γ and other Th1 type cytokines that are secreted during bacterial infection, while Th2 type cytokines inhibit autophagy (21). Conversely, autophagy can also influence cytokine signaling in myeloid cells. In particular, autophagy was shown to downregulate secretion of the inflammasome-associated cytokines IL-1β and IL-18 by murine and human macrophages (155–157) (Figure 3). In addition, IL-1β produced by activated autophagy-deficient macrophages can enhance their secretion of IL-23, thereby further potentiating inflammatory responses (157). The link between autophagy defects and excessive inflammasome activation was related to reactive oxygen species (ROS) production in response to mitochondrial stress (158–160), as well as defects in targeting of assembled inflammasomes, or components of the inflammasome pathway, for autophagosomal degradation (27, 161) (Figure 4). Recently, NFκB signaling was linked to autophagy-mediated silencing of inflammasomes in macrophages, as NFκB promoted the expression of the adaptor p62 that was needed for the removal of damaged mitochondria (162). In the absence of p62, signals from damaged mitochondria enhanced NLRP3 inflammasome activation (162). Consistent with these findings that autophagy functions as a key regulator of inflammasomes, in bone marrow chimeric mice with an Atg16l1-deficient hematopoietic compartment, increased production of inflammasome-dependent cytokines was associated with increased susceptibility to DSS-induced colitis (155). Furthermore, a recent study suggested that protective autophagy in MPs might be triggered through the activation of GCN2, a nutrient deprivation sensor kinase that was previously shown to facilitate antigen presentation by DCs through autophagy induction (163, 164). GCN2 promoted autophagy in intestinal MP during amino acid starvation or acute inflammation and this acted to limit ROS production and consequent inflammasome activation and thus had a protective effect on DSS colitis (164).
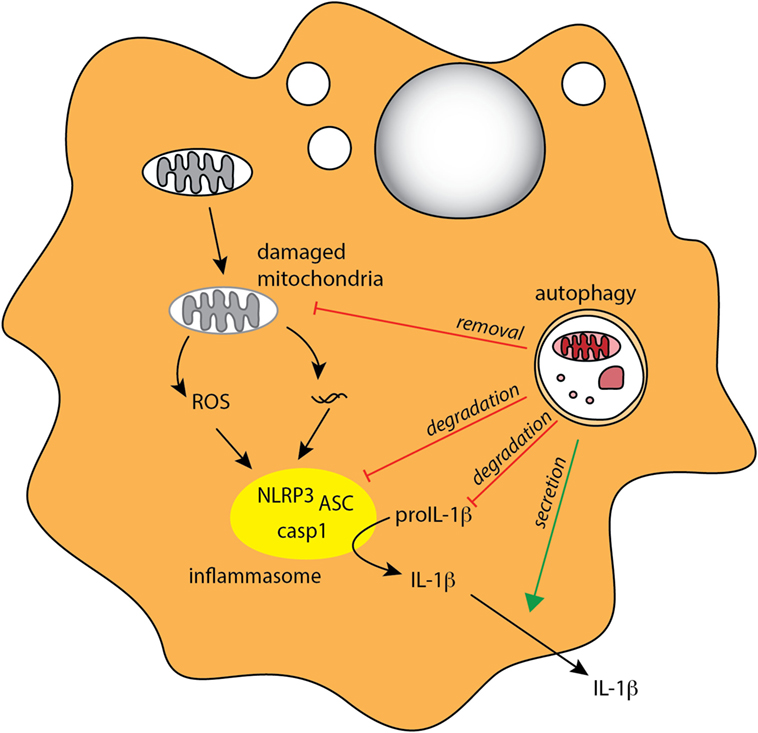
Figure 4. Autophagy regulates inflammasome-associated cytokine responses. Autophagy negatively regulates production of proinflammatory IL-1β in MPs through several mechanisms, indirectly through degradation of damaged mitochondria (mitophagy) to limit ROS production, and directly through degradation of inflammasome complexes and pro-IL-1β. Autophagy may also positively regulate IL-1β by mediating its unconventional extracellular secretion.
Although autophagy appears to negatively regulate inflammasome pathway at several stages, there is some evidence that, conversely, it might contribute to the secretion of IL-1β and IL-18 under some circumstances (165–167). IL-1β and IL-18 do not contain signal peptide and thus cannot access the classical secretory pathway; however, the mechanism of their unconventional secretion remains debatable. For instance, starvation-induced autophagy leads to increased IL-1β and IL-18 secretion in macrophages following inflammasome triggering, and this response was partially autophagy dependent (165). More recently, Zhang et al. proposed that autophagy-mediated secretion of IL-1β is mediated through sequestration of a fraction of this cytokine in the space between the membranes of the autophagosome, where it is protected from degradation (167).
Overall, these studies indicate that autophagy can both negatively and positively regulate inflammasome signaling (Figure 4) and suggest that this may depend on the timing and context of autophagy and inflammasome induction. It will now be important to fully understand what immunological clues dictate the location, and thus the fate of IL-1β and IL-18 within autophagosomes, and to investigate whether the tissue location of the MPs has an influence on how autophagy regulates inflammasome signaling.
Besides the crosstalk between autophagy and inflammasome signaling, autophagy also regulates type I IFN responses. In plasmacytoid DCs, autophagy was essential for IFN-α production in response to viral dsRNA, and this was attributed to the role of autophagy in delivering viral replication intermediates to endosomal TLR7 (147). Conversely, in some instances, autophagy appears to negatively regulate virus-sensing pathways by limiting signaling through RIG-1-like receptors (RLR) (168, 169). Additionally, autophagy was shown to enhance NFκB signaling in F4/80hi macrophages by selective degradation of the negative regulator A20 and this contributed to enhanced protection against Candida albicans infection (170). This is interesting, as autophagy was previously shown to limit NFκB signaling in intestinal epithelial cells (171) and activated T cells through selective degradation of Bcl-10 (29), suggesting that the impact of autophagy on NFκB activation could be cell type specific or context dependent. Additionally, it was recently demonstrated that autophagy-deficient macrophages show increased secretion of macrophage migration inhibitory factor (MIF) (172), a pleiotropic proinflammatory cytokine implicated in the pathology of IBD (173, 174), and that this increase was dependent on the mitochondrial ROS (172). Taken together, these studies indicate that the autophagy pathway intersects with other pathogen sensing and cellular stress responses to promote immune defense in MP populations. However, detailed analysis of the role of autophagy in the intestinal subsets of DCs and macrophages is lacking. Since mucosal MPs show some unique features, including hyporesponsiveness to PRR stimulation (134), it will be important to investigate the potential contribution of autophagy to these specific intestinal adaptations.
Antibody Production and Secretion in the Intestine
B cells are abundant within the GALT. Intestinal B cell development shows some unique features as LP-resident B cells appear to undergo V(D)J recombination and B cell receptor (BCR) editing (175). Early B cell development in the gut was promoted by commensals, suggesting involvement of microbiota-derived antigens in driving BCR editing (175), which might have implications for immunoglobulin diversification at mucosal sites and for tolerance against commensal antigens. Intestinal plasma cells can also acquire unique “innate like” properties that are dependent on microbiota stimulation, as IgA+ plasma cells can secrete TNFα and inducible nitric oxide synthase (iNOS) (176).
It is estimated that around 80% of the total antibody production takes place in the intestinal mucosa, making the gut the largest antibody-producing organ of the body (177). IgA constitutes the major antibody isotype produced and sIgA is the most abundant immunoglobulin in mucosal secretions (178). sIgA is a dimeric antibody that binds to the polymeric Ig receptor (pIgR) on the basolateral surface of IECs and is subsequently translocated across the epithelium and released into the lumen, where it interacts with various intestinal antigens, including self, dietary, and commensal antigens. This limits the access of commensal bacteria and soluble antigens to the intestinal epithelium and LP, and it appears that bacteria with higher potential to elicit local inflammatory responses can be distinguished on the basis of their high IgA coating (179, 180). Class switch recombination to IgA occurs mainly in the mLNs and PP through both T cell-dependent and T cell-independent mechanisms, and studies in GF mice established that commensal microbiota is strong inducers of IgA production (178, 179, 181). In the T cell-independent pathway, microbiota induces production of IgA through the modulation of IECs and MPs, which in turn secrete BAFF (B cell-activating factor), APRIL (a proliferation-inducing ligand), and TGF-β1, cytokines that promote IgA switching (178). Production of IgA in response to microbiota appears very flexible, as IgA specificity can rapidly change and adapt in response to alterations in microbiota composition (182, 183).
Pentameric IgM antibodies are also actively secreted into the intestinal lumen via the pIgR, have a similar function in shielding IECs from antigenic exposure, and are particularly important in newborns (177). There is also evidence that active transport across the epithelial layer takes place for IgG and IgE. In case of IgG antibodies, the neonatal Fc receptor (FcRn) is responsible for transcytosis across the IECs, and moreover, this transport is bidirectional, as IgG are able to bind antigen in the lumen and immune complexes may then be retrieved and released into LP, where they are proposed to provide antigen for DC sampling (184, 185). Mucosal sites, including the intestine, support isotope switching to IgE and indeed IgE is relatively abundant in the intestine (186, 187), with increased production being described in patients with food allergies (188, 189). IgE transcytosis across the intestinal epithelial barrier involves the low-affinity IgE-specific receptor CD23/FcRII, and, similar to IgG, also appears to be bidirectional, potentially resulting in antigen retrieval from the lumen to the intestinal LP (190–192). Active transport of IgE across the IECs to the lumen might have a particularly important role during helminth infections, as the concentration of IgE in the lumen after parasitic infection rapidly increases (193). However, the transport of antigen complexes from the lumen is thought to facilitate the rapid intestinal physiological changes that occur during allergic reactions to food antigens (187). Recent data demonstrated that IgE in the gut acts beyond driving immediate hypersensitivity reactions and mediates long-lasting immunomodulatory functions by enhancing the induction of proallergic Th2 cells and inhibiting Treg cell induction (194).
Autophagy Regulates Plasma Cell Responses
In contrast to what has been described for T lymphocytes (see below), autophagy seems largely dispensable for the development and maintenance of mature B lymphocytes in the periphery. Studies using mice with B cell-specific deletion of Atg5 or Atg7 (Cre expressed under the control of the CD19 promoter: Atg5ΔCD19 or Atg7ΔCD19 mice) indicated that the numbers of mature B cells and the ratios of marginal-zone B cells to follicular B cells are not affected when autophagy is lacking (195–197). Interestingly, while B-2 and B-1b populations were not affected by Atg5 or Atg7 deficiency, the B-1a B cell population in the peritoneal cavity was markedly reduced. It remains unclear why the development of this population of peripheral B-1a cells is uniquely sensitive to autophagy deficiency (195–197).
However, studies on antibody responses, plasma cell and memory B cell formation provided evidence of a role for autophagy in regulating these processes (Figure 3). After examining the capacity of autophagy-deficient B cells to produce immunoglobulins, Conway et al. reported decreased primary antibody responses to antigen immunization or following infection with Heligmosomoides polygyrus, along with defective plasma cell differentiation (196). However, in contrast, a recent report observed no defects in primary antibody responses from autophagy-deficient B cells after antigen immunization (197). Moreover, an extensive analysis of autophagy-deficient plasma cells revealed that although autophagy did not affect differentiation or proliferation of plasma cells, it was necessary to promote their long-term survival (195). Surprisingly, immunoglobulin production was, in fact, increased in Atg5-deficient plasma cells, a phenomenon attributed to a dysregulated ER stress pathway. Signs of elevated ER stress were observed in autophagy-deficient plasma cells, leading to increased Blimp-1 expression, which in turn resulted in increased IgH expression and immunoglobulin production (195). However, after in vivo challenge with a pneumococcal vaccine, antibody levels were reduced, as a result of decreased survival of autophagy-deficient plasma cells (195). As the choice of the antigen and adjuvant for immunization had a significant impact on the in vivo antibody responses in Atg5ΔCD19 mice, it is plausible that some of the discrepant observations were due to the different immunization regimes (195). It is noteworthy that a requirement for autophagy appears to be shared by distinct types of professional secretory cells with high protein synthesis, as Paneth cells, goblet cells, and plasma cells are particularly sensitive to perturbations in autophagy and ER stress pathways.
Recent studies on B cell responses after influenza virus infection showed that although autophagy was dispensable for initial memory B cell formation, the survival of memory B cells and secondary antibody responses after re-challenge were heavily dependent on autophagy, and this defect could be partially rescued by treatment with a ROS scavenger (197, 198). These results parallel studies on memory CD8+ T cell responses during viral infections where autophagy was also implicated in the late stages of memory cell formation (199, 200).
Overall, it appears that autophagy, while largely dispensable for mature B lymphocyte development, is necessary to maintain secondary, long-lasting antibody responses. However, one study reported decreased numbers of B cells in the intestinal LP and PP in Atg5ΔCD19 mice, indicating that intestinal B cells might have a higher dependence on autophagy compared with other peripheral B cells, although this has not been further investigated (196). Taking into account the marked differences displayed by the intestinal B cell compartment and the high constitutive demand for local antibody production in the intestine, it would be of interest to address the potential role of autophagy in homeostasis of mucosal B cells and plasma cells.
Autophagy Regulates T Cell Responses on Several Levels
Autophagy Affects Thymic Selection of T Cells
Generation of T cells occurs in the thymus and requires continuous trafficking of bone marrow-derived lymphoid progenitor cells (201). Thymic selection acts to generate T cells that have self-MHC restricted TCR and do not display pathological self-reactivity. Thymocytes with the capacity of interacting with self-peptides presented on MHC I and MHC II molecules expressed on thymic cortical epithelial cells (cTEC) are positively selected toward the CD8 and CD4 lineages, respectively, whereas thymocytes that fail to proceed through positive selection die by neglect (202). Negative selection then ensures deletion of thymocytes with potentially pathogenic specificity. During negative selection, thymocytes cease TCR rearrangement and migrate to the medulla where they interact with peptides expressed by thymic medullary epithelial cells (mTEC) and thymic antigen-presenting cells (APCs) (203). Intermediate affinity to self-peptide–MHC complexes promotes survival, whereas high affinity leads to the removal of such self-reactive clones by induction of apoptosis (clonal deletion).
Interestingly, autophagy has been implicated in shaping the thymic repertoire. Presentation of certain self-antigens on cTEC was promoted by autophagy, implying a role in positive selection (204). During negative selection, it appears that autophagy in mTEC is dispensable for abundant antigens, as it can be compensated by presentation by thymic APCs, but may be more important when antigen is present at lower doses (205). However, the physiological relevance of autophagy-associated antigen presentation during negative selection remains controversial, as autoimmunity development depended on the experimental model used; athymic mice which received an Atg5-deficient thymus developed autoimmunity (204), but this was not the case in mice where Atg7 was selectively deleted in thymic epithelium using a Cre-loxP approach (206). It is worth mentioning that several unconventional mucosal T cell subsets, such as invariant natural killer T (iNKT) cells, and CD8αα intraepithelial lymphocytes (IELs) undergo alternative thymic selection (207), and the impact of autophagy in the thymic epithelium on their selection is yet to be investigated.
The Intestinal Mucosa Has a Unique T Cell Composition
The intestinal LP and epithelium together contain the largest population of T lymphocytes in the body (208). Gut T cells are highly heterogeneous, and many populations are unique to the mucosa. T cells found at the basement membrane between enterocytes are classified as IELs, which are particularly abundant in the small intestine. Two major subtypes can be distinguished in mice: conventional IELs, which express CD4 or the CD8αβ heterodimer as well as an αβTCR, and unconventional IELs, which express the CD8αα homodimer and either a γδTCR or an αβTCR (208). Overall, γδ T cells constitute a large proportion of IELs (approximately 60%), while CD4+ T cells are underrepresented (209). Intestinal IELs not only regulate epithelial growth and homeostasis, for example, through secretion of TGF-β1 (209) but are also essential in protection against pathogens, as γδ T cells are an important source of IL-17A (210).
The intestinal LP harbors a significant population of CD4+ T cells, which predominantly express TCRαβ. Conventional CD8+ T cells are also present, although in lower frequencies, and these give rise to effector cytotoxic T lymphocytes (CTLs) that combat intracellular pathogens (211–213). In addition, small subsets of T cells that express an invariant TCR are also present in the intestinal mucosa (208). These subsets include mucosal-associated invariant T (MAIT) cells and iNKT cells. MAIT cells express a semi-invariant TCR that recognizes bacteria-derived vitamin B metabolites presented by the MHC class I-related protein (MR1) (214). MAIT cells contribute to protection against enteric bacteria, as they rapidly produce cytokines and exert cytolytic activity upon activation (214). iNKT cells, which in mice constitute approximately 0.5% of small intestinal LP lymphocytes, express an invariant form of the TCRαβ that is able to recognize lipid antigens presented by the CD1d molecule (215). As well as cytolytic activity, iNKT cells can also rapidly produce a spectrum of effector cytokines at an early stage in immune responses, including IFN-γ, IL-4, IL-10, IL-13, and IL-17A, allowing these cells to participate in a range of immune responses, including antimicrobial defense (215).
Commensal microbiota modulates the function of IELs for example, GF mice (or mice treated with antibiotics) have reduced numbers IL-17 producing γδ T cells (216). In addition, the antimicrobial response of γδ T cells can be triggered by a distinct subset of commensal bacteria following penetration of the epithelial barrier (217). The influence of commensals also extends to iNKT cells, as exposure to commensal microbiota during the neonatal period limits the accumulation of iNKT cells at mucosal sites, which otherwise can have detrimental effects in adult animals, such as increased susceptibility to asthma (218). Mechanistically, the inhibition of colonic iNKT cell development by commensal microbiota is at least in part mediated by the interaction of iNKT cells with commensal-derived inhibitory sphingolipids (219).
Naive T cells are maintained in a metabolic state that favors energy production over biosynthesis and rely on mitochondrial oxidative pathways for maximal energy generation, fueled predominantly by lipid and amino acid oxidation (220). Within the intestinal mucosa, the majority of the T cells display an activated/memory phenotype (211). Activation of T cells initiates multiple changes in their transcriptional and translational program, which go hand in hand with dynamic metabolic changes, matching bioenergetic and biosynthetic demands. Activation of T cells drives a rapid proliferative response, which drastically increases the demand for energy and building blocks for biosynthesis (220). During the initial growth phase, lipid oxidation is downregulated, and glycolytic, pentose phosphate, and glutaminolytic pathways increase (221). This initial metabolic shift is orchestrated by the transcription factors c-Myc, hypoxia-inducible factor-1α (HIF-1α), and the nuclear receptor estrogen-related receptor α (ERRα), leading to an increase in amino acids, nucleic acids, and lipid synthesis (222–224). Toward the end of an immune response, a small proportion of the T cells differentiate into memory T cells and revert back to lipid oxidation, maintaining increased capacity for efficient energy generation (224).
Memory T cells are formed from both CD4+ and CD8+ effector T cells, and it is now appreciated that this long-term immunity is provided by several distinct subsets of memory cells that can be distinguish, based on their location and effector functions, into central memory T cells, effector memory T cells, and tissue-resident memory T cells (TRM) (225). Mucosal tissues are enriched in TRM; in contrast to other memory cells TRM do not re-enter the circulation and are retained in the mucosa where they provide a rapid protection during a secondary local infection (226).
T Helper Cell Populations in the Gut
Upon activation, CD4+ T cells differentiate into subsets of T helper cells, which have traditionally been classified according to the expression of the lineage-specifying transcription factors (so-called master transcriptional regulators) and effector cytokine profiles. As such, Th1, Th2, Th17, and T follicular helper (Tfh) effector cells can be distinguished. Metabolic signals and the surrounding cytokine milieu greatly affect the differentiation process (227). As CD4+ T cells begin to proliferate and differentiate, metabolic programs support the commitment into separate lineages, with major roles for mTOR and AMPK in tailoring the metabolic adaptations of particular CD4+ T cell subsets (228, 229). Although this paradigm provides a useful framework for defining different functional CD4+ T cell responses, recent data suggest that it oversimplifies the dynamic interactions within the transcriptional network that orchestrates CD4+ T cell differentiation, which in turn provides a degree of functional and phenotypic plasticity within CD4+ T cell subsets (230, 231). Nevertheless, the functional specialization of CD4+ T cells generates effective immune responses that are tailored to meet particular infectious or inflammatory insults.
Th1 cells are characterized by production of the signature cytokine IFN-γ, but can also secrete TNF-α, GM-CSF, and lymphotoxin, and they are considered good producers of IL-2 (227). T-bet (T-box expressed in T cells) is the master transcription factor that orchestrates Th1 cell development (232). Metabolic sensors that favor differentiation toward Th1 lineage include signaling through mTORC1, which results in a strong engagement of glycolysis (220). Through the production of IFN-γ, Th1 cells are potent activators of macrophages and thus play a major role in the defense against intracellular pathogens such as Leishmania (233) and Toxoplasma (234, 235). However, aberrant Th1 responses have been implicated in several chronic inflammatory disorders, including type I diabetes (236), multiple sclerosis (237), and IBD (238).
Th2 cells are characterized by the production of IL-4, IL-5, IL-9, IL-13, and amphiregulin. IL-4 plays a crucial role in directing Th2 polarization (239). Other factors that promote Th2 differentiation include the alarmins TSLP, IL-33, and IL-25, which are released by epithelial cells in response to tissue injury and prime DCs and basophils to promote Th2 responses (240, 241). Th2 cell lineage commitment is orchestrated by the transcription factor Gata3, which is induced in response to IL-4 driven activation of STAT6 (242, 243). Although mTORC1 signaling is needed for Th2 cell lineage specification, Th2 cells are considered to be more reliant on mTORC2 in comparison to other T helper subsets (244, 245). Th2 responses are important in promoting tissue repair pathways (246), defense against large helminth parasites (247), resistance against toxins and venoms (248), and regulation of glucose homeostasis, adiposity, and thermogenesis (249, 250). However, the host protective functions of type 2 immunity are mirrored by detrimental effects when their activation is persistent or dysregulated. As such, type 2 responses can induce fibrosis, promote allergic diseases, including asthma and food allergies, and antagonize anti-tumor defense (241).
T follicular helper cells are becoming recognized as a separated lineage of CD4+ T cells that specialize in the provision of help for B cell responses. Tfh assistance is essential to induce maturation, isotype switching, and terminal differentiation of B cells (251), events that occur mainly within germinal centers (GC). As such, Tfh cells are essential for the production of most types of antibodies, although their role in IgE responses remains unclear (252). The transcriptional repressor B cell lymphoma 6 (Bcl-6) orchestrates Tfh lineage commitment and is both necessary and sufficient to drive Tfh differentiation (253–255). Not much is known about specific metabolic requirements of Tfh cells; however, recent evidence suggest that these cells have reduced mTORC1 activity and are less glycolytic compared with Th1 cells (256).
Th17 cells are characterized by production of the signature cytokines IL-17A, IL-17F, and IL-22 (257–261) and expression of IL-23R and the chemokine receptor CCR6 (262, 263). The transcription factor Rorγt is considered to be the master regulator of Th17 cells (264). A natural ligand for Rorγt – an intermediate of cholesterol biosynthesis pathway – has recently been identified (265, 266). The differentiation into Th17 cells requires TGF-β1 in the presence of proinflammatory cytokines, including IL-1β, and STAT3-activating cytokines, such as IL-6 or IL-21 (267), whereas IL-23 plays an essential role in sustaining Th17 differentiation and promoting their survival and acquisition of pathogenic effector potential (268–270). Similar to Th1 cells, Th17 cells require mTORC1 activation during differentiation, but are additionally dependent on HIF-1α activity and are thought to heavily rely on glycolysis, as blocking glycolysis with 2-deoxy-d-glucose (2-DG) inhibits Th17 cell differentiation (271, 272). Recently, fatty acid synthesis (FAS) was also shown to dictate the balance between Th17 and Treg differentiation, where de novo FAS promoted Th17 over Treg cell differentiation (273). Th17 cells are enriched at mucosal sites where they play a key role in protection against various extracellular pathogens, including fungal and bacterial infections (267, 274–276). However, Th17 cells have also been implicated in several chronic inflammatory and autoimmune disorders, including intestinal inflammation (267, 268, 277) and neurological disorders (278).
Commensal microbiota modulates abundance and function of intestinal Th cells, and this is well exemplified by the effect on Th17 cells. GF mice show reduced frequencies of intestinal Th17 cells, which can be restored upon colonization with the segmented filamentous bacteria (SFB), Gram-positive bacteria belonging to the Clostridiales genus (279, 280). How SFB promote effector T cell polarization and accumulation is still not completely understood, but it was proposed that SFB colonization increases levels of the acute-phase protein serum amyloid A (SAA), which conditioned intestinal MPs to promote Th17 cells (279). More recently, presentation of SFB antigens on MHC II molecules by intestinal DCs was implicated in SFB-specific Th17 cells differentiation (281, 282). Of note, it remains unknown whether equivalent single bacteria species able to promote Th17 cells exist in humans. Additionally, it was shown that Th17 cells could directly detect microbial-associated molecules through TLR2 and that this potentiated Th17 responses (283).
Regulatory T Cells in the Intestinal Mucosa
In comparison to the systemic immune compartment, the intestinal mucosa is significantly enriched in regulatory T cells and their non-redundant role in controlling intestinal inflammation is well documented (284, 285). The majority of these cells are Foxp3+ Treg cells. Foxp3 is a master transcriptional regulator of Treg cells, necessary for their development and maintenance (286). Treg cells can be generated in the thymus during development (tTreg cells), or in the periphery from conventional naive CD4+ T cells (pTreg cells). Differentiation of tTreg cells requires a transient TCR stimulation with a particular strength of TCR signaling following recognition of self-peptides, which corresponds to a TCR avidity between the one that dictates positive selection and the one that leads to negative selection (287). Induction of pTreg cells from naive CD4+ T cells in vivo occurs when antigen is presented under subimmunogenic or non-inflammatory conditions, during chronic inflammation, and in the setting of a tolerogenic microenvironment, which includes the intestinal LP (288). Indeed, the gastrointestinal tract is a preferential site of pTreg cell conversion; however, whether pTreg and tTreg have overlapping or distinct functions in maintaining gut homeostasis is an ongoing question (211).
The mechanisms used by Treg cells to suppress deleterious inflammatory responses have been extensively studied. They include production of regulatory cytokines (IL-10, TGF-β1, and IL-35), suppression by metabolic disruption, direct modulation of DC function, and cytolysis (289). In the context of intestinal homeostasis, IL-10 and TGF-β1 are of particular importance in enforcing tolerance and genetic deletion of IL-10, IL-10R, or impairment of the pathway results in microbiota driven intestinal inflammation in mice and humans (284, 285, 290).
Treg cells display specific adaptations that are tailored to the environmental context in which they operate. It was recently proposed that circulating Treg cells could be therefore divided into central, effector, and tissue-resident Treg cell populations (291). The existence of memory Treg cells has also been postulated (292–295). Tissue-resident Treg cells are long-term residents within various non-lymphoid organs, to which they adapt through transcriptional and metabolic reprograming (296). Potentially, each organ might have its own specific Treg cell population, and tissue-resident Treg cells have been described in skin, muscle, adipose tissue, placenta, and the intestine (296). Gut-resident Treg cells express chemokine receptors and adhesion molecules that facilitate gut homing, such as β7 family integrins and CCR9, expression of which is promoted by RA (121, 297), and G protein-coupled receptor 15 (GPR15) (298). Additionally, gut-resident Treg cells can be characterized by expression of the high affinity IL-2 receptor and the short-chain fatty acid (SCFA) receptor GPR43 (299). It is important to note that due to difficulties in distinguishing between effector and tissue-resident Treg cell populations the degree of plasticity between them remains unclear. Overall, Treg cells are thought to metabolically resemble memory T cells, in that they preferentially rely on lipid instead of glucose metabolism for energy generation; however, it remains largely unexplored whether particular subpopulations of Treg cells are metabolically distinct (220).
Commensals influence Foxp3+ Treg cell induction in the gut in several ways. For instance, the capsular polysaccharide A (PSA) of the Gram-negative anaerobic commensal Bacteroides fragilis was shown to promote IL-10 producing Foxp3+ Treg cells (300) through direct interaction of PSA with TLR2 on T lymphocytes (301). Furthermore, metabolites, such as SCFA, are emerging as key homeostatic signals provided by commensal microbiota to regulate local Treg cells. SCFA, such as butyrate and acetate, can act directly on mucosal Treg cells to promote their expansion. Mechanistically, butyrate appeared to promote pTreg cell induction by inhibiting histone deacetylases (HDAC). Butyrate-treated naive CD4+ T cells exhibited increased acetylation of the Foxp3 locus, including the key CNS1 enhancer region that is essential for pTreg differentiation (299, 302, 303). Of note, recent reports indicate that pTreg cells induced by the commensal microbiota antigens can be distinguished from other pTreg cells based on their expression of Rorγt (304, 305), suggesting an additional level of Treg cell specialization may exist in the gut.
Autophagy Regulates T Cell Survival
The first indication of the importance of the autophagy pathway for T lymphocyte homeostasis in vivo came from the study of Atg5−/− fetal liver chimeric mice where decreased numbers of thymic and splenic autophagy-deficient CD4+ and CD8+ T cells were reported (306). Several genetic models were subsequently employed to investigate a specific role for autophagy in T cell development. These studies investigated T cells within the thymus and in the secondary lymphoid organs, including spleen and lymph nodes. T cell-specific deletion of Atg3, Atg5, Atg7, Atg16l1, Vps34, or Beclin1 consistently showed decreased frequencies and numbers of CD4+ and CD8+ T in the secondary lymphoid organs, whereas thymic development was largely unperturbed (200, 307–312). However, the requirement for autophagy in thymic development seems to be dependent on the system that was used to generate T cell-specific deletion of autophagy genes. While mice where Cre recombinase expression was driven by the Lck promoter (Cre expression occurs at the double negative stage) showed a mild, but significant, reduction in thymocyte numbers (308, 313), mice where the CD4 promoter was used to drive Cre expression (resulting in excision during the later double positive stage) did not show any significant changes in thymocyte development (200, 307, 310, 312). One exception is NKT cells, which require autophagy during thymic development (309, 314, 315).
These studies also observed increased proportions of effector/memory phenotype (CD62low CD44hi) T cells among peripheral autophagy-deficient T cells, which was interpreted to be a result of decreased survival of naive T cells (308, 311). However, this phenotype might also occur as a result of lymphopaenia-induced proliferation (200). The role of autophagy in activated T cells has been predominantly studied in vitro. The question of when autophagy is activated in T cells remains controversial. While autophagy is known to be negatively regulated by mTORC1 signaling in many cell types, and therefore inversely correlates with cell proliferation (21), early in vitro studies indicated that TCR triggering induces autophagy in T lymphocytes and reported that chemical or genetic blockage of the autophagy pathway impaired T cell activation and proliferation (306, 308, 316, 317). Autophagy-deficient T cells also showed increased apoptosis during prolonged in vitro culture (308) or after activation (310, 312). Some of the studies also observed decreased production of effector cytokines by in vitro activated autophagy-deficient T cells, including IL-2, IL-17A, and IFNγ (309, 317). Conversely, another study reported increased IL-2 production by Atg7-deficient CD4+ T cells after TCR cross-linking (318). In several studies, these defects in autophagy-deficient T cells were linked to impaired organelle homeostasis, particularly mitochondria homeostasis, and were associated with an increase in ROS production (307, 308, 311). Increases in ER mass and changes in intracellular calcium signaling were also implicated in this impaired fitness and survival (318). However, whether this is indeed the mechanistic explanation for the decreased fitness of autophagy-deficient CD4+ T cells in vivo remains unclear, as other studies did not observe any increase in mitochondrial mass or ROS production (310, 319), and mitochondria were shown to be excluded from autophagosome degradation in activated wild-type CD4+ T cells (317). Further complication arises from the findings that although increased ROS can be detrimental for T cells (320), ROS production is increased after TCR triggering and is, in fact, required for T cell proliferation, particularly in CD8+ T cells (321–324). It was also proposed that imbalanced expression or accumulation of apoptosis-related proteins might contribute to the defective proliferation and survival of autophagy-deficient T cells. However, these results are difficult to interpret as increased levels of both proapoptotic (308–310) and anti-apoptotic proteins (Bcl-2, Mcl-1) (308–310) have been reported in autophagy-deficient T cells. Analysis of mice with T cell-specific deletion of Vps34 suggested that decreased levels of IL-7Rα expression on T cells might be involved, although this was not attributed to the cell-intrinsic effects of autophagy deficiency (307, 319). Treatment with the pan-caspase inhibitor zVAD could partially rescue the apoptotic phenotype of autophagy-deficient T cells (308, 310); however, it did not rescue the defects in T cell proliferation (309). Furthermore, it is worth pointing out that in addition to its role in T cell survival and proliferation, autophagy has also been reported to promote cell death in activated T cells under some circumstances (316, 325, 326).
Interestingly, autophagy has been directly linked to regulation of signaling cascades downstream of the TCR. For example, in activated, but not in naive T cells, autophagy was shown to selectively target Bcl-10 for degradation in a p62-dependent manner, limiting NFκB-dependent effector responses, including IL-2 secretion (29). NFκB signaling plays an important role in many aspects of activated T cell physiology, including entry into cell cycle (327), but the strength of NFκB signaling can also influence differentiation into distinct Th cell subsets; therefore, autophagy might contribute to these processes by regulating the NFκB pathway in activated T cells.
Recent reports identified a role of autophagy in the formation of memory CD8+ T cells during viral infections (199, 200, 328). These studies, which focused on in vivo responses of autophagy-deficient CD8+ T cells to influenza or lymphocytic choriomeningitis virus infections, revealed new aspects of autophagic regulation of T cells. Although distinct genetic approaches were used to generate mice with a selective autophagy deficiency in T cells, all observed that autophagy was dispensable during the early expansion phase of antigen-activated CD8+ T cells during viral infection (199, 200, 328). In addition, autophagy-deficient T cells did not show defects in effector cytokine production and were capable of controlling virus titers during the early phases of infection (199). However, activation of the autophagy pathway was shown to be crucial during the transition phase between late effector to memory T cells and mice with autophagy-deficient CD8+ T cells did not mount proper memory CD8+ T cell responses during secondary challenge (199, 200, 328). While the mechanism behind the requirement for autophagy in memory CD8+ T cell responses remains to be elucidated, comparison of metabolic profiles between WT and Atg7-deficient memory CD8+ T cells suggested that metabolic adaptation might be involved (199). In addition, CD8+ T cells from aged mice (200) and senescent human CD8+ T cells exhibited low levels of autophagy, which in human senescent cells was associated with high p38 kinase activity (329). Importantly, increasing autophagy levels was shown to boost memory CD8+ T cell responses after influenza vaccination in aged mice (200). Of note, it remains unknown whether autophagy is equally essential for the development of CD4+ T cell memory cells and whether autophagy plays any role in the formation of TRM cells, including mucosal TRM cells.
Overall, while it is clear that autophagy plays a crucial role in the maintenance of peripheral CD4+ and CD8+ T cells and memory CD8+ T cells (Figure 3), it is still not completely understood how autophagy influences different aspects of T cell physiology in naive and activated T cells. Methodological difficulties of monitoring autophagy, differences in the genetic models used, as well as differences between in vivo and in vitro stimuli might underlie some of the discrepancies observed. Indeed, autophagy is an essential homeostatic process for all eukaryotic cells, including T cells. Therefore, genetic deletion of essential autophagy genes early during T cell development, using CD4 or Lck promoter driven Cre-lox technology, can mediate major changes in autophagy-deficient T cells, including global alterations in gene expression (313). Arguably, this makes it difficult to study the function of essential autophagy genes in isolation on a particular process or step of T cell development. In order to avoid this complication, several groups attempted to use an inducible system to selectively knock-out autophagy genes at a chosen time point (311, 328), or used the granzyme B promoter to selectively drive Cre expression in mature CD8+ T cells (199). However, in the context of intestinal homeostasis, it is important to remember that in terms of revealing how polymorphisms in autophagy genes are linked to disease susceptibility, the system where autophagy is perturbed from the beginning of T cell development may be physiologically relevant.
Autophagy Regulates the Balance of Intestinal CD4+ T Cell Subsets
Although these studies investigated the role of several autophagy genes in T cell physiology, intestinal populations of T cells were not analyzed. We studied the role of the IBD susceptibility gene Atg16l1 in the intestinal T cell homeostasis and function (312). This study showed that autophagy pathway was essential for the maintenance of T cells in the small intestinal and colonic mucosa, as CD4+ and CD8+ T cell numbers were strongly reduced in the mice with selective Atg16l1-deficiency in the T cell compartment (312). Moreover, detailed examination of intestinal CD4+ T cell subsets revealed that Atg16l1-deficiency differentially affected distinct subsets of CD4+ T cells in the gut; it strongly compromised the Treg cell compartment, whereas the Th2 population was selectively enhanced. In addition, Th1 and Th17 cells, which are commonly implicated in intestinal pathology, were reduced in the gut of mice with Atg16l1-deficient T cells (Figure 5). These changes were most pronounced in the intestinal mucosa, suggesting different requirements for autophagy for the accumulation and survival of distinct subsets of CD4+ T cells within the intestinal environment. Aged mice with T cell-specific deletion of Atg16l1 developed spontaneous and progressive intestinal pathology, which was preceded by the production of Th2-associated antibodies toward dietary and commensal antigens (312). These aberrant type 2 responses resulted not only from a lack of sufficient Treg control but also from cell-intrinsic dysregulation, as T cell-intrinsic autophagy limited the expansion of Th2 cells (312).
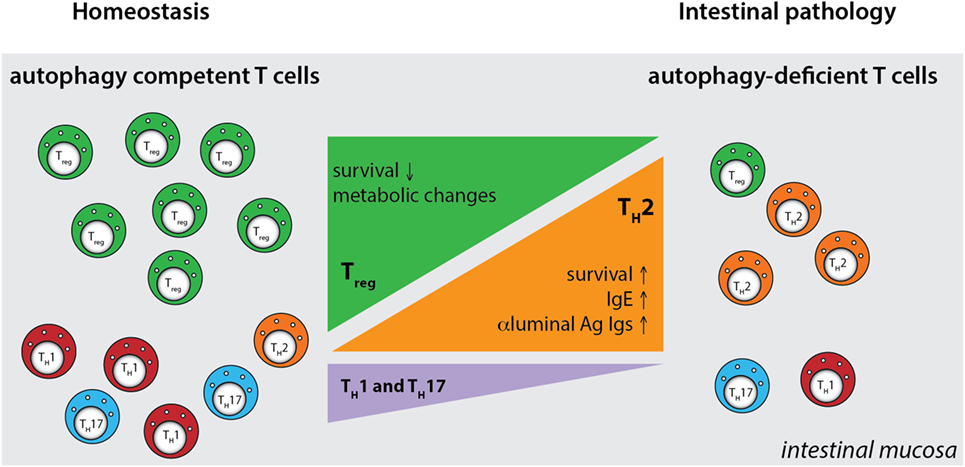
Figure 5. Defective autophagy alters the balance of the intestinal CD4+ T cell subsets. T cell-specific ablation of autophagy results in a strong decrease in the numbers of Treg cells and also reductions in Th1 and Th17 effector cells. Conversely, loss of autophagy promotes expansion of intestinal Th2 cells, leading to aberrant responses to luminal antigens and subsequent intestinal pathology. Ag - antigen.
By generating mice with a Treg cell-specific deletion of Atg16l1, we demonstrated that cell-intrinsic autophagy is indispensable for Treg cell maintenance in the periphery and thus for the control of effector T cell responses, as these mice developed severe systemic and gastrointestinal inflammation. These findings are consistent with an independent study, which reported that Treg cell-specific deletion of Atg5 or Atg7 led to the spontaneous development of severe multi-organ inflammation (330). Autophagy-deficient Treg cells exhibited marked phenotypical changes, including increased cell cycling, production of Th effector cytokines, and reduced Foxp3 expression, that were associated with increased activation of mTORC1 (312, 330). In addition, autophagy-deficient Treg cells showed a distinct metabolic profile, with increased glycolysis and reduced expression of genes involved in FAS/FAO (312, 330). Increased glycolysis could be a more general phenomenon observed in autophagy-deficient T cells, as others reported a similar glycolytic shift in autophagy-deficient CD8+ memory T cells (200). Based on recent evidence that metabolic changes are emerging as an important part of tissue-specific reprograming of tissue-resident Treg cells (291, 296), we predict that these metabolic perturbations introduced by autophagy deficiency strongly affect intestinal Treg cells, which rely on fatty acid metabolism, similar to memory T cells (Figure 6) (312). In contrast, Th2 cells seem resistant to these metabolic changes, presumably due to their ability to cope with prolonged high levels of glycolysis, perhaps explaining why this particular Th cell subset were not negatively affected by autophagy deficiency (312) (Figure 6).
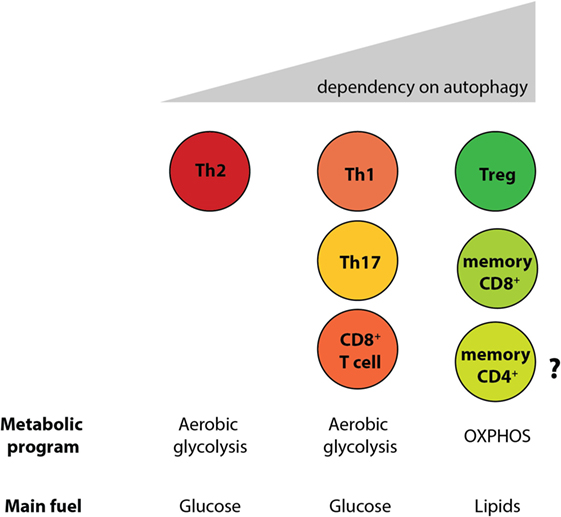
Figure 6. Differential reliance on autophagy within intestinal T cell subsets corresponds to their different metabolic profiles. Cell-intrinsic defects in autophagy negatively affect T cell subsets, with the exception of Th2 cells. Treg cells and memory CD8+ T cells, which rely on lipid metabolism for survival, were shown to be the most sensitive to autophagy perturbation. However, the role of autophagy CD4+ memory T cells formation has not yet been investigated.
In summary, autophagy differentially regulates survival of Treg cells and Th2 cells; T cell-intrinsic autophagy might facilitate metabolic adaptations that are required for the survival of intestinal Treg cells, while Th2 cell are resistant to metabolic perturbations introduced by autophagy deficiency. As polymorphisms in ATG16L1 and other autophagy genes have been linked to IBD susceptibility, these results identify a potential novel mechanism that links genetic susceptibility in the autophagy pathway to intestinal inflammation through dysregulation of mucosal T cell responses. Moreover, as defective Treg and increased Th2 responses at the mucosa are observed in food allergies and asthma, and since polymorphisms in the essential autophagy gene Atg5 have been implicated in asthma susceptibility (331, 332), these findings might also have implications for these conditions that affect mucosal tissues.
Conclusion and Perspectives
In recent years, researchers have employed the Cre-lox system to generate transgenic mice with conditional knock-out of autophagy genes to study the role of autophagy in a cell type-specific manner. While this is undoubtedly a powerful tool to look at autophagy functions in vivo, it is important to remember that these are rarely perfect, as few promoters show complete specificity for one cell type and off-target effects of autophagy gene deletion should be considered. Nevertheless, studies using distinct Cre lines, together with those using human cells with disease-associated autophagy mutations, have identified several key mechanisms through which autophagy can influence the functions of distinct cell populations in the gut (Table 1). However, some observations remain inconclusive and the molecular mechanisms through which autophagy controls such a range of functions in diverse cell types are not well defined (Table 1). It is important to point out that autophagy is a fundamental process in eukaryotic cells and complete inhibition or deletion of essential autophagy genes often results in strong perturbations in cellular physiology, including activation of compensatory or rescue pathways. Future studies may employ more refined approaches, such as using knock-in mice harboring disease-associated autophagy gene alleles.
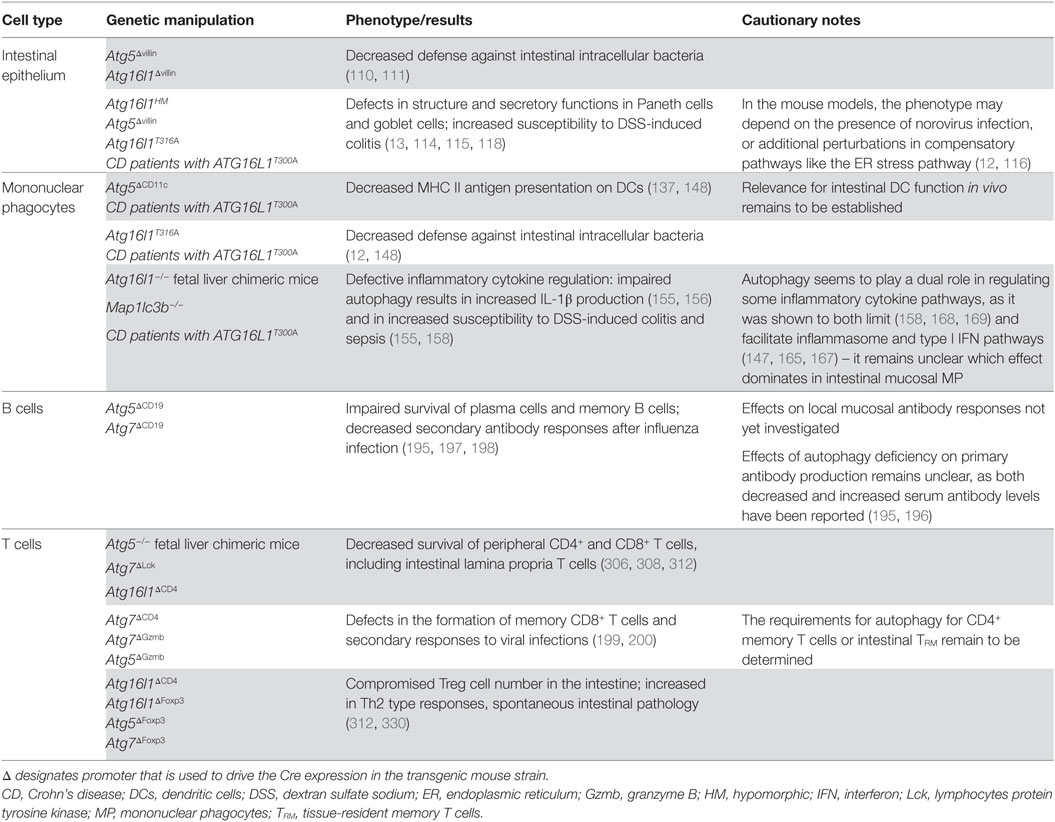
Table 1. Consequences of autophagy deficiency in the intestine.
The role of autophagy has been investigated for the main leukocyte subsets and we know a lot about how it regulates different aspects of their differentiation and function. However, the majority of these observations were made using leukocytes derived from secondary lymphoid tissues and extrapolation to the intestinal mucosa should be treated cautiously, as mucosal immune cells often display very distinct properties from their systemic counterparts. A good example of this is our discovery that enhanced Th2 responses in autophagy-deficient CD4+ T cells occur primarily within the intestinal mucosa. Emerging literature indicates that effector and memory T cell responses in tissues are dependent on metabolic adaptations that allow T cells to survive and function in environments with altered availability of nutrients and growth factors. We speculate that autophagy plays a key role in endowing T cells with the metabolic flexibility to adapt these challenges. Thus, the immune manifestations of autophagy deficiency depend not only on the cell type considered but also on the tissue context.
Many important issues still remain to be addressed to give a more complete picture of how autophagy regulates intestinal immune homeostasis. These include a comprehensive analysis of the effects of autophagy on unique populations of innate leukocytes that are present in the gut, such as ILCs and IELs. In addition, the influence of autophagy on CD4+ T cell memory formation and development of TRM cell responses in the mucosa has still to be determined. An improved understanding of the molecular mechanisms that make Th2 cells resistant to the effects of autophagy deficiency is also required.
Autophagy is an attractive therapeutic target and several autophagy modulating compounds are already in clinical trials for the treatment of various disorders (333). For instance, the autophagy-enhancing drug carbamazepine has been shown to ameliorate hepatic fibrosis in the mouse model of α1-antitrypsin deficiency liver cirrhosis, and this drug is currently in phase II of clinical trials (333, 334). Additionally, an autophagy-inducing agent has been shown to decrease pathology in a mouse model of chemically induced colitis (335, 336). The challenge, however, is to identify agents that can specifically induce autophagy with minimal side effects on other cellular processes. Indeed, considerable effort in the field of autophagy research is currently focused on finding small molecules that can induce autophagy in a very selective manner (337, 338). Interestingly, as some of the dietary-derived compounds, including RA (339) and vitamin D (340), have been shown to enhance autophagy, it is tempting to speculate that the use of such natural inducers could prove beneficial for treatment of intestinal inflammatory disorders.
Author Contributions
AK, JP, and KM prepared the manuscript text. AK and JP preparedthe figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Wellcome Trust (grant 102972 and 097112) and Medical Research Council (grant MR/K011898/1).
References
2. Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol (2003) 3:331–41. doi:10.1038/nri1057
3. Kabat AM, Srinivasan N, Maloy KJ. Modulation of immune development and function by intestinal microbiota. Trends Immunol (2014) 35:507–17. doi:10.1016/j.it.2014.07.010
4. Hill DA, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol (2010) 28:623–67. doi:10.1146/annurev-immunol-030409-101330
5. Pabst O, Mowat AM. Oral tolerance to food protein. Mucosal Immunol (2012) 5:232–9. doi:10.1038/mi.2012.4
6. Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature (2011) 474:298–306. doi:10.1038/nature10208
7. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol (2010) 28:573–621. doi:10.1146/annurev-immunol-030409-101225
8. Liu JZ, Anderson CA. Genetic studies of Crohn’s disease: past, present and future. Best Pract Res Clin Gastroenterol (2014) 28:373–86. doi:10.1016/j.bpg.2014.04.009
9. Van Limbergen J, Radford-Smith G, Satsangi J. Advances in IBD genetics. Nat Rev Gastroenterol Hepatol (2014) 11:372–85. doi:10.1038/nrgastro.2014.27
10. Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet (2007) 39:207–11. doi:10.1038/ng1954
11. Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet (2007) 39:596–604. doi:10.1038/ng2032
12. Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature (2014) 506:456–62. doi:10.1038/nature13044
13. Lassen KG, Kuballa P, Conway KL, Patel KK, Becker CE, Peloquin JM, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A (2014) 111:7741–6. doi:10.1073/pnas.1407001111
14. Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr (2007) 27:19–40. doi:10.1146/annurev.nutr.27.061406.093749
15. Reggiori F, Klionsky DJ. Autophagy in the eukaryotic cell. Eukaryot Cell (2002) 1:11–21. doi:10.1128/EC.01.1.11-21.2002
16. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res (2014) 24:24–41. doi:10.1038/cr.2013.168
17. Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature (2004) 432:1032–6. doi:10.1038/nature03029
18. Mizushima N. Autophagy: process and function. Genes Dev (2007) 21:2861–73. doi:10.1101/gad.1599207
19. Hurley JH, Schulman BA. Atomistic autophagy: the structures of cellular self-digestion. Cell (2014) 157:300–11. doi:10.1016/j.cell.2014.01.070
20. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol (2013) 13:722–37. doi:10.1038/nri3532
21. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469:323–35. doi:10.1038/nature09782
22. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy (2011) 7:279–96. doi:10.4161/auto.7.3.14487
23. Rogov V, Dotsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell (2014) 53:167–78. doi:10.1016/j.molcel.2013.12.014
24. Szeto J, Kaniuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, et al. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy (2006) 2:189–99. doi:10.4161/auto.2731
25. Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet (2002) 11:1107–17. doi:10.1093/hmg/11.9.1107
26. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem (2007) 282:24131–45. doi:10.1074/jbc.M702824200
27. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol (2012) 13:255–63. doi:10.1038/ni.2215
28. Chuang SY, Yang CH, Chou CC, Chiang YP, Chuang TH, Hsu LC. TLR-induced PAI-2 expression suppresses IL-1beta processing via increasing autophagy and NLRP3 degradation. Proc Natl Acad Sci U S A (2013) 110:16079–84. doi:10.1073/pnas.1306556110
29. Paul S, Kashyap AK, Jia W, He YW, Schaefer BC. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-kappaB. Immunity (2012) 36:947–58. doi:10.1016/j.immuni.2012.04.008
30. Iwata J, Ezaki J, Komatsu M, Yokota S, Ueno T, Tanida I, et al. Excess peroxisomes are degraded by autophagic machinery in mammals. J Biol Chem (2006) 281:4035–41. doi:10.1074/jbc.M512283200
31. Kim PK, Hailey DW, Mullen RT, Lippincott-Schwartz J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc Natl Acad Sci U S A (2008) 105:20567–74. doi:10.1073/pnas.0810611105
32. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol (2011) 12:9–14. doi:10.1038/nrm3028
33. Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys (2007) 462:245–53. doi:10.1016/j.abb.2007.03.034
34. Tal R, Winter G, Ecker N, Klionsky DJ, Abeliovich H. Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J Biol Chem (2007) 282:5617–24. doi:10.1074/jbc.M605940200
35. Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J (2008) 27:433–46. doi:10.1038/sj.emboj.7601963
36. Kissova I, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem (2004) 279:39068–74. doi:10.1074/jbc.M406960200
37. Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A (2007) 104:19500–5. doi:10.1073/pnas.0708818104
38. Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell (2009) 17:87–97. doi:10.1016/j.devcel.2009.06.013
39. Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol (2006) 4:e423. doi:10.1371/journal.pbio.0040423
40. Cebollero E, Reggiori F, Kraft C. Reticulophagy and ribophagy: regulated degradation of protein production factories. Int J Cell Biol (2012) 2012:182834. doi:10.1155/2012/182834
41. Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol (2008) 10:602–10. doi:10.1038/ncb1723
42. Kidane TZ, Sauble E, Linder MC. Release of iron from ferritin requires lysosomal activity. Am J Physiol Cell Physiol (2006) 291:445–55. doi:10.1152/ajpcell.00505.2005
43. Asano T, Komatsu M, Yamaguchi-Iwai Y, Ishikawa F, Mizushima N, Iwai K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol Cell Biol (2011) 31:2040–52. doi:10.1128/MCB.01437-10
44. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature (2009) 458:1131–5. doi:10.1038/nature07976
45. Kotoulas OB, Kalamidas SA, Kondomerkos DJ. Glycogen autophagy. Microsc Res Tech (2004) 64:10–20. doi:10.1002/jemt.20046
46. Rabinowitz JD, White E. Autophagy and metabolism. Science (2010) 330:1344–8. doi:10.1126/science.1193497
47. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol (2011) 27:107–32. doi:10.1146/annurev-cellbio-092910-154005
48. Hara T, Mizushima N. Role of ULK-FIP200 complex in mammalian autophagy: FIP200, a counterpart of yeast Atg17? Autophagy (2009) 5:85–7. doi:10.4161/auto.5.1.7180
49. Reggiori F, Shintani T, Nair U, Klionsky DJ. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy (2005) 1:101–9. doi:10.4161/auto.1.2.1840
50. Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy (2009) 5:649–62. doi:10.4161/auto.5.5.8249
51. Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell (2009) 20:1992–2003. doi:10.1091/mbc.E08-12-1249
52. Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene (2004) 23:9314–25. doi:10.1038/sj.onc.1208331
53. Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol (2007) 9:1102–9. doi:10.1038/ncb1007-1102
54. Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy (2010) 6:506–22. doi:10.4161/auto.6.4.11863
55. He C, Baba M, Cao Y, Klionsky DJ. Self-interaction is critical for Atg9 transport and function at the phagophore assembly site during autophagy. Mol Biol Cell (2008) 19:5506–16. doi:10.1091/mbc.E08-05-0544
56. Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem (2011) 80:125–56. doi:10.1146/annurev-biochem-052709-094552
57. Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol (2009) 335:1–32. doi:10.1007/978-3-642-00302-8_1
58. Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ (2013) 20:21–30. doi:10.1038/cdd.2012.72
59. Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature (1998) 395:395–8. doi:10.1038/26506
60. Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem (1998) 273:33889–92. doi:10.1074/jbc.273.51.33889
61. Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci (2003) 116:1679–88. doi:10.1242/jcs.00381
62. Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci (2004) 117:2805–12. doi:10.1242/jcs.01131
63. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J (2000) 19:5720–8. doi:10.1093/emboj/19.21.5720
64. Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J (2010) 29:1792–802. doi:10.1038/emboj.2010.74
65. Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem (2007) 282:37298–302. doi:10.1074/jbc.C700195200
66. Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep (2008) 9:859–64. doi:10.1038/embor.2008.163
67. Yoshimori T, Noda T. Toward unraveling membrane biogenesis in mammalian autophagy. Curr Opin Cell Biol (2008) 20:401–7. doi:10.1016/j.ceb.2008.03.010
68. Jiang P, Nishimura T, Sakamaki Y, Itakura E, Hatta T, Natsume T, et al. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell (2014) 25:1327–37. doi:10.1091/mbc.E13-08-0447
69. Takats S, Pircs K, Nagy P, Varga A, Karpati M, Hegedus K, et al. Interaction of the HOPS complex with syntaxin 17 mediates autophagosome clearance in Drosophila. Mol Biol Cell (2014) 25:1338–54. doi:10.1091/mbc.E13-08-0449
70. Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell (2012) 151:1256–69. doi:10.1016/j.cell.2012.11.001
71. Takats S, Nagy P, Varga A, Pircs K, Karpati M, Varga K, et al. Autophagosomal syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol (2013) 201:531–9. doi:10.1083/jcb.201211160
72. Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res (2010) 20:748–62. doi:10.1038/cr.2010.82
73. Fullgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol (2014) 15:65–74. doi:10.1038/nrm3716
74. Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell (2014) 159:1263–76. doi:10.1016/j.cell.2014.11.006
75. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149:274–93. doi:10.1016/j.cell.2012.03.017
76. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of ULK1. Nat Cell Biol (2011) 13:132–41. doi:10.1038/ncb2152
77. Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-ATG13-FIP200 complex required for autophagy. Mol Biol Cell (2009) 20:1981–91. doi:10.1091/mbc.E08-12-1248
78. Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem (2009) 284:12297–305. doi:10.1074/jbc.M900573200
79. Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol (2013) 15:406–16. doi:10.1038/ncb2708
80. Yuan HX, Russell RC, Guan KL. Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress-induced autophagy. Autophagy (2013) 9:1983–95. doi:10.4161/auto.26058
81. Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature (2010) 465:942–6. doi:10.1038/nature09076
82. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol (2012) 13:251–62. doi:10.1038/nrm3311
83. Bach M, Larance M, James DE, Ramm G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem J (2011) 440:283–91. doi:10.1042/BJ20101894
84. Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science (2011) 331:456–61. doi:10.1126/science.1196371
85. Kim J, Kim YC, Fang C, Russell RC, Kim JH, Fan W, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell (2013) 152:290–303. doi:10.1016/j.cell.2012.12.016
86. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell (2008) 30:214–26. doi:10.1016/j.molcel.2008.03.003
87. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell (2003) 115:577–90. doi:10.1016/S0092-8674(03)00929-2
88. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science (2011) 332:1429–33. doi:10.1126/science.1204592
89. Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol (2013) 14:283–96. doi:10.1038/nrm3565
90. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J (2012) 31:1095–108. doi:10.1038/emboj.2012.32
91. Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal (2012) 5:ra42. doi:10.1126/scisignal.2002790
92. Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy (2012) 8:903–14. doi:10.4161/auto.19653
93. Chauhan S, Goodwin JG, Chauhan S, Manyam G, Wang J, Kamat AM, et al. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol Cell (2013) 50:16–28. doi:10.1016/j.molcel.2013.01.024
94. Pott J, Hornef M. Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep (2012) 13:684–98. doi:10.1038/embor.2012.96
95. van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol (2009) 71:241–60. doi:10.1146/annurev.physiol.010908.163145
96. Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol (2011) 9:356–68. doi:10.1038/nrmicro2546
97. Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol (2010) 11:76–83. doi:10.1038/ni.1825
98. Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol (2012) 12:503–16. doi:10.1038/nri3228
99. Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature (2011) 469:415–8. doi:10.1038/nature09637
100. Shan M, Gentile M, Yeiser JR, Walland AC, Bornstein VU, Chen K, et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science (2013) 342:447–53. doi:10.1126/science.1237910
101. McDole JR, Wheeler LW, McDonald KG, Wang B, Konjufca V, Knoop KA, et al. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature (2012) 483:345–9. doi:10.1038/nature10863
102. Heazlewood C, Cook M, Eri R, Price G, Tauro S, Taupin D, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med (2008) 5:e54. doi:10.1371/journal.pmed.0050054
103. Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology (2006) 131:117–29. doi:10.1053/j.gastro.2006.04.020
104. Goto Y, Kiyono H. Epithelial barrier: an interface for the cross-communication between gut flora and immune system. Immunol Rev (2012) 245:147–63. doi:10.1111/j.1600-065X.2011.01078.x
105. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145:745–57. doi:10.1016/j.cell.2011.04.022
106. Nordlander S, Pott J, Maloy KJ. NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol (2014) 7:775–85. doi:10.1038/mi.2013.95
107. Song-Zhao GX, Srinivasan N, Pott J, Baban D, Frankel G, Maloy KJ. Nlrp3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol (2014) 7:763–74. doi:10.1038/mi.2013.94
108. Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature (2011) 469:543–7. doi:10.1038/nature09646
109. Harrison OJ, Srinivasan N, Pott J, Schiering C, Krausgruber T, Ilott NE, et al. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3 Treg cell function in the intestine. Mucosal Immunol (2015) 8(6):1226–36. doi:10.1038/mi.2015.13
110. Benjamin JL, Sumpter R Jr, Levine B, Hooper LV. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe (2013) 13:723–34. doi:10.1016/j.chom.2013.05.004
111. Conway KL, Kuballa P, Song JH, Patel KK, Castoreno AB, Yilmaz OH, et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology (2013) 145:1347–57. doi:10.1053/j.gastro.2013.08.035
112. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11:55–62. doi:10.1038/ni.1823
113. Nighot PK, Hu CA, Ma TY. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J Biol Chem (2015) 290:7234–46. doi:10.1074/jbc.M114.597492
114. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature (2008) 456:259–63. doi:10.1038/nature07416
115. Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell (2010) 141:1135–45. doi:10.1016/j.cell.2010.05.009
116. Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, et al. Paneth cells as a site of origin for intestinal inflammation. Nature (2013) 503:272–6. doi:10.1038/nature12599
117. Deuring JJ, Fuhler GM, Konstantinov SR, Peppelenbosch MP, Kuipers EJ, de Haar C, et al. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut (2014) 63:1081–91. doi:10.1136/gutjnl-2012-303527
118. Patel KK, Miyoshi H, Beatty WL, Head RD, Malvin NP, Cadwell K, et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J (2013) 32:3130–44. doi:10.1038/emboj.2013.233
119. Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat Rev Immunol (2008) 8:435–46. doi:10.1038/nri2335
120. Varol C, Zigmond E, Jung S. Securing the immune tightrope: mononuclear phagocytes in the intestinal lamina propria. Nat Rev Immunol (2010) 10:415–26. doi:10.1038/nri2778
121. Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY. Retinoic acid imprints gut-homing specificity on T cells. Immunity (2004) 21:527–38. doi:10.1016/j.immuni.2004.08.011
122. Johansson-Lindbom B, Svensson M, Pabst O, Palmqvist C, Marquez G, Forster R, et al. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J Exp Med (2005) 202:1063–73. doi:10.1084/jem.20051100
123. Jaensson E, Uronen-Hansson H, Pabst O, Eksteen B, Tian J, Coombes JL, et al. Small intestinal CD103+ dendritic cells display unique functional properties that are conserved between mice and humans. J Exp Med (2008) 205:2139–49. doi:10.1084/jem.20080414
124. Worthington JJ, Czajkowska BI, Melton AC, Travis MA. Intestinal dendritic cells specialize to activate transforming growth factor-beta and induce Foxp3+ regulatory T cells via integrin alphavbeta8. Gastroenterology (2011) 141:1802–12. doi:10.1053/j.gastro.2011.06.057
125. Paidassi H, Acharya M, Zhang A, Mukhopadhyay S, Kwon M, Chow C, et al. Preferential expression of integrin alphavbeta8 promotes generation of regulatory T cells by mouse CD103+ dendritic cells. Gastroenterology (2011) 141:1813–20. doi:10.1053/j.gastro.2011.06.076
126. Uematsu S, Fujimoto K, Jang MH, Yang BG, Jung YJ, Nishiyama M, et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing toll-like receptor 5. Nat Immunol (2008) 9:769–76. doi:10.1038/ni.1622
127. Mazzini E, Massimiliano L, Penna G, Rescigno M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1(+) macrophages to CD103(+) dendritic cells. Immunity (2014) 40:248–61. doi:10.1016/j.immuni.2013.12.012
128. Schulz O, Jaensson E, Persson EK, Liu X, Worbs T, Agace WW, et al. Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J Exp Med (2009) 206:3101–14. doi:10.1084/jem.20091925
129. Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol (2001) 2:361–7. doi:10.1038/86373
130. Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science (2005) 307:254–8. doi:10.1126/science.1102901
131. Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity (2011) 34:237–46. doi:10.1016/j.immuni.2011.01.016
132. Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G, et al. Ly6C hi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen-presenting cells. Immunity (2012) 37:1076–90. doi:10.1016/j.immuni.2012.08.026
133. Laffont S, Siddiqui KR, Powrie F. Intestinal inflammation abrogates the tolerogenic properties of MLN CD103+ dendritic cells. Eur J Immunol (2010) 40:1877–83. doi:10.1002/eji.200939957
134. Cerovic V, Bain CC, Mowat AM, Milling SW. Intestinal macrophages and dendritic cells: what’s the difference? Trends Immunol (2014) 35:270–7. doi:10.1016/j.it.2014.04.003
135. Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature (2008) 455:808–12. doi:10.1038/nature07240
136. Shaw MH, Kamada N, Kim YG, Nunez G. Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J Exp Med (2012) 209:251–8. doi:10.1084/jem.20111703
137. Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A, et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity (2010) 32:227–39. doi:10.1016/j.immuni.2009.12.006
138. Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M, et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A (2005) 102:7922–7. doi:10.1073/pnas.0501190102
139. Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity (2007) 26:79–92. doi:10.1016/j.immuni.2006.10.018
140. Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science (2005) 307:593–6. doi:10.1126/science.1104904
141. English L, Chemali M, Duron J, Rondeau C, Laplante A, Gingras D, et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol (2009) 10:480–7. doi:10.1038/ni.1720
142. Tey SK, Khanna R. Autophagy mediates transporter associated with antigen processing-independent presentation of viral epitopes through MHC class I pathway. Blood (2012) 120:994–1004. doi:10.1182/blood-2012-01-402404
143. Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal (2010) 3:ra42. doi:10.1126/scisignal.2000751
144. Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature (2007) 450:1253–7. doi:10.1038/nature06421
145. Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J (2008) 27:1110–21. doi:10.1038/emboj.2008.31
146. Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity (2007) 27:135–44. doi:10.1016/j.immuni.2007.05.022
147. Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science (2007) 315:1398–401. doi:10.1126/science.1136880
148. Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med (2010) 16:90–7. doi:10.1038/nm.2069
149. Irving AT, Mimuro H, Kufer TA, Lo C, Wheeler R, Turner LJ, et al. The immune receptor NOD1 and kinase RIP2 interact with bacterial peptidoglycan on early endosomes to promote autophagy and inflammatory signaling. Cell Host Microbe (2014) 15:623–35. doi:10.1016/j.chom.2014.04.001
150. Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature (2012) 482:414–8. doi:10.1038/nature10744
151. Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol (2014) 12:101–14. doi:10.1038/nrmicro3160
152. Starr T, Child R, Wehrly TD, Hansen B, Hwang S, Lopez-Otin C, et al. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe (2012) 11:33–45. doi:10.1016/j.chom.2011.12.002
153. Patel KK, Stappenbeck TS. Autophagy and intestinal homeostasis. Annu Rev Physiol (2013) 75:241–62. doi:10.1146/annurev-physiol-030212-183658
154. Baxt LA, Xavier RJ. Role of autophagy in the maintenance of intestinal homeostasis. Gastroenterology (2015) 149:553–62. doi:10.1053/j.gastro.2015.06.046
155. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature (2008) 456:264–8. doi:10.1038/nature07383
156. Plantinga TS, Crisan TO, Oosting M, van de Veerdonk FL, de Jong DJ, Philpott DJ, et al. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut (2011) 60:1229–35. doi:10.1136/gut.2010.228908
157. Peral de Castro C, Jones SA, Ni Cheallaigh C, Hearnden CA, Williams L, Winter J, et al. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J Immunol (2012) 189:4144–53. doi:10.4049/jimmunol.1201946
158. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12:222–30. doi:10.1038/ni.1980
159. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi:10.1038/nature09663
160. Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol (2013) 14:480–8. doi:10.1038/ni.2563
161. Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem (2011) 286:9587–97. doi:10.1074/jbc.M110.202911
162. Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, et al. NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell (2016) 164:896–910. doi:10.1016/j.cell.2015.12.057
163. Ravindran R, Khan N, Nakaya HI, Li S, Loebbermann J, Maddur MS, et al. Vaccine activation of the nutrient sensor GCN2 in dendritic cells enhances antigen presentation. Science (2014) 343:313–7. doi:10.1126/science.1246829
164. Ravindran R, Loebbermann J, Nakaya HI, Khan N, Ma H, Gama L, et al. The amino acid sensor GCN2 controls gut inflammation by inhibiting inflammasome activation. Nature (2016) 531(7595):523–7. doi:10.1038/nature17186
165. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J (2011) 30:4701–11. doi:10.1038/emboj.2011.398
166. Wang LJ, Huang HY, Huang MP, Liou W, Chang YT, Wu CC, et al. The microtubule-associated protein EB1 links AIM2 inflammasomes with autophagy-dependent secretion. J Biol Chem (2014) 289:29322–33. doi:10.1074/jbc.M114.559153
167. Zhang M, Kenny SJ, Ge L, Xu K, Schekman R. Translocation of interleukin-1 beta into a vesicle intermediate in autophagy-mediated secretion. Elife (2015) 4:e11205. doi:10.7554/eLife.11205
168. Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A (2009) 106:2770–5. doi:10.1073/pnas.0807694106
169. Lei Y, Wen HT, Yu YB, Taxman DJ, Zhang L, Widman DG, et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity (2012) 36:933–46. doi:10.1016/j.immuni.2012.03.025
170. Kanayama M, Inoue M, Danzaki K, Hammer G, He YW, Shinohara ML. Autophagy enhances NFkappaB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat Commun (2015) 6:5779. doi:10.1038/ncomms6779
171. Fujishima Y, Nishiumi S, Masuda A, Inoue J, Nguyen NM, Irino Y, et al. Autophagy in the intestinal epithelium reduces endotoxin-induced inflammatory responses by inhibiting NF-kappaB activation. Arch Biochem Biophys (2011) 506:223–35. doi:10.1016/j.abb.2010.12.009
172. Lee JP, Foote A, Fan H, de Castro CP, Lang T, Jones SA, et al. Loss of autophagy enhances MIF/macrophage migration inhibitory factor release by macrophages. Autophagy (2016) 12:907–16. doi:10.1080/15548627.2016.1164358
173. Hao NB, He YF, Luo G, Yong X, Zhang Y, Yang SM. Macrophage migration inhibitory factor polymorphism and the risk of ulcerative colitis and Crohn’s disease in Asian and European populations: a meta-analysis. BMJ Open (2013) 3:e003729. doi:10.1136/bmjopen-2013-003729
174. Yang J, Li Y, Zhang X. Meta-analysis of macrophage migration inhibitory factor (MIF) gene -173G/C polymorphism and inflammatory bowel disease (IBD) risk. Int J Clin Exp Med (2015) 8:9570–4.
175. Wesemann DR, Portuguese AJ, Meyers RM, Gallagher MP, Cluff-Jones K, Magee JM, et al. Microbial colonization influences early B-lineage development in the gut lamina propria. Nature (2013) 501:112–5. doi:10.1038/nature12496
176. Fritz JH, Rojas OL, Simard N, McCarthy DD, Hapfelmeier S, Rubino S, et al. Acquisition of a multifunctional IgA+ plasma cell phenotype in the gut. Nature (2012) 481:199–203. doi:10.1038/nature10698
177. Brandtzaeg P. Mucosal immunity: induction, dissemination, and effector functions. Scand J Immunol (2009) 70:505–15. doi:10.1111/j.1365-3083.2009.02319.x
178. Macpherson AJ, Geuking MB, Slack E, Hapfelmeier S, McCoy KD. The habitat, double life, citizenship, and forgetfulness of IgA. Immunol Rev (2011) 245:132–46. doi:10.1111/j.1600-065X.2011.01072.x
179. Pabst O. New concepts in the generation and functions of IgA. Nat Rev Immunol (2012) 12:821–32. doi:10.1038/nri3322
180. Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell (2014) 158:1000–10. doi:10.1016/j.cell.2014.08.006
181. Bunker JJ, Flynn TM, Koval JC, Shaw DG, Meisel M, McDonald BD, et al. Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity (2015) 43:541–53. doi:10.1016/j.immuni.2015.08.007
182. Hapfelmeier S, Lawson MA, Slack E, Kirundi JK, Stoel M, Heikenwalder M, et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science (2010) 328:1705–9. doi:10.1126/science.1188454
183. Lindner C, Thomsen I, Wahl B, Ugur M, Sethi MK, Friedrichsen M, et al. Diversification of memory B cells drives the continuous adaptation of secretory antibodies to gut microbiota. Nat Immunol (2015) 16:880–8. doi:10.1038/ni.3213
184. Yoshida M, Claypool SM, Wagner JS, Mizoguchi E, Mizoguchi A, Roopenian DC, et al. Human neonatal Fc receptor mediates transport of IgG into luminal secretions for delivery of antigens to mucosal dendritic cells. Immunity (2004) 20:769–83. doi:10.1016/j.immuni.2004.05.007
185. Pyzik M, Rath T, Lencer WI, Baker K, Blumberg RS. FcRn: the architect behind the immune and nonimmune functions of IgG and albumin. J Immunol (2015) 194:4595–603. doi:10.4049/jimmunol.1403014
186. Coeffier M, Lorentz A, Manns MP, Bischoff SC. Epsilon germ-line and IL-4 transcripts are expressed in human intestinal mucosa and enhanced in patients with food allergy. Allergy (2005) 60:822–7. doi:10.1111/j.1398-9995.2005.00782.x
187. Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol (2008) 8:205–17. doi:10.1038/nri2273
188. Belut D, Moneret-Vautrin DA, Nicolas JP, Grilliat JP. IgE levels in intestinal juice. Dig Dis Sci (1980) 25:323–32. doi:10.1007/BF01308055
189. Berin MC, Sampson HA. Food allergy: an enigmatic epidemic. Trends Immunol (2013) 34:390–7. doi:10.1016/j.it.2013.04.003
190. Yang PC, Berin MC, Yu LC, Conrad DH, Perdue MH. Enhanced intestinal transepithelial antigen transport in allergic rats is mediated by IgE and CD23 (FcepsilonRII). J Clin Invest (2000) 106:879–86. doi:10.1172/JCI9258
191. Bevilacqua C, Montagnac G, Benmerah A, Candalh C, Brousse N, Cerf-Bensussan N, et al. Food allergens are protected from degradation during CD23-mediated transepithelial transport. Int Arch Allergy Immunol (2004) 135:108–16. doi:10.1159/000080653
192. Baker K, Lencer WI, Blumberg RS. Beyond IgA: the mucosal immunoglobulin alphabet. Mucosal Immunol (2010) 3:324–5. doi:10.1038/mi.2010.15
193. Negrao-Correa D, Adams LS, Bell RG. Intestinal transport and catabolism of IgE: a major blood-independent pathway of IgE dissemination during a Trichinella spiralis infection of rats. J Immunol (1996) 157:4037–44.
194. Burton OT, Noval Rivas M, Zhou JS, Logsdon SL, Darling AR, Koleoglou KJ, et al. Immunoglobulin E signal inhibition during allergen ingestion leads to reversal of established food allergy and induction of regulatory T cells. Immunity (2014) 41:141–51. doi:10.1016/j.immuni.2014.05.017
195. Pengo N, Scolari M, Oliva L, Milan E, Mainoldi F, Raimondi A, et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat Immunol (2013) 14:298–305. doi:10.1038/ni.2524
196. Conway KL, Kuballa P, Khor B, Zhang M, Shi HN, Virgin HW, et al. ATG5 regulates plasma cell differentiation. Autophagy (2013) 9:528–37. doi:10.4161/auto.23484
197. Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, et al. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med (2014) 20:503–10. doi:10.1038/nm.3521
198. Chen M, Kodali S, Jang A, Kuai L, Wang J. Requirement for autophagy in the long-term persistence but not initial formation of memory B cells. J Immunol (2015) 194:2607–15. doi:10.4049/jimmunol.1403001
199. Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol (2014) 15:1152–61. doi:10.1038/ni.3025
200. Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, et al. Autophagy is a critical regulator of memory CD8(+) T cell formation. Elife (2014) 3:e03706. doi:10.7554/eLife.03706
201. Koch U, Radtke F. Mechanisms of T cell development and transformation. Annu Rev Cell Dev Biol (2011) 27:539–62. doi:10.1146/annurev-cellbio-092910-154008
202. Stritesky GL, Jameson SC, Hogquist KA. Selection of self-reactive T cells in the thymus. Annu Rev Immunol (2012) 30:95–114. doi:10.1146/annurev-immunol-020711-075035
203. Derbinski J, Schulte A, Kyewski B, Klein L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol (2001) 2:1032–9. doi:10.1038/ni723
204. Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature (2008) 455:396–400. doi:10.1038/nature07208
205. Aichinger M, Wu C, Nedjic J, Klein L. Macroautophagy substrates are loaded onto MHC class II of medullary thymic epithelial cells for central tolerance. J Exp Med (2013) 210:287–300. doi:10.1084/jem.20122149
206. Sukseree S, Mildner M, Rossiter H, Pammer J, Zhang CF, Watanapokasin R, et al. Autophagy in the thymic epithelium is dispensable for the development of self-tolerance in a novel mouse model. PLoS One (2012) 7:e38933. doi:10.1371/journal.pone.0038933
207. Bendelac A. Nondeletional pathways for the development of autoreactive thymocytes. Nat Immunol (2004) 5:557–8. doi:10.1038/ni0604-557
208. Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol (2014) 14:667–85. doi:10.1038/nri3738
209. Cheroutre H, Lambolez F, Mucida D. The light and dark sides of intestinal intraepithelial lymphocytes. Nat Rev Immunol (2011) 11:445–56. doi:10.1038/nri3007
210. Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity (2009) 31:321–30. doi:10.1016/j.immuni.2009.06.020
211. Shale M, Schiering C, Powrie F. CD4(+) T-cell subsets in intestinal inflammation. Immunol Rev (2013) 252:164–82. doi:10.1111/imr.12039
212. Ullrich R, Schieferdecker HL, Ziegler K, Riecken EO, Zeitz M. Gamma-delta-T-cells in the human intestine express surface-markers of activation and are preferentially located in the epithelium. Cell Immunol (1990) 128:619–27. doi:10.1016/0008-8749(90)90053-T
213. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol (2012) 12:749–61. doi:10.1038/nri3307
214. Ussher JE, Klenerman P, Willberg CB. Mucosal-associated invariant T-cells: new players in anti-bacterial immunity. Front Immunol (2014) 5:450. doi:10.3389/fimmu.2014.00450
215. Matsuda JL, Mallevaey T, Scott-Browne J, Gapin L. CD1d-restricted iNKT cells, the ‘Swiss-Army knife’ of the immune system. Curr Opin Immunol (2008) 20:358–68. doi:10.1016/j.coi.2008.03.018
216. Duan J, Chung H, Troy E, Kasper DL. Microbial colonization drives expansion of IL-1 receptor 1-expressing and IL-17-producing gamma/delta T cells. Cell Host Microbe (2010) 7:140–50. doi:10.1016/j.chom.2010.01.005
217. Ismail AS, Severson KM, Vaishnava S, Behrendt CL, Yu X, Benjamin JL, et al. Gammadelta intraepithelial lymphocytes are essential mediators of host-microbial homeostasis at the intestinal mucosal surface. Proc Natl Acad Sci U S A (2011) 108:8743–8. doi:10.1073/pnas.1019574108
218. Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science (2012) 336:489–93. doi:10.1126/science.1219328
219. An D, Oh SF, Olszak T, Neves JF, Avci FY, Erturk-Hasdemir D, et al. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell (2014) 156:123–33. doi:10.1016/j.cell.2013.11.042
220. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol (2013) 31:259–83. doi:10.1146/annurev-immunol-032712-095956
221. Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol (2012) 13:907–15. doi:10.1038/ni.2386
222. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi:10.1016/j.immuni.2011.09.021
223. Michalek RD, Gerriets VA, Nichols AG, Inoue M, Kazmin D, Chang CY, et al. Estrogen-related receptor-alpha is a metabolic regulator of effector T-cell activation and differentiation. Proc Natl Acad Sci U S A (2011) 108:18348–53. doi:10.1073/pnas.1108856108
224. Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med (2015) 212(9):1345–60. doi:10.1084/jem.20151159
225. Gebhardt T, Mueller SN, Heath WR, Carbone FR. Peripheral tissue surveillance and residency by memory T cells. Trends Immunol (2013) 34:27–32. doi:10.1016/j.it.2012.08.008
226. Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol (2016) 16:79–89. doi:10.1038/nri.2015.3
227. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol (2010) 28:445–89. doi:10.1146/annurev-immunol-030409-101212
228. Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol (2014) 14:435–46. doi:10.1038/nri3701
229. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186:3299–303. doi:10.4049/jimmunol.1003613
230. Wang C, Collins M, Kuchroo VK. Effector T cell differentiation: are master regulators of effector T cells still the masters? Curr Opin Immunol (2015) 37:6–10. doi:10.1016/j.coi.2015.08.001
231. O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science (2010) 327:1098–102. doi:10.1126/science.1178334
232. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell (2000) 100:655–69. doi:10.1016/S0092-8674(00)80702-3
233. Scott P, Pearce E, Cheever AW, Coffman RL, Sher A. Role of cytokines and CD4+ T-cell subsets in the regulation of parasite immunity and disease. Immunol Rev (1989) 112:161–82. doi:10.1111/j.1600-065X.1989.tb00557.x
234. Suzuki Y, Remington JS. The effect of anti-IFN-gamma antibody on the protective effect of Lyt-2+ immune T cells against toxoplasmosis in mice. J Immunol (1990) 144:1954–6.
235. Suzuki Y, Orellana MA, Schreiber RD, Remington JS. Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science (1988) 240:516–8. doi:10.1126/science.3128869
236. Trembleau S, Penna G, Bosi E, Mortara A, Gately MK, Adorini L. Interleukin 12 administration induces T helper type 1 cells and accelerates autoimmune diabetes in NOD mice. J Exp Med (1995) 181:817–21. doi:10.1084/jem.181.2.817
237. Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med (2004) 200:79–87. doi:10.1084/jem.20031819
238. Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL. Inhibition of Th1 responses prevents inflammatory bowel disease in SCID mice reconstituted with CD45RBhi CD4+ T cells. Immunity (1994) 1:553–62. doi:10.1016/1074-7613(94)90045-0
239. Jankovic D, Kullberg MC, Noben-Trauth N, Caspar P, Paul WE, Sher A. Single cell analysis reveals that IL-4 receptor/Stat6 signaling is not required for the in vivo or in vitro development of CD4(+) lymphocytes with a Th2 cytokine profile. J Immunol (2000) 164:3047–55. doi:10.4049/jimmunol.164.6.3047
240. Kitajima M, Lee HC, Nakayama T, Ziegler SF. TSLP enhances the function of helper type 2 cells. Eur J Immunol (2011) 41:1862–71. doi:10.1002/eji.201041195
241. Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol (2015) 15:271–82. doi:10.1038/nri3831
242. Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell (1997) 89:587–96. doi:10.1016/S0092-8674(00)80240-8
243. Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. Transcription factor GATA-3 is differentially expressed murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J Biol Chem (1997) 272:21597–603.
244. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol (2011) 12:295–303. doi:10.1038/ni.2005
245. Chapman NM, Chi H. mTOR links environmental signals to T cell fate decisions. Front Immunol (2014) 5:686. doi:10.3389/fimmu.2014.00686
246. Gause WC, Wynn TA, Allen JE. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat Rev Immunol (2013) 13:607–14. doi:10.1038/nri3476
247. Grencis RK. Immunity to helminths: resistance, regulation, and susceptibility to gastrointestinal nematodes. Annu Rev Immunol (2015) 33:201–25. doi:10.1146/annurev-immunol-032713-120218
248. Tsai M, Starkl P, Marichal T, Galli SJ. Testing the ‘toxin hypothesis of allergy’: mast cells, IgE, and innate and acquired immune responses to venoms. Curr Opin Immunol (2015) 36:80–7. doi:10.1016/j.coi.2015.07.001
249. Odegaard JI, Chawla A. Type 2 responses at the interface between immunity and fat metabolism. Curr Opin Immunol (2015) 36:67–72. doi:10.1016/j.coi.2015.07.003
250. Cheng LE, Locksley RM. Allergic inflammation – innately homeostatic. Cold Spring Harb Perspect Biol (2015) 7:a016352. doi:10.1101/cshperspect.a016352
251. Linterman MA, Liston A, Vinuesa CG. T-follicular helper cell differentiation and the co-option of this pathway by non-helper cells. Immunol Rev (2012) 247:143–59. doi:10.1111/j.1600-065X.2012.01121.x
252. Kemeny DM. The role of the T follicular helper cells in allergic disease. Cell Mol Immunol (2012) 9:386–9. doi:10.1038/cmi.2012.31
253. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity (2009) 31:457–68. doi:10.1016/j.immuni.2009.07.002
254. Johnston R, Poholek A, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325:1006–10. doi:10.1126/science.1175870
255. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science (2009) 325:1001–5. doi:10.1126/science.1176676
256. Ray JP, Staron MM, Shyer JA, Ho PC, Marshall HD, Gray SM, et al. The interleukin-2-mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity (2015) 43:690–702. doi:10.1016/j.immuni.2015.08.017
257. Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med (2003) 198:1951–7. doi:10.1084/jem.20030896
258. Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem (2003) 278:1910–4. doi:10.1074/jbc.M207577200
259. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med (2006) 203:2271–9. doi:10.1084/jem.20061308
260. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6:1133–41. doi:10.1038/ni1261
261. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6:1123–32. doi:10.1038/ni1254
262. Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med (2007) 204:2803–12. doi:10.1084/jem.20071397
263. Awasthi A, Riol-Blanco L, Jager A, Korn T, Pot C, Galileos G, et al. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol (2009) 182:5904–8. doi:10.4049/jimmunol.0900732
264. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi:10.1016/j.cell.2006.07.035
265. Hu X, Wang Y, Hao LY, Liu X, Lesch CA, Sanchez BM, et al. Sterol metabolism controls T(H)17 differentiation by generating endogenous RORgamma agonists. Nat Chem Biol (2015) 11:141–7. doi:10.1038/nchembio.1714
266. Santori FR, Huang PX, de Pavert SAV, Douglass EF, Leaver DJ, Haubrich BA, et al. Identification of natural ROR gamma ligands that regulate the development of lymphoid cells. Cell Metab (2015) 21:286–97. doi:10.1016/j.cmet.2015.01.004
267. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol (2009) 27:485–517. doi:10.1146/annurev.immunol.021908.132710
268. Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity (2010) 33:279–88. doi:10.1016/j.immuni.2010.08.010
269. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med (2005) 201:233–40. doi:10.1084/jem.20041257
270. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol (2009) 10:314–24. doi:10.1038/ni.1698
271. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146:772–84. doi:10.1016/j.cell.2011.07.033
272. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208:1367–76. doi:10.1084/jem.20110278
273. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20:1327–33. doi:10.1038/nm.3704
274. Huang WT, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis (2004) 190:624–31. doi:10.1086/422329
275. Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, et al. Differential roles of interleukin-17A and-17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity (2009) 30:108–19. doi:10.1016/j.immuni.2008.11.009
276. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med (2001) 194:519–27. doi:10.1084/jem.194.4.519
277. Kullberg MC, Jankovic D, Feng CG, Hue S, Gorelick PL, McKenzie BS, et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J Exp Med (2006) 203:2485–94. doi:10.1084/jem.20061082
278. Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science (2016) 351:933–9. doi:10.1126/science.aad0314
279. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139:485–98. doi:10.1016/j.cell.2009.09.033
280. Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity (2009) 31:677–89. doi:10.1016/j.immuni.2009.08.020
281. Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, et al. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity (2014) 40:594–607. doi:10.1016/j.immuni.2014.03.005
282. Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, et al. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature (2014) 510:152–6. doi:10.1038/nature13279
283. Reynolds JM, Pappu BP, Peng J, Martinez GJ, Zhang Y, Chung Y, et al. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity (2010) 32:692–702. doi:10.1016/j.immuni.2010.04.010
284. Izcue A, Coombes JL, Powrie F. Regulatory lymphocytes and intestinal inflammation. Annu Rev Immunol (2009) 27:313–38. doi:10.1146/annurev.immunol.021908.132657
285. Harrison OJ, Powrie FM. Regulatory T cells and immune tolerance in the intestine. Cold Spring Harb Perspect Biol (2013) 5:e018341. doi:10.1101/cshperspect.a018341
286. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi:10.1146/annurev.immunol.25.022106.141623
287. Li MO, Rudensky AY. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol (2016) 16:220–33. doi:10.1038/nri.2016.26
288. Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu Rev Immunol (2012) 30:733–58. doi:10.1146/annurev-immunol-020711-075043
289. Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8:523–32. doi:10.1038/nri2343
290. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med (2009) 361:2033–45. doi:10.1056/NEJMoa0907206
291. Liston A, Gray DH. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol (2014) 14:154–65. doi:10.1038/nri3605
292. Rosenblum MD, Gratz IK, Paw JS, Lee K, Marshak-Rothstein A, Abbas AK. Response to self antigen imprints regulatory memory in tissues. Nature (2011) 480:538–42. doi:10.1038/nature10664
293. Rowe JH, Ertelt JM, Xin LJ, Way SS. Pregnancy imprints regulatory memory that sustains anergy to fetal antigen. Nature (2012) 490:102–19. doi:10.1038/nature11462
294. Sanchez AM, Zhu JG, Huang XP, Yang YP. The development and function of memory regulatory T cells after acute viral infections. J Immunol (2012) 189:2805–14. doi:10.4049/jimmunol.1200645
295. Rosenblum MD, Way SS, Abbas AK. Regulatory T cell memory. Nat Rev Immunol (2016) 16:90–101. doi:10.1038/nri.2015.1
296. Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol (2013) 14:1007–13. doi:10.1038/ni.2683
297. Denning TL, Kim G, Kronenberg M. Cutting edge: CD4(+)CD25(+) for intestinal homing can regulatory T cells impaired prevent colitis. J Immunol (2005) 174:7487–91. doi:10.4049/jimmunol.174.12.7487
298. Kim SV, Xiang WKV, Kwak C, Yang Y, Lin XYW, Ota M, et al. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science (2013) 340:1456–9. doi:10.1126/science.1237013
299. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341:569–73. doi:10.1126/science.1241165
300. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A (2010) 107:12204–9. doi:10.1073/pnas.0909122107
301. Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, et al. The toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science (2011) 332:974–7. doi:10.1126/science.1206095
302. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature (2013) 500:232–6. doi:10.1038/nature12331
303. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5. doi:10.1038/nature12726
304. Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, et al. Mucosal immunology. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science (2015) 349:993–7. doi:10.1126/science.aaa9420
305. Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, et al. Mucosal immunology. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science (2015) 349:989–93. doi:10.1126/science.aac4263
306. Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med (2007) 204:25–31. doi:10.1084/jem.20061303
307. Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc Natl Acad Sci U S A (2012) 109:8670–5. doi:10.1073/pnas.1205305109
308. Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol (2009) 182:4046–55. doi:10.4049/jimmunol.0801143
309. Parekh VV, Wu L, Boyd KL, Williams JA, Gaddy JA, Olivares-Villagomez D, et al. Impaired autophagy, defective T cell homeostasis, and a wasting syndrome in mice with a T cell-specific deletion of Vps34. J Immunol (2013) 190:5086–101. doi:10.4049/jimmunol.1202071
310. Kovacs JR, Li C, Yang Q, Li G, Garcia IG, Ju S, et al. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ (2012) 19:144–52. doi:10.1038/cdd.2011.78
311. Jia W, He YW. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J Immunol (2011) 186:5313–22. doi:10.4049/jimmunol.1002404
312. Kabat AM, Harrison OJ, Riffelmacher T, Moghaddam AE, Pearson CF, Laing A, et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. Elife (2016) 5:e12444. doi:10.7554/eLife.12444
313. Stephenson LM, Miller BC, Ng A, Eisenberg J, Zhao Z, Cadwell K, et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy (2009) 5:625–35. doi:10.4161/auto.5.5.8133
314. Salio M, Puleston DJ, Mathan TSM, Shepherd D, Stranks AJ, Adamopoulou E, et al. Essential role for autophagy during invariant NKT cell development. Proc Natl Acad Sci U S A (2014) 111:5678–87. doi:10.1073/pnas.1413935112
315. Pei B, Zhao M, Miller BC, Vela JL, Bruinsma MW, Virgin HW, et al. Invariant NKT cells require autophagy to coordinate proliferation and survival signals during differentiation. J Immunol (2015) 194:5872–84. doi:10.4049/jimmunol.1402154
316. Li C, Capan E, Zhao Y, Zhao J, Stolz D, Watkins SC, et al. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal Cell death. J Immunol (2006) 177:5163–8. doi:10.4049/jimmunol.177.8.5163
317. Hubbard VM, Valdor R, Patel B, Singh R, Cuervo AM, Macian F. Macroautophagy regulates energy metabolism during effector T cell activation. J Immunol (2010) 185:7349–57. doi:10.4049/jimmunol.1000576
318. Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol (2011) 186:1564–74. doi:10.4049/jimmunol.1001822
319. McLeod IX, Zhou X, Li QJ, Wang F, He YW. The class III kinase Vps34 promotes T lymphocyte survival through regulating IL-7Ralpha surface expression. J Immunol (2011) 187:5051–61. doi:10.4049/jimmunol.1100710
320. Hildeman DA, Mitchell T, Teague TK, Henson P, Day BJ, Kappler J, et al. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity (1999) 10:735–44. doi:10.1016/S1074-7613(00)80072-2
321. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38:225–36. doi:10.1016/j.immuni.2012.10.020
322. Okoye I, Wang L, Pallmer K, Richter K, Ichimura T, Haas R, et al. T cell metabolism. The protein LEM promotes CD8(+) T cell immunity through effects on mitochondrial respiration. Science (2015) 348:995–1001. doi:10.1126/science.aaa7516
323. Chaudhri G, Hunt NH, Clark IA, Ceredig R. Antioxidants inhibit proliferation and cell surface expression of receptors for interleukin-2 and transferrin in T lymphocytes stimulated with phorbol myristate acetate and ionomycin. Cell Immunol (1988) 115:204–13. doi:10.1016/0008-8749(88)90174-8
324. Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med (2002) 195:59–70. doi:10.1084/jem.20010659
325. Bell BD, Leverrier S, Weist BM, Newton RH, Arechiga AF, Luhrs KA, et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci U S A (2008) 105:16677–82. doi:10.1073/pnas.0808597105
326. Feng CG, Zheng L, Jankovic D, Bafica A, Cannons JL, Watford WT, et al. The immunity-related GTPase Irgm1 promotes the expansion of activated CD4+ T cell populations by preventing interferon-gamma-induced cell death. Nat Immunol (2008) 9:1279–87. doi:10.1038/ni.1653
327. Vallabhapurapu S, Karin M. Regulation and function of NF-kappa B transcription factors in the immune system. Annu Rev Immunol (2009) 27:693–733. doi:10.1146/annurev.immunol.021908.132641
328. Schlie K, Westerback A, DeVorkin L, Hughson LR, Brandon JM, MacPherson S, et al. Survival of effector CD8+ T cells during influenza infection is dependent on autophagy. J Immunol (2015) 194:4277–86. doi:10.4049/jimmunol.1402571
329. Henson SM, Lanna A, Riddell NE, Franzese O, Macaulay R, Griffiths SJ, et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8(+) T cells. J Clin Invest (2014) 124:4004–16. doi:10.1172/JCI75051
330. Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol (2016) 17(3):277–85. doi:10.1038/ni.3365
331. Martin LJ, Gupta J, Jyothula SS, Butsch Kovacic M, Biagini Myers JM, Patterson TL, et al. Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PLoS One (2012) 7:e33454. doi:10.1371/journal.pone.0033454
332. Poon AH, Chouiali F, Tse SM, Litonjua AA, Hussain SNA, Baglole CJ, et al. Genetic and histologic evidence for autophagy in asthma pathogenesis. J Allergy Clin Immunol (2012) 129:569–71. doi:10.1016/j.jaci.2011.09.035
333. Jiang P, Mizushima N. Autophagy and human diseases. Cell Res (2014) 24:69–79. doi:10.1038/cr.2013.161
334. Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science (2010) 329:229–32. doi:10.1126/science.1190354
335. Moron B, Spalinger M, Kasper S, Atrott K, Frey-Wagner I, Fried M, et al. Activation of protein tyrosine phosphatase non-receptor type 2 by spermidine exerts anti-inflammatory effects in human THP-1 monocytes and in a mouse model of acute colitis. PLoS One (2013) 8:e73703. doi:10.1371/journal.pone.0073703
336. Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol (2009) 11:1305–14. doi:10.1038/ncb1975
337. Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature (2013) 494:201–6. doi:10.1038/nature11866
338. Shaw SY, Tran K, Castoreno AB, Peloquin JM, Lassen KG, Khor B, et al. Selective modulation of autophagy, innate immunity, and adaptive immunity by small molecules. ACS Chem Biol (2013) 8:2724–33. doi:10.1021/cb400352d
339. Isakson P, Bjoras M, Boe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood (2010) 116:2324–31. doi:10.1182/blood-2010-01-261040
Keywords: IBD, autophagy, ATG16L1, colitis, Treg cells, intestinal epithelial cells, metabolism, inflammasome
Citation: Kabat AM, Pott J and Maloy KJ (2016) The Mucosal Immune System and Its Regulation by Autophagy. Front. Immunol. 7:240. doi: 10.3389/fimmu.2016.00240
Received: 30 April 2016; Accepted: 07 June 2016;
Published: 22 June 2016
Edited by:
Ed C. Lavelle, Trinity College Dublin, IrelandCopyright: © 2016 Kabat, Pott and Maloy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agnieszka M. Kabat, agnieszka.kabat@path.ox.ac.uk;
Kevin J. Maloy, kevin.maloy@path.ox.ac.uk