 Thomas Clapes
Thomas Clapes Stylianos Lefkopoulos
Stylianos Lefkopoulos Eirini Trompouki
Eirini Trompouki- Department of Cellular and Molecular Immunology, Max Planck Institute of Immunobiology and Epigenetics, Freiburg, Germany
Hematopoietic stem cells (HSCs) are a rare population that gives rise to almost all cells of the hematopoietic system, including immune cells. Until recently, it was thought that immune cells sense inflammatory signaling and HSCs respond only secondarily to these signals. However, it was later shown that adult HSCs could directly sense and respond to inflammatory signals, resulting in a higher output of immune cells. Recent studies demonstrated that inflammatory signaling is also vital for HSC ontogeny. These signals are thought to arise in the absence of pathogens, are active during development, and indispensable for HSC formation. In contrast, during times of stress and disease, inflammatory responses can be activated and can have devastating effects on HSCs. In this review, we summarize the current knowledge about inflammatory signaling in HSC development and maintenance, as well as the endogenous molecular cues that can trigger inflammatory pathway activation. Finally, we comment of the role of inflammatory signaling in hematopoietic diseases.
Introduction
The processes of blood formation during development and its maintenance in adulthood have been intense topics of study for many years. Special attention is given to hematopoietic stem cells (HSCs) that can replenish the hematopoietic system and play pivotal roles during development and adult homeostasis. Given their multipotential and self-renewing capacities, HSCs are the key cell type in bone marrow (BM) transplantations to treat hematological diseases. Since some malignancies are the outcome of deregulation of the hematopoietic system, it is critical to understand how HSC homeostasis is maintained under steady state, but also under various stress situations. In this review, we will discuss the role of inflammatory cytokines in hematopoietic stem cell emergence but also during adulthood summarizing the current status. We will give a brief outline of the role of these signals in disease. The detailed ontogeny and molecular control of HSCs is beyond the scope of this review, but has been described in others (1–3).
Fairly and recently, inflammatory signaling and its role in HSC formation and maintenance has been of great interest for many researchers. For many years, it was believed that HSCs could not respond directly to these signals. Many groups have now shown that different cytokines and tonic inflammatory signaling are sensed by HSCs. These signals are required for the formation of HSCs during both mouse and zebrafish development. Similar cytokines are pivotal for maintaining HSC quiescence in adult organisms and regulating their differentiation in case of stress or disease.
Hematopoiesis is conserved between model organisms and two of them have been used extensively for studying both embryonic and adult hematopoiesis: zebrafish and mouse. During embryonic development, they both experience different hematopoietic waves (4, 5). The primitive wave is only transient and gives rise mainly to erythrocytes and myeloid cells, whereas the definitive wave gives rise to the actual HSCs through the conserved endothelial-to-hematopoietic transition (EHT) (6–9). In both organisms, EHT takes place in the aorta-gonad-mesonephros (AGM) region. Whether EHT in mice also takes place in other hematopoietic organs, such as in the placenta (10) or in the head (11–13), is still under question. Soon after generation, HSCs migrate to the caudal hematopoietic tissue (CHT) in zebrafish and to the fetal liver in mouse, where they amplify. Finally, HSCs reach the adult hematopoietic organs, namely the zebrafish kidney marrow and the mouse BM, where they remain quiescent and few of them actively replenish the hematopoietic system. However, in case of stress emergency, hematopoiesis arises and HSCs take the lead in reestablishing the balance of the hematopoietic system. All studies concerning hematopoiesis in those two organisms not only confirm some basic anatomical and functional equivalences between them but also a shared genetic program.
Interferons
Interferons (IFNs) are antiviral cytokines produced by host cells in response to pathogens (viruses, bacteria, and parasites) and tumor cells. Type I IFNs (IFN-α, IFN-β) are produced by a variety of cell types and signal through the IFN α/β receptor (IFNAR) on target cells. Type II IFN (IFN-γ) is released mainly by T-cells and NK-cells and signal via IFN-γ receptor (IFNγR) present on immune cells. Finally, type III IFNs include three IFN-λ (lambda) molecules, called IFN-λ1, IFN-λ2, and IFN-λ3 that signal through IL10R2 and IFNLR1 (14).
The Impact of Interferons on Embryonic Hematopoiesis
The zebrafish mutant crfb17−/− (ortholog of IFNGR1) demonstrated a marked reduction in HSCs budding from the ventral wall of the dorsal aorta, compared to their crfb17+/+ siblings. Consistently, morpholino-mediated knockdown of the same receptor, as well as ifng1-2 (15) reduced the expression of an important marker of HSPCs, runx1, at 33 hpf (hours post fertilization), indicating that IFN-γ signaling positively regulates HSCs. Notch signaling, a known regulator of EHT (16), was not sufficient to induce the EHT process in an ifng1-2-deficient background proving that ifng acts downstream of Notch (15). Interestingly, Sawamiphak et al. also demonstrated that IFN-γ expression in the AGM is dependent on blood flow (15), another important positive regulator of HSC emergence (17). Indeed, ifng1-2 overexpression partially rescued this defect (15), hinting to an interplay between IFN signaling and mechanical signals that regulate blood flow.
This correlation between IFN-γ signaling and HSC emergence seems to be conserved in mice (18). The numbers of HSCs in mouse embryos deficient in IFN-γ or IFN-γ receptor are decreased compared to wild type embryos. At the same time, transplantable HSCs are also decreased in embryos deficient for this receptor, all together showing an important role of this cytokine in HSC emergence. The conserved IFN effect on mammalian hematopoiesis is also supported by the fact that inflammatory signaling was found active in human fetal hematopoietic stem and progenitor cells (HSPCs) (18).
Apart from type II, type I IFNs can also regulate the process of embryonic hematopoiesis in both organisms, in a manner consistent with that of IFN-γ signaling. In particular, IFN-α in mice and its ortholog, IFN-ϕ, in zebrafish positively regulate the number of HSPCs during embryonic development (18), as embryos lacking IFN-α or -γ signaling harbor fewer HSPCs in the AGM. In another mouse study, it was shown that AGM HSCs exhibit lower levels of IFN-α than fetal liver HSCs, and this attribute can account for the lower engraftment potential of AGM HSCs. Treatment with IFN-α greatly enhances the repopulation capacity of AGM-HSCs. Mechanistically, adenine-thymine-rich interactive domain-3a (Arid3a) is responsible for the regulation of Stat1 and various IFN-responsive genes in developmental hematopoiesis (19).
Last, micro RNAs have been shown to regulate IFN-responsive genes and affect HSC development. mir-142-3p is essential for HSC emergence in both zebrafish and xenopus (20, 21). As shown in zebrafish, the main function of mir-142-3p is the downregulation of irf7 (20).
The Impact of Interferons on Adult Hematopoiesis
For decades, the role of IFNs, and in particular IFN-γ, has been quite controversial. In vitro studies on human cells reported that IFN-γ had generally a suppressive effect on hematopoietic progenitors (22–25), but could synergize with other growth factors, such as IL-3, IL-6, EPO, and IL-1b to stimulate HSPC proliferation (26–28). Colony-forming assays of human progenitor cells showed that all IFNs suppressed the formation of colony-forming unit-granulocyte, erythroid, macrophage, megakaryocyte (CFU-GEMM), and burst-forming unit-erythroid (BFU-E), but granulocyte-macrophage (CFU-GM) were mostly responsive to IFNγ and less to IFNα (29, 30). Interestingly, this regulation was also influenced by oxygen levels (31).
One of the first in vivo evidence demonstrating that IFNs could affect the adult hematopoietic progenitor compartment arose in a report studying lymphocytic choriomeningitis virus (LCMV) infection in mice. The authors observed that LCMV infection causes depletion of hematopoietic progenitors [colony-forming unit in the spleen (CFU-S)]. Interestingly, they demonstrated that BM aplasia following LCMV infection was abrogated in mice lacking both IFN-α/β receptors (32). Other reports have also intended to delineate the role of IFNs in vivo using mouse model. Sato et al. demonstrated that IFN-α treatment could induce the proliferation of HSCs (side population/LSK). Moreover, the authors showed that the interferon regulatory factor 2 (IRF2), a suppressor of type I IFN receptors, was responsible for maintaining HSC quiescence (33). Consistent with these findings, Essers et al. have shown that IFN-α could facilitate the exit of HSCs (LSK CD150+CD48−) from quiescence through activation of STAT-1 and AKT (34), and possibly by the upregulation of c-Myc protein (35). Furthermore, it was shown that the tonicity of the stimulation had a great impact on HSC repopulating capacity. Whereas acute and short-term (three doses) IFN-α treatment did not impair HSC activity, chronic exposure (eight doses for 2 weeks) to IFN-α greatly compromised HSC repopulation capacity in competitive transplantation assays (34). In depth studies of the acute and chronic exposure of HSCs to type I IFNs showed that, although brief exposure leads HSCs to exit quiescence, chronic exposure leads them back to quiescence. However, upon stress, when they are forced to enter cell cycle again (i.e., by transplantation), they activate a p53-dependent proapoptotic gene program (36), which would explain the suppressive effect of type-I IFNs on HSC activity.
Besides IFN-α, IFN-γ can also enhance the proliferation of HSPCs (LSK) in vitro and in vivo via STAT1 (37). Interestingly, murine BM cells cocultured with stromal cells overexpressing IFN-γ failed to reconstitute congenic recipient mice upon transplantation (38). Mice infected with Mycobaterium avium have a greater proportion of proliferating HSCs (LSK CD150+) due to increased IFN-γ signaling. Indeed, stimulation with IFN-γ alone could also mediate HSC proliferation in vivo. Moreover, IFN-γ-treated HSCs had decreased repopulating capacity compared to untreated HSCs upon in vivo transplantation assays (39). Similarly, mice infected with Ehrlichia muris also harbored increased HPSC (LSK) proliferation and reduction of long-term repopulating capacity (40). Interestingly, de Bruin et al. observed that IFN-γ impaired HSC maintenance by perturbing TPO signaling via SOCS-1. However, the authors did not find evidence for enhanced proliferation of HSCs, but rather that IFN-γ could modulate the expression of genes involved in HSC self-renewal, such as Cyclin D1 (41). Type I IFN can also regulate emergency megakaryopoiesis by upregulating megakaryocytic proteins in HSCs that already transcribe mRNAs of the megakaryocytic program (42).
Finally, ablation of the interferon-inducible GTPase Lrg-47 (Irgm1) inhibits baseline HSC proliferation and leads to severe impairment of HSCs in repopulation assays (43). Molecularly, it was proven that Irgm1 is a potent negative regulator of IFN response and that is how it exerts its action on HSCs (44).
Altogether, multiple recent reports in the literature prove that IFNs regulate HSC activity. However, it is still difficult to actually compare all these studies, as markers used to characterize HSCs differ (see Table 1). It is also important to note that some markers, such as Sca-1 (Ly6A), are upregulated following IFN-α stimulation (34). It is therefore critical to link HSC phenotype to their actual activity, by means of in vivo transplantation assays. Finally, it seems that both type I and II IFNs appear to have similar effects on HSCs. However, it is still unclear whether both types of IFN have redundant function in activating HSCs upon infection. It is likely that they are activated by specific cues, but concrete evidence is still lacking.
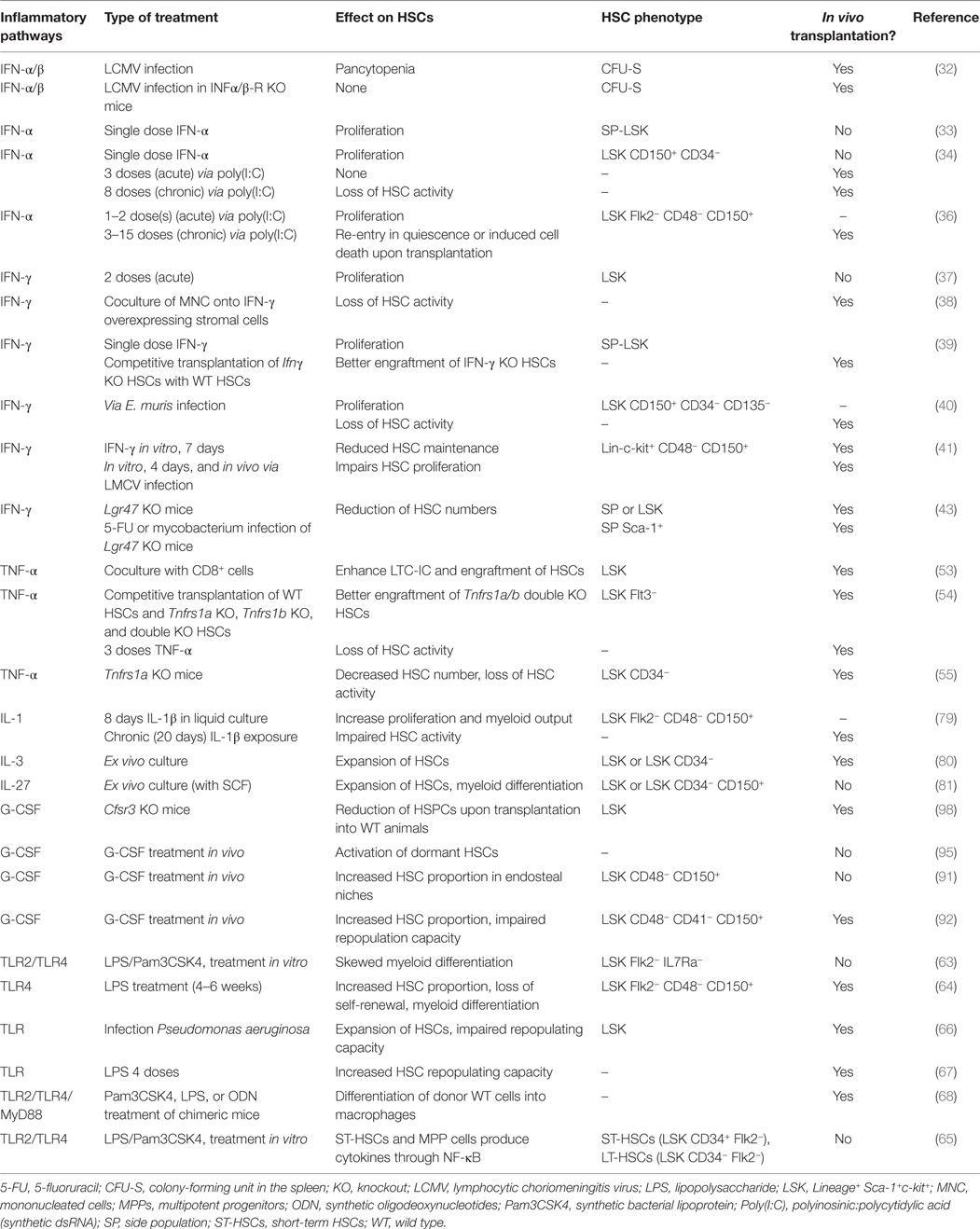
Table 1. Murine inflammatory pathways and their role in HSCs.
Tumor Necrosis Factors
Tumor necrosis factor alpha (TNFα) is a pro-inflammatory cytokine mainly produced by activated macrophages, lymphocytes, and endothelial cells (ECs). TNFα is synthesized as a prohormone and can be secreted after a cleavage process as a mature protein (45). Albeit it was first identified as a protein capable of inducing tumor cell necrosis (46), today it is also known that it functions as part of a number of different processes, ranging from cell apoptosis (47) to fever induction (48). TNFα signals through two different receptors, one of them being constitutively and almost ubiquitously expressed (p55 receptor, also known as TNFR1) and the other restricting its expression to hematopoietic cells (p75 receptor, also known as TNFR2) (49).
The Effect of TNFα on Embryonic Hematopoiesis
Recently, Espín-Palazón et al. clarified the role of TNFα in zebrafish embryonic hematopoiesis (50). Based on previous observations that this specific cytokine is necessary for embryonic blood vessel development (51) and the arterial origin of HSCs, Espín-Palazón et al. attempted to investigate a possible direct correlation between HSC emergence and TNFα signaling. By means of morpholino-mediated knockdown, at a dose that led to no vascular abnormalities, they showed that the number of cmyb+ cells in the dorsal aorta at 48 hpf was significantly reduced in tnfa- and tnfr2-, but not in tnfr1-deficient embryos (50). This indicates that TNFα signaling is required for HSC specification, and this effect is mediated through the hematopoietic-restricted Tnfr2. The source of TNFα was shown to be primitive neutrophils, answering the question about the role of these cells during early development, when their environment is relatively sterile.
Interestingly, ectopic provision of Notch signaling resulted in rescue of HSCs in both a tnfa- and a tnfr2-deficient background, thereby proving that Notch signals downstream of TNFα. Since the main mediator of TNFα is NF-κB, the authors asked whether NF-κB is required for HSC emergence. Blocking NF-κB resulted in loss of HSCs at 48 hpf and its function was suggested to lie downstream of Tnfr2, thus linking, for the first time, NF-κB with HSC emergence.
The absence of T lymphocytes by 4–5 dpf (days post fertilization) in a tnfa/tnfr2-deficient background, stresses the importance of TNF signaling not only in HSC emergence but also in maintenance of nascent HSC fate, exactly like IFN-γ does not only affect the EHT process but also has a long-term effect on HSC repopulating activity. Moreover, cooperative functions among pro-inflammatory cytokines during embryonic hematopoiesis might exist, since knockdown of both tnfa and ifng leads to a greater reduction of runx1 expression than the individual knockdown of either gene at 33 hpf (18). Interestingly, neither of the two cytokines was reported to have an influence on the primitive hematopoietic wave, implying that the role of primitive hematopoietic cells lies, at least partially, on the production of a variety of signaling molecules that will induce the subsequent definitive wave.
The Effect of TNFα on Adult Hematopoiesis
Pioneer studies in human cells have shown that TNFα treatment could inhibit the proliferation of hematopoietic progenitors in vitro (22, 25, 30). Dybedal et al. have shown that human HSPCs (CD34+ CD38−) exposed to TNFα could not repopulate NOD-SCID mice upon transplantation (52). Moreover, they have shown that TNFα promoted myeloid differentiation rather than apoptosis of HSPCs. Several in vivo studies in mice have clearly identified TNFα as an important physiologic regulator of hematopoiesis. However, some discrepancies still exist in the literature regarding the role of this cytokine in HSC activity. For instance, in vitro assays have shown that TNFα production by CD8+ cells enhanced the function of HSPCs, notably by suppressing apoptosis (53). Additionally, the authors reported that, upon transplantation, hematopoietic cells exposed to CD8+ produced-TNFα demonstrated a better engraftment (53). However, a recent study identified TNFα as a suppressor of HSC activity in vivo. The authors carefully analyzed the repopulating capacity of HSCs isolated from Tnfrs1a−/−, Tnfrs1b−/−, and double knockout mice. They observed that, although the proportion of HSCs (LSK Flk2−) was comparable to wild type animals, Tnfrs1a−/− or Tnfrs1b−/− HSCs had an increased repopulation capacity and increased self-renewal activity, as demonstrated by serial transplantation experiments (54). This phenotype was even more pronounced in double knockout animals. Furthermore, they demonstrated that treatment of wild type mice with TNFα (three doses) was sufficient to suppress HSC proliferation and repopulating capacity. Nevertheless, it seems that the effect of TNFα may differ with age. Indeed, Rebel et al. noticed that the percentage of BM HSCs (LSK CD34−) was fourfold decreased in older (6 months) Tnfrs1a−/− mice. The impaired self-renewal activity of Tnfrs1a−/− HSCs was confirmed after secondary transplantation assays (55).
As stressed in this section, the effects of TNFα on HSCs are somehow complex. The comparison of the proportion of HSCs from one study to another is hampered by the use of different markers to characterize the HSC pool (see Table 1). However, it is clear that TNFα might act as a suppressor of HSC activity in vivo, and that this role might change in older mice. Therefore, it seems that a baseline TNFα signaling is crucial for the maintenance of HSC function through aging, providing perhaps adequate stimulation for proper HSC cell division, and hence, maintenance.
In addition to TNFα, other TNF-associated molecules have been implicated to HSC survival. Depletion of the receptor-interacting serine/threonine-protein kinase 1 (RIPK1) stimulates pro-inflammatory cytokines and results in hematopoietic cell death that is partially rescued by simultaneous deletion of Ripk3. Fetal liver cells from these mice fail to reconstitute lethally irradiated recipients (56). The NF-κB-associated deubiquitinase CYLD together with its substrate TRAF2 regulate HSC cycling and promote dormancy (57).
Although the role of TNF, especially during HSC maintenance and stress, is still somewhat contradictory, these studies represent a perfect example of how a classic inflammatory signaling can play multiple roles during development and adulthood. The next step is to tightly titrate both the dosage and the timing of TNF to better understand its implication in HSC biology.
Toll-Like Receptors
Toll-like receptors (TLRs) are a family of transmembrane pattern recognition receptors that recognize a wide variety of pathogen- and danger-associated molecular patterns (PAMPs/DAMPs). TLRs are located either on the plasma membrane or in endosomes, and signal through either MyD88-dependent or TRIF-dependent pathways to activate NF-κB, IRF7, or IRF3 and induce the expression of pro-inflammatory cytokines (58, 59).
Toll-Like Receptor Influence on Embryonic Hematopoiesis
Regarding embryonic hematopoiesis in zebrafish, it was shown by He et al. that both tlr4bb and myd88 morphants demonstrated a reduced expression of the HSPC marker runx1 at both 24 and 36 hpf and of the T cell marker rag1 at 4 dpf, compared to their wild type siblings. To confirm their observations, they used the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 technology and targeted gene-specific promoters involved in TLR4–MyD88 signaling. This caused a transcriptional repression of several molecules involved in the pathway and led to a remarkably decreased expression of runx1, while the number of cmyb+kdrl+ cells in the CHT was markedly lower compared to wild type embryos, all together depicting the importance of TLR4 signaling in HSPC emergence, in a MyD88-dependent manner (60).
Notably, morpholino-mediated knockdown of ikbaa, the cytoplasmic inhibitor of NF-κB, was able to rescue HSPCs in tlr4bb and myd88-deficient embryos, revealing that the impact of TLR4–MyD88 axis on HSPC emergence is mediated via NF-κB activation. Moreover, Notch signaling, which had already been reported to integrate with TLR signaling (61) and act upstream of Runx1 to contribute to HSPC specification (62), was proven to be impaired in the AGM region of TLR4–MyD88-deficient embryos, as reflected by the decreased expression of its target genes. Endothelial-specific Notch overexpression managed to rescue HSPCs in these embryos, showing that TLR4–MyD88–NF-κB-mediated inflammatory signaling is upstream of endothelial Notch signaling and that they, in tandem, regulate HSPC emergence during zebrafish embryonic development.
In the same study, the effect of TLR4–MyD88–NF-κB-mediated inflammatory signaling was shown to be conserved in mouse. Immunofluorescence and qRT-PCR analysis of the AGM region of tlr4−/− mice, demonstrated a decreased expression of Runx1 at E10.5, cmyb, Il1b, and Il6 (NF-κB targets), as well as Notch target genes. Importantly, transplantation assays aiming to evaluate HSC activity, showed that 5/6 recipients transplanted with cells deriving from Tlr4+/− embryos were reconstituted after 2 months, while only 1/6 recipients transplanted with cells deriving from Tlr4−/− embryos was reconstituted (60).
Toll-Like Receptor Influence on Adult Hematopoiesis
Recent studies reveal that TLR ligands can exert a direct effect on HSC activation and hematopoietic potential. Nagai et al. have found that murine HSCs (LSK Flk2− IL7Ra−) express TLR-2 and TLR-4 and functionally respond to receptor stimulation in vitro (LPS and Pan3CSK4, respectively) resulting in increased myeloid differentiation and enhanced cell cycling via MyD88 (63). The same group addressed the consequences of in vivo exposure to LPS. Mice injected with LPS for 4–6 weeks harbored a higher proportion of HSCs (LSK Flk2− CD48− CD150+) compared to control mice. Moreover, upon competitive transplantation assays, HSCs activity of LPS-treated mice showed a skewing toward myeloid differentiation and impaired self-renewal capacity as compared to untreated mice (64). Interestingly, the authors also observed that LPS-treated HSCs developed some features of aged HSCs, i.e., CD150+ expansion and lack of both CD86 (B7.2) and CD18 (Integrin β-2) (64). In a recent study, Zhao et al. have shown that HSPCs respond to TLR stimulation by producing (via NF-κB activation) cytokines to regulate myeloid differentiation (65). Another study of severe sepsis reported that, although Pseudomonas aeruginosa caused the expansion of HSPCs (LSK) and impaired repopulating capacity, they observed a block of myeloid differentiation (66). Another group, however, suggested a different impact of TLR signaling on HSCs. They described that short-term treatment with higher-dose LPS (four doses of 35 μg, every 2 days) led to increased HSC repopulating capacity with no lineage bias (67). Thus, the dose and duration of TLR stimulation may have different outcomes on HSC activity, mainly promoting HSPC cycling and enhancing myeloid differentiation. However, despite the fact that HSPCs express TLRs, it was not clear yet whether direct recognition of pathogens by HSPCs, or changes in the microenvironment during infection were responsible for the changes in hematopoiesis. To tackle this question, Megías et al. have used an elegant experimental approach. They isolated wild type HSCs (LSK IL7Ra−) and transplanted them into lethally irradiated Tlr2−/−, Tlr4−/−, or MyD88−/− recipient mice, which where then challenged with TLR agonists (Pam3CSK4, LPS, or ODN, respectively). As recipient mice could not recognize the TLR agonist injected, secondary effects of cytokines or soluble factors secreted by the microenvironment were excluded. The authors observed that wild type donor cells rapidly differentiated into macrophages, supporting the initial idea that direct stimulation of HSPCs via TLRs occurs in vivo (68). Other TLRs might very well be expressed also by HSCs. It was notably shown that TLR-9 transcripts (predominantly expressed by lymphoid progenitors) were detected in HSCs (Lin−Rag1GFP−c-kithiSca1+Thy1.1lo) (69). Last, it will be interesting to use such TLR knockout mice, in order to determine whether TLR signaling also contributes to the maintenance of HSCs in absence of infection.
Interleukins
Interleukins (ILs) are secreted proteins produced by various immune cell types, ranging from macrophages to T lymphocytes, but also other cell types, like ECs, and their role is to orchestrate the immune system responses (70). Among all members, IL-1 and IL-3 have been shown to also play a role in hematopoiesis. The correlation between IL-1 and hematopoiesis emerged by the expression of this cytokine and its receptor, IL-1RI, in HSPCs of the BM (71–73). After early studies published from the lab of Hal Broxmeyer, it became evident that IL-1 would play synergistic roles with other factors to promote the proliferation and survival of more committed progenitors, in both mouse and humans (74, 75).
Effects of Interleukins During Embryonic Hematopoiesis
In zebrafish, it has been reported that overexpression of il1b can rescue the HSPC emergence defect in TLR4-deficient embryos, while downregulation of il1b expression in an NF-κB-deficient background can be rescued by knocking down ikbaa. Therefore, IL-1β signaling appears to be downstream of the TLR4–MyD88–NF-κB-mediated regulation of HSPC emergence (60).
When Elaine Dzierzak and her group tackled the role of IL-1 in mouse embryonic development of HSCs, they discovered that IL-1R1 was detected in cells of the ventral part of the dorsal aortas from E11 onward. Il1r1−/− embryos demonstrated higher percentage of myeloid progenitor cells (CFU-G and CFU-M), indicating that IL-1RI-mediated signaling in the AGM region limits the growth of these particular progenitors. Upon transplantation, they observed a slight decrease in repopulating cells isolated from Il1r1−/− embryos. However, the fact that the majority of IL-1-signaling molecules were not detected in the AGM at E10 indicates that IL-1 probably does participate in HSC emergence (76).
Since IL-3 was known to be a Runx1 target, the same group also attempted to decipher the role of IL-3 in mouse embryonic hematopoiesis (77). The expression of the cytokine and its receptor was detected in major embryonic vessels at E11. Functional experiments divulged that, even if Runx1+/− embryos have fewer HSCs in the AGM region (78), IL-3 treatment managed to rescue (and even amplify) the number of HSCs as shown by transplantation experiments, suggesting that IL-3 signaling acts downstream of Runx1 (77).
Effects of Interleukins on Adult Hematopoiesis
Recently, the role of chronic exposure of HSCs to IL-1 was delineated in a study by Pietras et al. (79). This study showed that IL-1 exposure of HSCs leads to the activation of a Pu1-dependent program, which in turn results in HSC cycling and massive differentiation toward myeloid cells. In addition, HSCs lose their repopulation capacity. However, these results are transient, since withdrawal of IL-1 restores a normal phenotype (79). Although conflicting evidence exists, IL-3 was also shown to be capable of supporting HSC expansion and long-term multilineage repopulating capacity (80). Finally, a recent report showed that IL-27 exposure leads to expansion of HSPCs (LSK) while simultaneously promoting their differentiation toward myeloid cells (81).
It is evident that ILs play new exciting roles in the biology of HSCs, however, more studies are needed to clearly dissect the role of this family. It would be interesting to see how different ILs or combinations of different family members interact and affect HSC development and maintenance.
Granulocyte Colony-Stimulating Factor
Granulocyte colony-stimulating factor (G-CSF) is a glycoprotein that signals through its cognate receptor, granulocyte colony-stimulating factor receptor (G-CSFR). G-CSF plays important roles in hematopoiesis, notably by inducing the production of granulocytes in response to infection (82). G-CSFR is mainly expressed in hematopoietic cells, and its stimulation leads to activation of downstream intracellular signaling cascades, including the JAK/STAT/SOCS pathway [reviewed in Ref. (83)]. Besides its role in granulopoiesis, G-CSF is used for clinical purposes to mediate HSPC mobilization from the BM into the circulation. Indeed, early reports have shown that G-CSF significantly increased the number of HSPCs in the circulation (84). This potential was confirmed by others (85), and G-CSF has been adopted as a source of HSC for transplantations, ever since.
Effects of G-CSF During Embryonic Hematopoiesis
Initial studies to investigate the role of G-CSF during embryonic development were conducted in zebrafish. The authors showed that in a context of emergency granulopoietic response (LPS stimulation), Gcsf signaling was critical to increase the number of neutrophils in both AGM and CHT regions. However, knockdown of gcsfr did not impair definitive hematopoiesis (86). In a later study though, Hall and colleagues observed that emergency granulopoiesis (induced by bacterial infection) led to an increase in HSC numbers (87). This increase in HSCs was more likely due to increased cell division rather than increased EHT, and induced in a C/ebpb-driven Nos2a-dependent manner (87). This proliferative signal was confirmed by gain-of-function experiments. Both G-CSFR ligands (Gcsfa and Gscfb) caused the expansion of runx1+ HSPCs at 24 hpf. Conversely, morpholino-mediated knockdown of gcsfr, gcsfa, or gcsfb significantly reduced the number of runx1+ HSPCs (88). Moreover, it seems that HSPCs in the CHT were also greatly increased by enhanced Gcsf signaling at 48 hpf, indicating that both ligands play a role in the expansion of HSCs in the zebrafish embryo (88). Unlike in the zebrafish model, there is a lack of evidence in the literature for the involvement of G-CSF or G-CSFR (Csfr3) in promoting HSC expansion during embryonic development in mice.
Effects of G-CSF on Adult Hematopoiesis
Both Gcsf−/− and Csfr3−/− animals are viable, despite having reduced number of circulating neutrophils (89, 90). G-CSF administration in vivo was reported to increase the proportion of HSCs (LSK CD150+ CD48−) in the central marrow, but not in endosteal niches in the BM (91). More recently, Schuettpelz and colleagues confirmed the increase in the pool of HSCs (LSK CD150+ CD48− CD41−) in the BM upon G-CSF treatment, together with loss of long-term repopulating activity due to induction of TLR signaling in HSCs. Interestingly, loss of TLR signaling or suppression of commensal flora (via antibiotic treatment or housing in a germ-free environment), mitigates the G-CSF-mediated expansion of HSCs (92).
Granulocyte colony-stimulating factor is also largely used as a mobilizing agent, notably by targeting cells composing the HSC niche, and critical for HSC maintenance (93). It is important to note that HSC mobilization by G-CSF is abolished in Csfr3−/− mice (94) and G-CSF administration is capable of activating dormant HSCs in vivo (95). However, chimeric mice harboring mixtures of Csf3r+/+ and Csfr3−/− hematopoietic cells have normal mobilization. This emphasizes that G-CSFR expression in HSCs is not required for G-CSF-mediated mobilization, and probably acts in trans on HSCs. This study concluded that G-CSFR expression on HSCs or stromal cells is not important for mobilization. Rather another unknown population of G-CSFR expressing cells is responsible for acting on HSC mobilization (94). Interestingly the Manz lab showed that at least in situations of systemic bacterial stress, Tlr4-expressing ECs are the source of G-CSF and contribute to emergency myelopoiesis (96, 97). The baseline numbers of HPSCs in Csfr3−/− mice was estimated to be similar to Csfr3+/+ controls, although the authors used long-term culture-initiating cells (LTC-ICs) to draw this conclusion. However, by competitive transplantation assays, the authors noticed a marked reduction of Csfr3−/− HSPCs (LSK) frequencies in recipient mice (98).
Macrophage Colony-Stimulating Factor
Macrophage colony-stimulating factor (M-CSF) is a growth factor that is crucial for the proliferation, differentiation, and survival of monocytes and macrophages but also progenitor cells in the BM. Combination of human or mouse G-CSF, GM-CSG together, or with addition of IL3 led to increased cycling and elevated numbers of hematopoietic progenitors in vitro (99–101). The lab of Michael Sieweke brought evidence that M-CSF could exert a function directly on HSCs. Based on the observation that the monocyte/macrophage transcription factor MafB was highly enriched in HSCs (LSK CD34−Flk2−), but not in downstream progenitors, Sarrazin and colleagues investigated the effect of MafB deficiency on HSCs. MafB−/− HSCs were more sensitive to M-CSF treatment, and as a result, more prone to skewing toward myeloid differentiation (102). In a recent study, it was shown that M-CSF could directly induce endogenous PU.1 protein (a transcription factor promoting myelomonocytic differentiation), in single HSCs in vivo and stimulates a reversible, PU.1-dependent myeloid differentiation (103).
Inflammation and Hematological Diseases
Based on the numerous connections between inflammation and hematopoiesis, it should be unsurprising that deregulation of this interplay plays a role in hematopoietic diseases. Below, we will briefly discuss some examples of HSC-associated diseases, and how inflammation affects the emergence, progression, or the outcome of the disease.
Myelodysplastic Syndromes
Myelodysplastic syndromes (MDS) (104) are a heterogeneous group of blood diseases mainly characterized by ineffective hematopoiesis, generic blood dysplasia, and a tendency toward leukemic transformation. The pathophysiology lying behind the progression of such diseases is extremely variable and, therefore, classified in a generic way, involving causes related to intrinsic abnormalities in myeloid progenitors and causes related to extrinsic alterations in the BM microenvironment (104, 105). Although the precise connection is still being elucidated, several studies have correlated MDS with inflammatory disorders. In a recent study, 4.4% of MDS patients presented also autoimmune manifestations (106). In addition, it was reported that inflammatory bowel disease patients were diagnosed with MDS. Both these studies suggest an association between MDS and inflammation (107). However, it is still not clear whether inflammation precedes MDS or vice versa.
Toll-like receptor signaling is one of the pathways identified as a connection between inflammation and MDS. In particular, a number of TLRs and several signal transducers involved in this pathway have been found overexpressed in a high percentage of MDS patients (108). For example TLR2 and TLR9 are overexpressed in all MDS subtypes. Chronic TLR stimulation in mice induces phenotypes very similar to MDS (64). Among all TLRs that were found upregulated (109), Wei et al. reported a somatic mutation of TLR2, namely TLR2-F217S, present in 11% of MDS patients and connected with amplified NF-κB activation, a transcription factor that had already been associated with the disease (108).
NF-κB was one of the factors expected to participate in those diseases due to its important role in HSC proliferation and differentiation (110). Inhibition of its transcriptional activity was reported to result in apoptosis in both healthy and MDS BM progenitors, indicating its high significance in the maintenance and progression of the disease (111–113). Although induction of NF-κB alone is not sufficient to cause MDS, deregulation of NF-κB in the non-hematopoietic compartment can cause a myeloproliferative disorder (114).
Apart from NF-κB, TLRs are also known to induce IFN production. In MDS patients, IFN secretion levels were found severely elevated, which seems to strongly impair hematopoietic progenitor differentiation, for instance, erythroid differentiation (115) and reduce their long-term repopulating activity (116). Many other cytokines are induced by TLR signaling and also play a role in MDS. For example, TLR4 contributes to elevated TNFα levels in MDS patients (117).
The BM microenvironment plays a crucial role in MDS and is also the source or the substrate of many cytokines produced. MDS have been strongly associated with a specific cell type known as the myeloid-derived suppressor cells (MDSCs), which are mostly known to accumulate in cancer patients and suppress effector T cell response, thereby constituting one of the greatest defenses of a tumor against the host’s immune system (118, 119). These cells were also found notably increased and accumulated in the BM of MDS patients, where they have been proposed to mediate their effect mainly via secretion of several cytokines (e.g., IL-10) and other inflammatory effectors [e.g., reactive oxygen species (ROS)] leading to DNA instability and constantly active inflammation, which creates a permissive environment for the development of myelodysplasia (120).
Inherited Bone Marrow Failure
Bone marrow failure (BMF) syndromes include a group of blood disorders that can be either acquired or inherited and they are characterized by diverse kinds of defects, involving all hematopoietic lineages and lead to cytopenia. Inherited BMF syndromes have been associated with increased risks of developing MDS, acute myeloid leukemia (AML), or even solid tumors (121). This group of rare genetic disorders involves Fanconi anemia (FA), Diamond–Blackfan anemia (DBA), dyskeratosis congenita (DC), and Shwachman–Diamond syndrome (SDS). Similar to MDS, BMF syndromes have also been correlated with inflammatory signals, and this correlation is better studied in FA (122).
During the course of this disease, a cluster of 16 genes (FANC genes) coding for DNA repair proteins are genetically defective (123–125). The prominent role of FA proteins is to respond to DNA damage and DNA replication stress and lead to DNA repair (123–125). Thus, FA is a genomic instability syndrome that can lead to BMF or leukemia. Patients have been often reported to demonstrate elevated plasma or intracellular levels of TNFα (126, 127) that affects the production and secretion of other cytokines. Imbalance between different cytokines like IL-6 and GM-CSF affects the BM microenvironment (128). These alterations can cause deregulation of cellular homeostasis and are crucial for the progression of the disease (129–132).
Apart from mediating its role through regulation of other cytokine secretion, however, TNFα is also known to induce ROS, which represent another important factor for the disease. Several FA patients cannot detoxify superoxide anions, which leads to elevated oxidative stress. Indeed, Zhang et al. showed that, in a Fancc-deficient background, TNFα-injected mice had high levels of oxidative stress, indicating the significant role of this FA protein in oxidative stress inhibition and suggesting an interplay between FA proteins and TNFα in the disease progression (133).
Several studies have proven that one of the reasons why patients experience the TNFα upregulation is TLR signaling. In particular, FancA- and FancC-deficient mononuclear phagocytes, as well as mononuclear phagocytes deriving from FA patients, showed upregulated levels of TLR8 and its canonical downstream effectors, which is known to lead to increased TNF production (134). On the other hand, TLR8 signaling also activates IFN (116), a cytokine that has also been found upregulated in FA patients (131), linking inflammatory signaling with this disease. Finally Walter et al. showed that repeated activation of dormant HSCs due to infection or constant blood loss, leads to DNA damage. Thus HSCs lacking an intact FA DNA repair pathway cannot overcome constant stress and, as a result, the hematopoietic system collapses completely (135).
Leukemia
Leukemias are a group of blood cancers defined by an increased number of abnormal white blood cells. Leukemias can be either chronic or acute and can be roughly divided in four different categories: AML, chronic myeloid leukemia (CML), acute lymphoblastic leukemia (ALL), and chronic lymphoblastic leukemia (CLL), based on whether the origin of the transformed cell is myeloid or lymphoid, but also on the characteristics of the disease initiation and progression. Although these leukemias mainly refer to differentiated hematopoietic populations, AML and CML arise from HSCs. CLL can be derived from more differentiated cells but accumulating evidence shows that they may also originate, at least partly, from HSCs (136).
The significance of inflammation in leukemia progression and maintenance is not unexpected, given its known role in hematopoiesis. Apart from the correlation between inflammation and MDS or BMF, which might result in leukemic transformation, it is known today that inflammatory pathways are directly connected with blood cancers (137–139).
One of the leukemic diseases for which the connection and dependence on inflammation has been well studied is AML. AML is the most common type of acute leukemia in adults. AML phenotypes can be divided in several subtypes and is generally characterized by the presence of hyperproliferative myeloid cells (140). TNFα and IFNγ have been shown to suppress clonogenic growth and promote differentiation in blast cells from AML patients (141). Studies on AML patients showed that the canonical NF-κB pathway is constitutively active in AML cells (137). Kurokawa and his group recently showed that the NF-κB pathway was active in leukemia-initiating cells (LICs) isolated from several murine myeloid leukemia models, as well as AML patients (142). Treatment with IκB-SR, a super repressor of NF-κB, led to a significant growth delay of murine myeloid leukemic cells. Additionally, the leukemia progression after transplantation of these cells into irradiated mice was delayed, suggesting that NF-κB is a contributing factor to the disease progression, but not to the survival and fate maintenance of the leukemic cells. Analysis of published microarray-based gene expression profiles and comparison between human or murine LICs and normal HSPCs (143–145) hinted at TNFα as the factor that maintains the constitutively active NF-κB pathway. Indeed, when LICs were treated with a TNFα-neutralizing antibody, p65 nuclear translocation was markedly impaired, indicating that constitutive NF-κB activation is dependent on TNFα signaling. Furthermore, Tnf-deficient cells were significantly impaired in their potential to initiate leukemia, but NF-κB inhibition alone was not sufficient to inhibit leukemic progression. Thus, TNF may exert its function through other factors besides NF-κB (142). Additionally CML, ALL, and CLL cells also exhibit elevated NF-κB levels indicating a potential role of inflammatory signaling in this disease (146–150).
Other inflammatory cytokines are also prominent in leukemia (151). CLL patients exhibit elevated levels of IFN-γ, IL6, IL8, and IL10 (152–154) that can be produced by either CLL cells or from cells of the microenvironment. Many mutations on inflammatory mediators also exist in CLL patients. For example, MYD88, a mediator of toll receptor signaling, is mutated in 3–10% of patients (155, 156).
It is evident that inflammation and leukemia are heavily interconnected. However we still do not know clearly the order of events and how inflammation affects disease progression.
Discussion and Future Directions
In summary, it is by now clear that inflammatory signaling arises as a novel regulator of HSCs emergence and maintenance, both under sterile conditions and in case of stress and disease. Studies in development and during homeostasis provide evidence that HSCs are affected by alteration of different cytokines (Figure 1). Excellent reviews referring to the subject of inflammatory signaling in hematopoiesis can also complement the reading of this review (157–163). However, many questions are still unanswered and two major points include (a) the interaction of HSCs with other cells like niche and immune cells (158) and (b) the ligands that lead to inflammatory response and their source.
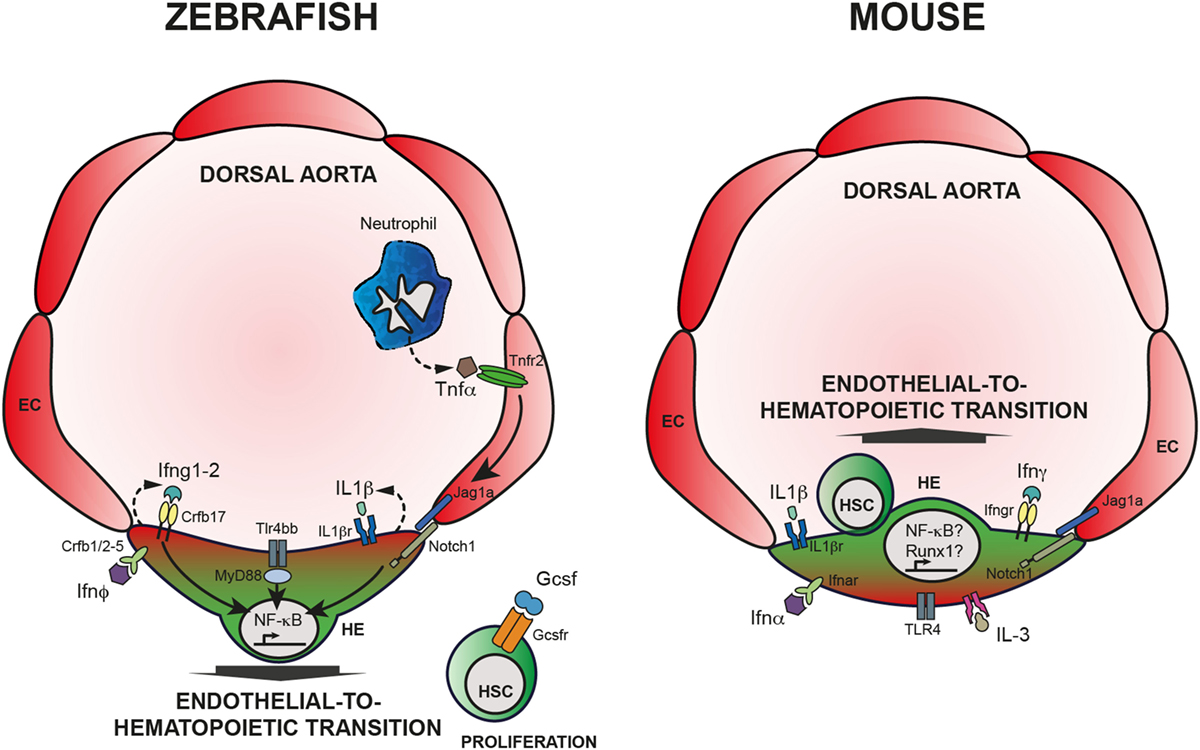
Figure 1. Inflammatory signaling is required for HSC emergence. Left panel: HSC specification in zebrafish embryos. Neutrophils produce TNFα that binds Tnfr2, expressed by endothelial cells (EC). Activation of Tnfr2 leads to upregulation of jag1a and activation of Notch signaling in the hemogenic endothelium (HE), ultimately leading to the activation of NF-κB. Alternatively, Tlr4/Myd88 also induce NF-κB, crucial for HSC specification throughout endothelial-to-hematopoietic transition. Downstream of Notch1 activation, Ifng participates in HSC specification in a cell autonomous fashion. IL-1β signaling, downstream of the TLR4–MyD88–NF-κB axis has also an impact on HSC emergence as does Ifn-ϕ. Finally, Gcsf acts on newly generated HSCs and contributes to HSC proliferation; however, the mechanism is still unclear. Right panel: HSC specification in mouse embryos. Contrary to zebrafish embryos, it is still unclear how inflammatory pathways are being regulated in the developing embryo, or what are the sources of inflammatory cytokines. Nevertheless, it is clear that IFN-α, IFN-γ, as well as Il-1β, IL-3, and TLR4 are required for proper HSC specification, possibly engaging Notch1 signaling and Runx1 and/or NF-κB.
Diverse kinds of cells constitute the hematopoietic niche like osteoblasts, mesenchymal stem cells, ECs, and others (164, 165). For example, mesenchymal stem cells participate in the interplay with HSCs but can also interact with immune cells and adopt both pro- and anti-inflammatory roles (166–170). Immune cells have been found integral for HSCs both during development but also in adulthood. Primitive neutrophils provide crucial cytokines that are imperative for HSC formation during development (50). Macrophages play prominent roles during HSC formation in development but are also actively participating in the BM microenvironment (36, 171, 172). In depth characterization of all these interactions is needed to get a full overview of the interplay between cell populations.
The population of the niche and immune cells discussed above can actually be the source of different cytokines that will act on respective receptors on HSCs both during development and maintenance. Communication with other sources like commensal flora could also provide a source of stimulation for HSCs (92). Thus, most data to date support the hypothesis that the inflammatory ligands that affect HSCs are produced from various sources of the microenvironment. However, another hypothesis could hint to the existence of endogenous ligands that can play autoregulatory roles in HSCs. Lately, it has been shown in many systems that the epigenetic status of a cell plays crucial role in the expression of endogenous repetitive elements like endogenous retroviral elements. Members of the RIG-I-like receptors respond to this upregulation and can mediate an IFN response (173–175). It would be interesting if such a mechanism is used to fine tune the inflammatory milieu of the hematopoietic system.
Another aspect that needs further investigation is the consequence of acute versus chronic inflammatory stress on HSCs. Although many predicted that chronic stress will lead to HSC exhaustion, Pietras et al. showed that HSCs re-enter quiescence in conditions of chronic stress (36). However, upon a second challenge, these HSCs are much more vulnerable. Could an epigenetic memory, newly established on the challenged HSCs, lead them to such a reaction?
Finally, understanding the inflammatory pathways in a disease context is of crucial importance, since this knowledge has often provided us with the potential to target these pathways and treat the diseases. To name just few examples, specific inhibition of TLR2 and its downstream effectors showed significant results in MDS patients since it induced differentiation, apoptosis, and impaired the clonal potential of MDS cells. OPN-305, an antibody against TLR2, is entering phase II clinical trials and holds promise for MDS treatment (176). Monoclonal antibodies against CD33 [gemtuzumab ozogamicin (177)] and bispecific antibodies like AMG330 (anti CD33 and CD3) are being explored for AML therapy (178). Both these antibodies redirect cytolytic effector T cells against leukemic cells.
In short, inflammatory signaling has a diverse and multifaceted role in many aspects of HSC biology from the generation of HSCs to their expansion and maintenance, response to stress, and disease. Various groups have contributed to our understanding of these non-immune roles of inflammatory agents, but many questions remain still unanswered.
Author Contributions
TC, SL, and ET wrote the manuscript; TC designed the figure and the table.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to apologize to all scientists that have contributed to this subject but are not mentioned in this review due to space limitations. The authors would like to thank Teresa V. Bowman (Albert Einstein College of Medicine, USA) for critical reading of the manuscript and useful discussions.
Funding
All the authors are supported by Max Planck Gesellschaft and precisely Max Planck Institute of Immunobiology and Epigenetics, Freiburg, Germany. ET is supported by a Marie Curie Career Integration grant (631432).
References
1. Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med (2014) 20:833–46. doi:10.1038/nm.3647
2. Akunuru S, Geiger H. Aging, clonality, and rejuvenation of hematopoietic stem cells. Trends Mol Med (2016) 22:701–12. doi:10.1016/j.molmed.2016.06.003
3. Göttgens B. Regulatory network control of blood stem cells. Blood (2015) 125:2614–20. doi:10.1182/blood-2014-08-570226
4. Dzierzak E, Speck NA. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat Immunol (2008) 9:129–36. doi:10.1038/ni1560
5. Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell (2008) 132:631–44. doi:10.1016/j.cell.2008.01.025
6. Boisset J-C, van Cappellen W, Andrieu-Soler C, Galjart N, Dzierzak E, Robin C. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature (2010) 464:116–20. doi:10.1038/nature08764
7. Kissa K, Herbomel P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature (2010) 464:112–5. doi:10.1038/nature08761
8. Bertrand JY, Chi NC, Santoso B, Teng S, Stainier DYR, Traver D. Haematopoietic stem cells derive directly from aortic endothelium during development. Nature (2010) 464:108–11. doi:10.1038/nature08738
9. Lam EYN, Hall CJ, Crosier PS, Crosier KE, Flores MV. Live imaging of Runx1 expression in the dorsal aorta tracks the emergence of blood progenitors from endothelial cells. Blood (2010) 116:909–14. doi:10.1182/blood-2010-01-264382
10. Rhodes KE, Gekas C, Wang Y, Lux CT, Francis CS, Chan DN, et al. The emergence of hematopoietic stem cells is initiated in the placental vasculature in the absence of circulation. Cell Stem Cell (2008) 2:252–63. doi:10.1016/j.stem.2008.01.001
11. Li Z, Lan Y, He W, Chen D, Wang J, Zhou F, et al. Mouse embryonic head as a site for hematopoietic stem cell development. Cell Stem Cell (2012) 11:663–75. doi:10.1016/j.stem.2012.07.004
12. Li Z, Vink CS, Mariani SA, Dzierzak E. Subregional localization and characterization of Ly6aGFP-expressing hematopoietic cells in the mouse embryonic head. Dev Biol (2016) 416:34–41. doi:10.1016/j.ydbio.2016.05.031
13. Iizuka K, Yokomizo T, Watanabe N, Tanaka Y, Osato M, Takaku T, et al. Lack of phenotypical and morphological evidences of endothelial to hematopoietic transition in the murine embryonic head during hematopoietic stem cell emergence. PLoS One (2016) 11:e0156427. doi:10.1371/journal.pone.0156427.s005
14. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol (2003) 4:69–77. doi:10.1038/ni875
15. Sawamiphak S, Kontarakis Z, Stainier DYR. Interferon gamma signaling positively regulates hematopoietic stem cell emergence. Dev Cell (2014) 31:640–53. doi:10.1016/j.devcel.2014.11.007
16. Butko E, Pouget C, Traver D. Complex regulation of HSC emergence by the Notch signaling pathway. Dev Biol (2016) 409:129–38. doi:10.1016/j.ydbio.2015.11.008
17. North TE, Goessling W, Peeters M, Li P, Ceol C, Lord AM, et al. Hematopoietic stem cell development is dependent on blood flow. Cell (2009) 137:736–48. doi:10.1016/j.cell.2009.04.023
18. Li Y, Esain V, Teng L, Xu J, Kwan W, Frost IM, et al. Inflammatory signaling regulates embryonic hematopoietic stem and progenitor cell production. Genes Dev (2014) 28:2597–612. doi:10.1101/gad.253302.114
19. Kim PG, Canver MC, Rhee C, Ross SJ, Harriss JV, Tu HC, et al. Interferon-α signaling promotes embryonic HSC maturation. Blood (2016) 128:204–16. doi:10.1182/blood-2016-01-689281
20. Nimmo R, Ciau-Uitz A, Ruiz-Herguido C, Soneji S, Bigas A, Patient R, et al. MiR-142-3p controls the specification of definitive hemangioblasts during ontogeny. Dev Cell (2013) 26:237–49. doi:10.1016/j.devcel.2013.06.023
21. Lu X, Li X, He Q, Gao J, Gao Y, Liu B, et al. miR-142-3p regulates the formation and differentiation of hematopoietic stem cells in vertebrates. Cell Res (2013) 23:1356–68. doi:10.1038/cr.2013.145
22. Broxmeyer HE, Williams DE, Lu L, Cooper S, Anderson SL, Beyer GS, et al. The suppressive influences of human tumor necrosis factors on bone marrow hematopoietic progenitor cells from normal donors and patients with leukemia: synergism of tumor necrosis factor and interferon-gamma. J Immunol (1986) 136:4487–95.
23. Maciejewski J, Selleri C, Anderson S, Young NS. Fas antigen expression on CD34+ human marrow cells is induced by interferon gamma and tumor necrosis factor alpha and potentiates cytokine-mediated hematopoietic suppression in vitro. Blood (1995) 85:3183–90.
24. Snoeck HW, Van Bockstaele DR, Nys G, Lenjou M, Lardon F, Haenen L, et al. Interferon gamma selectively inhibits very primitive CD342+CD38- and not more mature CD34+CD38+ human hematopoietic progenitor cells. J Exp Med (1994) 180:1177–82. doi:10.1084/jem.180.3.1177
25. Selleri C, Sato T, Anderson S, Young NS, Maciejewski JP. Interferon-gamma and tumor necrosis factor-alpha suppress both early and late stages of hematopoiesis and induce programmed cell death. J Cell Physiol (1995) 165:538–46. doi:10.1002/jcp.1041650312
26. Kawano Y, Takaue Y, Hirao A, Abe T, Saito S, Matsunaga K, et al. Synergistic effect of recombinant interferon-gamma and interleukin-3 on the growth of immature human hematopoietic progenitors. Blood (1991) 77:2118–21.
27. Caux C, Moreau I, Saeland S, Banchereau J. Interferon-gamma enhances factor-dependent myeloid proliferation of human CD34+ hematopoietic progenitor cells. Blood (1992) 79(10):2628–35.
28. Brugger W, Möcklin W, Heimfeld S, Berenson RJ, Mertelsmann R, Kanz L. Ex vivo expansion of enriched peripheral blood CD34+ progenitor cells by stem cell factor, interleukin-1 beta (IL-1 beta), IL-6, IL-3, interferon-gamma, and erythropoietin. Blood (1993) 81:2579–84.
29. Broxmeyer HE, Lu L, Platzer E, Feit C, Juliano L, Rubin BY. Comparative analysis of the influences of human gamma, alpha and beta interferons on human multipotential (CFU-GEMM), erythroid (BFU-E) and granulocyte-macrophage (CFU-GM) progenitor cells. J Immunol (1983) 131:1300–5.
30. Lu L, Welte K, Gabrilove JL, Hangoc G, Bruno E, Hoffman R, et al. Effects of recombinant human tumor necrosis factor alpha, recombinant human gamma-interferon, and prostaglandin E on colony formation of human hematopoietic progenitor cells stimulated by natural human pluripotent colony-stimulating factor, pluripoietin alpha, and recombinant erythropoietin in serum-free cultures. Cancer Res (1986) 46:4357–61.
31. Broxmeyer HE, Cooper S, Rubin BY, Taylor MW. The synergistic influence of human interferon-gamma and interferon-alpha on suppression of hematopoietic progenitor cells is additive with the enhanced sensitivity of these cells to inhibition by interferons at low oxygen tension in vitro. J Immunol (1985) 135:2502–6.
32. Binder D, Fehr J, Hengartner H, Zinkernagel RM. Virus-induced transient bone marrow aplasia: major role of interferon-alpha/beta during acute infection with the noncytopathic lymphocytic choriomeningitis virus. J Exp Med (1997) 185:517–30. doi:10.1084/jem.185.3.517
33. Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon–dependent exhaustion. Nat Med (2009) 15:696–700. doi:10.1038/nm.1973
34. Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature (2009) 458:904–8. doi:10.1038/nature07815
35. Ehninger A, Boch T, Uckelmann H, Essers MA, Mudder K, Sleckman BP, et al. Posttranscriptional regulation of c-Myc expression in adult murine HSCs during homeostasis and interferon-induced stress response. Blood (2014) 123:3909–13. doi:10.1182/blood-2013-10-531038
36. Pietras EM, Lakshminarasimhan R, Techner J-M, Fong S, Flach J, Binnewies M, et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J Exp Med (2014) 211:245–62. doi:10.4161/cc.6.5.3921
37. Zhao X, Ren G, Liang L, Ai PZ, Zheng B, Tischfield JA, et al. Brief report: interferon-gamma induces expansion of Lin(-)Sca-1(+)C-Kit(+) Cells. Stem Cells (2010) 28:122–6. doi:10.1002/stem.252
38. Yu JM, Emmons RV, Hanazono Y, Sellers S, Young NS, Dunbar CE. Expression of interferon-gamma by stromal cells inhibits murine long-term repopulating hematopoietic stem cell activity. Exp Hematol (1999) 27:895–903. doi:10.1016/S0301-472X(99)00009-0
39. Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN. Nature (2010) 465:793–7. doi:10.1038/nature09135
40. MacNamara KC, Jones M, Martin O, Winslow GM. Transient activation of hematopoietic stem and progenitor cells by IFNγ during acute bacterial infection. PLoS One (2011) 6:e28669. doi:10.1371/journal.pone.0028669
41. de Bruin AM, Demirel O, Hooibrink B, Brandts CH, Nolte MA. Interferon-γ impairs proliferation of hematopoietic stem cells in mice. Blood (2013) 121:3578–85. doi:10.1182/blood-2012-05-432906
42. Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H, et al. Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell (2015) 17:422–34. doi:10.1016/j.stem.2015.07.007
43. Feng CG, Weksberg DC, Taylor GA, Sher A, Goodell MA. The p47 GTPase Lrg-47 (Irgm1) links host defense and hematopoietic stem cell proliferation. Cell Stem Cell (2008) 2:83–9. doi:10.1016/j.stem.2007.10.007
44. King KY, Baldridge MT, Weksberg DC, Chambers SM, Lukov GL, Wu S, et al. Irgm1 protects hematopoietic stem cells by negative regulation of IFN signaling. Blood (2011) 118:1525–33. doi:10.1182/blood-2011-01-328682
45. Sherry B, Jue DM, Zentella A, Cerami A. Characterization of high molecular weight glycosylated forms of murine tumor necrosis factor. Biochem Biophys Res Commun (1990) 173:1072–8. doi:10.1016/S0006-291X(05)80895-2
46. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A (1975) 72:3666–70. doi:10.1073/pnas.72.9.3666
47. Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ (2003) 10:45–65. doi:10.1038/sj.cdd.4401189
48. Netea MG, Kullberg BJ. Circulating cytokines as mediators of fever. Clin Infect Dis (2000) 31:S178–84. doi:10.1086/317513
49. Naudé PJW, Boer den JA, Luiten PGM, Eisel ULM. Tumor necrosis factor receptor cross-talk. FEBS J (2011) 278:888–98. doi:10.1111/j.1742-4658.2011.08017.x
50. Espín-Palazón R, Stachura DL, Campbell CA, García-Moreno D, Del Cid N, Kim AD, et al. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell (2014) 159:1070–85. doi:10.1016/j.cell.2014.10.031
51. Espín R, Roca FJ, Candel S, Sepulcre MP, González-Rosa JM, Alcaraz-Pérez F, et al. TNF receptors regulate vascular homeostasis in zebrafish through a caspase-8, caspase-2 and P53 apoptotic program that bypasses caspase-3. Dis Model Mech (2013) 6:383–96. doi:10.1242/dmm.010249
52. Dybedal I, Bryder D, Fossum A, Rusten LS, Jacobsen SE. Tumor necrosis factor (TNF)-mediated activation of the p55 TNF receptor negatively regulates maintenance of cycling reconstituting human hematopoietic stem cells. Blood (2001) 98:1782–91. doi:10.1182/blood.V98.6.1782
53. Rezzoug F, Huang Y, Tanner MK, Wysoczynski M, Schanie CL, Chilton PM, et al. TNF-α is critical to facilitate hemopoietic stem cell engraftment and function. J Immunol (2008) 180:49–57. doi:10.4049/jimmunol.180.1.49
54. Pronk CJH, Veiby OP, Bryder D, Jacobsen SEW. Tumor necrosis factor restricts hematopoietic stem cell activity in mice: involvement of two distinct receptors. J Exp Med (2011) 208:1563–70. doi:10.1073/pnas.93.9.4040
55. Rebel VI, Hartnett S, Hill GR, Lazo-Kallanian SB, Ferrara JL, Sieff CA. Essential role for the p55 tumor necrosis factor receptor in regulating hematopoiesis at a stem cell level. J Exp Med (1999) 190:1493–504. doi:10.1084/jem.190.10.1493
56. Roderick JE, Hermance N, Zelic M, Simmons MJ, Polykratis A, Pasparakis M, et al. Hematopoietic RIPK1 deficiency results in bone marrow failure caused by apoptosis and RIPK3-mediated necroptosis. Proc Natl Acad Sci U S A (2014) 111:14436–41. doi:10.1073/pnas.1409389111
57. Tesio M, Tang Y, Müdder K, Saini M, Paleske von L, Macintyre E, et al. Hematopoietic stem cell quiescence and function are controlled by the CYLD–TRAF2–p38MAPK pathway. J Exp Med (2015) 212:525–38. doi:10.1016/j.stem.2011.07.003
58. Brieger A, Rink L, Haase H. Differential regulation of TLR-dependent MyD88 and TRIF signaling pathways by free zinc ions. J Immunol (2013) 191:1808–17. doi:10.4049/jimmunol.1301261
59. Medzhitov R. Approaching the asymptote: 20 years later. Immunity (2009) 30:766–75. doi:10.1016/j.immuni.2009.06.004
60. He Q, Zhang C, Wang L, Zhang P, Ma D, Lv J, et al. Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood (2015) 125:1098–106. doi:10.1182/blood-2014-09-601542
61. Hu X, Chung AY, Wu I, Foldi J, Chen J, Ji JD, et al. Integrated regulation of toll-like receptor responses by Notch and interferon-gamma pathways. Immunity (2008) 29:691–703. doi:10.1016/j.immuni.2008.08.016
62. Burns CE, Traver D, Mayhall E, Shepard JL, Zon LI. Hematopoietic stem cell fate is established by the Notch-Runx pathway. Genes Dev (2005) 19:2331–42. doi:10.1101/gad.1337005
63. Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S, et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity (2006) 24:801–12. doi:10.1016/j.immuni.2006.04.008
64. Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q, et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J Immunol (2011) 186:5367–75. doi:10.4049/jimmunol.1003438
65. Zhao JL, Ma C, O’Connell RM, Mehta A, DiLoreto R, Heath JR, et al. Conversion of danger signals into cytokine signals by hematopoietic stem and progenitor cells for regulation of stress-induced hematopoiesis. Cell Stem Cell (2014) 14:445–59. doi:10.1016/j.stem.2014.01.007
66. Rodriguez S, Chora A, Goumnerov B, Mumaw C, Goebel WS, Fernandez L, et al. Dysfunctional expansion of hematopoietic stem cells and block of myeloid differentiation in lethal sepsis. Blood (2009) 114:4064–76. doi:10.1182/blood-2009-04-214916
67. Takizawa H, Regoes RR, Boddupalli CS, Bonhoeffer S, Manz MG. Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. J Exp Med (2011) 208:273–84. doi:10.1084/jem.20101643
68. Megías J, Yáñez A, Moriano S, O’Connor J-E, Gozalbo D, Gil M-L. Direct toll-like receptor-mediated stimulation of hematopoietic stem and progenitor cells occurs in vivo and promotes differentiation toward macrophages. Stem Cells (2012) 30:1486–95. doi:10.1002/stem.1110
69. Welner RS, Pelayo R, Nagai Y, Garrett KP, Wuest TR, Carr DJ, et al. Lymphoid precursors are directed to produce dendritic cells as a result of TLR9 ligation during herpes infection. Blood (2008) 112:3753–61. doi:10.1182/blood-2008-04-151506
70. Brocker C, Thompson D, Matsumoto A, Nebert DW, Vasiliou V. Evolutionary divergence and functions of the human interleukin (IL) gene family. Hum Genomics (2010) 5:30–55. doi:10.1186/1479-7364-5-1-30
71. Fibbe WE, Goselink HM, Van Eeden G, Van Damme J, Billiau A, Voogt PJ, et al. Proliferation of myeloid progenitor cells in human long-term bone marrow cultures is stimulated by interleukin-1 beta. Blood (1988) 72:1242–7.
72. Heimfeld S, Hudak S, Weissman I, Rennick D. The in vitro response of phenotypically defined mouse stem cells and myeloerythroid progenitors to single or multiple growth factors. Proc Natl Acad Sci U S A (1991) 88:9902–6. doi:10.1073/pnas.88.21.9902
73. McKinstry WJ, Li CL, Rasko JE, Nicola NA, Johnson GR, Metcalf D. Cytokine receptor expression on hematopoietic stem and progenitor cells. Blood (1997) 89:65–71.
74. Hangoc G, Williams DE, Falkenburg JH, Broxmeyer HE. Influence of IL-1 alpha and -1 beta on the survival of human bone marrow cells responding to hematopoietic colony-stimulating factors. J Immunol (1989) 142:4329–34.
75. Williams DE, Broxmeyer HE. Interleukin-1 alpha enhances the in vitro survival of purified murine granulocyte-macrophage progenitor cells in the absence of colony-stimulating factors. Blood (1988) 72:1608–15.
76. Orelio C, Haak E, Peeters M, Dzierzak E. Interleukin-1-mediated hematopoietic cell regulation in the aorta-gonad-mesonephros region of the mouse embryo. Blood (2008) 112:4895–904. doi:10.1182/blood-2007-12-123836
77. Robin C, Ottersbach K, Durand C, Peeters M, Vanes L, Tybulewicz V, et al. An unexpected role for IL-3 in the embryonic development of hematopoietic stem cells. Dev Cell (2006) 11:171–80. doi:10.1016/j.devcel.2006.07.002
78. Cai Z, de Bruijn MFTR, Ma X, Dortland B, Luteijn T, Downing RJ, et al. Haploinsufficiency of AML1 affects the temporal and spatial generation of hematopoietic stem cells in the mouse embryo. Immunity (2000) 13:423–31. doi:10.1016/S1074-7613(00)00042-X
79. Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol (2016) 18:607–18. doi:10.1038/ncb3346
80. Bryder D, Jacobsen SE. Interleukin-3 supports expansion of long-term multilineage repopulating activity after multiple stem cell divisions in vitro. Blood (2000) 96:1748–55.
81. Furusawa J-I, Mizoguchi I, Chiba Y, Hisada M, Kobayashi F, Yoshida H, et al. Promotion of expansion and differentiation of hematopoietic stem cells by interleukin-27 into myeloid progenitors to control infection in emergency myelopoiesis. PLoS Pathog (2016) 12:e1005507. doi:10.1371/journal.ppat.1005507.s013
82. Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor. Blood (1991) 78:2791–808.
83. O’Sullivan LA, Liongue C, Lewis RS, Stephenson SEM, Ward AC. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol Immunol (2007) 44:2497–506. doi:10.1016/j.molimm.2006.11.025
84. Dührsen U, Villeval JL, Boyd J, Kannourakis G, Morstyn G, Metcalf D. Effects of recombinant human granulocyte colony-stimulating factor on hematopoietic progenitor cells in cancer patients. Blood (1988) 72:2074–81.
85. Molineux G, Pojda Z, Hampson IN, Lord BI, Dexter TM. Transplantation potential of peripheral blood stem cells induced by granulocyte colony-stimulating factor. Blood (1990) 76:2153–8.
86. Liongue C, Hall CJ, O’Connell BA, Crosier P, Ward AC. Zebrafish granulocyte colony-stimulating factor receptor signaling promotes myelopoiesis and myeloid cell migration. Blood (2009) 113:2535–46. doi:10.1182/blood-2008-07-171967
87. Hall CJ, Flores MV, Oehlers SH, Sanderson LE, Lam EY, Crosier KE, et al. Infection-responsive expansion of the hematopoietic stem and progenitor cell compartment in zebrafish is dependent upon inducible nitric oxide. Cell Stem Cell (2012) 10:198–209. doi:10.1016/j.stem.2012.01.007
88. Stachura DL, Svoboda O, Campbell CA, Espín-Palazón R, Lau RP, Zon LI, et al. The zebrafish granulocyte colony-stimulating factors (Gcsfs): 2 paralogous cytokines and their roles in hematopoietic development and maintenance. Blood (2013) 122:3918–28. doi:10.1182/blood-2012-12-475392
89. Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood (1994) 84:1737–46.
90. Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity (1996) 5:491–501. doi:10.1016/S1074-7613(00)80504-X
91. Grassinger J, Williams B, Olsen GH, Haylock DN, Nilsson SK. Granulocyte colony stimulating factor expands hematopoietic stem cells within the central but not endosteal bone marrow region. Cytokine (2012) 58:218–25. doi:10.1016/j.cyto.2012.01.014
92. Schuettpelz LG, Borgerding JN, Christopher MJ, Gopalan PK, Romine MP, Herman AC, et al. G-CSF regulates hematopoietic stem cell activity, in part, through activation of toll-like receptor signaling. Leukemia (2014) 28:1851–60. doi:10.1038/leu.2014.68
93. Greenbaum AM, Link DC. Mechanisms of G-CSF-mediated hematopoietic stem and progenitor mobilization. Leukemia (2011) 25:211–7. doi:10.1038/leu.2010.248
94. Liu F, Poursine-Laurent J, Link DC. Expression of the G-CSF receptor on hematopoietic progenitor cells is not required for their mobilization by G-CSF. Blood (2000) 95:3025–31.
95. Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell (2008) 135:1118–29. doi:10.1016/j.cell.2008.10.048
96. Boettcher S, Gerosa RC, Radpour R, Bauer J, Ampenberger F, Heikenwalder M, et al. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood (2014) 124:1393–403. doi:10.1182/blood-2014-04-570762
97. Boettcher S, Ziegler P, Schmid MA, Takizawa H, van Rooijen N, Kopf M, et al. Cutting edge: LPS-induced emergency myelopoiesis depends on TLR4-expressing non hematopoietic cells. The Journal of Immunology (2012) 188:5824–8. doi:10.4049/jimmunol.1103253
98. Richards MK, Liu F, Iwasaki H, Akashi K, Link DC. Pivotal role of granulocyte colony-stimulating factor in the development of progenitors in the common myeloid pathway. Blood (2003) 102:3562–8. doi:10.1182/blood-2003-02-0593
99. Broxmeyer HE, Williams DE, Hangoc G, Cooper S, Gillis S, Shadduck RK, et al. Synergistic myelopoietic actions in vivo after administration to mice of combinations of purified natural murine colony-stimulating factor 1, recombinant murine interleukin 3, and recombinant murine granulocyte/macrophage colony-stimulating factor. Proc Natl Acad Sci U S A (1987) 84:3871–5. doi:10.1073/pnas.84.11.3871
100. Williams DE, Straneva JE, Cooper S, Shadduck RK, Waheed A, Gillis S, et al. Interactions between purified murine colony-stimulating factors (natural CSF-1, recombinant GM-CSF, and recombinant IL-3) on the in vitro proliferation of purified murine granulocyte-macrophage progenitor cells. Exp Hematol (1987) 15:1007–12.
101. Broxmeyer HE, Williams DE, Cooper S, Hangoc G, Ralph P. Recombinant human granulocyte-colony stimulating factor and recombinant human macrophage-colony stimulating factor synergize in vivo to enhance proliferation of granulocyte-macrophage, erythroid, and multipotential progenitor cells in mice. J Cell Biochem (1988) 38:127–36. doi:10.1002/jcb.240380207
102. Sarrazin S, Mossadegh-Keller N, Fukao T, Aziz A, Mourcin F, Vanhille L, et al. MafB restricts M-CSF-dependent myeloid commitment divisions of hematopoietic stem cells. Cell (2009) 138:300–13. doi:10.1016/j.cell.2009.04.057
103. Mossadegh-Keller N, Sarrazin S, Kandalla PK, Espinosa L, Stanley ER, Nutt SL, et al. M-CSF instructs myeloid lineage fate in single haematopoietic stem cells. Nature (2013) 497:239–43. doi:10.1038/nature12026
104. Ganan-Gomez I, Wei Y, Starczynowski DT, Colla S, Yang H, Cabrero-Calvo M, et al. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia (2015) 29:1458–69. doi:10.1038/leu.2015.69
105. Yang L, Qian Y, Eksioglu E, Epling-Burnette PK, Wei S. The inflammatory microenvironment in MDS. Cell Mol Life Sci (2015) 72:1959–66. doi:10.1007/s00018-015-1846-x
106. Williamson BT, Foltz L, Leitch HA. Autoimmune syndromes presenting as a paraneoplastic manifestation of myelodysplastic syndromes: clinical features, course, treatment and outcome. Hematol Rep (2016) 8(2):6480. doi:10.4081/hr.2016.6480
107. Wang Z, Zhou Y, Liu Y. Concurrent inflammatory bowel disease and myelodysplastic syndrome: report of nine new cases and a review of the literature. Dig Dis Sci (2008) 53:1929–32. doi:10.1007/s10620-007-0090-0
108. Wei Y, Dimicoli S, Bueso-Ramos C, Chen R, Yang H, Neuberg D, et al. Toll-like receptor alterations in myelodysplastic syndrome. Leukemia (2013) 27:1832–40. doi:10.1038/leu.2013.180
109. Kuninaka N, Kurata M, Yamamoto K, Suzuki S, Umeda S, Kirimura S, et al. Expression of toll-like receptor 9 in bone marrow cells of myelodysplastic syndromes is down-regulated during transformation to overt leukemia. Exp Mol Pathol (2010) 88:293–8. doi:10.1016/j.yexmp.2010.01.009
110. Zhao C, Xiu Y, Ashton J, Xing L, Morita Y, Jordan CT, et al. Noncanonical NF-κB signaling regulates hematopoietic stem cell self-renewal and microenvironment interactions. Stem Cells (2012) 30:709–18. doi:10.1002/stem.1050
111. Fabre C, Carvalho G, Tasdemir E, Braun T, Adès L, Grosjean J, et al. NF-κB inhibition sensitizes to starvation-induced cell death in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene (2007) 26:4071–83. doi:10.1038/sj.onc.1210187
112. Pyatt DW, Stillman WS, Yang Y, Gross S, Zheng JH, Irons RD. An essential role for NF-kappaB in human CD34(+) bone marrow cell survival. Blood (1999) 93:3302–8.
113. Braun T, Carvalho G, Coquelle A, Vozenin M-C, Lepelley P, Hirsch F, et al. NF-kappaB constitutes a potential therapeutic target in high-risk myelodysplastic syndrome. Blood (2006) 107:1156–65. doi:10.1182/blood-2005-05-1989
114. Rupec RA, Jundt F, Rebholz B, Eckelt B, Weindl GN, Herzinger T, et al. Stroma-mediated dysregulation of myelopoiesis in mice lacking IκBα. Immunity (2005) 22:479–91. doi:10.1016/j.immuni.2005.02.009
115. Felli N, Pedini F, Zeuner A, Petrucci E, Testa U, Conticello C, et al. Multiple members of the TNF superfamily contribute to IFN – mediated inhibition of erythropoiesis. J Immunol (2005) 175:1464–72. doi:10.4049/jimmunol.175.3.1464
116. Yang K, Puel A, Zhang S, Eidenschenk C, Ku C-L, Casrouge A, et al. Human TLR-7-, -8-, and -9-mediated induction of IFN-alpha/beta and -lambda is IRAK-4 dependent and redundant for protective immunity to viruses. Immunity (2005) 23:465–78. doi:10.1016/j.immuni.2005.09.016
117. Maratheftis CI, Andreakos E, Moutsopoulos HM, Voulgarelis M. Toll-like receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes. Clin Cancer Res (2007) 13:1154–60. doi:10.1158/1078-0432.CCR-06-2108
118. Chen X, Eksioglu EA, Zhou J, Zhang L, Djeu J, Fortenbery N, et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J Clin Invest (2013) 123:4595–611. doi:10.1172/JCI67580
119. Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother (2006) 55:237–45. doi:10.1007/s00262-005-0048-z
120. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
121. Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev (2010) 24:101–22. doi:10.1016/j.blre.2010.03.002
122. Wilson DB, Link DC, Mason PJ, Bessler M. Inherited bone marrow failure syndromes in adolescents and young adults. Ann Med (2014) 46:353–63. doi:10.3109/07853890.2014.915579
123. Lopez-Martinez D, Liang C-C, Cohn MA. Cellular response to DNA interstrand crosslinks: the Fanconi anemia pathway. Cell Mol Life Sci (2016) 73:3097–114. doi:10.1007/s00018-016-2218-x
124. Michl J, Zimmer J, Tarsounas M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J (2016) 35:909–23. doi:10.15252/embj.201693860
125. Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys (2014) 43:257–78. doi:10.1146/annurev-biophys-051013-022737
126. Schultz JC, Shahidi NT. Tumor necrosis factor-alpha overproduction in Fanconi’s anemia. Am J Hematol (1993) 42:196–201. doi:10.1002/ajh.2830420211
127. Du W, Erden O, Pang Q. TNF-α signaling in Fanconi anemia. Blood Cells Mol Dis (2014) 52:2–11. doi:10.1016/j.bcmd.2013.06.005
128. Du W, Adam Z, Rani R, Zhang X, Pang Q. Oxidative stress in Fanconi anemia hematopoiesis and disease progression. Antioxid Redox Signal (2008) 10:1909–21. doi:10.1089/ars.2008.2129
129. de Cremoux P, Gluckman E, Podgorniak MP, Menier C, Thierry D, Calvo F, et al. Decreased IL-1 beta and TNF alpha secretion in long-term bone marrow culture supernatant from Fanconi’s anaemia patients. Eur J Haematol (1996) 57:202–7. doi:10.1111/j.1600-0609.1996.tb01364.x
130. Stark R, Andre C, Thierry D, Cherel M, Galibert F, Gluckman E. The expression of cytokine and cytokine receptor genes in long-term bone marrow culture in congenital and acquired bone marrow hypoplasias. Br J Haematol (1993) 83:560–6. doi:10.1111/j.1365-2141.1993.tb04691.x
131. Dufour C, Corcione A, Svahn J, Haupt R, Poggi V, Béka’ssy AN, et al. TNF-α and IFN-γ are overexpressed in the bone marrow of Fanconi anemia patients and TNF-α suppresses erythropoiesis in vitro. Blood (2003) 102:2053–9. doi:10.1182/blood-2003-01-0114
132. Rosselli F, Sanceau J, Wietzerbin J, Moustacchi E. Abnormal lymphokine production: a novel feature of the genetic disease Fanconi anemia. I. Involvement of interleukin-6. Hum Genet (1992) 89:42–8. doi:10.1007/BF00207040
133. Zhang X, Sejas DP, Qiu Y, Williams DA, Pang Q. Inflammatory ROS promote and cooperate with the Fanconi anemia mutation for hematopoietic senescence. J Cell Sci (2007) 120:1572–83. doi:10.1242/jcs.003152
134. Vanderwerf SM, Svahn J, Olson S, Rathbun RK, Harrington C, Yates J, et al. TLR8-dependent TNF-(alpha) overexpression in Fanconi anemia group C cells. Blood (2009) 114:5290–8. doi:10.1182/blood-2009-05-222414
135. Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature (2015) 520:549–52. doi:10.1038/nature14131
136. Fabbri G, Dalla-Favera R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat Rev Cancer (2016) 16:145–62. doi:10.1038/nrc.2016.8
137. Guzman ML, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, et al. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood (2001) 98:2301–7. doi:10.1182/blood.V98.8.2301
138. Riether C, Schürch CM, Ochsenbein AF. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ (2015) 22:187–98. doi:10.1038/cdd.2014.89
139. Gasparini C, Celeghini C, Monasta L, Zauli G. NF-κB pathways in hematological malignancies. Cell Mol Life Sci (2014) 71:2083–102. doi:10.1007/s00018-013-1545-4
140. Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin (2012) 62:220–41. doi:10.3322/caac.21149
141. Geissler K, Tricot G, Leemhuis T, Walker E, Broxmeyer HE. Differentiation-inducing effect of recombinant human tumor necrosis factor alpha and gamma-interferon in vitro on blast cells from patients with acute myeloid leukemia and myeloid blast crisis of chronic myeloid leukemia. Cancer Res (1989) 49:3057–62.
142. Kagoya Y, Yoshimi A, Kataoka K, Nakagawa M, Kumano K, Arai S, et al. Positive feedback between NF-κB and TNF-α promotes leukemia-initiating cell capacity. J Clin Invest (2014) 124:528–42. doi:10.1172/JCI68101DS1
143. Kvinlaug BT, Chan W-I, Bullinger L, Ramaswami M, Sears C, Foster D, et al. Common and overlapping oncogenic pathways contribute to the evolution of acute myeloid leukemias. Cancer Res (2011) 71:4117–29. doi:10.1158/0008-5472.CAN-11-0176
144. Theilgaard-Mönch K, Boultwood J, Ferrari S, Giannopoulos K, Hernandez-Rivas JM, Kohlmann A, et al. Gene expression profiling in MDS and AML: potential and future avenues. Leukemia (2011) 25:909–20. doi:10.1038/leu.2011.48
145. Gentles AJ, Plevritis SK, Majeti R, Alizadeh AA. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA (2010) 304:2706–15. doi:10.1001/jama.2010.1862
146. Kirchner D, Duyster J, Ottmann O, Schmid RM, Bergmann L, Munzert G. Mechanisms of Bcr-Abl-mediated NF-kappaB/Rel activation. Exp Hematol (2003) 31:504–11. doi:10.1016/S0301-472X(03)00069-9
147. Vilimas T, Mascarenhas J, Palomero T, Mandal M, Buonamici S, Meng F, et al. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat Med (2007) 13:70–7. doi:10.1038/nm1524
148. Liu Z, Hazan-Halevy I, Harris DM, Li P, Ferrajoli A, Faderl S, et al. STAT-3 activates NF-kappaB in chronic lymphocytic leukemia cells. Mol Cancer Res (2011) 9:507–15. doi:10.1158/1541-7786.MCR-10-0559
149. Xu J, Zhou P, Wang W, Sun A, Guo F. RelB, together with RelA, sustains cell survival and confers proteasome inhibitor sensitivity of chronic lymphocytic leukemia cells from bone marrow. J Mol Med (2014) 92:77–92. doi:10.1007/s00109-013-1081-6
150. Mineva ND, Rothstein TL, Meyers JA, Lerner A, Sonenshein GE. CD40 ligand-mediated activation of the de novo RelB NF-kappaB synthesis pathway in transformed B cells promotes rescue from apoptosis. J Biol Chem (2007) 282:17475–85. doi:10.1074/jbc.M607313200
151. Rozovski U, Keating MJ, Estrov Z. Targeting inflammatory pathways in chronic lymphocytic leukemia. Crit Rev Oncol Hematol (2013) 88:655–66. doi:10.1016/j.critrevonc.2013.07.011
152. Mahadevan D, Choi J, Cooke L, Simons B, Riley C, Klinkhammer T, et al. Gene expression and serum cytokine profiling of lowstage CLL identify WNT/PCP, Flt-3L/Flt-3, and CXCL9/CXCR3 as regulators of cell proliferation, survival, and migration. Hum Genomics Proteomics (2009) 2009:1–12. doi:10.1182/blood-2006-02-005488
153. Fayad L, Keating MJ, Reuben JM, O’Brien S, Lee BN, Lerner S, et al. Interleukin-6 and interleukin-10 levels in chronic lymphocytic leukemia: correlation with phenotypic characteristics and outcome. Blood (2001) 97:256–63. doi:10.1182/blood.V97.1.256
154. Yoon J-Y, Lafarge S, Dawe D, Lakhi S, Kumar R, Morales C, et al. Association of interleukin-6 and interleukin-8 with poor prognosis in elderly patients with chronic lymphocytic leukemia. Leuk Lymphoma (2012) 53:1735–42. doi:10.3109/10428194.2012.666662
155. Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med (2011) 365:2497–506. doi:10.1056/NEJMoa1109016
156. Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature (2011) 475:101–5. doi:10.1038/nature10113
157. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-γ on hematopoiesis. Blood (2014) 124:2479–86. doi:10.1182/blood-2014-04-568451
158. King KY, Goodell MA. Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nat Rev Immunol (2011) 11:685–92. doi:10.1038/nri3062
159. Schuettpelz LG, Link DC. Regulation of hematopoietic stem cell activity by inflammation. Front Immunol (2013) 4:204. doi:10.3389/fimmu.2013.00204/abstract
160. He Q, Liu F. Unexpected role of inflammatory signaling in hematopoietic stem cell development. Curr Opin Hematol (2016) 23:18–22. doi:10.1097/MOH.0000000000000197
161. Mirantes C, Passegué E, Pietras EM. Pro-inflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Exp Cell Res (2014) 329:248–54. doi:10.1016/j.yexcr.2014.08.017
162. Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood (2012) 119:2991–3002. doi:10.1182/blood-2011-12-380113
163. Espín-Palazón R, Traver D. The NF-κB family: key players during embryonic development and HSC emergence. Exp Hematol (2016) 44:519–27. doi:10.1016/j.exphem.2016.03.010
164. Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature (2014) 505:327–34. doi:10.1038/nature12984
165. Boulais PE, Frenette PS. Making sense of hematopoietic stem cell niches. Blood (2015) 125:2621–9. doi:10.1182/blood-2014-09-570192
166. Pevsner-Fischer M, Morad V, Cohen-Sfady M, Rousso-Noori L, Zanin-Zhorov A, Cohen S, et al. Toll-like receptors and their ligands control mesenchymal stem cell functions. Blood (2007) 109:1422–32. doi:10.1182/blood-2006-06-028704
167. Liotta F, Angeli R, Cosmi L, Filì L, Manuelli C, Frosali F, et al. Toll-like receptors 3 and 4 are expressed by human bone marrow-derived mesenchymal stem cells and can inhibit their T-cell modulatory activity by impairing Notch signaling. Stem Cells (2008) 26:279–89. doi:10.1634/stemcells.2007-0454
168. Hwa Cho H, Bae YC, Jung JS. Role of toll-like receptors on human adipose-derived stromal cells. Stem Cells (2006) 24:2744–52. doi:10.1634/stemcells.2006-0189
169. Tomchuck SL, Zwezdaryk KJ, Coffelt SB, Waterman RS, Danka ES, Scandurro AB. Toll-like receptors on human mesenchymal stem cells drive their migration and immunomodulating responses. Stem Cells (2008) 26:99–107. doi:10.1634/stemcells.2007-0563
170. Shi C, Jia T, Méndez-Ferrer S, Hohl TM, Serbina NV, Lipuma L, et al. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity (2011) 34:590–601. doi:10.1016/j.immuni.2011.02.016
171. Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood (2010) 116:4815–28. doi:10.1182/blood-2009-11-253534
172. Travnickova J, Tran Chau V, Julien E, Mateos-Langerak J, Gonzalez C, Lelièvre E, et al. Primitive macrophages control HSPC mobilization and definitive haematopoiesis. Nat Commun (2015) 6:6227. doi:10.1038/ncomms7227
173. Zeng M, Hu Z, Shi X, Li X, Zhan X, Li X-D, et al. MAVS, cGAS, and endogenous retroviruses in T-independent B cell responses. Science (2014) 346:1486–92. doi:10.1126/science.346.6216.1486
174. Roulois D, Yau HL, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell (2015) 162:961–73. doi:10.1016/j.cell.2015.07.056
175. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell (2015) 162:974–86. doi:10.1016/j.cell.2015.07.011
176. Reilly M, Miller RM, Thomson MH, Patris V, Ryle P, McLoughlin L, et al. Randomized, double-blind, placebo-controlled, dose-escalating phase I, healthy subjects study of intravenous OPN-305, a humanized anti-TLR2 antibody. Clin Pharmacol Ther (2013) 94:593–600. doi:10.1038/clpt.2013.150
177. Castaigne S, Pautas C, Terré C, Raffoux E, Bordessoule D, Bastie J-N, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet (2012) 379:1508–16. doi:10.1016/S0140-6736(12)60485-1
Keywords: inflammatory signaling, hematopoiesis, zebrafish, mouse, development, stress, disease
Citation: Clapes T, Lefkopoulos S and Trompouki E (2016) Stress and Non-Stress Roles of Inflammatory Signals during HSC Emergence and Maintenance. Front. Immunol. 7:487. doi: 10.3389/fimmu.2016.00487
Received: 23 September 2016; Accepted: 21 October 2016;
Published: 07 November 2016
Edited by:
Takayuki Yoshimoto, Tokyo Medical University, JapanReviewed by:
Julien Y. Bertrand, University of Geneva, SwitzerlandMartijn Nolte, Sanquin, Netherlands
Hal Broxmeyer, Indiana University School of Medicine, USA
Copyright: © 2016 Clapes, Lefkopoulos and Trompouki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eirini Trompouki, trompouki@ie-freiburg.mpg.de
†Thomas Clapes and Stylianos Lefkopoulos contributed equally to this work.