 Abhishek Saxena
Abhishek Saxena
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 December 2016
Sec. Vaccines and Molecular Therapeutics
Volume 7 - 2016 | https://doi.org/10.3389/fimmu.2016.00580
This article is part of the Research Topic Antibody Fc engineering: Towards better therapeutics View all 11 articles
Today, monoclonal immunoglobulin gamma (IgG) antibodies have become a major option in cancer therapy especially for the patients with advanced or metastatic cancers. Efficacy of monoclonal antibodies (mAbs) is achieved through both its antigen-binding fragment (Fab) and crystallizable fragment (Fc). Fab can specifically recognize tumor-associated antigen (TAA) and thus modulate TAA-linked downstream signaling pathways that may lead to the inhibition of tumor growth, induction of tumor apoptosis, and differentiation. The Fc region can further improve mAbs’ efficacy by mediating effector functions such as antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, and antibody-dependent cell-mediated phagocytosis. Moreover, Fc is the region interacting with the neonatal Fc receptor in a pH-dependent manner that can slow down IgG’s degradation and extend its serum half-life. Loss of the antibody Fc region dramatically shortens its serum half-life and weakens its anticancer effects. Given the essential roles that the Fc region plays in the modulation of the efficacy of mAb in cancer treatment, Fc engineering has been extensively studied in the past years. This review focuses on the recent advances in therapeutic Fc engineering that modulates its related effector functions and serum half-life. We also discuss the progress made in aglycosylated mAb development that may substantially reduce the cost of manufacture but maintain similar efficacies as conventional glycosylated mAb. Finally, we highlight several Fc engineering-based mAbs under clinical trials.
Monoclonal antibodies (mAbs) can target tumors through specific recognition of tumor-associated antigens and subsequent recruitment of effector elements including macrophages, dendritic cells, natural killer (NK) cells, T-cells, and the complement pathway components (1). Such recruitments are achieved by interactions among the immunoglobulin gamma (IgG)-crystallizable fragment (Fc) and the immune cell receptors like Fcγ receptors (FcγRs) and the complement protein C1q of the complement system (2–4). These interactions lead to the activation of immune cells for enhanced antibody-dependent cellular cytotoxicity (ADCC)/antibody-dependent cell-mediated phagocytosis (ADCP), formation of the membrane attack complex, and more efficient presentation of antigen to the dendritic cells (1). Through a recycling mechanism, the neonatal Fc receptor (FcRn) prolongs the half-life of mAbs in a pH-dependent interaction with the Fc region (5). The schematic of overall IgG structure and its binding regions with FcγRs, C1q, and FcRn is depicted in Figure 1.
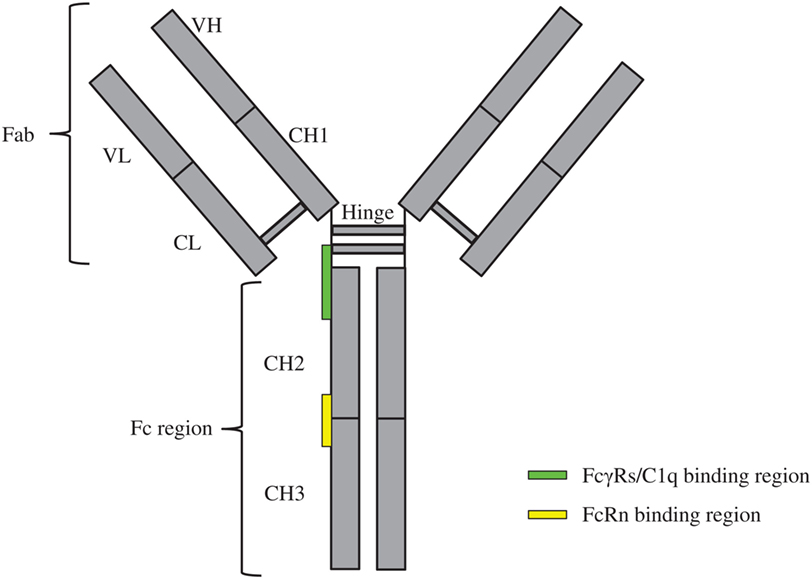
Figure 1. Schematics of immunoglobulin gamma overall structure and its binding regions with FcγRs, C1q, and FcRn. The constituent heavy [VH, CH1, hinge, CH2, and CH3 (gray)] and light chains [VL and CL (gray)] linked by inter-chain disulfide bonds are shown. The site at which FcγRs/C1q interacts with the crystallizable fragment (Fc) region is located in the lower hinge-upper CH2 (green rectangle); the site at which FcRn interacts with the Fc region is located in the interface of CH2–CH3 (yellow rectangle).
The FcγRs, consisting of FcγRI (CD64), FcγRII (CD32), and FcγRIII (CD16) classes, are heterogeneous in terms of their cellular expression and Fc binding affinities (1, 6). FcγRI binds to the Fc region with KD ~10−8–10−9 M and is expressed on mononuclear phagocytes, dendritic cells, and IFN-γ-activated neutrophils (1, 7, 8). FcγRII binds to the Fc region with relatively lower affinity (KD ~10−7 M) and exists in five isoforms; among them, activating (FcγRIIa, harboring an immunoreceptor tyrosine-based activation motif on neutrophils) or inhibitory (FcγRIIb, harboring an immunoreceptor tyrosine-based inhibitory motif predominantly on B-lymphocytes) are critical for immune regulation (1, 7). FcγRIII, expressed in two isoforms, binds the Fc region with the lowest affinities (KD ~10−5 M) (1, 7). Among these, FcγRIIIa has a moderate Fc binding allele (V158) and a low binding allele (F158), and is expressed on NK cells, macrophages, and T-cell subsets and activates NK and T cell-mediated ADCC response (1, 6, 7); FcγRIIIb is exclusively present on neutrophils and lacks signal generation capacity (1, 7).
Crystal structures of Fc in complex with FcγRI (9) (Figure 2A), Fc in complex with FcγRII (10) (Figure 2B), and Fc in complex with FcγRIII (11) (Figure 2C) reveal that the FcγRs’ interaction sites on Fc are all located within the lower hinge-upper heavy chain constant domain 2 (CH2). Furthermore, the binding affinity of Fc region to FcγRs also varies with the IgG subclasses (12).
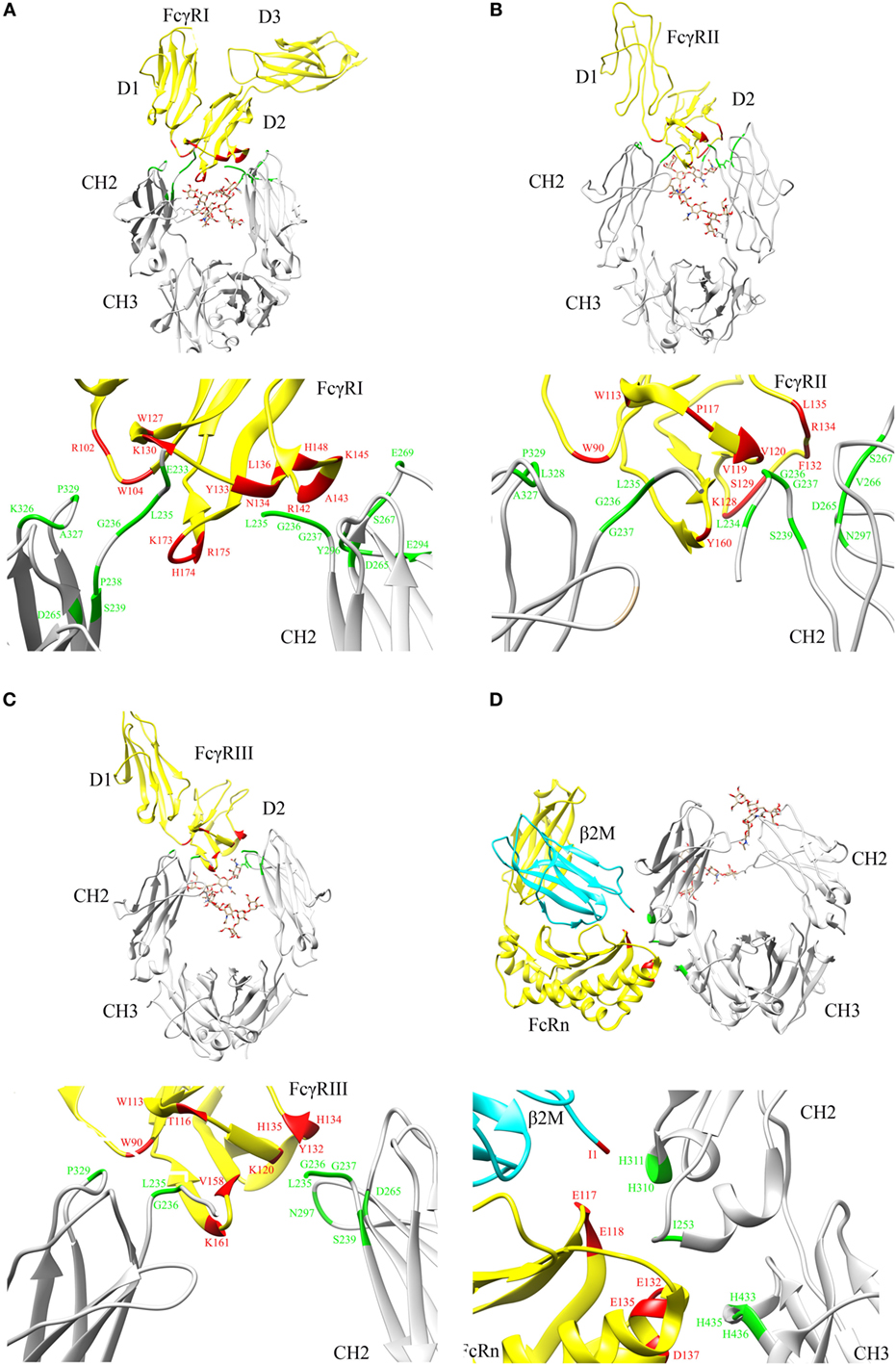
Figure 2. Crystal structures illustrating crystallizable fragment (Fc) interactions with FcγRs and FcRn. Representative structures are shown for (A) Fc–FcγRI cocrystals [PDB: 4W4O (9)], (B) Fc–FcγRII cocrystals [PDB: 3RY6 (10)], (C) Fc–FcγRIII cocrystals [PDB: 1T89 (11)], and (D) Fc–FcRn cocrystals [PDB: 1I1A (13)] with β2 microglobulin (β2M) domain shown in cyan. The Fc region and FcγRs are represented by gray and yellow color, respectively (A–D). N297 glycans within the CH2 domain are shown in stick model. The critical binding regions are highlighted in the upper part of each panel; region from the Fc fragment in green, region from the FcγRs, FcRn, and β2M in red. The lower part of each panel shows the detailed residues, which are involved in the interactions between Fc and its binding partners.
The C1q is a multisubunit protein of the complement system (3). It uses one of its six heads to establish a low-affinity (~10−6 M) interaction with the lower hinge-upper CH2 domain of the Fc region (3). Crystal structure of the human C1q head revealed that it is assembled with a heterotrimer globular architecture (14). Though the molecular basis of how the C1q head recognizes the Fc region is not known at the atomic resolution, Schneider and Zacharias (15) proposed a working model of C1q in complex with Fc based on known experimental data, docking, and molecular dynamics simulation. According to this model, upon initial weak interaction between the C1q head and the Fc region, IgGs can aggregate while recognizing “multiple epitopes” on the antigen surface and thus give many C1q molecules an opportunity to bind to their Fc regions, which enhance the “cumulative affinity” to ~10−9 M (3, 15). This leads to the deposition of complement component 3 (C3b) on the target cell and ultimate formation of the membrane attack complex that disrupts the lipid bilayer of the target cell, promotes cytolysis, and completes complement dependent cytotoxicity (CDC) (16, 17).
The pharmacokinetic profiles of antibodies vary among subclasses and are related to the structural features of the Fc region (18, 19). It is known that the serum half-life of IgG subclasses (IgG1, IgG2, and IgG4) is ~23 days as compared to 2–6 days for IgG3 and other Ig classes (18, 19). The Fc region spanning the interface of CH2 and CH3 domains interacts with the FcRn in the placenta, liver, mammary glands, and adult intestine to regulate IgG homeostasis and deliver maternal IgG across the placenta to the fetus (5). This interaction is favored by an acidic environment of the endosome after IgG is pinocytosed and thus IgG is protected from lysosomal degradation (20). The endocytosed IgG is then recycled to the cell surface and released into the blood stream at an alkaline pH, thereby maintaining the sufficient IgG serum half-life for proper immune functions and desired therapeutic efficacies (20). Recently, the endothelial and hematopoietic cells are identified as the major sites associated with FcRn expression and their critical role in IgG homeostasis (20–23).
Based on site-directed mutagenesis of Fc region and design of a hybrid Fc heterodimer harboring one half of Fc wild type (WT) and one half of Fc mutant, Kim and coworkers identified the key Fc residues involved in the FcRn interaction and proposed a preliminary model that one Fc hinge homodimer bound with two FcRn molecules (24, 25). Shortly thereafter, Burmeister and coworkers reported high resolution crystal structure of FcRn alone at 2.2 Å and low resolution crystal structure of Fc in complex with FcRn at 6.5 Å (26, 27). Structural analysis and biophysical data confirmed that one Fc homodimer binds with two FcRn molecules (26–29). Later, Martin and coworkers reported a high resolution crystal structure of Fc in complex with FcRn at 2.8 Å (13). This structure clearly shows the key residues involved in Fc and FcRn interactions and reveals the pH-dependent binding mechanism (13) (Figure 2D).
Evidence demonstrates the presence of oligosaccharides, attached to the N297 residue within the CH2 domain Asn-X-Ser/Thr glycosylation motif of Fc region, is essential in maintaining the Fc conformation and mediating its interactions with FcγRs (FcγRI, FcγRIIa, FcγRIIb, and FcγRIIIa) and C1q, but not FcRn (30–41). The glycan moiety is formed by two N-linked biantennary oligosaccharide chains consisting of a core heptasaccharide [N-acetylglucosamine (GlcNAc) and mannose (Man)] but occurrence of other residues like terminal N-acetlyneuraminic acid, galactose (Gal), bisecting N-acetylglucosamine (GlcNAc), and fucose (Fuc) have also been reported (1, 42, 43). Additionally, 5–17 and 2–7% of IgG structures could be monosialylated and disialylated, respectively (1, 44). This imparts a significant complexity and heterogeneity to therapeutic IgG molecules when expressed in mammalian cells, which can affect the therapeutic profile of IgG (30). On the other hand, the presence of bisected N-acetylglucosamine structures in rituximab, a purer glycoform with lesser heterogeneity, leads to an efficient engagement of FcγRIII and increases ADCC activity against CD20+ cells by up to 20-fold (30, 45). Similarly, non-fucosylated glycoform of Herceptin produced in engineered Chinese hamster ovary (CHO) cell line (LEC13) can enhance ADCC via FcγRIII engagement by up to 50-fold (30, 46).
However, mAb-associated glycan heterogeneity poses several key challenges (30, 33, 45–51) including (1) difficulties in developing therapeutic mAbs with glycan composition similar to naturally occurring human IgG1, (2) difficulties in controlling glycan heterogeneity, (3) lengthier development time to construct cell lines producing glycan homogeneity, (4) lengthier IgG production time and higher manufacturing cost in mammalian cells as compared to that in E. coli or yeast-based expression systems, (5) dominance of particular glycoforms that can affect effector functions of IgG molecules, and (6) difficulties in separating various glycoforms generated from mammalian cells. Alternatively, development of aglycosylated mAbs with similar efficacy as glycosylated counterpart but lower manufacturing cost has attracted great efforts in the past decade.
In this review, we focus on the recent progress in therapeutic Fc engineering-associated effector functions (ADCC, ADCP, and CDC) and pharmacokinetics. The mutations known to induce profound effects on Fc interaction with FcγRs, C1q, and FcRn are summarized (see Table 1). We also briefly describe the advances in aglycosylated mAb development. Finally, we highlight clinical trials of several mAbs developed from relevant Fc engineering.
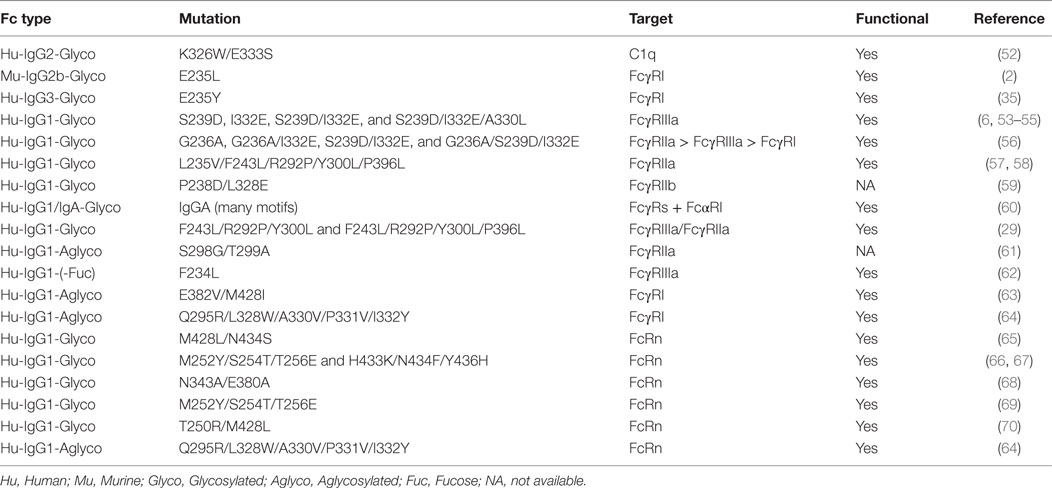
Table 1. Tabulation of the Fc mutations known to mediate a profound effect on antibody effector functions and immunoglobulin gamma homeostasis.
To develop more effective antibodies with desired ADCC, ADCP, and CDC activities, various strategies including site-directed mutagenesis, alanine scanning, structure-based computational design, and directed evolution technologies are employed.
The Fc amino acid residues that confer improved binding to FcγRs/C1q and enhanced immune response were initially characterized by site-directed mutagenesis studies. The earliest described mutations were discovered by scanning residues to isolate non-binders while focusing on the conserved residues. Fc residues (E318, K320, and L322) in the mouse IgG2b-Fc region were identified as the C1q binding site (3). However, the relevance of E318 and K320 was challenged in human Fc–C1q interaction (71). Novel residues (D270, K322, P329, and P331) were proposed for normal C1q binding on human Fc (71). This finding underscores the interspecies differences in such molecular interactions that may show a different effect in preclinical models. Furthermore, an IgG1 isotype of rituximab carrying K326W/E333S mutations was shown to have fivefold more binding to C1q (52) and the same motif, when transferred to the IgG2 isotype (poor complement activator) of rituximab, increased the cell lysis by fivefold (52). Next, a single mutation from E to L at position 235 of the mouse IgG2b-Fc region proposed it to be the “major determinant” for FcγRI binding (with ~100-fold increased affinity to human monocyte FcγRI) (2). Additionally, using a mouse–human chimeric antibody, amino acids at position 234 and 237 were shown to mainly influence the interaction with FcγRII. Based on these observations, FcγRI and FcγRII were proposed to recognize an overlapping but non-identical site on the Fc region (35).
Alanine scanning mutagenesis of selected Fc residues resulted in many variants with altered binding to specific FcγRs, which was also reflected in their ability to promote ADCC. Activating FcγRIIIa mutations improved ADCC by 100% (68). Furthermore, mutants based on the activating or suppressing effect on FcγRs were categorized into different classes. Among these, IgG1 mutations A327Q/P329A (interact with FcγRI), D265A/S267A/H268A/D270A/K326A/S337A (interact with FcγRIIa), and T256A/K290A/S298A/E333A/K334A (interact with FcγRIIIa) promoted high-affinity interactions (68).
Computational optimization of the Fc region by creating a single (S239D or I332E), double (S239D/I332E), and triple mutations (S239D/I332E/A330L) improved the affinity against human FcγRIIIaV158/F158 allele by up to 169-fold (6). The mutations favoring Fc binding to activating (FcγRIIIa) receptor over the inhibitory (FcγRIIb) receptor are important to develop IgGs with better activating to inhibitory capacity (IIIa:IIb ratio), which was monitored using surface plasmon resonance. These mutations showed up to ninefold improvement in IIIa:IIb ratio and contributed to more than twofold enhancement in ADCC/ADCP activity, and the S239D/I332E double mutant significantly depleted CD20+ B cells in vivo compared to WT IgG (6). The same Fc mutations also enhanced in vitro ADCC/ADCP activity against lymphoma cell lines and directly translated into a more effective treatment of lymphoproliferative diseases when incorporated into anti-CD19/CD40 mAbs (53, 54). Furthermore, it was shown that a change from glycine to alanine at residue 236 can shift the immune balance toward activating FcγRIIa relative to inhibitory FcγRIIb (56). The coupling of G236A to either I332E or S239D/I332E had dual beneficial effect as these mutants not only improve FcγRIIa:FcγRIIb ratio but also enhance binding to FcγRIIIa by ~6- to 31-fold (56). These mutants had significantly improved NK cell-mediated ADCC and macrophage-mediated ADCP activity (56).
In addition, “shuffled variants” of anti-CD20/CD57 antibody were constructed by grafting the CH1/hinge and CH3 carboxyl-terminal of IgG1 into the Fc of IgG3 to retain both the ADCC activity from IgG1 and the CDC activity from IgG3 (72). It is known that IgG1 is the most potent ADCC activator, while IgG3 has highest potency to recruit complement system (72). Therefore, IgG1 and IgG3 Fc regions can complement one another to maximize the immune effector response. These variants with chimeric CH regions showed ~25–60% increase in ADCC and CDC activity compared to WT of IgG1 and IgG3 molecules (72). Furthermore, the CDC activity of humanized anti-CD20 IgG1 (ocrelizumab) was increased by ~23-fold while retaining normal IgG1 ADCC by combining a triple mutant (S267E/H268F/S324T) with earlier reported G236A/I332E in the CH2 domain (73).
Multiple mutations (L235V/F243L/R292P/Y300L/P396L) in the trastuzumab Fc region (MGAH22) increased the potency against low Her2-expressing cells via low-affinity FcγRIIIaF158 engagement (57). The same Fc motif was applied to the MGA271 mAb (anti-CD276), which targets B7-H3+ tumor cells and resulted in an increased binding to FcγRIIIa, enhanced ADCC, and potent antitumor activity in a renal cell carcinoma/bladder cancer xenograft mouse model (58). Recently, the immune activating potential of IgA via FcαRI engagement was exploited by developing IgG and IgA hybrid molecules “IgGA” through substituting α1 loop residues of CHγ12/3 region with CHα12/3 (60). The “IgGA” hybrid trastuzumab mediated an enhanced ADCC/ADCP activity against Her2 overexpressing cells and destroyed up to 50% SkBr3 breast cancer cells (via ADCC) and MDA-MB-453 cells (via ADCP) (60). Similarly, “IgGA” hybrid rituximab lysed ~70% of the CD20+ calcein-AM-loaded Raji tumor cells when compared to the WT counterparts (60).
A negative selection strategy was applied using yeast surface display to enrich Fc mutants exhibiting selective high affinity to FcγRIIIa (29). Among these isolates, F243L was predicted to make a direct contact with the carbohydrate portion, which can “influence sialylation and affect the quaternary structure” of the Fc domain (29). Additionally, R292P partially reduced the binding to FcγRIIa, while Y300L, in combination with other mutations (F243L/R292P/V305I/P396L), showed an ~10-fold less KD, 100-fold enhanced ADCC activity, and potency in a xenograft mouse model of ovarian and breast cancer (29). Furthermore, an Fc variant with three changes (F243L/R292P/Y300L) was also effective in increasing the rate of cytolysis by ~100-fold (29). In another report, human IgG1 Fc variants were generated by an error-prone PCR and ribosome display to select high-affinity aglycosylated binders to human FcγRIIIa using a solution phase method (62). The isolated Fc mutant (F243L) lacked Fuc residues in most oligosaccharide chains and exhibited an improved FcγRIIIaV158/F158 binding and enhanced ADCC as compared to WT Fc (62). Recently, an anti-EGFR antibody (S239D/I332E) was noted to elicit mononuclear cell-mediated ADCC via FcγRIIIa engagement and at the same time showed impaired polymorphonuclear cell (PMN)-mediated ADCC due to the engagement of FcγRIIIb, a highly homologous isoform to FcγRIIIa (55). The inability of FcγRIIIb to activate immune signaling in such a scenario can be overcome by imparting high-affinity binding to FcγRIIa, which can enhance both NK cell- and PMN mediated ADCC (55). These observations highlight that Fc engineering toward related FcγRs needs to be tailored specifically to achieve desirable immune effects.
Along with the efforts to engineer Fc regions for enhanced effector functions, attempts have been made to improve antibody pharmacokinetics. Clearly, enhanced Fc–FcRn interaction at acidic pH can extend IgG’s serum half-life and positively regulate its homeostasis, which may benefit patients by greater therapeutic efficacy, less frequent dosing, and lower cost burden. Alanine scanning and display/directed evolution are commonly used techniques to identify favorable Fc mutants that can strengthen Fc–FcRn pH-dependent interactions.
A human Fc variant (N434A), isolated by alanine scanning of all solvent-exposed residues, showed fourfold increased binding to FcRn at pH 6.0 (68), which later, when studied in cynomolgus monkeys, showed a twofold extension of IgG serum half-life confirming the modulation of pharmacokinetics (74). Such an interaction at pH 6.0 prolongs IgG availability in serum, which correlates with the therapeutic effectiveness as demonstrated by the improved antitumor activity of IgG-Fc mutant (M428L/N434S from the CH3 domain) in a human FcRn transgenic mouse model (65).
A phage displayed antibody library approach was employed with the aim of isolating high-affinity binders against FcRn at pH 6.0. Random mutations were created at residues T252, T254, and T256, which are proximal to the IgG–FcRn interaction site, and binders were selected in a solution phase against FcRn at pH 6.0 and eluted by PBS at pH 7.4 (66). The Fc mutants generated in this manner were able to bind both human and rat FcRn with high affinity at pH 6.0; however, these variants (M252Y/S254T/T256E from the CH2 domain and H433K/N434F/Y436H from the CH3 domain) also showed tighter binding to mouse FcRn at pH 7.4 that decreased the serum IgG concentration in a mouse model (67). Yeung and coworkers (74) reported a similar observation that an Fc mutant (N434W), though possessing an ~80-fold enhanced affinity to FcRn at both acidic and neutral pH, did not stabilize serum IgG due to the loss in pH selectivity. These results suggest that high-affinity binding to the receptor at neutral pH can compromise the “beneficiary effect” of increased affinity at pH 6.0. Later, a humanized anti-respiratory syncytial virus antibody (MEDI-524) with a triple mutant Fc (M252Y/S254T/T256E from the CH2 domain) was reported to have a 10-fold increase in a pH-dependent way toward FcRn and about 4-fold improvement in serum half-life in cynomolgus monkey (69). Similarly, a high-affinity Fc variant (T250R/M428L) bound FcRn selectively at pH 6.0 and accounted for a 2.8-fold lesser degradation of serum IgG2 (70) and IgG1 (75) in rhesus monkey. Accumulating studies highlight that to prolong IgG serum half-life, pH-dependent FcRn affinity has to be maintained.
Of note, Grevys and coworkers recently analyzed known IgG-Fc mutants, which show enhanced FcRn pH-dependent affinity and extended serum half-life (M252Y/S254T/T256E from the CH2 domain and M428L/N434S from the CH3 domain), for their effects on ADCC, ADCP, and CDC (65, 69, 76). Surprisingly, they found that both mutants showed reduced effector functions with regards to ADCC, ADCP, and CDC. More interestingly, they found one previously known mutant (H433K/N434F from the CH3 domain), which showed reduced FcRn pH-dependent affinity and shortened serum half-life, displayed enhanced effector functions in ADCP, CDC, and ADCC (76, 77). These findings highlight that, though the interaction region of Fc with FcγRs and C1q (lower hinge region-CH2 domain) is distant from its interaction with FcRn (CH2–CH3 domain interface), it is still possible that these mutants can trigger a long range effect to other part of the Fc region in an unknown mechanism.
The demand for therapeutic antibodies is high, and it has been estimated that 8,000 kg of clinical grade mAbs were produced in 2013 (78). Currently, ~50% of the clinical grade biologics are produced in mammalian cells like CHO, mouse myeloma cell lines NSO and SP2/0 (30, 78–80).
However, the inherent glycan heterogeneity of mAbs when expressed in mammalian cell systems can cause high production cost and variations of mAb functions from batch to batch. Recently, great efforts have been invested in developing aglycosylated mAbs as an alternative. Ideally, aglycosylated mAbs would be as efficient as or even better than its glycosylated peers in mediating effector functions for antigen or target cell clearance.
To develop aglycosylated IgGs as alternatives, Sazinsky and coworkers constructed three small subsets of saturation substitutions covering the Asn-X-Ser/Thr glycan motif of the Fc C′/E loop and displayed these libraries on the yeast cell surface (61). After selection against FcγRIIa by fluorescence-activated cell sorting (FACS), a double Fc mutant isolate (S298G/T299A) in an aglycosylated form showed threefold stronger binding as compared to the WT and variants with single mutation, which indicates that the glycosylation of N297 is not a strict requirement for the interaction of Fc with FcγRIIa (61). In contrast, the indispensable role of asparagine at position 297 was demonstrated in the backdrop of N297Q, N297D, or N297A mutation, which abolished the double mutant (S298G/T299A) binding to FcγRIIa (61). Moreover, this dual mutant is functional in vivo in that murine platelet clearance is as efficient as that of WT mAb. Based on the modeling of Fc dual mutants in complex with FcγRIIa, the N297 residue of an aglycosylated IgG can make a hydrogen bond with the S126 residue of FcγRIIa (61, 81). Furthermore, such interaction may be strengthened by a bridging water molecule present in an unbound FcγRII crystal (61, 81). However, this dual mutant showed 10-fold reduced FcγRI binding and no binding to both FcγRIII and C1q proteins. The inability of aglycosylated mAbs to bind with C1q and thus activate CDC can limit their application in treating hospital acquired microbial infections among cancer patients where complement activity plays an important role (82).
In another major breakthrough, bacterial display and FACS was used to isolate Fc variants displaying increased binding and specificity to FcγRI (63). One such aglycosylated variant called “Fc5” (E382V/M428I) was incorporated into trastuzumab. The trastuzumab-Fc5 variant bound selectively to the FcγRI with nanomolar range affinity and promoted monocyte-derived dendritic cell-dependent lysis of SkBr3 breast cancer cells that overexpress Her2. (63). The three-dimensional structure of human aglycosylated Fc domain suggests a greater “conformational flexibility of the CH2-CH3 domain interface,” as compared to the glycosylated counterpart (83). Additional mutations (Q295R/L328W/A330V/P331V/I332Y) in trastuzumab-Fc5 variant (63) increased the affinity for FcγRI by ~120-fold and retained pH-dependent FcRn binding and function (64). Other mutants were also reported with specific binding to FcγRI and without compromising the pH-dependent FcRn binding (84).
It is worth mentioning that removal of appended glycans, though does not affect IgG’s solubility, binding affinities to FcγRs, and in vivo half-life but often compromises IgG related CDC, lowers its thermostability and increases its aggregation at the low pH (85–87).
A number of mAbs harboring various modifications in the Fc region are being investigated in different clinical trial stages (see Table 2 for Fc engineering-based mAbs being tested in clinics). These molecules broadly fall into three categories (1) with enhanced effector response to treat cancer and infectious diseases, (2) capable of inhibiting immune activation to treat inflammatory diseases, and (3) new class of aglycosylated mAbs with either inert or active-immune function.
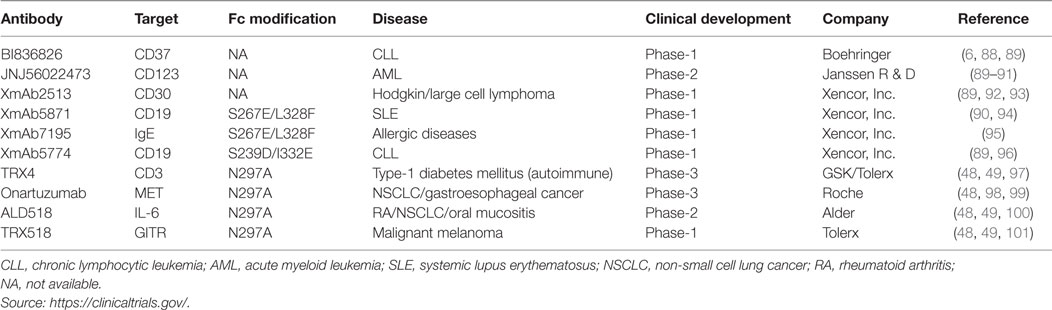
Table 2. Fc-engineered antibody candidates under clinical evaluation.
The Fc variants capable of inducing enhanced ADCC are being tested in many antibody candidates. Anti-CD37 antibody (BI836826; Boehringer) against B cell malignancies is currently under phase-1 trial for the treatment of chronic lymphocytic leukemia (CLL) (6, 88, 89). This is a mouse–human chimeric antibody, which targets tetraspanin CD37 and shows high proapoptotic activity against malignant B cells via enhanced ADCC. Using human CD37 transgenic mice, a single dose of BI836826 was demonstrated to reduce peripheral B cells (88) and efficacious in suppressing tumor growth in Ramos mouse model of human B-cell lymphoma (88). Furthermore, therapeutic efficacy of a surrogate Fc-engineered antibody against macaque CD37 has also been demonstrated in cynomolgus monkey (88). A fully humanized anti-CD123 antibody (JNJ-56022473; Janssen R & D) targeting overexpressed interleukin-3 receptor α-chain is being tested in acute myeloid leukemia patients (89–91). The Fc fragment of JNJ-56022473 has been engineered for enhanced NK cell-mediated ADCC. The molecule efficiently reduced the growth of the patient-derived acute myelogenous leukemia xenografts in bone marrow and peripheral organs and increased the survival in animal models (91). Similarly, a humanized anti-CD30 antibody (XmAb2513; Xencor) with enhanced binding to FcγRIIIa is being evaluated in the treatment of CD30+ Hodgkin’s lymphoma (HL) patients who had previously received two or more therapies (89, 92, 93). It has been shown to be safely administered and biologically active in relapsed, refractory HL subjects and reduces tumor in a majority of patients (92, 93). Another interesting molecule is an anti-CD19 antibody (XmAb5774; Xencor), which induces potent NK cell-mediated ADCC/ADCP response against CLL (89, 96). The antibody is known to get internalized in primary CLL cells and induces a modest toxicity (96).
Antibodies harboring Fc mutations that can suppress the immune response are being tested for the treatment of inflammatory diseases. The immunosuppressive version of anti-CD19 antibody (XmAb5871; Xencor) binds inhibitory FcγRIIb with ~430-fold enhanced affinity and efficiently depletes CD19+ B-cells in systemic lupus erythematosus (SLE) patients (90, 94). The depletion of CD19+ B-cells correlates with the strong inhibition of B-cell receptor-induced calcium mobilization among healthy volunteers and SLE patients (94). The same Fc fragment has been engineered into a humanized anti-IgE antibody (XmAb7195; Xencor) for the treatment of allergies (95). This antibody prevents the binding of IgE to its high-affinity IgE receptor (FcϵRI) that is present on basophils and mast cells and is useful in the treatment of allergic asthma (95). The XmAb7195 has 5- and ~430-fold higher affinity for human IgE and FcγRIIb, respectively, and therefore is effective in inhibiting IgE production and plasma cell differentiation (95).
Finally, aglycosylated IgG molecules have recently been shown to have therapeutic properties (48, 49, 61, 63), and a few of them are undergoing clinical testing. An anti-CD3 antibody (TRX4; Tolerx) incorporating the N297A mutation suppresses pathogenic T-cells in type-1 diabetes (T1D) patients (48, 49, 97) and is being evaluated in phase-3 trials. The antibody downregulates pathogenic T-cells while restoring the normal activity of T-regulatory cells and thereby inhibits autoimmune mediated T1D (97).
It is widely accepted that hepatocyte growth factor (HGF) binding to receptor tyrosine kinase MET aggravates malignancy in a variety of cancers (98). Therefore, an aglycosylated anti-MET antibody (Onartuzumab; Roche) is being evaluated in phase-3 trials to inhibit the binding of HGF for treating lung and gastroesophageal cancers (49, 98, 99). This is an E. coli-derived humanized, affinity-matured antibody, which engages MET, thereby inhibiting HGF binding and receptor phosphorylation in HGF-dependent tumor models (98). Similarly, an aglycosylated mAb (ALD518; Alder) targeting IL-6 is being tested in phase-2 for a variety of diseases including rheumatoid arthritis, non-small cell lung cancer (NSCLC), and oral mucositis (48, 49, 100). The antibody was developed to inhibit proinflammatory cytokine IL-6 in oncogenic niches, which can otherwise lead to a cancer. The mAb ALD518 is reported to be well tolerated in phase-1 and -2 studies and ameliorated NSCLC-related anemia and cachexia (100). Another aglycosylated mAb (TRX518; Tolerx) is currently in phase-1 trials to treat malignant myeloma (48, 49, 101). TRX518 mAb recognizes the glucocorticoid-induced tumor necrosis factor receptor on regulatory and effector T-cells, B-cells, NK cells, and antigen-presenting cells to enhance effector T-cell response and inhibits T-regulatory cell-mediated suppression (101). Furthermore, the efficacy of TRX518 mAb in reducing tumor burden and increased survival rates has been demonstrated in mouse and non-human primate models (101).
These studies highlight that Fc engineering has played important roles in developing antibodies with desirable properties and functions, and the ongoing clinical studies can give valuable information on the efficacy of the Fc-engineered mAbs, as compared to their existing peers.
Crystallizable fragment engineering has made substantial progress in the identification of new mutant(s) that can enhance effector functions and improve pharmacokinetics of mAbs for cancer treatment. Two major notions have emerged during the past decades’ efforts. First, the ratio of human activating FcγRs (FcγRI, FcγRIIa, and FcγRIIIa) and inhibitory FcγR (FcγRIIb) has to be taken into account during Fc engineering design. Beneficial effect can be achieved when Fc mutant(s) show higher selectivity and binding affinity toward activating FcγRs, as compared to the inhibitory FcγR. Second, the synergistic effects of improved ADCC, ADCP, and CDC of Fc region could increase the potency in cancer treatment.
Excitingly, aglycosylated mAb, expressed in bacteria and yeast, have been found to possess similar properties as glycosylated mAb with regards to FcγRs binding and serum half-life. However, efforts are still needed to improve its thermostability, solubility, and its binding affinity to C1q and CDC activity before it can really compete with its glycosylated peer for cancer therapy. On the other hand, the inability of aglycosylated mAbs to bind to C1q may have beneficial effects when CDC activity is not required such as for treatment of autoimmune diseases where CDC is chronically and pathologically activated (102). Similarly, aglycosylated mAbs may have advantages over glycosylated counterparts when only selective activation of FcγRs is desired such as activation of FcγRI (63) or FcγRIIa (84) to stimulate tumor cell killing.
DW conceived the topic; AS and DW wrote the manuscript; and DW revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by National Natural Science Foundation of China (Grant No: 81572698) to DW.
1. Jefferis R, Lund J, Pound JD. IgG-Fc-mediated effector functions: molecular definition of interaction sites for effector ligands and the role of glycosylation. Immunol Rev (1998) 163:59–76. doi:10.1111/j.1600-065X.1998.tb01188.x
2. Duncan AR, Woof JM, Partridge LJ, Burton DR, Winter G. Localization of the binding site for the human high-affinity Fc receptor on IgG. Nature (1988) 332:563–4. doi:10.1038/332563a0
3. Duncan AR, Winter G. The binding site for C1q on IgG. Nature (1988) 332:738–40. doi:10.1038/332738a0
4. Rayner LE, Hui GK, Gor J, Heenan RK, Dalby PA, Perkins SJ. The solution structures of two human IgG1 antibodies show conformational stability and accommodate their C1q and FcgammaR ligands. J Biol Chem (2015) 290:8420–38. doi:10.1074/jbc.M114.631002
5. Ghetie V, Ward ES. Multiple roles for the major histocompatibility complex class I-related receptor FcRn. Annu Rev Immunol (2000) 18:739–66. doi:10.1146/annurev.immunol.18.1.739
6. Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A (2006) 103:4005–10. doi:10.1073/pnas.0508123103
7. Ghirlando R, Keown MB, Mackay GA, Lewis MS, Unkeless JC, Gould HJ. Stoichiometry and thermodynamics of the interaction between the Fc fragment of human IgG1 and its low-affinity receptor Fc.gamma.RIII. Biochemistry (1995) 34:13320–7. doi:10.1021/bi00041a007
8. Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol (2001) 19:275–90. doi:10.1146/annurev.immunol.19.1.275
9. Kiyoshi M, Caaveiro JM, Kawai T, Tashiro S, Ide T, Asaoka Y, et al. Structural basis for binding of human IgG1 to its high-affinity human receptor FcγRI. Nat Commun (2015) 6:6866. doi:10.1038/ncomms7866
10. Ramsland PA, Farrugia W, Bradford TM, Sardjono CT, Esparon S, Trist HM, et al. Structural basis for Fc gammaRIIa recognition of human IgG and formation of inflammatory signaling complexes. J Immunol (2011) 187:3208–17. doi:10.4049/jimmunol.1101467
11. Radaev S, Motyka S, Fridman WH, Sautes-Fridman C, Sun PD. The structure of a human type III Fcgamma receptor in complex with Fc. J Biol Chem (2001) 276:16469–77. doi:10.1074/jbc.M100350200
12. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood (2009) 113:3716–25. doi:10.1182/blood-2008-09-179754
13. Martin WL, West AP, Gan L, Bjorkman PJ. Crystal structure at 2.8 Å of an FcRn/heterodimeric Fc complex. Mol Cell (2001) 7:867–77. doi:10.1016/s1097-2765(01)00230-1
14. Gaboriaud C, Juanhuix J, Gruez A, Lacroix M, Darnault C, Pignol D, et al. The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J Biol Chem (2003) 278:46974–82. doi:10.1074/jbc.M307764200
15. Schneider S, Zacharias M. Atomic resolution model of the antibody Fc interaction with the complement C1q component. Mol Immunol (2012) 51:66–72. doi:10.1016/j.molimm.2012.02.111
16. Sarma JV, Ward PA. The complement system. Cell Tissue Res (2011) 343:227–35. doi:10.1007/s00441-010-1034-0
17. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol (2014) 5:520. doi:10.3389/fimmu.2014.00520
18. Firan M, Bawdon R, Radu C, Ober RJ, Eaken D, Antohe F, et al. The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of gamma-globulin in humans. Int Immunol (2001) 13:993–1002. doi:10.1093/intimm/13.8.993
19. Medesan C, Matesoi D, Radu C, Ghetie V, Ward ES. Delineation of the amino acid residues involved in transcytosis and catabolism of mouse IgG1. J Immunol (1997) 158:2211–7.
20. Pyzik M, Rath T, Lencer WI, Baker K, Blumberg RS. FcRn: the architect behind the immune and nonimmune functions of IgG and albumin. J Immunol (2015) 194:4595–603. doi:10.4049/jimmunol.1403014
21. Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol (2007) 179:4580–8. doi:10.4049/jimmunol.179.7.4580
22. Montoyo HP, Vaccaro C, Hafner M, Ober RJ, Mueller W, Ward ES. Conditional deletion of the MHC class I-related receptor FcRn reveals the sites of IgG homeostasis in mice. Proc Natl Acad Sci U S A (2009) 106:2788–93. doi:10.1073/pnas.0810796106
23. Zhu X, Meng G, Dickinson BL, Li X, Mizoguchi E, Miao L, et al. MHC class I-related neonatal Fc receptor for IgG is functionally expressed in monocytes, intestinal macrophages, and dendritic cells. J Immunol (2001) 166:3266–76. doi:10.4049/jimmunol.166.5.3266
24. Kim JK, Tsen MF, Ghetie V, Ward ES. Localization of the site of the murine IgG1 molecule that is involved in binding to the murine intestinal Fc receptor. Eur J Immunol (1994) 24:2429–34. doi:10.1002/eji.1830241025
25. Kim JK, Tsen MF, Ghetie V, Ward ES. Identifying amino acid residues that influence plasma clearance of murine IgG1 fragments by site-directed mutagenesis. Eur J Immunol (1994) 24:542–8. doi:10.1002/eji.1830240308
26. Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature (1994) 372:379–83. doi:10.1038/372379a0
27. Burmeister WP, Gastinel LN, Simister NE, Blum ML, Bjorkman PJ. Crystal structure at 2.2 A resolution of the MHC-related neonatal Fc receptor. Nature (1994) 372:336–43. doi:10.1038/372336a0
28. Huber AH, Kelley RF, Gastinel LN, Bjorkman PJ. Crystallization and stoichiometry of binding of a complex between a rat intestinal Fc receptor and Fc. J Mol Biol (1993) 230:1077–83. doi:10.1006/jmbi.1993.1220
29. Stavenhagen JB, Gorlatov S, Tuaillon N, Rankin CT, Li H, Burke S, et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res (2007) 67:8882–90. doi:10.1158/0008-5472.CAN-07-0696
30. Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol Prog (2005) 21:11–6. doi:10.1021/bp040016j
31. Wright A, Morrison SL. Effect of glycosylation on antibody function: implications for genetic engineering. Trends Biotechnol (1997) 15:26–32. doi:10.1016/S0167-7799(96)10062-7
32. Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J Mol Biol (2003) 325:979–89. doi:10.1016/s0022-2836(02)01250-0
33. Jefferis R. Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol Sci (2009) 30:356–62. doi:10.1016/j.tips.2009.04.007
34. Jefferis R, Lund J. Interaction sites on human IgG-Fc for FcgammaR: current models. Immunol Lett (2002) 82:57–65. doi:10.1016/S0165-2478(02)00019-6
35. Lund J, Winter G, Jones PT, Pound JD, Tanaka T, Walker MR, et al. Human Fc gamma RI and Fc gamma RII interact with distinct but overlapping sites on human IgG. J Immunol (1991) 147:2657–62.
36. Lund J, Toshiyuki T, Noriko T, Sarmay G, Yoji A, Jefferis R. A protein structural change in aglycosylated IgG3 correlates with loss of huFcγR1 and hufcγR111 binding and/or activation. Mol Immunol (1990) 27:1145–53. doi:10.1016/0161-5890(90)90103-7
37. Medesan C, Radu C, Kim JK, Ghetie V, Ward ES. Localization of the site of the IgG molecule that regulates maternofetal transmission in mice. Eur J Immunol (1996) 26:2533–6. doi:10.1002/eji.1830261038
38. Mimura Y, Sondermann P, Ghirlando R, Lund J, Young SP, Goodall M, et al. Role of oligosaccharide residues of IgG1-Fc in Fc gamma RIIb binding. J Biol Chem (2001) 276:45539–47. doi:10.1074/jbc.M107478200
39. Radaev S, Sun PD. Recognition of IgG by Fcgamma receptor. The role of Fc glycosylation and the binding of peptide inhibitors. J Biol Chem (2001) 276:16478–83. doi:10.1074/jbc.M100351200
40. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol (2007) 7:715–25. doi:10.1038/nri2155
41. Walker MR, Lund J, Thompson KM, Jefferis R. Aglycosylation of human IgG1 and IgG3 monoclonal antibodies can eliminate recognition by human cells expressing FcγRI and/or FcγRII receptors. Biochem J (1989) 259:347–53. doi:10.1042/bj2590347
42. Jefferis R, Lund J, Mizutani H, Nakagawa H, Kawazoe Y, Arata Y, et al. A comparative study of the N-linked oligosaccharide structures of human IgG subclass proteins. Biochem J (1990) 268:529–37. doi:10.1042/bj2680529
43. Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr Opin Immunol (2008) 20:471–8. doi:10.1016/j.coi.2008.06.007
44. Routier FH, Hounsell EF, Rudd PM, Takahashi N, Bond A, Hay FC, et al. Quantitation of the oligosaccharides of human serum IgG from patients with rheumatoid arthritis: a critical evaluation of different methods. J Immunol Methods (1998) 213:113–30. doi:10.1016/S0022-1759(98)00032-5
45. Davies J, Jiang L, Pan LZ, LaBarre MJ, Anderson D, Reff M. Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FC gamma RIII. Biotechnol Bioeng (2001) 74:288–94. doi:10.1002/bit.1119.abs
46. Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem (2002) 277:26733–40. doi:10.1074/jbc.M202069200
47. Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov (2009) 8:226–34. doi:10.1038/nrd2804
48. Ju MS, Jung ST. Aglycosylated full-length IgG antibodies: steps toward next-generation immunotherapeutics. Curr Opin Biotechnol (2014) 30:128–39. doi:10.1016/j.copbio.2014.06.013
49. Jung ST, Kang TH, Kelton W, Georgiou G. Bypassing glycosylation: engineering aglycosylated full-length IgG antibodies for human therapy. Curr Opin Biotechnol (2011) 22:858–67. doi:10.1016/j.copbio.2011.03.002
50. Raju TS. Assessing Fc glycan heterogeneity of therapeutic recombinant monoclonal antibodies using NP-HPLC. Methods Mol Biol (2013) 988:169–80. doi:10.1007/9781627033275_10
51. Kubota T, Niwa R, Satoh M, Akinaga S, Shitara K, Hanai N. Engineered therapeutic antibodies with improved effector functions. Cancer Sci (2009) 100:1566–72. doi:10.1111/j.1349-7006.2009.01222.x
52. Idusogie EE, Wong PY, Presta LG, Gazzano-Santoro H, Totpal K, Ultsch M, et al. Engineered antibodies with increased activity to recruit complement. J Immunol (2001) 166:2571–5. doi:10.4049/jimmunol.166.4.2571
53. Horton HM, Bernett MJ, Pong E, Peipp M, Karki S, Chu SY, et al. Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res (2008) 68:8049–57. doi:10.1158/0008-5472.CAN-08-2268
54. Horton HM, Bernett MJ, Peipp M, Pong E, Karki S, Chu SY, et al. Fc-engineered anti-CD40 antibody enhances multiple effector functions and exhibits potent in vitro and in vivo antitumor activity against hematologic malignancies. Blood (2010) 116:3004–12. doi:10.1182/blood-2010-01-265280
55. Derer S, Glorius P, Schlaeth M, Lohse S, Klausz K, Muchhal U, et al. Increasing FcgammaRIIa affinity of an FcgammaRIII-optimized anti-EGFR antibody restores neutrophil-mediated cytotoxicity. MAbs (2014) 6:409–21. doi:10.4161/mabs.27457
56. Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther (2008) 7:2517–27. doi:10.1158/1535-7163.MCT-08-0201
57. Nordstrom JL, Gorlatov S, Zhang W, Yang Y, Huang L, Burke S, et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcgamma receptor binding properties. Breast Cancer Res (2011) 13:R123. doi:10.1186/bcr3069
58. Loo D, Alderson RF, Chen FZ, Huang L, Zhang W, Gorlatov S, et al. Development of an Fc-enhanced anti-B7-H3 monoclonal antibody with potent antitumor activity. Clin Cancer Res (2012) 18:3834–45. doi:10.1158/1078-0432.CCR-12-0715
59. Mimoto F, Katada H, Kadono S, Igawa T, Kuramochi T, Muraoka M, et al. Engineered antibody Fc variant with selectively enhanced FcgammaRIIb binding over both FcgammaRIIa(R131) and FcgammaRIIa(H131). Protein Eng Des Sel (2013) 26:589–98. doi:10.1093/protein/gzt022
60. Kelton W, Mehta N, Charab W, Lee J, Lee CH, Kojima T, et al. IgGA: a “cross-isotype” engineered human Fc antibody domain that displays both IgG-like and IgA-like effector functions. Chem Biol (2014) 21:1603–9. doi:10.1016/j.chembiol.2014.10.017
61. Sazinsky SL, Ott RG, Silver NW, Tidor B, Ravetch JV, Wittrup KD. Aglycosylated immunoglobulin G1 variants productively engage activating Fc receptors. Proc Natl Acad Sci U S A (2008) 105:20167–72. doi:10.1073/pnas.0809257105
62. Stewart R, Thom G, Levens M, Guler-Gane G, Holgate R, Rudd PM, et al. A variant human IgG1-Fc mediates improved ADCC. Protein Eng Des Sel (2011) 24:671–8. doi:10.1093/protein/gzr015
63. Jung ST, Reddy ST, Kang TH, Borrok MJ, Sandlie I, Tucker PW, et al. Aglycosylated IgG variants expressed in bacteria that selectively bind FcgammaRI potentiate tumor cell killing by monocyte-dendritic cells. Proc Natl Acad Sci U S A (2010) 107:604–9. doi:10.1073/pnas.0908590107
64. Jung ST, Kang TH, Kim D-il. Engineering an aglycosylated Fc variant for enhanced FcγRI engagement and pH-dependent human FcRn binding. Biotechnol Bioprocess Eng (2014) 19:780–9. doi:10.1007/s12257-013-0432-z
65. Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, et al. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol (2010) 28:157–9. doi:10.1038/nbt.1601
66. Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, et al. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotechnol (1997) 15:637–40. doi:10.1038/nbt0797-637
67. Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, et al. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol (2002) 169:5171–80. doi:10.4049/jimmunol.169.9.5171
68. Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem (2001) 276:6591–604. doi:10.1074/jbc.M009483200
69. Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem (2006) 281:23514–24. doi:10.1074/jbc.M604292200
70. Hinton PR, Johlfs MG, Xiong JM, Hanestad K, Ong KC, Bullock C, et al. Engineered human IgG antibodies with longer serum half-lives in primates. J Biol Chem (2004) 279:6213–6. doi:10.1074/jbc.C300470200
71. Idusogie EE, Presta LG, Gazzano-Santoro H, Totpal K, Wong PY, Ultsch M, et al. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J Immunol (2000) 164:4178–84. doi:10.4049/jimmunol.164.8.4178
72. Natsume A, In M, Takamura H, Nakagawa T, Shimizu Y, Kitajima K, et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res (2008) 68:3863–72. doi:10.1158/0008-5472.CAN-07-6297
73. Moore GL, Chen H, Karki S, Lazar GA. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs (2010) 2:181–9. doi:10.4161/mabs.2.2.11158
74. Yeung YA, Leabman MK, Marvin JS, Qiu J, Adams CW, Lien S, et al. Engineering human IgG1 affinity to human neonatal Fc receptor: impact of affinity improvement on pharmacokinetics in primates. J Immunol (2009) 182:7663–71. doi:10.4049/jimmunol.0804182
75. Hinton PR, Xiong JM, Johlfs MG, Tang MT, Keller S, Tsurushita N. An engineered human IgG1 antibody with longer serum half-life. J Immunol (2006) 176:346–56. doi:10.4049/jimmunol.176.1.346
76. Grevys A, Bern M, Foss S, Bratlie DB, Moen A, Gunnarsen KS, et al. Fc engineering of human IgG1 for altered binding to the neonatal Fc receptor affects Fc effector functions. J Immunol (2015) 194:5497–508. doi:10.4049/jimmunol.1401218
77. Vaccaro C, Bawdon R, Wanjie S, Ober RJ, Ward ES. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. Proc Natl Acad Sci U S A (2006) 103:18709–14. doi:10.1073/pnas.0606304103
78. Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs (2015) 7:9–14. doi:10.4161/19420862.2015.989042
79. Naso MF, Tam SH, Scallon BJ, Raju TS. Engineering host cell lines to reduce terminal sialylation of secreted antibodies. MAbs (2010) 2:519–27. doi:10.4161/mabs.2.5.13078
80. Chu L, Robinson DK. Industrial choices for protein production by large-scale cell culture. Curr Opin Biotechnol (2001) 12:180–7. doi:10.1016/S0958-1669(00)00197-X
81. Maxwell KF, Powell MS, Hulett MD, Barton PA, McKenzie IF, Garrett TP, et al. Crystal structure of the human leukocyte Fc receptor, Fc gammaRIIa. Nat Struct Biol (1999) 6:437–42. doi:10.1038/8241
82. Steele RW. Managing infection in cancer patients and other immunocompromised children. Ochsner J (2012) 12:202–10.
83. Borrok MJ, Jung ST, Kang TH, Monzingo AF, Georgiou G. Revisiting the role of glycosylation in the structure of human IgG Fc. ACS Chem Biol (2012) 7:1596–602. doi:10.1021/cb300130k
84. Jung ST, Kelton W, Kang TH, Ng DT, Andersen JT, Sandlie I, et al. Effective phagocytosis of low Her2 tumor cell lines with engineered, aglycosylated IgG displaying high FcgammaRIIa affinity and selectivity. ACS Chem Biol (2013) 8:368–75. doi:10.1021/cb300455f
85. Latypov RF, Hogan S, Lau H, Gadgil H, Liu D. Elucidation of acid-induced unfolding and aggregation of human immunoglobulin IgG1 and IgG2 Fc. J Biol Chem (2012) 287:1381–96. doi:10.1074/jbc.M111.297697
86. Alsenaidy MA, Kim JH, Majumdar R, Weis DD, Joshi SB, Tolbert TJ, et al. High-throughput biophysical analysis and data visualization of conformational stability of an IgG1 monoclonal antibody after deglycosylation. J Pharm Sci (2013) 102:3942–56. doi:10.1002/jps.23730
87. Hristodorov D, Fischer R, Joerissen H, Muller-Tiemann B, Apeler H, Linden L. Generation and comparative characterization of glycosylated and aglycosylated human IgG1 antibodies. Mol Biotechnol (2013) 53:326–35. doi:10.1007/s12033-012-9531-x
88. Heider KH, Kiefer K, Zenz T, Volden M, Stilgenbauer S, Ostermann E, et al. A novel Fc-engineered monoclonal antibody to CD37 with enhanced ADCC and high proapoptotic activity for treatment of B-cell malignancies. Blood (2011) 118:4159–68. doi:10.1182/blood-2011-04-351932
89. Sondermann P, Szymkowski DE. Harnessing Fc receptor biology in the design of therapeutic antibodies. Curr Opin Immunol (2016) 40:78–87. doi:10.1016/j.coi.2016.03.005
90. Horton HM, Chu SY, Ortiz EC, Pong E, Cemerski S, Leung IW, et al. Antibody-mediated coengagement of FcgammaRIIb and B cell receptor complex suppresses humoral immunity in systemic lupus erythematosus. J Immunol (2011) 186:4223–33. doi:10.4049/jimmunol.1003412
91. Lee EM, Yee D, Busfield SJ, McManus JF, Cummings N, Vairo G, et al. Efficacy of an Fc-modified anti-CD123 antibody (CSL362) combined with chemotherapy in xenograft models of acute myelogenous leukemia in immunodeficient mice. Haematologica (2015) 100:914–26. doi:10.3324/haematol.2014.113092
92. Blum KA, Smith M, Fung H, Zalevsky J, Combs D, Ramies DA, et al. Phase I study of an anti-CD30 Fc engineered humanized monoclonal antibody in Hodgkin lymphoma (HL) or anaplastic large cell lymphoma (ALCL) patients: safety, pharmacokinetics (PK), immunogenicity, and efficacy. J Clin Oncol (2009) 27:8531.
93. Kumar A, Blum KA, Fung HC, Smith MR, Foster PA, Younes A. A phase 1 dose-escalation study of XmAb(R) 2513 in patients with relapsed or refractory Hodgkin lymphoma. Br J Haematol (2015) 168:902–4. doi:10.1111/bjh.13152
94. Chu SY, Vostiar I, Karki S, Moore GL, Lazar GA, Pong E, et al. Inhibition of B cell receptor-mediated activation of primary human B cells by coengagement of CD19 and FcgammaRIIb with Fc-engineered antibodies. Mol Immunol (2008) 45:3926–33. doi:10.1016/j.molimm.2008.06.027
95. Chu SY, Horton HM, Pong E, Leung IW, Chen H, Nguyen DH, et al. Reduction of total IgE by targeted coengagement of IgE B-cell receptor and FcgammaRIIb with Fc-engineered antibody. J Allergy Clin Immunol (2012) 129:1102–15. doi:10.1016/j.jaci.2011.11.029
96. Awan FT, Lapalombella R, Trotta R, Butchar JP, Yu B, Benson DM Jr, et al. CD19 targeting of chronic lymphocytic leukemia with a novel Fc-domain-engineered monoclonal antibody. Blood (2010) 115:1204–13. doi:10.1182/blood-2009-06-229039
97. Aronson R, Gottlieb PA, Christiansen JS, Donner TW, Bosi E, Bode BW, et al. Low-dose otelixizumab anti-CD3 monoclonal antibody DEFEND-1 study: results of the randomized phase III study in recent-onset human type 1 diabetes. Diabetes Care (2014) 37:2746–54. doi:10.2337/dc13-0327
98. Merchant M, Ma X, Maun HR, Zheng Z, Peng J, Romero M, et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc Natl Acad Sci U S A (2013) 110:E2987–96. doi:10.1073/pnas.1302725110
99. Park HI, Yoon HW, Jung ST. The highly evolvable antibody Fc domain. Trends Biotechnol (2016) 34:895–908. doi:10.1016/j.tibtech.2016.04.005
100. Bayliss TJ, Smith JT, Schuster M, Dragnev KH, Rigas JR. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin Biol Ther (2011) 11:1663–8. doi:10.1517/14712598.2011.627850
101. Rosenzweig M, Ponte J, Apostolou I, Doty D, Guild J, Slavonic M, et al. Development of TRX518, an aglycosyl humanized monoclonal antibody (Mab) agonist of huGITR. J Clin Oncol (2010) 28:e13028.
Keywords: antibody Fc region, ADCC, CDC, ADCP, serum half-life, aglycosylated antibody, FcRn, cancer therapy
Citation: Saxena A and Wu D (2016) Advances in Therapeutic Fc Engineering – Modulation of IgG-Associated Effector Functions and Serum Half-life. Front. Immunol. 7:580. doi: 10.3389/fimmu.2016.00580
Received: 12 July 2016; Accepted: 24 November 2016;
Published: 12 December 2016
Edited by:
Tianlei Ying, Fudan University, ChinaReviewed by:
Pierre Guermonprez, King’s College London, UKCopyright: © 2016 Saxena and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Donghui Wu, wudh@shanghaitech.edu.cn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.