 Andrea Majoros1
Andrea Majoros1 Ekaterini Platanitis1
Ekaterini Platanitis1 Elisabeth Kernbauer-Hölzl1
Elisabeth Kernbauer-Hölzl1 Felix Rosebrock1
Felix Rosebrock1 Mathias Müller2
Mathias Müller2 Thomas Decker1*
Thomas Decker1*
- 1Department of Microbiology, Immunobiology and Genetics, Max F. Perutz Laboratories, University of Vienna, Vienna, Austria
- 2Institute of Animal Breeding and Genetics, University of Veterinary Medicine Vienna, Vienna, Austria
Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signal transduction mediates cytokine responses. Canonical signaling is based on STAT tyrosine phosphorylation by activated JAKs. Downstream of interferon (IFN) receptors, activated JAKs cause the formation of the transcription factors IFN-stimulated gene factor 3 (ISGF3), a heterotrimer of STAT1, STAT2 and interferon regulatory factor 9 (IRF9) subunits, and gamma interferon-activated factor (GAF), a STAT1 homodimer. In recent years, several deviations from this paradigm were reported. These include kinase-independent JAK functions as well as extra- and intranuclear activities of U-STATs without phosphotyrosines. Additionally, transcriptional control by STAT complexes resembling neither GAF nor ISGF3 contributes to transcriptome changes in IFN-treated cells. Our review summarizes the contribution of non-canonical JAK–STAT signaling to the innate antimicrobial immunity imparted by IFN. Moreover, we touch upon functions of IFN pathway proteins beyond the IFN response. These include metabolic functions of IRF9 as well as the regulation of natural killer cell activity by kinase-dead TYK2 and different phosphorylation isoforms of STAT1.
Introduction
Since their discovery in the late 1950s (1), interferons (IFNs) have been assigned various functions that extend far beyond the initially observed antiviral activity. Three families of IFNs have been described and are known as type I (IFN-I, mainly IFNα/β), type II (IFN-II or IFNγ), and type III (IFN-III or IFNλ). In canonical IFN signaling, all types of IFNs produce a transcriptionally active signal transducer and activator of transcription 1 (STAT1) through receptor-bound Janus kinase (JAK)-mediated phosphorylation of tyrosine (Y) 701. The IFNγ receptor employs JAK1 and JAK2 to phosphorylate exclusively STAT1, causing its homodimerization. STAT1 dimers, also called gamma interferon-activated factor (GAF), translocate to the nucleus and promote gene expression by binding to gamma interferon-activated site (GAS) of interferon-stimulated genes (ISG). On the other hand, stimulation with IFN-I or IFN-III leads to TYK2- and JAK1-mediated phosphorylation of STAT1 and STAT2. After forming heterodimers, these two proteins associate with interferon regulatory factor 9 (IRF9) to form a transcriptionally active IFN-stimulated gene factor 3 (ISGF3) that controls gene expression by binding to interferon-stimulated response elements (ISRE) in a different set of ISGs (2, 3). Although a large majority of IFN-induced gene expression is mediated by canonical pathways, it has become clear that the components of these pathways are able to exert non-canonical activity: tyrosine kinase-independent action of JAKs, transcriptional complexes other than ISGF3 and GAF, and pathways building on U-STATs that are not phosphorylated on tyrosine. This review will focus on these non-canonical functions of JAK–STAT signaling components.
IFNs and Their Role in Resistance to Viruses and Bacteria
As major components of the innate immune system against viral infections, all type I interferons (IFN-I) stimulate cell-autonomous antiviral activity (4). In addition, they increase cellular immunity through contributions to natural killer (NK) and T cell activation (5). IFN-I act as modulators of cellular immunity by selectively enhancing clonal expansion and survival of CD8+ T cells (6), directing the immune response toward Th1-dominace (7) and activating NK cells (8).
In the context of antibacterial defense, genes activated by IFN-I enhance inflammation and the death of infected cells (9). In addition, they impact on cells at the interface of the innate and adaptive immune systems, such as macrophages and dendritic cells, to increase antigen presentation and trigger the adaptive response (10). Immunostimulatory activities of IFN-I also contribute to immunosurveillance against cancer (11). On the other hand, their proinflammatory activity renders them driving forces behind a group of interferonopathies, such as the Aicardi-Goutieres syndrome (12, 13).
While IFN-I are generally protective against viral infections, they can be both friend and foe in the defense of bacterial pathogens (14, 15). For example, IFN-I exert protective effects in the case of Chlamydia pneumoniae (16), Legionella pneumophila (17), Salmonella typhimurium (18), and both group A and group B Streptococcus infections (19–21). However, in infections with Listeria monocytogenes, Francisella tularensis, and Mycobacterium tuberculosis, production of IFN-I is associated with decreased innate immunity (22–27).
Major target cells of IFNγ are macrophages and T cells. Many of the genes induced by IFNγ are transcription factors, which amplify the transcriptional response and, as in Th cells, influence cell differentiation (28–30). IFNγ is particularly important in macrophage biology, where it provides cell-autonomous antimicrobial activity through the upregulation of microbicidal gene products (31). Further, impact of IFNγ on macrophage activation results from its ability to synergize with or to antagonize the effects of different cytokines, growth factors, and pathogen-associated molecular pattern-signaling pathways (e.g., TNFα, IL-4, CSF-1, IFNα/β, LPS, and CpG DNA). Through these mechanisms, IFNγ activates macrophages to express antimicrobial and antitumor effects. It upregulates chemokines and adhesion molecules, directing cells to the sites of inflammation. In the adaptive immunity, IFNγ plays an important role in Th1 responses, repressing the development of Th2 and Th17 T cell responses (30) and acting directly on B cells to promote class switching from IgG2 to IgG3 (32). Mice and humans deficient in IFNγ or IFNGR1 show a decrease in natural resistance to bacterial, parasitic, and viral infections (33–35). Mice and cells lacking IFNγ display compromised tumor rejection, underlining its importance in tumor surveillance (11).
Discovered in the year 2003, IFN-III, better known as IFNλ, are the most recently described members of the IFN family (36). Although signaling through a different receptor complex with IFNλR/IL10R2 chains, IFNλ also stimulate formation of the ISGF3 complex. Given the similarities between the IFN-I and IFNλ signaling pathways, some of the non-canonical signals described below for IFN-I may apply to IFNλ as well. While IFNλ produce similar biological changes of their target cells as IFN-I, including antiviral, antiproliferative, and antitumor activity, the key to different organismic responses to IFN-III lies in their receptor distribution, which is prevalent on cells of epithelial origin (37). In line with that, defects in IFN-III production or signaling cause reduced innate immunity to viral pathogens replicating in epithelia of the lung and the gut. Mice lacking the IFN-I receptor are resistant to all IFN-I subtypes, but retain their sensitivity to IFN-III (38). However, studies performed in IFNAR1−/−, IL-28Rα−/−, and IFNAR1/IL-28Rα double knockout mice show that, in primary airway epithelia, upon influenza infection, IFN-I and IFN-III mediate parallel amplification loops that lead to the induction of fully overlapping groups of ISGs (39).
Kinase-Independent JAK Activity
Janus kinases are non-receptor tyrosine kinases which have essential roles in cytokine and growth factor signaling (40, 41). There are four different JAKs (JAK1, JAK2, JAK3, and TYK2) that cross-phosphorylate and activate each other when ligand-associated cytokine receptor chains come in close proximity. Canonical, i.e., kinase-dependent functions of JAKs include tyrosine phosphorylation of receptor chains and of STATs at a single tyrosine residue near the C-terminal end. STAT phosphorylation is thought to require SH2 domain-mediated docking to the modified receptor chains, consistent with impaired phosphorylation at mutant receptors lacking the critical tyrosine for JAK-mediated phosphorylation (2).
Recently, reports studying kinase-inactive mutants of TYK2 and JAK2 suggest that these proteins exert functions not requiring their kinase activity and have important non-canonical roles. Elegant studies in mouse models with kinase-dead enzymes allow the demonstration of kinase-independent functions under physiologic conditions and complement studies in human and murine cell lines. Furthermore, description of naturally occurring mutations in JAKs in the human population broadens our understanding of the multifaceted role of JAKs in health and disease.
TYK2
TYK2 is involved in a large number of cytokine signaling cascades as it associates with the IFN-I (IFNAR), IL-12Rβ1, IL-10R2, gp130, and IL13α1 receptor (42). Early work in human cell lines has demonstrated the important function of TYK2 in IFN-I signaling (43–46), as human cells fail to respond to IFNα in absence of TYK2. Furthermore, such cells display reduced IFNAR expression at the cell surface. Interestingly, this is a consequence of a non-canonical role of TYK2. The scaffold TYK2, but not its kinase or pseudokinase domains are needed for surface expression of IFNAR in human 11,1 cells (47). Specifically, TYK2 masks a tyrosine-based motif found in IFNAR, thereby shielding the receptor from endocytosis and preventing the binding of an enzyme (AP2), which leads to ubiquitin-dependent internalization (48). In this context, TYK2-receptor association requires neither ligand nor ubiquitination. Preventing receptor degradation is a species-specific TYK2 activity, consistent with the lack of the Tyr-based motif in the murine IFNAR. However, TYK2 is not essential for IFN-I signaling in murine cells, and residual IFN-I activity is detected in the absence of this JAK (49, 50).
Mice with a targeted mutation of a critical lysine residue in the ATP-binding pocket of the TYK2 kinase domain (K923E) express a kinase-dead enzyme (51). Kinase-dead TYK2 shows a strongly reduced half-life owing to increased turnover via autophagosomal degradation. The reduced levels of TYK2K923E do not increase IFN-induced STAT activation above the level seen in the Tyk2−/− animals. Consistently, TYK2K923E mice are more susceptible to viral infections, comparable to Tyk2−/− mice. In contrast, TYK2K923E expression partially rescued the defect in natural killer cell (NK-cell) maturation and tumor killing that accompanies TYK2 deficiency (Table 1). At present, this first observation of a kinase-independent in vivo function of TYK2 cannot be assigned to a defined NK signaling pathway. Other attributes of activated NK cells like IL-12 synthesis or activating receptor-stimulated production of IFNγ rely on TYK2 kinase activity (52).

Table 1. An overview of non-canonical Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signaling by components of interferon (IFN) pathways.
Ligation of the IFNAR causes TYK2-dependent activation of the phosphoinositide 3 kinase (PI3K) pathway. Reportedly, this occurs without a need for TYK2 kinase activity (45). Moreover, in murine pro B cells, a part of TYK2’s functions in mitochondrial respiration is retained in absence of its kinase activity (53). The cells show a drastic defect in basal oxygen consumption and steady-state cellular ATP levels in the absence of TYK2, which can be reversed after transfection of wild-type or a kinase-dead mutant of TYK2 into these cells. In contrast, the kinase activity of TYK2 is required for other functions of mitochondria like complex I-mediated respiration and the induction of apoptosis after IFNβ treatment. TYK2 has also been linked to the energy expenditure of cells, to the regulation of lipid metabolism, differentiation of brown adipose tissue, and obesity (54–56). In human obese patients and obese mice, decreased TYK2 levels are associated with increased obesity. This effect is regulated via Stat3 signaling and prolonged stability of the transcriptional coactivator PRDM16, a master regulator of brown adipose tissue (55). The interaction of TYK2 with STAT3 is most probably a non-canonical event, as tyrosine phosphorylation of STAT3 does not require TYK2. Therefore, the kinase must provide another kind of mechanistic input, either indirectly through other pathways or through another modification of STAT3.
The Tyk2 gene displays many different SNPs in the human population, and GWAS studies have linked mutations in Tyk2 to autoimmune diseases like systemic lupus erythematosus, multiple sclerosis, Crohn’s disease, psoriasis, type I diabetes, endometriosis-related infertility, primary biliary cirrhosis, and rheumatoid arthritis (22–25, 57–68). The first patient described with Tyk2 mutation suffered from hyper-IgE syndrome (HIES) and presented with viral, bacterial, and mycobacterial infections. The role of TYK2 in several cytokine signaling pathways leads to these diverse susceptibilities, explained in part by a bias toward Th2 immunity and defective IL-12 and IFN-I pathways (26). Complementing this study, Kreins et al. described seven different patients from four different ethnic backgrounds with different mutations in Tyk2 (27). All patients examined so far had mutations, deletions, or substitutions in the Tyk2 gene which ultimately led to a premature stop codon and no expression of Tyk2 protein. This group of patients did not present HIES. However, similar to the first patient, they suffered from widespread mycobacterial and viral infections. Using microarray analysis, Kreins et al. demonstrated that, similar to TYK2-deficient mice, responses of the patient’s cells to various cytokines like IL-12, IFN-I, IL-23, and IL-10 are greatly reduced, but not completely abolished. This observation, made by the use of a sensitive detection method, might resolve the apparent discrepancy regarding the partial or absolute requirement of TYK2 for cytokine signaling in murine versus human cells. Another study described disease-associated human TYK2 variants, which are catalytically impaired, but able to rescue signaling in response to IFN-I, IL-6, and IL-10 in vitro (69). The authors proposed a model for receptors associating with more than one JAK according to which only one JAK needs to be catalytically active in order to convey signals, as long as the second JAK functions as a scaffold.
JAK2
JAK2 is involved in many biological processes, including the growth control, survival, and differentiation particularly of hematopoietic cells. Accordingly, the widespread use of this kinase is reflected by the embryonic lethality of homozygous deletion (70, 71). The kinase activity of JAK2 is also implicated in various lymphoid and myeloid leukemias in which chromosomal translocation generates a hyperactive kinase (72–74). A scaffold function of JAK2 is suggested by the finding that the N-terminal domain of JAK2 alone is sufficient to enhance surface expression of the Epo receptor (EpoR) (75). Like Tyk2, mice expressing kinase-dead JAK2 were generated and resulted in the discovery of kinase-independent functions. Frenzel et al. generated a mouse model expressing a dominant-negative, kinase-inactive JAK2 by mutating residues in the C-terminal kinase domain (W1038G, E1046R) (76). This mouse mimics the complete loss of JAK2 as homozygous embryos die in utero. Heterozygous mice containing this dominant-negative form of Jak2 did not show any hematopoietic abnormalities, and it seems that one intact copy of Jak2 can compensate for the loss of kinase activity of the other, inactive form. Keil et al. generated mice with inactive JAK2 by mutation of the activation loop tyrosines [JAK2YY1007/1008FF—(77)]. Similar to the mouse described by Frenzel et al., homozygous JAK2YY1007/1008FF alleles caused embryonic lethality and defective EpoR signaling, whereas heterozygous mice appeared phenotypically normal. However, the study revealed a kinase-independent scaffolding function of JAK2 for the heteromeric IFN-gamma receptor (IFNGR) complex. Importantly, JAK2YY1007/1008FF mediated the cell surface expression of the IFNGR indistinguishable from wild-type JAK2. Likewise, the recruitment of JAK1 to the receptor was normal. Contrasting Jak2-deficient cells, JAK1 alone was able to partly compensate for the loss of JAK2 signaling by phosphorylating STAT1 and inducing genes in response to IFNγ.
STAT1-Independent Resistance to Viruses and Bacteria—The Role of STAT2 and IRF9
Experiments performed in cells and mice lacking functional STAT1 revealed its indispensable role in the IFN signaling pathway. To date, mice lacking STAT1 have been challenged with at least 27 different pathogens and proved to be highly susceptible to most of the viruses, with exception of (+) single-strand RNA dengue virus (DENV) and the (−) single-strand RNA measles virus (MV). These mice are also highly susceptible to intracellular bacteria, such as Listeria monocytogenes and Mycobacterium tuberculosis, and parasites such as Toxoplasma gondii and Leishmania major (78). Patients with complete autosomal recessive STAT1 deficiency succumb to lethal mycobacterial and viral infections. Heterozygous autosomal recessive STAT1 deficiency also causes impaired IFN responses, but with milder clinical symptoms and more positive prognosis. Patients with autosomal dominant STAT1 loss-of-function mutations suffer from mycobacterial diseases and show an impaired response to IFNγ and IL-27 (78, 79). However, the majority of patients with inborn errors of STAT1 show autosomal dominant gain-of-function mutations leading to an unexpectedly broad clinical phenotype, including mucocutaneous candidiasis and autoimmune disorders (80, 81). In addition, some heterozygous de novo acquired mutations of the Stat1 gene, affecting coiled-coil and DNA-binding domains, can be associated with progressive combined immunodeficiency. This kind of Stat1 mutations lead to reduction in overall Stat1 expression and signaling responses and are ultimately fatal due to overwhelming infections and inflammation (82).
While these findings emphasize the critical importance of STAT1 in IFN responses, studies in mice support the idea that residual IFN-dependent activity against some pathogens remains in absence of STAT1. Initial evidence for transcriptional responses to IFNs through non-canonical STAT complexes was provided by studies in Stat1−/− mice infected with Sendai virus, MCMV, or DENV, showing that STAT1-independent responses to IFNs provide some level of resistance against these pathogens (83–85). In contrast, the combined loss of IFNγ and type I IFN receptors or of STAT1 and STAT2 results in early death of infected mice (86, 87). Further, consistent with STAT1-independent responses to IFN, Hahm et al. demonstrated that infection of hematopoietic bone marrow cells with MV or LCMV impaired the maturation of dendritic cells in an IFNβ-dependent and Stat2-dependent, but STAT1-independent, manner (88). Additional studies revealed that STAT2 was required for IFN-I-induced expression of a set of ISGs independently of STAT1 (85, 89, 90). Despite the inability to form ISGF3 complexes, Stat1−/− mice and cells could still induce a subset of ISGs in response to the DENV infection (87). This response was absent in compound Stat1/2−/− mice, directing the main attention toward STAT2 as a component of STAT1-independent ISG expression in vivo. STAT2 homodimers alone are known to bind DNA very poorly, owing to the lack of a functional DNA-binding domain (91). This suggests that one or more additional components must contribute to STAT1-independent ISG regulation. IRF9, the DNA-binding subunit of the ISGF3 complex is an obvious candidate. The formation of STAT2–IRF9 complexes after IFN-I stimulation had been proposed by several authors based on studies addressing ISG expression in HEK293 cells with overexpressed STAT2 and IRF9 (91), in U3A cells that lack STAT1 (90), and in Hep3B cells (89). In addition, STAT2 was shown to have a STAT1-independent role in the ability of IFNβ and TNFα to synergistically stimulate the expression of Duox2 NADPH oxidase in epithelial cell lines [(92), reviewed in detail in Ref (93, 94); Figure 1]. More recently, the potential of STAT2/IRF9 complexes to stimulate ISG emerged from studies addressing the role of type I IFN in bacterial infections.
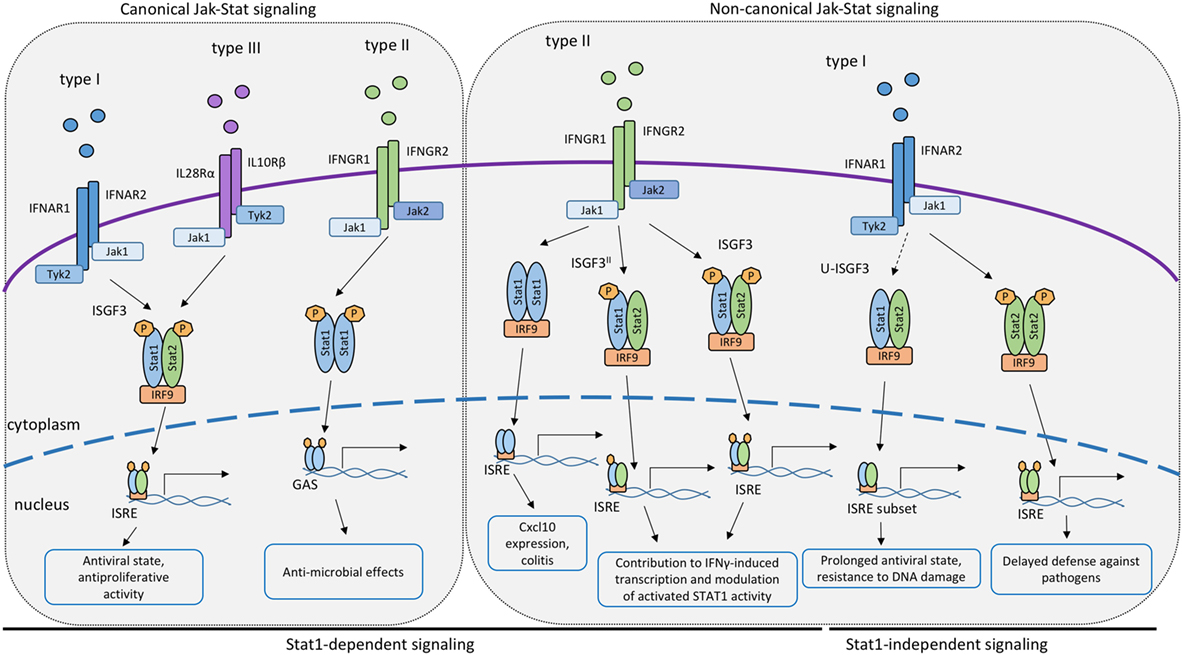
Figure 1. Canonical and non-canonical STAT signaling by the IFN receptors. Proposed roles of STAT or STAT/interferon regulatory factor (IRF9) complexes participating in signal transduction and transcriptional activation by the receptors of IFN-I, IFN-II or IFN-III. Complexes containing IRF9 associate with interferon-stimulated response elements promoter sequences whereas dimerized STAT1 binds to GAS elements.
As mentioned above, type I IFNs are secreted in response to many bacterial pathogens, such as Listeria monocytogenes, Francisella tularensis, Legionella pneumophila, and others (14, 95–97). The impact of IFN-I on bacterial growth in mammalian hosts is variable, as described in detail elsewhere (14). In case of Legionella pneumophila, IFN-I inhibits its ability to grow inside macrophages. The growth inhibitory effect was retained in cells isolated from Stat1−/−, Stat2−/−, or Stat3−/− single knockout animals (17). In contrast, macrophages from compound Stat1/2−/− macrophages were not able to limit the bacterial growth after IFN-I stimulation and had higher bacterial loads than single knockouts of STAT1 or STAT2, pinpointing redundant functions of STAT1 and STAT2 in defense against this bacterial pathogen (98). Studies addressing the molecular mechanism of STAT1-independent ISG expression showed that IFN-I stimulated a delayed activation of STAT2 in Stat1−/− cells. Together with IRF9, STAT2 formed a complex able to bind to the ISRE sequence. Data in Stat1−/− macrophages further demonstrated prolonged JAK activation by the IFN-I receptor and a slow but steady accumulation of both STAT2 and pYSTAT2. The data were consistent with a model according to which a threshold of STAT2 phosphorylation needs to be overcome for the formation of transcriptionally active STAT2–IRF9 complexes and for delayed stimulation of ISG transcription (98, 99). This hypothesis has subsequently been validated in cells derived from mice expressing a STAT1Y701F mutant (99). It is further consistent with reports showing that STAT2 constitutively associates with IRF9 (100) and that this preassociation may be required for rapid generation of the ISGF3 in response to IFN signals in wt cells (101). All the findings together strongly suggest that STAT2–IRF9-stimulated ISG expression could serve as a backup or a support mechanism of defense against pathogens that impede STAT1 signaling or serve to integrate the responses to IFN-I and TNFα (17, 84, 87, 92, 98).
Novel Aspects of IFNγ Signaling by Canonical and Non-Canonical STAT Complexes
Canonical signaling by the IFNγ receptor causes the formation of GAF, the STAT1 homodimer. GAF activates gene transcription by associating with its cognate DNA-binding sequence, the GAS (2, 102). As in the case of IFN-γ, the reality of transcriptional responses to IFNγ adds complexity. In part, this is due to STAT1 modification. The implications of Y701 phosphorylation as a dimerization signal are undisputed. Similarly, the enhancement of IFNγ-induced gene expression through phosphorylation of the C-terminal S727 and increased association with histone acetylase complexes are well documented (103, 104). Sumoylation of K703 was first described by the group of Curt Horvath (105). The implications of this modification for STAT1 activation and activity were initially unclear (105, 106), but subsequent studies, particularly those in cells derived from mice expressing SUMOylation-defective STAT1, clearly linked SUMOylation to decreased IFNγ responsiveness (107, 108). Mechanistically, SUMOylation interferes with the conjugation of a phosphate at the proximal Y701 and increases nuclear tyrosine dephosphorylation by increasing STAT1’s solubility. In absence of the SUMO modification, STAT1 molecules assemble into an insoluble, phosphatase-resistant paracrystalline array (108, 109). Other than modification, the ability of STAT1 dimers to interact on DNA is essential for the expression of a large fraction of IFNγ-induced genes. This surprising finding emerged from studies in cells and mice expressing a STAT1F77A mutation that inhibits polymerization of promoter-bound STAT1 dimers (110). Strikingly, responsiveness to type I IFN, hence the activity of the ISGF3 complex, was unaffected by the STAT1F77A mutation.
Further variety is introduced to the IFNγ pathway by association between STAT1 and other proteins, i.e., non-canonical complexes (Figure 1). Already in the 1990s, Ifit2, a classical ISRE-regulated gene, was found to be induced by IFNγ in a STAT1- and IRF9-dependent, but STAT2-independent manner (111), suggesting that transcription factors containing both STAT1 and IRF9 are able to control IFNγ-responsive genes. Affirmative observations were made for the expression of the Cxcl10 gene in IFNγ-stimulated 2fTGH cells (112). More recently, work from our lab identified an important role for STAT1/IRF9 in the context of a murine colitis model (113). The Cxcl10 gene, known to contribute to colitogenic inflammation, was shown to be induced downstream of the IFNγ receptor in a STAT1/IRF9-dependent fashion, but independently of STAT2. Molecular analysis confirmed that STAT1/IRF9 complexes form in response to IFNγ and associate with ISRE sequences of enhancer regions 1 and 2 of the Cxcl10 gene promoter.
Other than STAT1/IRF9, STAT2 was proposed to contribute to IFNγ-induced transcription (Figure 1). The extent to which this occurs is unclear, owing in part to the fact that the lack of STAT2 reduces STAT1 levels in some cell types, resulting in a mixed Stat1/Stat2−/− phenotype. For example, Stat2−/− fibroblasts express little STAT1 and show impaired inhibition of vesicular stomatitis virus replication when treated with IFNγ (114). In support of a direct role for STAT2 in the IFNγ response, its tyrosine phosphorylation was reported in a study using IFNγ-treated wild-type mouse primary embryonic fibroblasts. This caused the formation of the ISGF3 transcription factor (115). Similar observations were made by Zimmerman and colleagues in MEFs (116). Stimulation of human lung epithelial cells with IFNγ triggered early and delayed peaks of STAT1 Y701 phosphorylation (117). The delayed peak corresponded to the formation of an ISGF3 complex, named ISGF3II, that contained Y701-phosphorylated STAT1 and IRF9, in addition to STAT2 that remained unphosphorylated on Y690. The idea of a STAT2 contribution to the delayed IFNγ response is in line with our recent finding that expression of the Cxcl10 gene is decreased specifically at later stages of the IFNγ response in Stat2−/− macrophages (113). Consistently, we found STAT2 in association with the two Cxcl10 promoter ISREs at the delayed stage of the IFNγ response of wild-type macrophages. Of note, the same study shows that some genes with ISGF3 binding sites did not respond at any time to IFNγ in Stat2−/− macrophages. It appears possible, therefore, that both ISGF3 and ISGF3II complexes contribute to IFNγ-induced transcription in a gene- and stage-specific manner. Of note, however, a stimulatory activity of ISGF3 and ISGF3II complexes is challenged by an entirely different perspective on the role of STAT2 (118). Ho and colleagues show a strong, N-terminal association between unphosphorylated STAT2 and STAT1. This association persists during the IFNγ response and prevents a fraction of STAT1 molecules to enter the nucleus (Figure 2). Consistently, mutations disrupting the N-terminal contacts increase the transcriptional IFNγ response. The study thus presents STAT2 as a moderator of IFNγ-activated STAT1. All findings together support both positive and negative regulation of IFNγ-induced transcription by STAT2. Our studies addressing the Cxcl10 gene suggest that this dual function of STAT2 may represent early and late phases of the transcriptional response to IFNγ, with initial repression being followed by stimulatory activity (110).
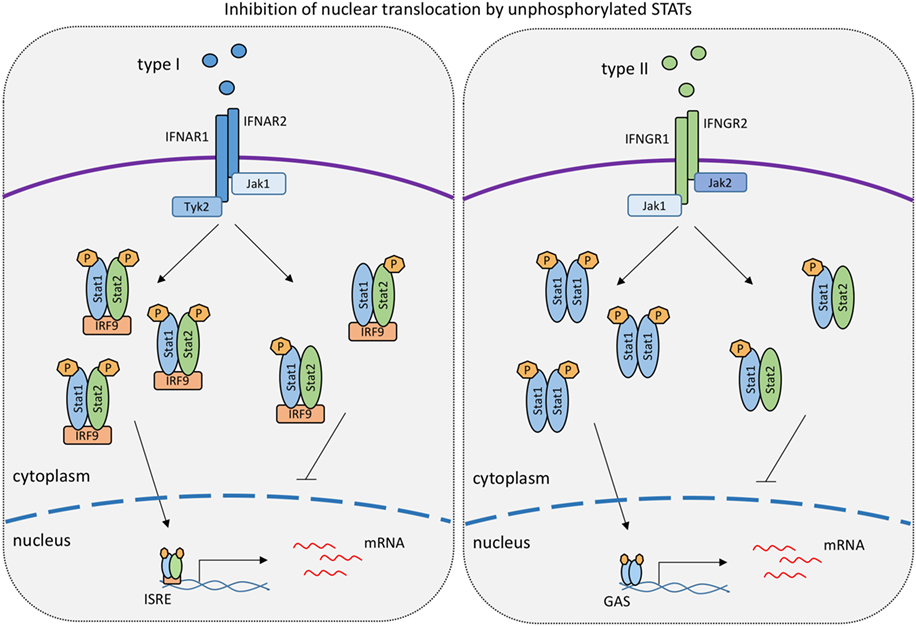
Figure 2. Levels of unphosphorylated signal transducer and activator of transcriptions (STATs) determine the strength of responses to type I IFN and IFNγ. The model is based on work published in references 99 and 118 showing that unphosphorylated STAT1 binds to tyrosine-phosphorylated STAT2 and vice versa. Such hemiphosphorylated STAT dimers are incapable of nuclear translocation (118). In the IFNγ response, unphosphorylated STAT2 thus lowers the formation and nuclear translocation of tyrosine-phoshorylated STAT1 dimers (118). Conversely, unphosphorylated STAT1 inhibits the nuclear translocation of tyrosine-phosphorylated STAT2 in the type I IFN response (99).
U-STAT Contribution to the IFN Response
According to the original JAK–STAT paradigm, there is a strict correlation between STAT activity and their tyrosine phosphorylation. Defying this notion, numerous reports have meanwhile assigned important tasks to U-STATs lacking a phosphate at the critical tyrosine residue. U-STAT activities include control of organelle metabolism and function in mitochondria or the Golgi apparatus [STAT1, STAT2, and STAT3 and STAT6 in the biology of mitochondria (119–125); STAT5 in the Golgi apparatus (125, 126)]. U-STAT1 was required for TNF-mediated apoptosis of U3A cells. This activity required the protein to be phosphorylated at the C-terminal S727 (127). To address potential functions of U-STAT1 in the immune system, we and our collaborators generated mice expressing a STAT1Y701F mutant and compared immune responses of these animals with Stat1−/− mice (99). Apart from a modest gain of function in antibacterial immunity described below, a notable difference was observed in NK cells. Whereas STAT1 deficiency led to a severe loss in NK cytotoxicity, this is partially retained in Stat1Y701F mice (128). In contrast, U-STAT1 did not rescue the NK maturation defect observed in Stat1−/− mice. Localization to the NK-target cell interface hints at a potential role of U-STAT1 at the immunological synapse. NK cells also demonstrate a further non-canonical activity of STAT1 related to its second phosphorylation site, the C-terminal S727. This site is a target for both p38MAPK and the S/T kinase CDK8 (103, 129–131). Whereas in macrophages or fibroblasts S727 phosphorylation increases the IFNγ-induced expression of a subset of STAT1 target genes, CDK8-mediated S727 phosphorylation in NK cells restricts their cytotoxicity (132). Speculatively, the synaptic pool of STAT1 may be the relevant target, as Stat1S727A mutation in NK cells had little impact on their gene expression. In summary, NK cells reveal several non-canonical activities not only for STAT1, but, as described above, also for TYK2 (Table 1). The integration of these activities in cellular signaling networks remains a future challenge.
In addition to cytoplasm or organelle-based roles, nuclear functions were reported involving U-STATs as either gene repressors or activators (133). For example, in Drosophila, STAT92E has been shown to associate with heterochromatin protein 1 (HP1) in cells lacking JAK activity, thus maintaining the structure of heterochromatin and gene repression (134, 135). Likewise, mammalian U-STAT5A reportedly binds to HP1α, stabilizing the heterochromatin in a similar fashion and repressing the genes involved in cancer development (136). In a more recent publication, mouse U-STAT5 was shown to actively repress the transcriptional program required for megakaryocytic differentiation by preventing the binding of the transcriptional activator EGR, acting as a partial antagonist of biological activity of phosphorylated STAT5 (137). U-STAT3 was shown to compete with IκB for binding to unphosphorylated NFκB, translocating to the nucleus and participating in the activation of a subset of NFκB-dependent genes (138). U-STAT6, in cooperation with p300, was suggested to bind to a consensus STAT6 binding site in the promoter of the Cox-2 gene, regulating its constitutive expression (139). The mechanism by which the unphosphorylated STATs enter the nucleus remains to be explored. It is most likely related to the ability of unphosphorylated STATs to shuttle between nucleus and cytoplasm (140). While the nuclear export rate usually localizes most U-STATs in the cytoplasm at steady state, it is conceivable that some of these are trapped in the nucleus by DNA or chromatin association. Other possibilities include the association with non-STAT transcription factors like IRF1, or direct contact with the nuclear pore complex (141–143).
The link between U-STATs and the IFN response was made by George Stark and colleagues. The group initially demonstrated an association of U-STAT1 and IRF1 with the partially overlapping interferon consensus sequence 2 and GAS sites in the promoter of the ISG Lmp2. This constitutive interaction resulted in expression of the Lmp2 gene in U3A cells (141). Additional studies, performed in cells overexpressing subunits of the ISGF3 complex, support the concept that U-STATs prolong the expression of a distinct subset of ISG (144) (Figure 3). The authors hypothesize that this occurs as a result of the accumulation of newly synthesized STAT1 and STAT2 following early, canonical type I IFN signaling. According to this hypothesis, prolonged exposure of cells to IFN-I and accumulation of STAT1, STAT2, and IRF9 causes formation of an unphosphorylated ISGF3 complex (U-ISGF3), which in turn maintains the expression of a subset of ISGs that increase resistance to viruses and DNA damage (145).
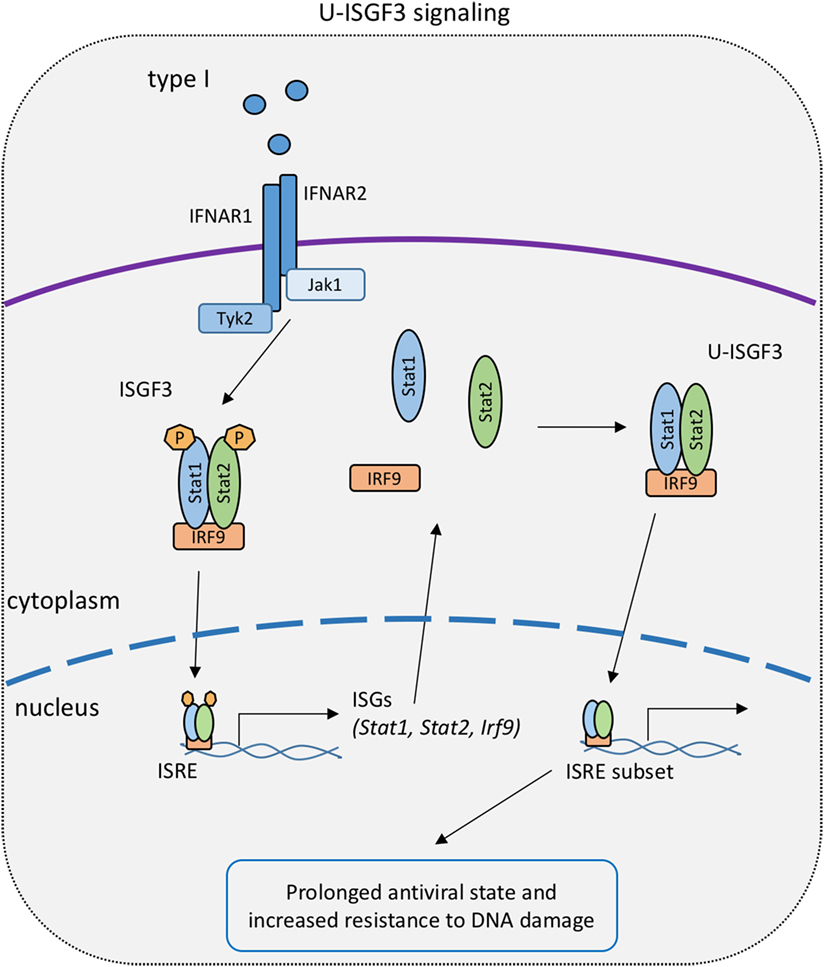
Figure 3. U-STAT signaling in the type I IFN response. Early canonical signaling causes the upregulation of IFN-stimulated gene factor 3 (ISGF3) subunits and the subsequent formation of an ISGF3 complex with unphosphorylated signal transducer and activator of transcriptions 1 and 2 [unphosphorylated ISGF3 complex (U-ISGF3)]. U-ISGF3 stimulates a subset of interferon-stimulated genes to prolong the transcriptional response to IFN-I.
We attempted to test the U-STAT1 model in Stat1Y701F mice (99). Indeed, some gain of function was noted when cells and animals expressing mutant STAT1 were infected with the bacterial pathogen Listeria monocytogenes and compared to STAT1-deficient counterparts. However, Starks U-STAT model could not be tested in these mice owing to the lack of an early, phosphotyrosine-based response to IFN-I that causes an increase of ISGF3 components (Figure 3). In fact, Stat1Y701F mutation caused a drastic decrease in basal levels of STAT1 in cells and animals, due to the lack of tonic signaling by the IFN-I receptor (99, 146). Thus, improved animal models expressing increased U-STAT amounts are needed to collect in vivo evidence for their function.
Of interest, the low amounts of U-STAT1 expressed in Stat1Y701F mice acted as suppressors of the delayed STAT1-independent, STAT2-dependent expression of ISGs after IFNβ stimulation (see above). This was a consequence of preventing cytoplasmic, tyrosine-phosphorylated STAT2 from entering the nucleus. The data suggest that hemiphosphorylated STAT dimers do not show the enhanced nuclear translocation of the fully phosphorylated dimers. This notion is in line with the above-mentioned data from the Vinkemeier lab that demonstrate the inability of hemiphosphorylated dimers of wild-type STATs to enter the cell nucleus (118). Therefore, relative ratios of STAT1 and STAT2 and their phosphorylated isoforms may be an important determinant of nuclear signaling by the IFN receptors (Figure 2).
Functions of IRF9 Beyond IFN Signaling
The studies described above assign an important function of IRF9 to both IFN-I and IFNγ signaling. In this paragraph, we briefly describe some observations linking IRF9 to different diseases, either protecting from or exacerbating pathology. Notably, although IRF9 is an immune regulator, these data demonstrate additional mechanisms that may either link the immune system with these diseases or reflect IRF9 activities unrelated to the immune system. In most cases, the link to IFN signaling remains to be determined. The studies raise the possibility that IRF9 is capable of interacting with other transcription factors to fulfill a different set of functions (147).
Overexpression of IRF9 has been observed in breast and uterine tumors, where it provides resistance to microtubule-disrupting agents through transcriptional activation of ISGs in a STAT1- and STAT2-independent manner (148). In this situation, the ability of IL-6 to act as an inducer of IRF9 may be of importance, as shown for human prostate cancers (149). The role of IRF proteins in adipocyte biology connects the immune response with metabolic regulation (150, 151). In obese mice IRF7 is increased (152), while the expression of IRF3 (153) and IRF9 (152) is decreased. Thus, IRFs respond differently to overnutrition stress. In line with this, mice lacking IRF7 show improved hepatic insulin sensitivity and protection from local and systemic inflammation during high-fat diet (152). In contrast, IRF3 and IRF9 play a protective role in high-fat diet-induced obesity (153, 154). IRF9 was shown to interact with peroxisome proliferator-activated receptor α to regulate gene expression in the liver (154). Their target genes are mostly involved in lipid metabolism, thus attenuating insulin resistance in obese mice. This observation suggests a key role for IRF9 in metabolic functions.
Cardiac hypertrophy and pathological remodeling are hallmarks of cardiomyopathy associated with many pathological stressors. Recent reports found that IRF3, IRF7, and IRF9 protect against cardiac hypertrophy (155–157). In murine disease models, IRF9 binds myocardin, an activator of the transcription factor serum response factor (SRF), thereby inhibiting SRF activation and associated proliferative response. Consistently, an aggravated cardiac hypertrophy occurs in Irf9−/− mice (157). Contrasting its protective effect in cardiac hypertrophy, upregulation of IRF9 during myocardial ischemia–reperfusion (I/R) injury contributes to cardiomyocyte death and inflammation through the Sirt1–p53 axis. In this context, the downregulation of the deacetylase SIRT1 by IRF9 promotes apoptotic signaling through p53 (158). Similarly, IRF9 overexpression leads to cell death signaling in neurons in context of a cerebral ischemic stroke. Hence, IRF9 deficiency mitigates neurological deficits upon stroke (159). In arteria, IRF9 mediates neointima formation, a scar that forms upon vascular injury. IRF9 overexpression increases, and its deficiency decreases the proliferation and migration of vascular smooth muscle cells (VSMCs). As in case of cardiomyocytes, IRF9 suppresses SIRT1 by directly binding to an ISRE in the SIRT1 promoter. Thereby, IRF9 prevents the suppression of AP-1 transactivation by SIRT1. AP-1 induces a vascular injury response pathway that promotes VSMC proliferation in the context of neointima formation (160). Although critical roles of IRF9 in immunity, metabolism, and disease have been revealed, many questions regarding the mechanisms by which IRF9 interconnects such a variety of pathways still remain.
Concluding Remarks
The overwhelming biological impact of canonical JAK–STAT pathways is undisputed. However, as happens often in biological sciences, long shadows of paradigmatic signaling systems obscure alternative installations of their components for distinct purposes. Recent years of JAK–STAT research have begun to uncover some of these undogmatic events and establish them as non-canonical pathways side-by-side with the canonical ones. This provides food for thoughts about the evolution of JAK–STAT pathways, the emergence of non-canonical and canonical functions. Studies in Dictyostelium suggest that STATs evolved without the necessity for tyrosine phosphorylation or the ability to activate transcription (161). Thus, it is tempting to speculate that what we now perceive as a deviation from the canonical pathways is in reality closer to the primordial STAT function that had nothing to do with cytokines or the genes they activate. Although this review provides a very brief overview, we hope it allows readers to get an idea of the many different ways by which non-canonical JAK–STAT pathways are established in the mammalian immune system. Their analysis is far from easy, as results from straightforward experimental approaches are likely to be dominated by the pathway’s canonical output. In spite of this, future research with an open eye for the unexpected holds the promise of new fascinating insights into the many facets of JAKs and STATs in our immune system.
Author Contributions
AM, EP, EK-H, FR, MM, and TD wrote the review. AM compiled graphics and table.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Work in TD’s laboratory is supported by the Austrian Science Fund (FWF) through grants P25186-B22 (to TD) and SFB F61 (to MM and TD).
References
1. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci (1957) 147:258–67. doi:10.1098/rspb.1957.0048
2. Levy DE, Darnell JE Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol (2002) 3:651–62. doi:10.1038/nrm909
3. Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov (2007) 6:975–90. doi:10.1038/nrd2422
4. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi:10.1146/annurev-immunol-032713-120231
5. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev (2004) 202:8–32. doi:10.1111/j.0105-2896.2004.00204.x
6. Tough DF, Zhang X, Sprent J. An IFN-gamma-dependent pathway controls stimulation of memory phenotype CD8+ T cell turnover in vivo by IL-12, IL-18, and IFN-gamma. J Immunol (2001) 166:6007–11. doi:10.4049/jimmunol.166.10.6007
7. Brinkmann V, Geiger T, Alkan S, Heusser CH. Interferon alpha increases the frequency of interferon gamma-producing human CD4+ T cells. J Exp Med (1993) 178:1655–63. doi:10.1084/jem.178.5.1655
8. Biron CA. Interferons α and β as immune regulators – a new look. Immunity (2001) 14:661–4. doi:10.1016/S1074-7613(01)00154-6
9. Rauch I, Müller M, Decker T. The regulation of inflammation by interferons and their STATs. JAKSTAT (2013) 2:e23820. doi:10.4161/jkst.23820
10. Coccia EM, Battistini A. Early IFN type I response: learning from microbial evasion strategies. Semin Immunol (2015) 27:85–101. doi:10.1016/j.smim.2015.03.005
11. Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol (2006) 6:836–48. doi:10.1038/nri1961
12. Crow YJ, Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat Rev Immunol (2015) 15:429–40. doi:10.1038/nri3850
13. Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med (2016) 213:2527–38. doi:10.1084/jem.20161596
14. Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol (2005) 5:675–87. doi:10.1038/nri1684
15. Trinchieri G. Type I interferon: friend or foe? J Exp Med (2010) 207:2053–63. doi:10.1084/jem.20101664
16. Buß C, Opitz B, Hocke AC, Lippmann J, van Laak V, Hippenstiel S, et al. Essential role of mitochondrial antiviral signaling, IFN regulatory factor (IRF)3, and IRF7 in Chlamydophila pneumoniae-mediated IFN-β response and control of bacterial replication in human endothelial cells. J Immunol (2010) 184:3072–8. doi:10.4049/jimmunol.0902947
17. Plumlee CR, Lee C, Beg AA, Decker T, Shuman HA, Schindler C. Interferons direct an effective innate response to Legionella pneumophila infection. J Biol Chem (2009) 284:30058–66. doi:10.1074/jbc.M109.018283
18. Freudenberg MA, Merlin T, Kalis C, Chvatchko Y, Stubig H, Galanos C. Cutting edge: a murine, IL-12-independent pathway of IFN-gamma induction by gram-negative bacteria based on STAT4 activation by Type I IFN and IL-18 signaling. J Immunol (2002) 169:1665–8. doi:10.4049/jimmunol.169.4.1665
19. Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol (2007) 178:3126–33. doi:10.4049/jimmunol.178.5.3126
20. Mancuso G, Gambuzza M, Midiri A, Biondo C, Papasergi S, Akira S, et al. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol (2009) 10:587–94. doi:10.1038/ni.1733
21. Castiglia V, Piersigilli A, Ebner F, Janos M, Goldmann O, Damböck U, et al. Type I Interferon signaling prevents IL-1β-driven lethal systemic hyperinflammation during invasive bacterial infection of soft tissue. Cell Host Microbe (2016) 19:375–87. doi:10.1016/j.chom.2016.02.003
22. Couturier N, Bucciarelli F, Nurtdinov RN, Debouverie M, Lebrun-Frenay C, Defer G, et al. Tyrosine kinase 2 variant influences T lymphocyte polarization and multiple sclerosis susceptibility. Brain (2011) 134:693–703. doi:10.1093/brain/awr010
23. Dyment DA, Cader MZ, Chao MJ, Lincoln MR, Morrison KM, Disanto G, et al. Exome sequencing identifies a novel multiple sclerosis susceptibility variant in the TYK2 gene. Neurology (2012) 79:406–11. doi:10.1212/WNL.0b013e3182616fc4
24. Liu JZ, Almarri MA, Gaffney DJ, Mells GF, Jostins L, Cordell HJ, et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat Genet (2012) 44:1137–41. doi:10.1038/ng.2395
25. Peluso C, Christofolini DM, Goldman CS, Mafra FA, Cavalcanti V, Barbosa CP, et al. TYK2 rs34536443 polymorphism is associated with a decreased susceptibility to endometriosis-related infertility. Hum Immunol (2013) 74:93–7. doi:10.1016/j.humimm.2012.09.007
26. Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity (2006) 25:745–55. doi:10.1016/j.immuni.2006.09.009
27. Kreins AY, Ciancanelli MJ, Okada S, Kong X-F, Ramírez-Alejo N, Kilic SS, et al. Human TYK2 deficiency: mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med (2015) 212:1641–62. doi:10.1084/jem.20140280
28. Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol (1997) 15:563–91. doi:10.1146/annurev.immunol.15.1.563
29. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol (2004) 75:163–89. doi:10.1189/jlb.0603252
30. Lazarevic V, Glimcher LH, Lord GM. T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol (2013) 13:777–89. doi:10.1038/nri3536
31. MacMicking JD. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol (2012) 12:367–82. doi:10.1038/nri3210
32. Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol (1997) 15:749–95. doi:10.1146/annurev.immunol.15.1.749
33. Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, et al. Immune response in mice that lack the interferon-gamma receptor. Science (1993) 259:1742–5. doi:10.1126/science.8456301
34. Dalton DK, Pitts Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science (1993) 259:1739–42. doi:10.1126/science.8456300
35. Casanova J-L, Abel L. The human model: a genetic dissection of immunity to infection in natural conditions. Nat Rev Immunol (2004) 4:55–66. doi:10.1038/nri1264
36. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol (2003) 4:69–77. doi:10.1038/ni875
37. Donnelly RP, Kotenko SV. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res (2010) 30:555–64. doi:10.1089/jir.2010.0078
38. Uzé G, Monneron D. IL-28 and IL-29: newcomers to the interferon family. Biochimie (2007) 89:729–34. doi:10.1016/j.biochi.2007.01.008
39. Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML, et al. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog (2013) 9:e1003773. doi:10.1371/journal.ppat.1003773
40. Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev (2009) 228:273–87. doi:10.1111/j.1600-065X.2008.00754.x
41. O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med (2015) 66:311–28. doi:10.1146/annurev-med-051113-024537
42. Strobl B, Stoiber D, Sexl V, Mueller M. Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front Biosci (Landmark Ed) (2011) 16:3224. doi:10.2741/3908
43. Velazquez L, Fellous M, Stark GR, Pellegrini S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell (1992) 70:313–22. doi:10.1016/0092-8674(92)90105-L
44. Gauzzi MC, Velazquez L, McKendry R, Mogensen KE, Fellous M, Pellegrini S. Interferon-alpha-dependent activation of Tyk2 requires phosphorylation of positive regulatory tyrosines by another kinase. J Biol Chem (1996) 271:20494–500. doi:10.1074/jbc.271.34.20494
45. Rani MR, Leaman DW, Han Y, Leung S, Croze E, Fish EN, et al. Catalytically active TYK2 is essential for interferon-beta-mediated phosphorylation of STAT3 and interferon-alpha receptor-1 (IFNAR-1) but not for activation of phosphoinositol 3-kinase. J Biol Chem (1999) 274:32507–11. doi:10.1074/jbc.274.45.32507
46. Rani MR, Gauzzi C, Pellegrini S, Fish EN, Wei T, Ransohoff RM. Induction of beta-R1/I-TAC by interferon-beta requires catalytically active TYK2. J Biol Chem (1999) 274:1891–7. doi:10.1074/jbc.274.4.1891
47. Ragimbeau J, Dondi E, Alcover A, Eid P, Uzé G, Pellegrini S. The tyrosine kinase Tyk2 controls IFNAR1 cell surface expression. EMBO J (2003) 22:537–47. doi:10.1093/emboj/cdg038
48. Kumar KGS, Varghese B, Banerjee A, Baker DP, Constantinescu SN, Pellegrini S, et al. Basal ubiquitin-independent internalization of interferon alpha receptor is prevented by Tyk2-mediated masking of a linear endocytic motif. J Biol Chem (2008) 283:18566–72. doi:10.1074/jbc.M800991200
49. Karaghiosoff M, Neubauer H, Lassnig C, Kovarik P, Schindler H, Pircher H, et al. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity (2000) 13:549–60. doi:10.1016/S1074-7613(00)00054-6
50. Shimoda K, Kato K, Aoki K, Matsuda T, Miyamoto A, Shibamori M, et al. Tyk2 plays a restricted role in IFNα signaling, although it is required for IL-12-mediated T cell function. Immunity (2000) 13:561–71. doi:10.1016/S1074-7613(00)00055-8
51. Prchal-Murphy M, Semper C, Lassnig C, Wallner B, Gausterer C, Teppner-Klymiuk I, et al. TYK2 kinase activity is required for functional type I interferon responses in vivo. PLoS One (2012) 7:e39141. doi:10.1371/journal.pone.0039141
52. Prchal-Murphy M, Witalisz-Siepracka A, Bednarik KT, Putz EM, Gotthardt D, Meissl K, et al. In vivo tumor surveillance by NK cells requires TYK2 but not TYK2 kinase activity. Oncoimmunology (2015) 4:e1047579. doi:10.1080/2162402X.2015.1047579
53. Potla R, Koeck T, Wegrzyn J, Cherukuri S, Shimoda K, Baker DP, et al. Tyk2 tyrosine kinase expression is required for the maintenance of mitochondrial respiration in primary Pro-B lymphocytes. Mol Cell Biol (2006) 26:8562–71. doi:10.1128/MCB.00497-06
54. Vogl C, Flatt T, Fuhrmann B, Hofmann E, Wallner B, Stiefvater R, et al. Transcriptome analysis reveals a major impact of JAK protein tyrosine kinase 2 (Tyk2) on the expression of interferon-responsive and metabolic genes. BMC Genomics (2010) 11:199. doi:10.1186/1471-2164-11-199
55. Derecka M, Gornicka A, Koralov SB, Szczepanek K, Morgan M, Raje V, et al. Tyk2 and Stat3 regulate brown adipose tissue differentiation and obesity. Cell Metab (2012) 16:814–24. doi:10.1016/j.cmet.2012.11.005
56. Mostafavi S, Yoshida H, Moodley D, LeBoité H, Rothamel K, Raj T, et al. Parsing the interferon transcriptional network and its disease associations. Cell (2016) 164:564–78. doi:10.1016/j.cell.2015.12.032
57. Sigurdsson S, Nordmark G, Göring HH, Lindroos K, Wiman AC, Sturfelt G, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet (2005) 76:528–37. doi:10.1086/428480
58. Graham DSC, Akil M, Vyse TJ. Association of polymorphisms across the tyrosine kinase gene, TYK2 in UK SLE families. Rheumatology (2007) 46:927–30. doi:10.1093/rheumatology/kel449
59. Ban M, Goris A, Lorentzen AR, Baker A, Mihalova T, Ingram G, et al. Replication analysis identifies TYK2 as a multiple sclerosis susceptibility factor. Eur J Hum Genet (2009) 17:1309–13. doi:10.1038/ejhg.2009.41
60. Hellquist A, Järvinen TM, Koskenmies S, Zucchelli M, Orsmark-Pietras C, Berglind L, et al. Evidence for genetic association and interaction between the TYK2 and IRF5 genes in systemic lupus erythematosus. J Rheumatol (2009) 36:1631–8. doi:10.3899/jrheum.081160
61. Sato K, Shiota M, Fukuda S, Iwamoto E, Machida H, Inamine T, et al. Strong evidence of a combination polymorphism of the tyrosine kinase 2 gene and the signal transducer and activator of transcription 3 gene as a DNA-based biomarker for susceptibility to Crohn’s disease in the Japanese population. J Clin Immunol (2009) 29:815–25. doi:10.1007/s10875-009-9320-x
62. Suarez-Gestal M, Calaza M, Gonzalez A. Lack of interaction between systemic lupus erythematosus-associated polymorphisms in TYK2 and IRF5. J Rheumatol (2010) 37:676–7. doi:10.1186/ar2990
63. Wang K, Zhang H, Kugathasan S, Annese V, Bradfield JP, Russell RK, et al. Diverse genome-wide association studies associate the IL12/IL23 pathway with Crohn disease. Am J Hum Genet (2009) 84:399–405. doi:10.1016/j.ajhg.2009.01.026
64. Franke A, McGovern DPB, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet (2010) 42:1118–25. doi:10.1038/ng.717
65. Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet (2012) 44:1341–8. doi:10.1038/ng.2467
66. Johnson BA, Wang J, Taylor EM, Caillier SJ, Herbert J, Khan OA, et al. Multiple sclerosis susceptibility alleles in African Americans. Genes Immun (2010) 11:343–50. doi:10.1038/gene.2009.81
67. Mero I-L, Lorentzen AR, Ban M, Smestad C, Celius EG, Aarseth JH, et al. A rare variant of the TYK2 gene is confirmed to be associated with multiple sclerosis. Eur J Hum Genet (2010) 18:502–4. doi:10.1038/ejhg.2009.195
68. Wallace C, Smyth DJ, Maisuria-Armer M, Walker NM, Todd JA, Clayton DG. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat Genet (2010) 42:68–71. doi:10.1038/ng.493
69. Li Z, Gakovic M, Ragimbeau J, Eloranta M-L, Rönnblom L, Michel F, et al. Two rare disease-associated Tyk2 variants are catalytically impaired but signaling competent. J Immunol (2013) 190:2335–44. doi:10.4049/jimmunol.1203118
70. Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell (1998) 93:385–95. doi:10.1016/S0092-8674(00)81167-8
71. Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell (1998) 93:397–409. doi:10.1016/S0092-8674(00)81168-X
72. Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffé M, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science (1997) 278:1309–12. doi:10.1126/science.278.5341.1309
73. Lacronique V, Boureux A, Monni R, Dumon S, Mauchauffé M, Mayeux P, et al. Transforming properties of chimeric TEL-JAK proteins in Ba/F3 cells. Blood (2000) 95:2076–83.
74. Carron C, Cormier F, Janin A, Lacronique V, Giovannini M, Daniel MT, et al. TEL-JAK2 transgenic mice develop T-cell leukemia. Blood (2000) 95:3891–9.
75. Huang LJ, Constantinescu SN, Lodish HF. The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Mol Cell (2001) 8:1327–38. doi:10.1016/S1097-2765(01)00401-4
76. Frenzel K, Wallace TA, McDoom I, Xiao HD, Capecchi MR, Bernstein KE, et al. A functional Jak2 tyrosine kinase domain is essential for mouse development. Exp Cell Res (2006) 312:2735–44. doi:10.1016/j.yexcr.2006.05.004
77. Keil E, Finkenstädt D, Wufka C, Trilling M, Liebfried P, Strobl B, et al. Important scaffold function of the Janus kinase 2 uncovered by a novel mouse model harboring a Jak2 activation loop mutation. Blood (2013) 123:520–9. doi:10.1182/blood-2013-03-492157
78. Boisson-Dupuis S, Kong X-F, Okada S, Cypowyj S, Puel A, Abel L, et al. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol (2012) 24:1–15. doi:10.1016/j.coi.2012.04.011
79. Casanova J-L, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity (2012) 36:515–28. doi:10.1016/j.immuni.2012.03.016
80. Boisson B, Quartier P, Casanova J-L. Immunological loss-of-function due to genetic gain-of-function in humans: autosomal dominance of the third kind. Curr Opin Immunol (2015) 32:90–105. doi:10.1016/j.coi.2015.01.005
81. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood (2016) 127:3154–64. doi:10.1182/blood-2015-11-679902
82. Sharfe N, Nahum A, Newell A, Dadi H, Ngan B, Pereira SL, et al. Fatal combined immunodeficiency associated with heterozygous mutation in STAT1. J Allergy Clin Immunol (2014) 133:807–17. doi:10.1016/j.jaci.2013.09.032
83. Gil MP, Bohn E, O’Guin AK, Ramana CV, Levine B, Stark GR, et al. Biologic consequences of Stat1-independent IFN signaling. Proc Natl Acad Sci U S A (2001) 98:6680–5. doi:10.1073/pnas.111163898
84. Shresta S, Sharar KL, Prigozhin DM, Snider HM, Beatty PR, Harris E. Critical roles for both STAT1-dependent and STAT1-independent pathways in the control of primary dengue virus infection in mice. J Immunol (2005) 175:3946–54. doi:10.4049/jimmunol.175.6.3946
85. George CX, Das S, Samuel CE. Organization of the mouse RNA-specific adenosine deaminase Adar1 gene 5’-region and demonstration of STAT1-independent, STAT2-dependent transcriptional activation by interferon. Virology (2008) 380:338–43. doi:10.1016/j.virol.2008.07.029
86. Johnson AJ, Roehrig JT. New mouse model for dengue virus vaccine testing. J Virol (1999) 73:783–6.
87. Perry ST, Buck MD, Lada SM, Schindler C, Shresta S. STAT2 mediates innate immunity to Dengue virus in the absence of STAT1 via the type I interferon receptor. PLoS Pathog (2011) 7:e1001297. doi:10.1371/journal.ppat.1001297
88. Hahm B, Trifilo MJ, Zuniga EI, Oldstone MB. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity (2005) 22:247–57. doi:10.1016/j.immuni.2005.01.005
89. Sarkis PTN, Ying S, Xu R, Yu X-F. STAT1-independent cell type-specific regulation of antiviral APOBEC3G by IFN-alpha. J Immunol (2006) 177:4530–40. doi:10.4049/jimmunol.177.7.4530
90. Lou Y-J, Pan X-R, Jia P-M, Li D, Xiao S, Zhang Z-L, et al. IRF-9/STAT2 [corrected] functional interaction drives retinoic acid-induced gene G expression independently of STAT1. Cancer Res (2009) 69:3673–80. doi:10.1158/0008-5472.CAN-08-4922
91. Bluyssen HAR, Levy DE. Stat2 is a transcriptional activator that requires sequence specific contacts provided by stat1 and p48 for stable interaction with DNA. J Biol Chem (1997) 272:4600–5. doi:10.1074/jbc.272.7.4600
92. Fink K, Martin L, Mukawera E, Chartier S, De Deken X, Brochiero E, et al. IFNβ/TNFα synergism induces a non-canonical STAT2/IRF9-dependent pathway triggering a novel DUOX2 NADPH oxidase-mediated airway antiviral response. Cell Res (2013) 23:673–90. doi:10.1038/cr.2013.47
93. Fink K, Grandvaux N. STAT2 and IRF9: beyond ISGF3. JAKSTAT (2013) 2:e27521. doi:10.4161/jkst.27521
94. Blaszczyk K, Nowicka H, Kostyrko K, Antonczyk A, Wesoly J, Bluyssen HAR. The unique role of STAT2 in constitutive and IFN-induced transcription and antiviral responses. Cytokine Growth Factor Rev (2016) 29:71–81. doi:10.1016/j.cytogfr.2016.02.010
95. Stockinger S, Materna T, Stoiber D, Bayr L, Steinborn R, Kolbe T, et al. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J Immunol (2002) 169:6522–9. doi:10.4049/jimmunol.169.11.6522
96. Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J Exp Med (2007) 204:987–94. doi:10.1084/jem.20062665
97. Tam MA, Rydström A, Sundquist M, Wick MJ. Early cellular responses to Salmonella infection: dendritic cells, monocytes, and more. Immunol Rev (2008) 225:140–62. doi:10.1111/j.1600-065X.2008.00679.x
98. Abdul-Sater AA, Majoros A, Plumlee CR, Perry S, Gu A-D, Lee C, et al. Different STAT transcription complexes drive early and delayed responses to type I IFNs. J Immunol (2015) 195:210–6. doi:10.4049/jimmunol.1401139
99. Majoros A, Platanitis E, Szappanos D, Cheon H, Vogl C, Shukla P, et al. Response to interferons and antibacterial innate immunity in the absence of tyrosine-phosphorylated STAT1. EMBO Rep (2016) 17:e201540726. doi:10.15252/embr.201540726
100. Martinez-Moczygemba M, Gutch MJ, French DL, Reich NC. Distinct STAT structure promotes interaction of STAT2 with the p48 subunit of the interferon-alpha-stimulated transcription factor ISGF3. J Biol Chem (1997) 272:20070–6. doi:10.1074/jbc.272.32.20070
101. Kraus TA, Lau JF, Parisien J-P, Horvath CM. A hybrid IRF9-STAT2 protein recapitulates interferon-stimulated gene expression and antiviral response. J Biol Chem (2003) 278:13033–8. doi:10.1074/jbc.M212972200
102. Decker T, Lew DJ, Mirkovitch J, Darnell JEJ. Cytoplasmic activation of GAF, an IFN-gamma-regulated DNA-binding factor. EMBO J (1991) 10:927–32.
103. Varinou L, Ramsauer K, Karaghiosoff M, Kolbe T, Pfeffer K, Muller M, et al. Phosphorylation of the Stat1 transactivation domain is required for full-fledged IFN-gamma-dependent innate immunity. Immunity (2003) 19:793–802. doi:10.1016/S1074-7613(03)00322-4
104. Ramsauer K, Farlik M, Zupkovitz G, Seiser C, Kroger A, Hauser H, et al. Distinct modes of action applied by transcription factors STAT1 and IRF1 to initiate transcription of the IFN-gamma-inducible gbp2 gene. Proc Natl Acad Sci U S A (2007) 104:2849–54. doi:10.1073/pnas.0610944104
105. Rogers RS, Horvath CM, Matunis MJ. SUMO modification of STAT1 and its role in PIAS-mediated inhibition of gene activation. J Biol Chem (2003) 278:30091–7. doi:10.1074/jbc.M301344200
106. Song L, Bhattacharya S, Yunus AA, Lima CD, Schindler C. Stat1 and SUMO modification. Blood (2006) 108:3237–44. doi:10.1182/blood-2006-04-020271
107. Ungureanu D, Vanhatupa S, Kotaja N, Yang J, Aittomaki S, Jänne OA, et al. PIAS proteins promote SUMO-1 conjugation to STAT1. Blood (2003) 102:3311–3. doi:10.1182/blood-2002-12-3816
108. Begitt A, Droescher M, Knobeloch K-P, Vinkemeier U. SUMO conjugation of STAT1 protects cells from hyperresponsiveness to IFNγ. Blood (2011) 118:1002–7. doi:10.1182/blood-2011-04-347930
109. Droescher M, Begitt A, Marg A, Zacharias M, Vinkemeier U. Cytokine-induced paracrystals prolong the activity of signal transducers and activators of transcription (STAT) and provide a model for the regulation of protein solubility by small ubiquitin-like modifier (SUMO). J Biol Chem (2011) 286:18731–46. doi:10.1074/jbc.M111.235978
110. Begitt A, Droescher M, Meyer T, Schmid CD, Baker M, Antunes F, et al. STAT1-cooperative DNA binding distinguishes type 1 from type 2 interferon signaling. Nat Immunol (2014) 15:168–76. doi:10.1038/ni.2794
111. Bluyssen HA, Muzaffar R, Vlieststra RJ, van der Made AC, Leung S, Stark GR, et al. Combinatorial association and abundance of components of interferon-stimulated gene factor 3 dictate the selectivity of interferon responses. Proc Natl Acad Sci U S A (1995) 92:5645–9. doi:10.1073/pnas.92.12.5645
112. Majumder S, Zhou LZ, Chaturvedi P, Babcock G, Aras S, Ransohoff RM. p48/STAT-1alpha-containing complexes play a predominant role in induction of IFN-gamma-inducible protein, 10 kDa (IP-10) by IFN-gamma alone or in synergy with TNF-alpha. J Immunol (1998) 161:4736–44.
113. Rauch I, Rosebrock F, Hainzl E, Heider S, Majoros A, Wienerroither S, et al. Noncanonical effects of IRF9 in intestinal inflammation: more than type I and type III interferons. Mol Cell Biol (2015) 35:2332–43. doi:10.1128/MCB.01498-14
114. Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity (2000) 13:795–804. doi:10.1016/S1074-7613(00)00077-7
115. Matsumoto M, Tanaka N, Harada H, Kimura T, Yokochi T, Kitagawa M, et al. Activation of the transcription factor ISGF3 by interferon-gamma. Biol Chem (1999) 380:699–703. doi:10.1515/BC.1999.087
116. Zimmermann A, Trilling M, Wagner M, Wilborn M, Bubic I, Jonjic S, et al. Cytomegaloviral protein reveals a dual role for STAT2 in IFN-{gamma} signaling and antiviral responses. J Exp Med (2005) 201:1543–53. doi:10.1084/jem.20041401
117. Morrow AN, Schmeisser H, Tsuno T, Zoon KC. A novel role for IFN-stimulated gene factor 3II in IFN-γ signaling and induction of antiviral activity in human cells. J Immunol (2011) 186:1685–93. doi:10.4049/jimmunol.1001359
118. Ho J, Pelzel C, Begitt A, Mee M, Elsheikha HM, Scott DJ, et al. STAT2 is a pervasive cytokine regulator due to its inhibition of STAT1 in multiple signaling pathways. PLoS Biol (2016) 14:e2000117. doi:10.1371/journal.pbio.2000117
119. Bourke LT, Knight RA, Latchman DS, Stephanou A, McCormick J. Signal transducer and activator of transcription-1 localizes to the mitochondria and modulates mitophagy. JAKSTAT (2013) 2:e25666. doi:10.4161/jkst.25666
120. Sisler JD, Morgan M, Raje V, Grande RC, Derecka M, Meier J, et al. The signal transducer and activator of transcription 1 (STAT1) inhibits mitochondrial biogenesis in liver and fatty acid oxidation in adipocytes. PLoS One (2015) 10:e0144444. doi:10.1371/journal.pone.0144444
121. Shahni R, Cale CM, Anderson G, Osellame LD, Hambleton S, Jacques TS, et al. Signal transducer and activator of transcription 2 deficiency is a novel disorder of mitochondrial fission. Brain (2015) 138:2834–46. doi:10.1093/brain/awv182
122. Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science (2009) 324:1713–6. doi:10.1126/science.1171721
123. Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, et al. Function of mitochondrial Stat3 in cellular respiration. Science (2009) 323:793–7. doi:10.1126/science.1164551
124. Szczepanek K, Lesnefsky EJ, Larner AC. Multi-tasking: nuclear transcription factors with novel roles in the mitochondria. Trends Cell Biol (2012) 22:429–37. doi:10.1016/j.tcb.2012.05.001
125. Sehgal PB. Non-genomic STAT5-dependent effects at the endoplasmic reticulum and Golgi apparatus and STAT6-GFP in mitochondria. JAKSTAT (2013) 2(4):e24860. doi:10.4161/jkst.24860
126. Lee JE, Yang Y-M, Liang F-X, Gough DJ, Levy DE, Sehgal PB. Nongenomic STAT5-dependent effects on Golgi apparatus and endoplasmic reticulum structure and function. Am J Physiol Cell Physiol (2012) 302:C804–20. doi:10.1152/ajpcell.00379.2011
127. Kumar A, Commane M, Flickinger TW, Horvath CM, Stark GR. Defective TNF-alpha-induced apoptosis in STAT1-null cells due to low constitutive levels of caspases. Science (1997) 278:1630–2. doi:10.1126/science.278.5343.1630
128. Putz EM, Majoros A, Gotthardt D, Prchal-Murphy M, Zebedin-Brandl EM, Fux DA, et al. Novel non-canonical role of STAT1 in natural killer cell cytotoxicity. Oncoimmunology (2016) 5:e1186314. doi:10.1080/2162402X.2016.1186314
129. Wen Z, Zhong Z, Darnell JEJ. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell (1995) 82:241–50. doi:10.1016/0092-8674(95)90311-9
130. Kovarik P, Stoiber D, Eyers PA, Menghini R, Neininger A, Gaestel M, et al. Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen- activated protein kinase whereas IFN-gamma uses a different signaling pathway. Proc Natl Acad Sci U S A (1999) 96:13956–61. doi:10.1073/pnas.96.24.13956
131. Bancerek J, Poss ZC, Steinparzer I, Sedlyarov V, Pfaffenwimmer T, Mikulic I, et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity (2013) 38:250–62. doi:10.1016/j.immuni.2012.10.017
132. Putz EM, Gotthardt D, Hoermann G, Csiszar A, Wirth S, Berger A, et al. CDK8-mediated STAT1-S727 phosphorylation restrains NK cell cytotoxicity and tumor surveillance. Cell Rep (2013) 4:437–44. doi:10.1016/j.celrep.2013.07.012
133. Decker T. Emancipation from transcriptional latency: unphosphorylated STAT5 as guardian of hematopoietic differentiation. EMBO J (2016) 35(6):555–7. doi:10.15252/embj.201693974
134. Brown S, Zeidler MP. Unphosphorylated STATs go nuclear. Curr Opin Genet Dev (2008) 18:455–60. doi:10.1016/j.gde.2008.09.002
135. Shi S, Larson K, Guo D, Lim SJ, Dutta P, Yan SJ, et al. Drosophila STAT is required for directly maintaining HP1 localization and heterochromatin stability. Nat Cell Biol (2008) 10:489–96. doi:10.1038/ncb1713
136. Hu X, Dutta P, Tsurumi A, Li J, Wang J, Land H, et al. Unphosphorylated STAT5A stabilizes heterochromatin and suppresses tumor growth. Proc Natl Acad Sci U S A (2013) 110:10213–8. doi:10.1073/pnas.1221243110
137. Park HJ, Li J, Hannah R, Biddie S, Leal-Cervantes AI, Kirschner K, et al. Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. EMBO J (2015) 35:580–94. doi:10.15252/embj.201592383
138. Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev (2007) 21:1396–408. doi:10.1101/gad.1553707
139. Cui X, Zhang L, Luo J, Rajasekaran A, Hazra S, Cacalano N, et al. Unphosphorylated STAT6 contributes to constitutive cyclooxygenase-2 expression in human non-small cell lung cancer. Oncogene (2007) 26:4253–60. doi:10.1038/sj.onc.1210222
140. Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol (2006) 6:602–12. doi:10.1038/nri1885
141. Chatterjee-Kishore M, Wright KL, Ting JP, Stark GR. How Stat1 mediates constitutive gene expression: a complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J (2000) 19:4111–22. doi:10.1093/emboj/19.15.4111
142. Meyer T, Gavenis K, Vinkemeier U. Cell type-specific and tyrosine phosphorylation-independent nuclear presence of STAT1 and STAT3. Exp Cell Res (2002) 272:45–55. doi:10.1006/excr.2001.5405
143. Marg A, Shan Y, Meyer T, Meissner T, Brandenburg M, Vinkemeier U. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distribution of latent Stat1. J Cell Biol (2004) 165:823–33. doi:10.1083/jcb.200403057
144. Cheon H, Stark GR. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc Natl Acad Sci U S A (2009) 106:9373–8. doi:10.1073/pnas.0903487106
145. Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, et al. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J (2013) 32:2751–63. doi:10.1038/emboj.2013.203
146. Gough DJ, Messina NL, Clarke CJP, Johnstone RW, Levy DE. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity (2012) 36:166–74. doi:10.1016/j.immuni.2012.01.011
147. Suprunenko T, Hofer MJ. The emerging role of interferon regulatory factor 9 in the antiviral host response and beyond. Cytokine Growth Factor Rev (2016) 29:35–43. doi:10.1016/j.cytogfr.2016.03.002
148. Luker KE, Pica CM, Schreiber RD, Piwnica-Worms D. Overexpression of IRF9 confers resistance to antimicrotubule agents in breast cancer cells. Cancer Res (2001) 61:6540–7.
149. Erb HH, Langlechner RV, Moser PL, Handle F, Casneuf T, Verstraeten K, et al. IL6 sensitizes prostate cancer to the antiproliferative effect of IFNα2 through IRF9. Endocr Relat Cancer (2013) 20:677–89. doi:10.1530/ERC-13-0222
150. Hotamisligil GS. Inflammation and metabolic disorders. Nature (2006) 444:860–7. doi:10.1038/nature05485
151. Eguchi J, Yan Q-W, Schones DE, Kamal M, Hsu C-H, Zhang MQ, et al. Interferon regulatory factors are transcriptional regulators of adipogenesis. Cell Metab (2008) 7:86–94. doi:10.1016/j.cmet.2007.11.002
152. Wang X-A, Zhang R, Zhang S, Deng S, Jiang D, Zhong J, et al. Interferon regulatory factor 7 deficiency prevents diet-induced obesity and insulin resistance. Am J Physiol Endocrinol Metab (2013) 305:E485–95. doi:10.1152/ajpendo.00505.2012
153. Wang X-A, Zhang R, She Z-G, Zhang X-F, Jiang D-S, Wang T, et al. Interferon regulatory factor 3 constrains IKKβ/NF-κB signaling to alleviate hepatic steatosis and insulin resistance. Hepatology (2014) 59:870–85. doi:10.1002/hep.26751
154. Wang X-A, Zhang R, Jiang D, Deng W, Zhang S, Deng S, et al. Interferon regulatory factor 9 protects against hepatic insulin resistance and steatosis in male mice. Hepatology (2013) 58:603–16. doi:10.1002/hep.26368
155. Lu J, Bian Z-Y, Zhang R, Zhang Y, Liu C, Yan L, et al. Interferon regulatory factor 3 is a negative regulator of pathological cardiac hypertrophy. Basic Res Cardiol (2013) 108:326. doi:10.1007/s00395-012-0326-9
156. Jiang D-S, Liu Y, Zhou H, Zhang Y, Zhang X-D, Zhang X-F, et al. Interferon regulatory factor 7 functions as a novel negative regulator of pathological cardiac hypertrophy. Hypertension (2014) 63:713–22. doi:10.1161/HYPERTENSIONAHA.113.02653
157. Jiang D-S, Luo Y-X, Zhang R, Zhang X-D, Chen H-Z, Zhang Y, et al. Interferon regulatory factor 9 protects against cardiac hypertrophy by targeting myocardin. Hypertension (2014) 63:119–27. doi:10.1161/HYPERTENSIONAHA.113.02083
158. Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature (2014) 517:89–93. doi:10.1038/nature13801
159. Chen H-Z, Guo S, Li Z-Z, Lu Y, Jiang D-S, Zhang R, et al. A critical role for interferon regulatory factor 9 in cerebral ischemic stroke. J Neurosci (2014) 34:11897–912. doi:10.1523/JNEUROSCI.1545-14.2014
160. Zhang S-M, Zhu L-H, Chen H-Z, Zhang R, Zhang P, Jiang D-S, et al. Interferon regulatory factor 9 is critical for neointima formation following vascular injury. Nat Commun (2014) 5:5160. doi:10.1038/ncomms6160162
Keywords: signal transduction, JAK–STAT, non-canonical, interferon, innate immunity
Citation: Majoros A, Platanitis E, Kernbauer-Hölzl E, Rosebrock F, Müller M and Decker T (2017) Canonical and Non-Canonical Aspects of JAK–STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 8:29. doi: 10.3389/fimmu.2017.00029
Received: 28 September 2016; Accepted: 09 January 2017;
Published: 26 January 2017
Edited by:
Claudia U. Duerr, McGill University, CanadaReviewed by:
Laura Eugenia Velazquez, French Institute of Health and Medical Research, FranceUwe Vinkemeier, University of Nottingham, UK
Copyright: © 2017 Majoros, Platanitis, Kernbauer-Hölzl, Rosebrock, Müller and Decker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Decker, thomas.decker@univie.ac.at