 Maryann P. Platt1
Maryann P. Platt1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 21 April 2017
Sec. Multiple Sclerosis and Neuroimmunology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.00442
This article is part of the Research TopicInduction of Central Nervous System Disease by the Adaptive Immune ResponseView all 12 articles
Antibodies against neuronal receptors and synaptic proteins are associated with autoimmune encephalitides (AE) that produce movement and psychiatric disorders. In order to exert their pathological effects on neural circuits, autoantibodies against central nervous system (CNS) targets must gain access to the brain and spinal cord by crossing the blood–brain barrier (BBB), a tightly regulated gateway formed by endothelial cells lining CNS blood vessels. To date, the pathogenic mechanisms that underlie autoantibody-triggered encephalitic syndromes are poorly understood, and how autoantibodies breach the barrier remains obscure for almost all AE syndromes. The relative importance of cellular versus humoral immune mechanisms for disease pathogenesis also remains largely unexplored. Here, we review the proposed triggers for various autoimmune encephalopathies and their animal models, as well as basic structural features of the BBB and how they differ among various CNS regions, a feature that likely underlies some regional aspects of autoimmune encephalitis pathogenesis. We then discuss the routes that antibodies and immune cells employ to enter the CNS and their implications for AE. Finally, we explore future therapeutic strategies that may either preserve or restore barrier function and thereby limit immune cell and autoantibody infiltration into the CNS. Recent mechanistic insights into CNS autoantibody entry indicate promising future directions for therapeutic intervention beyond current, short-lived therapies that eliminate circulating autoantibodies.
Antibody-mediated central nervous system (CNS) autoimmunity, the hallmark of several autoimmune encephalitis (AE) syndromes that produce movement and psychiatric disorders, is initiated when antibodies recognize neuronal receptors or synaptic proteins as foreign proteins (1–3). Antibodies directed against neuronal antigens can arise in the periphery when target proteins are expressed ectopically on tumor cells. AE has also been linked to various infectious triggers, including bacteria, viruses, fungi, and other parasites. However, an underlying infection is the causative agent for only a small fraction of all AE cases; in many cases, the triggers are either unknown or unidentified infectious agents (4, 5). While the evidence supporting various microbial triggers is scant, some autoimmune encephalopathies have well-established infectious links. For example, untreated Group A Streptococcus (S. pyogenes) infections cause autoimmune sequela in many target tissues such as the heart or CNS, manifested as either rheumatic fever or Sydenham’s chorea (SC), respectively (6–8). Several recent reviews have highlighted the clinical and mechanistic features for many of these currently accepted autoimmune encephalitides and the aberrant autoimmunity/CNS axis (4, 5, 9–15). Here, we review infectious and non-infectious triggers for AE and discuss findings from animal models of AE related to blood–brain barrier (BBB) integrity, immune cell infiltration into the CNS, and neuronal circuit dysfunction that provide useful avenues to improve diagnosis (e.g., clinical assays and imaging techniques). We will then outline routes for antibody and immune cell entry into the CNS, with a focus on the predominant pathways leading to BBB breakdown that allows entry of autoantibodies into the CNS. Finally, we will briefly review current therapies for selected AEs and propose future treatment options aimed at preventing autoantibody entry. Recent advances point to an underlying autoimmune etiology that may be relevant for many movement and neuropsychiatric diseases (2, 4, 16). Thus, elucidating the mechanisms for autoantibody access to the CNS may provide a wider spectrum of treatment options for patients with these complex and puzzling disorders.
Autoimmunity against brain targets is inherently mysterious, because traditional thinking holds that the immune system minimally surveys the CNS compared to other organs. Non-CNS self-antigens are selected against to maintain tolerance; however, CNS antigens have been thought to be excluded from immune monitoring (17). Although this thinking has been recently challenged with the identification of both glymphatic and lymphatic circulation in the CNS (18, 19), re-exposure of the immune system to brain antigens, or the presence of an outside trigger that causes production of cross-reactive autoantibodies, is a crucial step in CNS autoimmune disease.
Perhaps the earliest identified CNS autoimmune disease is linked to an infectious trigger: untreated Group A Streptococcus (GAS or S. pyogenes) infections can in some cases give rise to SC, in which antibodies directed against a streptococcal surface protein cross-react with brain antigens (20). While SC is characterized by movement difficulties (chorea) affecting both gait and the tone of large muscle groups, more recently, a group of children within an SC cohort displaying both prominent psychiatric symptoms and fine choreiform movements prompted the recognition of a new syndrome: pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) (21). Both SC and PANDAS are part of a group of basal ganglia autoimmune encephalopathies (BGE) for which the humoral adaptive immune response (i.e., autoantibodies) plays an important role in disease pathogenesis, as described in human patients as well as rodent models (3, 16). In most cases, a recent GAS exposure or infection can be identified in afflicted children and subsequent exposures typically prompt an acute exacerbation of symptoms (22). Anti-GAS titers correlate with symptom severity in many but not all SC and PANDAS cases (23). Anti-neuronal autoantibodies that erroneously recognize dopamine D1R/D2R receptors or other neuronal targets in the basal ganglia have been identified in sera from patients with SC or PANDAS, and they respond positively to immune therapies such as intravenous immunoglobulin (IVIg) or plasmapheresis, consistent with an autoimmune mechanism (7, 16, 21, 24–30). The autoimmune mimicry hypothesis, namely that antibodies generated from an aberrant humoral immune response to S. pyogenes infections recognize host-specific proteins due to epitope similarity, has been proposed to underlie the secondary sequela in BGE (20, 31). However, this hypothesis assumes that BBB permeability is impaired to allow antibody entry into the CNS, because BGE occurs in the absence of brain infection.
Infections by Campylobacter jejuni and, in rare cases, by other bacteria (32, 33), induce Guillain–Barré syndrome (GBS) and the atypical Guillain–Barré-related diseases [Miller Fisher syndrome (MFS) and Bickerstaff brainstem encephalitis (BBE)], whose symptoms lie on a continuum with traditional GBS including prickling, weaknesses in extremities, motor deficits, and pain (34–36). While these diseases are caused by the same autoantibodies against gangliosides (GD3, GQ1b, GM1, or GT1a), GBS and MFS affect peripheral nerves whereas BBE affects primarily the CNS (37, 38). Blood vessels in peripheral nerves are protected by a blood–nerve barrier (BNB) that has some similarities to the BBB (39–41). Although the BNB can be disrupted by autoantibodies present in sera from patients with multifocal motor neuropathy (42), this review is primarily focused on autoantibody entry into the CNS across the BBB rather than PNS across the BNB.
Viruses have been proposed to initiate some autoimmune encephalopathies. In systemic lupus erythematosus (SLE), autoantibodies cross-reacting with Epstein–Barr nuclear antigen-1 and the 60 kDa Ro protein target a variety of organs, including the CNS (31). Anti-Ro antibodies are frequently generated and detected early in clinical SLE, making them attractive candidates for an initiating autoantigen. Other viruses implicated in neuropsychiatric disease include influenza, herpes virus-1 and -2, Epstein–Barr virus, and bornavirus (43, 44). Herpes simplex encephalitis has been linked to subsequent development of NMDAR encephalitis in some cases (2, 9). Notably, the majority of viral triggers are hypothesized to create a pro-inflammatory state that “primes” the immune system, including CNS-resident immune cells termed microglia, to become overactive leading to an autoimmune response against the CNS (2, 45). This contrasts with the molecular mimicry hypothesis proposed for S. pyogenes-induced BGE, in which antibodies directed against bacterial surface antigens cross-react directly with self-antigens (20, 31).
In contrast to molecular mimicry, production of antibodies against a brain antigen can occur in the periphery when a tumor cell expresses surface proteins found in the brain. Tumor masses are known to express a wide variety of non-tissue-specific surface proteins, including neuronal antigens. The original cohort of NMDA receptor encephalitis (NMDARE) patients contained young women bearing ovarian teratomas that express NMDAR, thus initiating peripheral immune activation (46). Subsequent BBB permeability by an unknown mechanism would then allow NMDAR antibodies to target glutamatergic synapses also containing this receptor. However, tumors are not present in all cases of NMDARE, and indeed, the tumor rate is approximately 50% for such patients (47). Anti-GAD65 antibodies, which are the putative culprits underlying cerebellar ataxia, stiff person syndrome, and Batten’s disease (3), are also associated with neoplasms that aberrantly express GAD65, albeit at lower frequency than those producing NMDAR antibodies (48, 49). Finally, serum antibodies targeting the voltage-gated potassium channel (VGKC) complex may be also linked to tumors. These include antibodies against leucine-rich glioma-inactivated 1 (LGI1), contactin-associated protein 2 (Caspr2), and contactin 2 as well as those recognizing the entire VGKC protein complex. IgGs targeting the VGKC complex have been found in sera (50, 51) and cerebrospinal fluid (CSF) (51) from patients with both limbic encephalitis and Morvan syndrome (VGKC complex encephalitis, peripheral nerve hyperexcitability) (52), and Morvan syndrome has been linked to thymoma in 37% of patients (53). Conversely, anti-VGKC complex antibodies are present in sera from 32% of patients with verified thymoma, the majority of whom develop myasthenia gravis, which targets acetylcholine receptors on muscles that are supplied by blood vessels lacking a tight endothelial barrier (54). Since only a minority of patients with VGKC complex antibodies have co-occurring thymoma, this suggests that other mechanisms underlie development of these autoantibodies, similar to NMDARE. In addition, the clinical features accompanying patients with VGKC antibodies are highly variable (52). VGKC antibodies may, therefore, be useful biomarkers for inflammatory neurological disease in general and are potentially associated with the presence of other autoantibodies or immune cell infiltration into the CNS (52).
Numerous rodent studies have expanded our understanding of the molecular events involved in antibody generation and pathogenicity for several varieties of AE, shedding light on the mechanisms of immune cell infiltration and barrier breakdown. Rodent models for NMDARE, BGE, GAD65 encephalitis, BBE, and Morvan syndrome vary widely in both their methods of antigen exposure and the disease parameters recapitulated (see Table 1 for summary). A majority of the studies surveyed here use animal models in which disease is initiated either by exposure to an initial trigger (bacteria or virus) in the periphery or by direct infusion into the CNS of sera or purified autoantibodies isolated from patients, thereby circumventing the BBB.
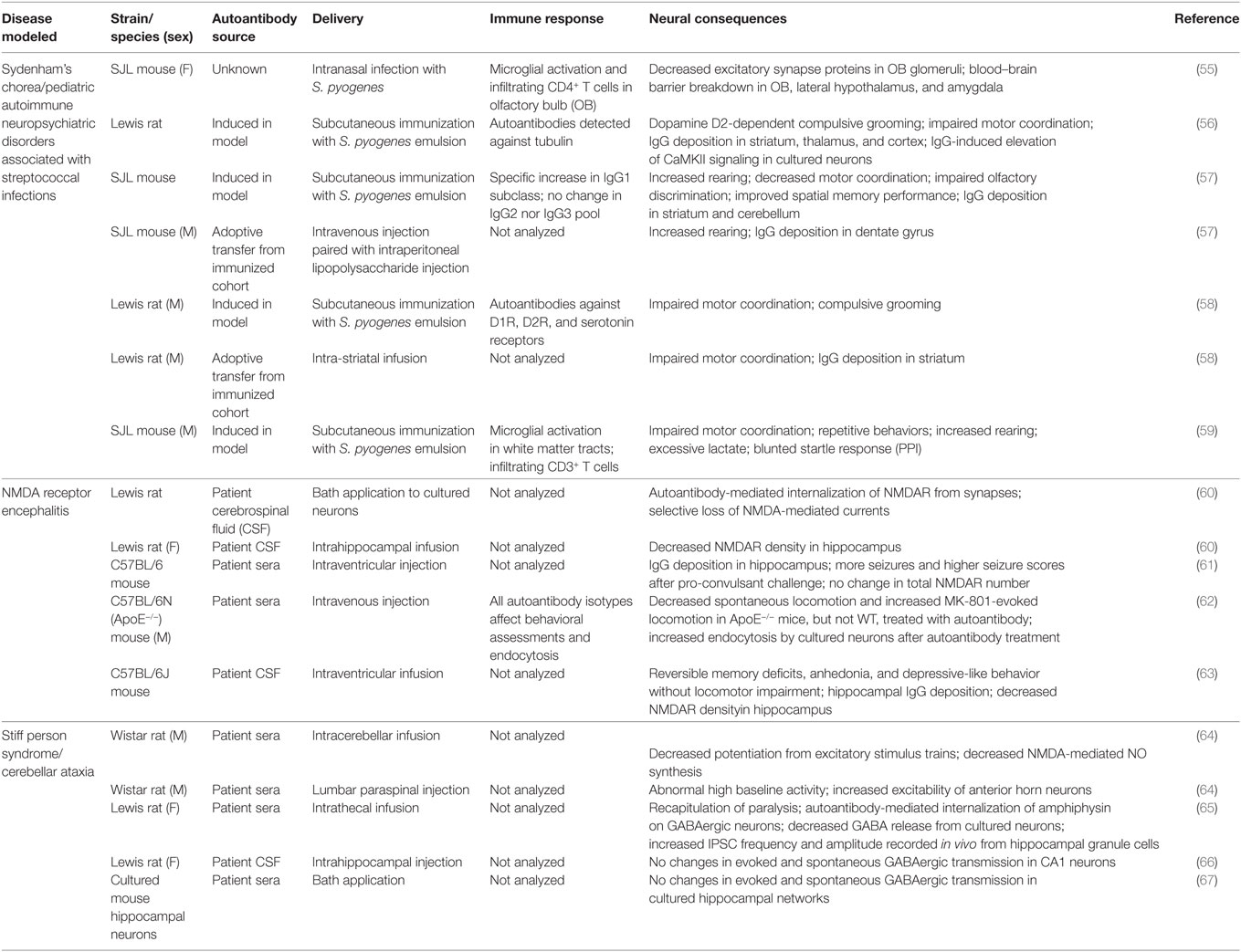
Table 1. Summary of published rodent models for several autoimmune encephalitides.
Animal models for post-streptococcal BGE have been focused on demonstrating the ability of GAS to prime development of an autoimmune reaction by stimulating adaptive cellular and humoral immune responses. In the mouse, intranasal (i.n.) infections with live bacteria polarize T cells located in the nasal-associated lymphoid tissue (NALT, the mouse structural analog of human tonsils and adenoids) toward a Th17 phenotype, a T cell subtype that is both essential for mucosal immune protection against bacteria but also strongly implicated in many autoimmune diseases (Figure 1A). Multiple i.n. S. pyogenes infections strengthen this Th17 immune response, largely due to induction of IL-6 and TGF-β1, which are two pro-inflammatory cytokines essential for Th17 differentiation (68). IL-6 is essential for clearance of bacteria after i.n. infection, via generation of Th17 cells; IL-6−/− mice are capable of generating a Th1 immune response to an i.n. bacterial challenge but cannot control infection (69). This model has been used to demonstrate that repeated i.n. infections with S. pyogenes induce migration of GAS-specific Th17 cells and other T cell subtypes from the nasal epithelium to the olfactory bulb (OB) (Figure 2), where sensory axons make connections with projection interneurons to form the neural circuitry essential for odor discrimination, as well as to other CNS regions (55). The presence of Streptococcus-specific Th17 cells in the CNS after repeated i.n. infections increases the permeability of capillaries in several CNS regions, including the OB, amygdala, and hypothalamus, thereby enabling deposition of serum IgGs and potential anti-CNS autoantibodies. This is largely due to disruption in the organization of tight junction (TJ)-associated proteins, which control an essential aspect of BBB function (55) (see below for a more detailed discussion). The intranasal model produces profound changes in olfactory neural circuitry by reducing vGluT2 expression and thus excitatory input at the presynaptic terminals of olfactory sensory axons and perturbing the excitatory/inhibitory balance within the primary olfactory circuit (55). This model of post-S. pyogenes autoimmunity demonstrates a central role for the cellular adaptive immune response (e.g., bacterial-specific Th17 cells in the CNS) in disrupting BBB function, thus promoting entry of antibodies into the CNS and inducing changes in synaptic signaling. Although such a cellular adaptive immune response has not been identified to date in the nervous systems of children suffering from BGE, S. pyogenes-specific Th17 cells can be found in the tonsils of human patients (55), making Th17 lymphocytes a potential causative agent in either initiation or persistence of BGE disease pathogenesis.
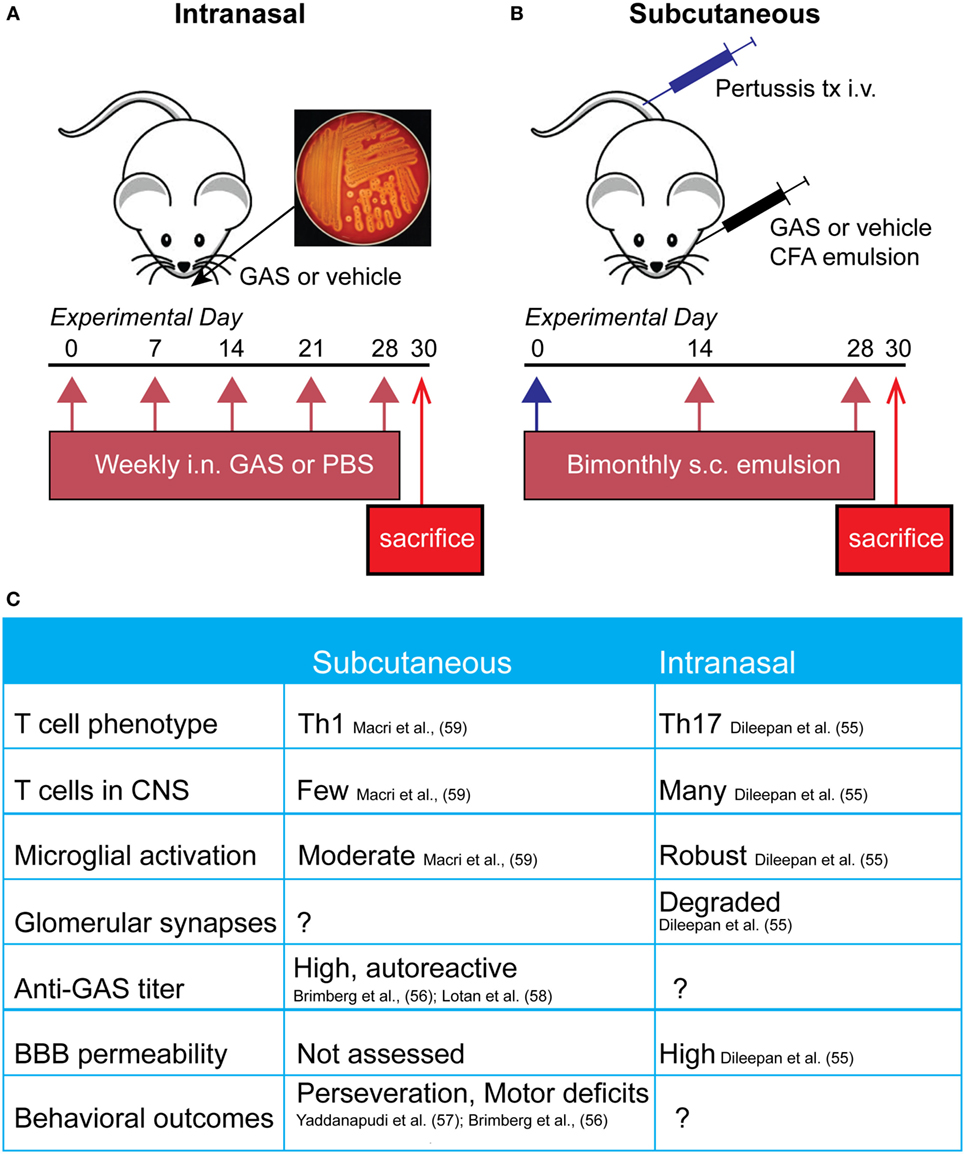
Figure 1. Comparison of mouse pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS)/Sydenham’s chorea (SC) models. (A) Schematic representing the initiation of the intranasal model, where mice receive live bacteria intranasally once a week for 5 weeks prior to sacrifice. (B) Subcutaneous GAS exposure involves adjuvant and antigen exposure three times, every 2 weeks, following an initial boost with intravenous pertussis toxin. (C) Comparison of immune, neural, and behavioral outcomes after each route of GAS exposure. Investigators have used either subcutaneous or intranasal routes to induce an immune response against S. pyogenes [Group A Streptococcus (GAS)] in efforts to understand the mechanisms underlying the behavioral and motor symptoms characteristic of PANDAS and SC patients. The former route necessitates opening the blood–brain barrier (BBB) artificially using B. pertussis toxin, whereas the latter features intranasal inhalation of live bacteria to trigger a Th17 response in nasal tissue that is directly communicated to the brain along the olfactory nerve.
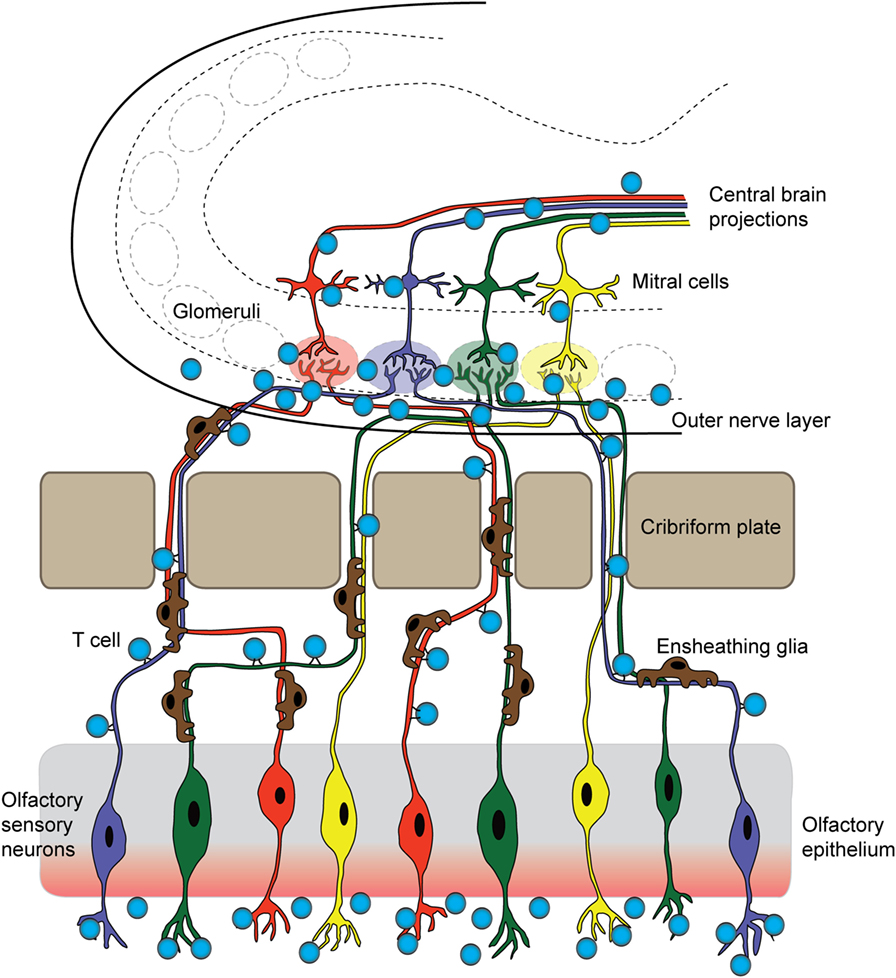
Figure 2. T cells originating in the nose infiltrate the brain parenchyma. In a mouse model for pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections, T cells first arise in the nasal-associated lymphoid tissue and olfactory epithelium at the site of a latent S. pyogenes infection. These cells then respond to chemotactic cues release by olfactory ensheathing glia to accompany sensory axons into the brain. Once there, infiltrating T cells release inflammatory cytokines and chemokines, damaging synapses within olfactory glomeruli and breaking down tight junctions of olfactory bulb capillaries. These T cells may then move centrally, against the rostral migratory stream and toward the SVZ, and exit through the ventricles, or continue following the projections of olfactory mitral/tufted neurons.
A second group of rodent models for BGE employs subcutaneous immunization with an antigenic target (bacterial homogenate) plus complete Freund’s adjuvant to activate the immune system, in conjunction with agents (i.e., B. pertussis toxin) that open the BBB to provide access to brain targets (Figure 1B). In this model, mice and rats develop a strong humoral immune response toward S. pyogenes and show behavioral abnormalities. Specifically, GAS-immunized rodents display increased rearing and decreased locomotion, as well as increased repetitive and perseverative behaviors, impaired pre-pulse inhibition, and reduced concentrations of serotonin in the prefrontal cortex as compared to controls (56, 57, 59, 70). Moreover, adoptive transfer of serum IgGs from S. pyogenes-immunized mice to naive recipient mice, or direct infusion of sera into rat brains, recapitulates some of the behavioral deficits in recipient rodents, whereas no effects were observed after adoptive transfer of IgG-depleted serum (57, 58). Histological examination of brain tissue revealed antibody deposition in the deep cerebellar nuclei and hippocampus in mice and the striatum, cortex, and thalamus in rats (56, 57, 70). Serum IgG isolated from immunized rodents recognizes both cerebellar targets and human D1/D2 dopamine receptors by either western blotting or ELISA (56, 57). There is a high variability among the mouse and rat BGE models. This may reflect differences in key immune-related genes between species, especially those related to T cell function or within the MHC locus for antigen presentation and immune cell stimulation. All studies used inbred animals (Lewis rats and SJL or C57BL/6 mice); therefore, intra-strain variability is low in these highly variable regions; however, differences between strains and species may be more pronounced. It is also possible that differences observed in mouse and rat studies are due to variability in their humoral immune response to GAS or differences in immunization protocols. Taken together, subcutaneous animal models for BGE have provided useful information regarding the humoral immune response after bacterial infection (i.e., the presence of antibodies directed against GAS and CNS) and demonstrate a clear link between S. pyogenes exposure and behavioral abnormalities (see Figure 1C for comparison of intranasal and subcutaneous models). However, these are somewhat artificial models for immune system activation, because human GAS infections occur primarily by the i.n. route. Moreover, subcutaneous animal models for BGE leave unanswered an important question for disease pathogenesis, namely how these autoantibodies can penetrate the CNS since the BBB is artificially opened (71).
Most animal models for NMDARE involve infusing serum antibodies from acutely ill patients into the rodent brain in an attempt to replicate behavioral symptoms of the human disease. Infusion of autoantibodies from NMDARE patients into the lateral ventricles of mice causes impaired recognition memory after 10 days of infusion, which is reversible after antibody washout (63). Histologically, patient IgG binding is strongest in the hippocampus, which has a very high concentration of NMDARs, supporting the conclusion that internalization of NMDAR in this region may underlie memory deficits with minimal effects on aggression, locomotion, and anxiety-like behavior (61, 63). Intraventricular delivery of NMDAR antibodies also decreases seizure thresholds after administration of the convulsant pentylenetetrazol, resulting in more frequent and stronger seizures in NMDAR antibody-recipient mice (61). These data mirror the course of human disease, because NMDARE patients frequently present with seizures in conjunction with memory loss, hallucinations, and anxiety (72). Using the electroencephalograph (EEG), NMDARE patients show unusual delta rhythmic activity with superimposed beta or gamma activity (72). Similar spike events are also detectable using EEG during seizures in mice after NMDAR antibody infusion. Electrophysiological changes are also apparent in hippocampal neurons after intrahippocampal infusion of anti-NMDAR antibodies (73). Patient-derived NMDAR antibodies decrease NMDAR-dependent hippocampal long-term potentiation (LTP) in a manner similar to antibodies directed against extracellular domains of the NR2 or NR1 subunits of the NMDAR. After anti-NMDAR antibody treatment, dentate granule cells show smaller evoked responses to stimulation, increased spike thresholds after EPSP, impaired LTP, and decreased NMDAR densities in postsynaptic areas (60). In summary, there is strong evidence that antibody binding to the NMDAR leads to cross-linking and internalization but not necessarily destruction of the receptor, causing deficits in synaptic transmission that include LTP. NMDAR antibody deposition is strongest in the hippocampus after intraventricular infusion, indicating that deficits in perforant pathway may be the most prominent in this animal model for NMDARE. However, this model bypasses the BBB. In an elegant series of experiments, Hammer and colleagues showed that the BBB efficiently excluded NMDAR antibodies from the hippocampus if delivered intravenously (i.v.) in healthy mice. However, in ApoE−/− mice that have a defective BBB, intravenously injected anti-NMDAR antibodies can access brain antigens, and mutant mice injected i.v. with anti-NMDAR antibodies from patient sera show decreased locomotion as compared to those treated with control sera (62). Therefore, there is a clear need to develop animal models for NMDARE that incorporate mechanisms that disrupt BBB integrity to more closely mirror the human syndrome.
Autoantibodies against proteins involved in GABAergic transmission, including anti-glutamic acid decarboxylase (GAD) 65, anti-GAD67, and anti-amphiphysin, are the putative autoantibodies for cerebellar ataxia, stiff person syndrome, and Batten’s disease (3, 74). Animal models using intraventricular infusion of anti-GAD65 antibodies isolated from patients have provided conflicting results. Bath application of such antibodies causes increased IPSP frequencies in cultured hippocampal neurons (75). Cerebellar infusion of anti-GAD65 antibodies in rodents also leads to reduced GABA synthesis in cerebellar basket cell terminals, resulting in disinhibition of Purkinje cells. However, hippocampal infusion of anti-GAD65 antibodies in vivo yields no changes in synaptic transmission assessed with physiological measurements in hippocampal slice preparations, whereas anti-NMDAR antibodies are sufficient to induce defective LTP (66). It is possible that anti-GAD65 antibodies do not gain access to their intracellular antigen after infusion in the hippocampus, or that pathogenic antibodies are in fact directed against a different antigen. Antibodies against amphiphysin rather than GAD65 have been shown to induce stiff-person syndrome-like symptoms in rats (65). Methodological differences and debate about pathogenicity of GAD65 antibodies has complicated the interpretation of these studies, because GAD65 antibodies are also prevalent, albeit at lower concentrations, in type 1 diabetic patients who have no neurological abnormalities. Careful screening of patient antibody samples is, therefore, necessary for anti-GAD65 collections, in order to exclude the likely non-pathogenic antibodies that are present in patients with diabetes.
There are few studies on the CNS effects of anti-GQ1b antibodies, the pathogenic basis for BBE, which hinders our understanding of this disease; however, there have been some reports on their binding location and disease mechanism. Anti-GQ1b antibodies were shown to induce complement deposition; the combination of antibody and complement is necessary to disrupt neuromuscular junction function in vivo (76). Binding of the antibody-complement complex induces local Ca2+ flux into neurons in vitro, which in turn alters mitochondrial function and causes hydrogen peroxide production in addition to neuronal excitation (76). Mechanistically, this provides an interesting perspective on GBS and related syndromes, which lie at the intersection of innate and adaptive immune mechanisms. However, other than lesions identified by MRI in human MFS and BBE patients, there has been no investigation into how anti-GQ1b antibodies enter the CNS to affect brain function (77, 78).
The VGKC complex contains a signaling protein tetramer as well as several scaffolding proteins. Early identification of antigenic targets had attributed antigenicity to the potassium channel Kv1.1/1.2 itself, while more recent work argues that the antibody targets are more likely the associated scaffold proteins (LGI1, Caspr2, and contactin-2) that precipitated along with the channel during the original identification (79). Most investigation into VGKC antibodies has used rodent tissue or cultured cells to test for reactivity of patient antibodies that are harvested from CSF or serum. Sera from both AE and neuromyotonia patients react with rat (80) and mouse (81) hippocampal sections, as well as with the potassium channels Kv1.1 and Kv1.2 transfected into HeLa cells (80), but not with Caspr2−/− mouse hippocampal sections (81). One rodent study used adoptive transfer of patient plasma or purified IgG into mice to address how these antibodies alter neuronal transmission in peripheral nerves (82). After repeated injections with patient IgG, mice showed no clinical symptoms but had increased quantal size and potassium-dependent increases in EPP amplitude in peripheral nerves as compared to control mice (82). While there has been much progress in identifying the target antigen of these antibodies, many questions about their pathogenicity and CNS access remain unanswered.
Epithelial cells within the choroid plexus form a tight barrier termed the BCSFB that restricts diffusion of serum proteins and immune cells from the more leaky blood vessels of this tissue into the CSF (83). The molecular composition of TJs within the BSCFB is less well understood than those comprising the BBB. However, several key junctional proteins that regulate paracellular permeability across epithelial cells of the choroid plexus are present at high levels in both embryonic and adult stages, suggesting that this barrier matures early during development (84). The critical function of these junctions is to create a physical barrier to paracellular diffusion, allowing cells to become polarized with distinct luminal and abluminal components. In addition, epithelial cells of the BCSFB also express specialized transporter proteins to allow transit of certain plasma proteins across this barrier (84, 85). The BCSFB is less tightly regulated than its brain counterpart and can become more permeable to immune cells or antibodies during disease states. Th17 lymphocytes cross the BCSFB several days prior to BBB damage, in order to enter the CNS and initiate their immune attack during experimental autoimmune encephalitis (EAE), a mouse model for human multiple sclerosis (86). This entry appears to be an essential step in initiating the CNS immune response, because CCR6 receptor-deficient Th17 cells that cannot cross the BCSFB do not induce EAE (86). In mouse models for SLE, the cell adhesion molecules vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1) show elevated expression in the choroid epithelium and promote large amounts of cellular infiltrates (T and B cells) in the choroid (87, 88). The role of such infiltrating immune cells is still debated, but the BCSFB epithelium is clearly activated and disrupted by increased cytokine expression (87–90). However, the role of BSCFB during AE remains unexplored. It is possible that either B cells or antibodies may cross this barrier more efficiently than the BBB to enter the CSF, but data from either in vitro studies or animal models are currently lacking (Figure 3A).
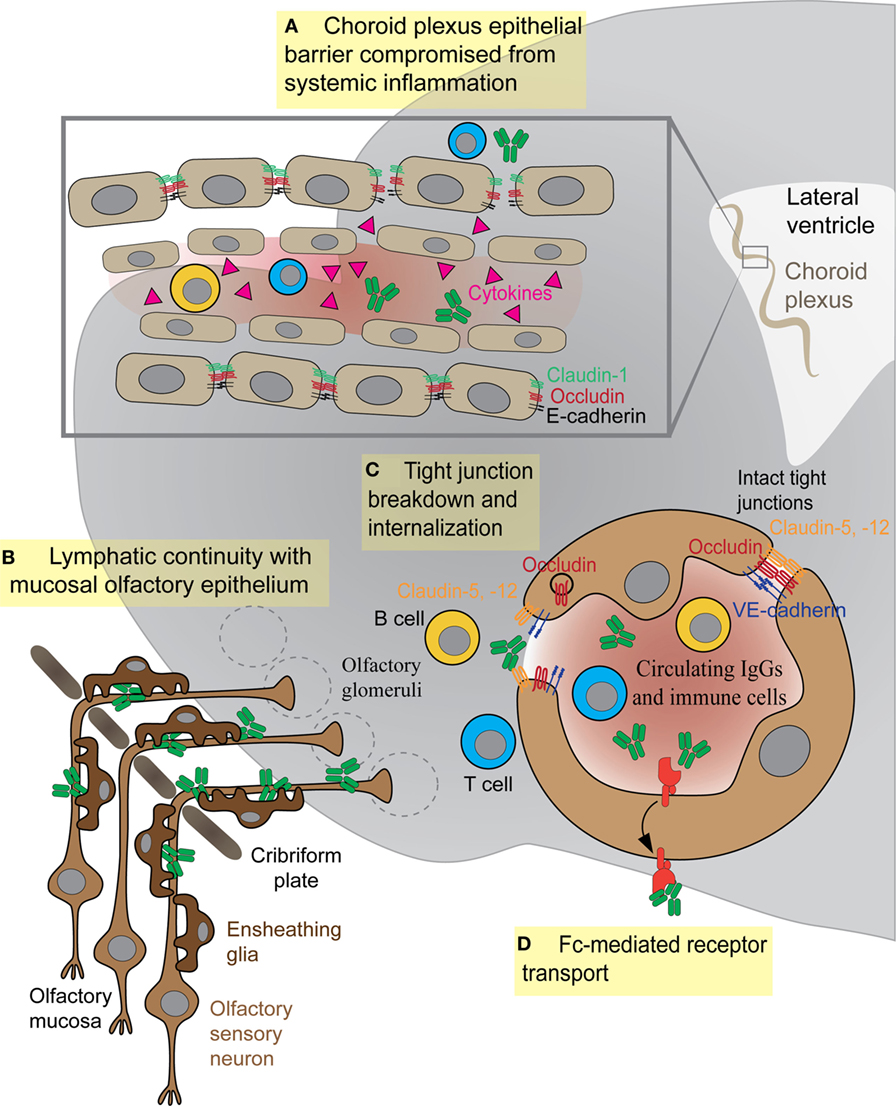
Figure 3. Antibody and immune cell access to the brain parenchyma via four distinct routes. (A) Systemic cytokines break down tight junctions (TJs) within the brain–cerebrospinal fluid barrier to allow central nervous system (CNS) access of antibodies or immune cells. (B) Olfactory ensheathing glia facilitate transport of IgGs or immune cells along sensory axons exiting the olfactory mucosa. (C) Inflammatory cytokines in the bloodstream damage TJs between endothelial cells, thus allowing antibodies or immune cells (T or B cells) to enter the CNS. (D) Fc receptor directionality reverses, shuttling IgG from vessels into brain parenchyma as in systemic lupus erythematosus.
The proximity of the OB to the nasal mucosa makes it both a vulnerable niche to insult and infection of the brain and an attractive option for delivery of therapeutics that cannot otherwise traverse the BBB (91, 92). Many viruses and bacteria co-opt this olfactory route for brain infection, and more than 40 substances have been shown to enter the brain by this means (93, 94). Viruses, such as Venezuelan equine encephalitis virus, initially infiltrate the CNS via olfactory axons, then induce BBB permeability throughout the brain to promote a second wave of infection, as virion particles enter the brain from the circulation via the damaged barrier (95). The neurotrophic Nipah virus also infects hamster olfactory sensory axons, travels into the OB, and disseminates from olfactory processing centers like the olfactory tubercle before spreading throughout the brain (96). After an initial injury to nasal mucosa, S. aureus infects olfactory sensory neurons and spreads to the OB via the olfactory axons within 6 h (97). In addition to intranasal pathogens, the olfactory route has been successfully used to deliver drugs, large proteins, and stem cells into the brain (91), largely due to rapid diffusion via cerebral perivascular spaces (98). Why is the olfactory route so amenable for CNS delivery? This is due to several factors: (1) diffusion across the heavily vascularized nasal mucosa that also contains lymphatic vessels, (2) direct transport by olfactory sensory axons into the OB, and (3) direct transport by the trigeminal nerve into the brainstem. Although the nasal mucosa is highly vascularized, the mean capillary density and relative permeability to hydrophilic macromolecule tracers is significantly greater in the respiratory epithelium of the nose than in olfactory sensory regions (99). Thus, sensory regions of the nasal mucosa may provide easier access to the brain due to their relatively slower clearance rates into the bloodstream (99). We have recently shown that GAS-specific Th17 lymphocytes and other T cell subtypes generated in the olfactory cavity use the sensory axon route to enter the CNS in an animal model for BGE (Figures 2 and 3B). Because T lymphocytes are primarily found in the outer nerve and glomerular layers within the OB, where incoming sensory axons form synaptic connections with projection neurons, we propose that T cells travel along the sensory axon route into the CNS (55). However, it is currently unclear whether GAS-specific T cells passively move into the brain along these sensory axons or are actively recruited into the brain by resident antigen-processing macrophages, microglia, or the olfactory ensheathing glia, the last of which are known to phagocytose bacterial debris. Olfactory ensheathing glia guide olfactory sensory axons toward their targets in the OB and thus may also serve this role for GAS-specific T cells. Once in the brain, Th17 cells likely persist due to high levels of CCR6 and LFA-1 signals, which are required for CD4+ T cells to remain in the CNS in the EAE model. The olfactory route is also used by immune cells to exit the CNS through the OB and drain into the deep cervical lymph nodes, in order to dampen anti-CNS immune responses in the periphery (100, 101). There are, however, no data on whether antibodies use the olfactory route to gain access to the CNS from sites of infection in the olfactory mucosa, especially for BGE associated with i.n. GAS infections.
The BBB achieves its selective permeability to proteins and immune cells by the presence of (1) TJs that prevent paracellular diffusion of small molecules and immune cells between endothelial cells, (2) very few endocytotic vesicles that restrict movement of large molecules through the transcellular pathway, and (3) transporters that shuttle select nutrients between the blood and the brain (Figure 3C) (102). The junctional transmembrane proteins claudin-3, -5, -12, and occludin are expressed at the barrier and interact in a homotypic manner to form paracellular pores that restrict diffusion between cells (Figure 3C) (102). Endocytotic caveolae in the CNS endothelium provide an essential route for receptor-mediated transcytosis (102). This process requires caveolin-1 (Cav-1), a transmembrane protein expressed at low levels within CNS blood vessels. Cav-1 levels increase during BBB breakdown following ischemic stroke, when enhanced transcytosis initiates BBB dysfunction (103). Healthy BBB vasculature also has low levels of leukocyte adhesion molecules such as VCAM-1, ICAM-1, and ICAM-2, which are upregulated on CNS vessels during neuroinflammation to promote T lymphocyte trafficking into the parenchyma (104, 105).
Just as capillaries in the CNS possess unique characteristics as compared to systemic capillaries, the BBB may be differentially porous throughout the CNS. Several studies have suggested that endothelial barriers in the brain and spinal cord are functionally different. Blood–brain and blood–spinal cord barriers differ in expression of some BBB-specific proteins and their functional permeability. Endothelial cells in the spinal cord have decreased expression of adherens junction (VE-cadherin; β-catenin) and TJ (occludin, ZO-1) proteins, as well as increased permeability to small molecular weight tracers, compared to brain endothelium (106). Endothelial cells in these two CNS regions also differ in their affinities for immune cells. Encephalitogenic T cells in the cervical spinal cord rapidly arrest on the vasculature without crawling, whereas they do not display this behavior on brain blood vessels (104). Higher expression of sphingosine 1-phosphate receptors in spinal cord ECs may facilitate preferential immune cell infiltration as compared to other CNS regions during EAE (107). Within the CNS, blood vessels in the OB and neurogenic niches also have a relatively higher permeability as compared to other CNS regions (excepting the circumventricular organs that have leaky blood vessels); blood vessels in these regions become highly permeable, in particular following viral infections (108–110), and may predispose them to infiltration of immune cells or antibodies in addition to viral particles.
Blood–brain barrier function can be impaired by several factors including inflammatory cytokines, hormones (e.g., epinephrine), and substances of abuse (e.g., alcohol, cocaine, and methamphetamine) (31, 102, 111). Several inflammatory cytokines including IL-1β, TNF-α, CCL-2, and IL-17A, which are present in the CNS or blood during neuroinflammation, affect the stability of the BBB by either degrading TJ proteins, modifying their phosphorylation states, or affecting the turnover rate (Figure 3C). IL-1β indirectly destabilizes the BBB by inducing expression of matrix metalloproteinase-9 and VEGF, two factors that promote degradation of Claudin-5, Occludin, and ZO-1 (112, 113). TNF-α also induces formation of gaps between cell junctions by upregulating transcription of NF-κB and myosin light chain kinase, a factor known to internalize TJ proteins via caveolae (114, 115). The chemokine CCL-2 produced by macrophages also enhances BBB permeability by inducing phosphorylation of occludin and claudin-5 and promoting their endocytosis via caveolae (116). IL-17 and IL-22 produced by Th17 cells in EAE/MS are also known to disrupt endothelial cell TJs (117). Although these cytokines have been shown to disrupt barrier function during neuroinflammation, it is unclear whether they are present in AE. Recent cytokine profiling of CSF from patients with viral and autoimmune encephalitis revealed that several Th1 cytokines, such TNF-α, IFN-γ, CXCL9, and CXCL10, are elevated during viral encephalitis, but not in AE samples. In contrast, IL-6, a cytokine essential for development of Th17 cells, is elevated in all CNS autoimmune disorders (11). The predominant mechanism for BBB disruption in our animal model for S. pyogenes-induced BGE appears to be disruption of endothelial TJs via formation of gaps and protrusions that enhance BBB permeability and promote entry of autoantibodies into the CNS (55). Although we cannot exclude other factors such as cytokines released by activated microglia/macrophages that are present in the CNS after multiple i.n. GAS infections (55), or the ability of GAS antibodies to break down endothelial cell junctions, the high degree of correlation between the presence of bacterial-specific Th17 cells and BBB leakage suggests that IL-17 likely contributes to BBB dysfunction in a similar fashion as in EAE/MS (55, 115). S. pyogenes-specific Th17 cells are present in the tonsils of human patients (55); however, it remains to be shown whether they are important for human disease pathogenesis.
B cells can arrest along endothelial cell walls using the same adhesion mechanisms as T cells (VLA-4- and ICAM-1-mediated arrest and adhesion). In addition, B cells can infiltrate into the CNS, by extravasation through disrupted TJs, reminiscent of T cell entry across the BBB (118–120). However, the number of B cells that has been reported to present in the CNS following histological analyses of patients with GBS, BBE, MFS, or any of the animal models of AE discussed above is indeed extremely small compared to the number of T cells (77). B cells can infiltrate the inflamed CNS in MS and SLE patients and form ectopic lymphoid structures, or cellular aggregates outside germinal centers, which reinforce the immune response locally (90, 121–123). In MS, B cells within cellular aggregates present antigens to T cells, contribute to epitope spreading, and produce antibodies that are detectable as oligoclonal bands in the CSF collected from patients (123). However, studies using mouse models of SLE have produced confounding results as to whether B cells that are resident within the CNS secrete autoantibodies (89, 124). In addition, there is scant evidence for B cell infiltration during AE. It is possible that these diseases are mediated by locally infiltrating B or plasma cells (125), but firm evidence for CNS-resident B cell populations is still lacking.
Endothelial cells are also highly vulnerable to endotoxins secreted by infectious agents. Both antigenic surface proteins on Gram-positive bacteria and lipopolysaccharide from Gram-negative bacteria change endothelial barrier properties (126). Environmental toxins and food additives can also increase BBB permeability, as evidenced by serum protein leakage after bis(tributyltin) oxide exposure (127). Finally, the gut microbiome may also affect the function of the barrier (128). Given the presence of many anti-neuronal autoantibodies in circulation, transient BBB permeability from any of these sources, or reactivated Fc receptor-mediated transcytosis through blood vessels (Figure 3D), would provide opportunistic access to brain targets. Pathological antibodies may also enhance BBB permeability, promote production of inflammatory cytokines, and stimulate adhesion and migration of immune cells across the CNS. For example, anti-NR2 glutamate receptor antibodies (anti-NR2) derived from patients with SLE promote expression of both VCAM-1 and ICAM-1 and increase the production of IL-6 in brain endothelium that promotes BBB inflammation and changes to its permeability (129). Antibacterial antibodies can also change endothelial barrier permeability; however it is unknown whether this mechanism mediates antibody entry into the CNS during AE. Furthermore, rare cases of GBS with CNS lesions provide insight into how the same bacterial infection may cause central as opposed to peripheral autoimmunity (77, 78). CNS lesions in the brain (77, 78) and spinal cord (77), as well as CSF pleiocytosis or excess protein (130), together with the presence of immune cell infiltration in both CNS regions (77) indicate immune involvement in GBS infections of the CNS. Active forebrain lesions in the patient described by Okumura are compelling evidence for a BBB breach (78). Cytokine-producing CD4+ and CD8+ immune cells in the CNS were mostly confined to dense clusters in the meninges and perivascular cuffs, but a population of CD4+ and CD8+ T cells were found in the brainstem with a very few B cells intermixed in perivascular immune cell infiltrates (77). Therefore, brain lesions or T cell infiltration into perivascular spaces may be important steps in breaching the BBB and thereby providing antibody access to the brainstem, for certain GBS subtypes.
The treatments currently employed for BGE focus on immunotherapy to weaken the autoimmune response. Screening for cancer is essential in ruling out a neoplastic syndrome; if a tumor is present, surgical interventions are effective in alleviating symptoms for 75% of NMDAR encephalitis cases. In cases without a tumor, first-line treatment with immunotherapy using high doses of corticosteroids, plasma exchange, and/or IVIg improves patient outcomes in two-thirds of cases. Cases that do not respond to primary immunotherapy receive treatment with Rituximab (a monoclonal antibody that neutralizes B cells, thereby halting antibody production) or cyclophosphamide (a potent immunosuppressant) (72). EEG monitoring and antiepileptic treatments are frequently necessary in AE; seizures are common with encephalitides of autoimmune and infectious origin (13) and may trigger BBB leakage due to release of the potent inflammatory cytokines IL-1 and TNF-α from dying neurons. Controlling further seizures is paramount to stave off permanent damage, especially for pediatric patients.
Encephalitis with an infectious trigger such as S. pyogenes warrants primary treatment with antibiotics to eliminate the latent infection (131). First-line treatment for SC or PANDAS, both of which are associated with streptococcal infections, centers on eliminating autoantibodies from circulation by means of plasma exchange or plasma apheresis, coupled with corticosteroid treatment. Plasma exchange shows promise as a treatment option, but IVIg has only a minor effect (29, 30). A recent double-blind clinical study of IVIg for 35 PANDAS patients found no improvement compared to placebo after two rounds of IVIg (132). Plasmapheresis is a beneficial, if invasive, treatment option, with an average improvement of 65%, 6 months after treatment (30). Indeed, many immunotherapies recommended for AE require several weeks or months to show effectiveness.
Outcomes are generally good in younger patients, including children. Young adult and pediatric autoimmune encephalitis patients have a recovery rate of up to 80%, although improvements continue slowly for up to 2 years. In patients who recovered from NMDAR encephalitis, the relapse rate is around 25%, so yearly screening for tumors is recommended after recovery (133). After surgical excision of tumors, if warranted, following up by treatment with immunotherapy leads to generally good outcomes for NMDAR encephalitis. PANDAS patients typically also recover after treatment and learn to manage their exposure to S. pyogenes. Some patients continue prophylactic antibiotics to minimize such exposure, sometimes for several years after their most recent relapse.
For any type of AE, studying BBB function during both disease initiation and periods of remission would greatly clarify the role of the barrier over the course of disease and help inform treatment options. MRI with gadolinium enhancement is a standard tool for MS diagnosis and disease monitoring (102, 134) and is a sensitive diagnostic tool for GBS. Based on our studies on the animal model for S. pyogenes-induced BGE, we argue that MRI with gadolinium enhancement may reveal latent BBB damage in AE patients or foci of BBB damage (135–137). Our working hypothesis that barrier breakdown is a crucial step during AE pathogenesis would, therefore, be strengthened by evidence from patient MRI with gadolinium enhancement or careful analysis of BBB integrity using intravenous fluorescently labeled low- or high-molecular-weight tracers in animal models for other types of AE (55).
Future treatment avenues may rely on modulating BBB permeability using biological or chemical therapeutics. One exciting option is reactivation of signaling pathways that normally function to form the BBB during development, in order to repair the dysfunctional barrier during disease. The barrier properties of CNS vessels develop prenatally in most CNS regions for both mice and humans with the exception of the retina, which vascularizes postnatally in mice. Wnt/β-catenin signaling promotes both CNS angiogenesis and BBB formation by production of Wnts from neural progenitors (138–142). Wnts also induce expression of some EC-specific proteins, including the glucose transporter Glut-1 and several TJ components (138–142). Following angiogenesis, Hedgehog signaling is required for acquisition of barrier properties in CNS ECs, including expression of TJ proteins occludin and claudin-5 (142, 143). Mature blood vessels continue to respond to Wnt and Shh cues, indicating that these pathways are active in maintaining EC barrier properties (139, 142, 143). Recently, Wnt signaling was reported to be upregulated in both EAE and human MS lesions, correlating with increased neuronal Wnt3 expression and TJ breakdown. Reactivation of Wnt signaling in EAE may serve to repair the damaged BBB in inflammation. Inhibition of Wnt signaling hastened disease progression and resulted in increased numbers of CD4+ T cells in the CNS (105). Chemical modulators of Wnt signaling have been validated in vitro and in rodent models of neuroinflammation (144, 145). Translation of such therapies to clinical use could repair the damaged barrier, translating to improved clinical outcomes in AE patients by limiting influx of blood-borne immune cells, cytokines, and/or antibodies.
Autoimmunity in the CNS remains confoundingly complex, in both etiology and treatment. While triggers for AE are defined in some cases, frequently, no clear infectious or cancerous cause can be found. BBB integrity clearly plays a role in disease development, and more research on differential barrier permeability over the course of disease would resolve many questions about autoantibody entry and pathogenesis. Animal models for these diseases have the potential to uncover new routes of antibody entry that are not apparent from patient imaging studies. Indeed, infectious triggers themselves may have the inherent ability to disrupt the BBB to allow access to brain antigens. By approaching AE from several angles, a more complete picture of pathogenesis, contributions to symptom exacerbations, and recommended treatment options will emerge.
MP, DA, and TC conceived of the topic and wrote the manuscript, and MP prepared figures and figure legends.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Holly and Mark Kerslake from Newport Equities LLC for their generous financial support that made much of our work on the animal model for S. pyogenes-induced BGE possible.
MP, DA, and TC are supported by grants from the NIH (NHLBI R01 HL116995-01, NIMH R56 MH109987-01A1, and NCATS CTSA CaMPR-BASIC at CUMC) and the International OCD Foundation. DA is partially supported by an unrestricted gift from John. F. Castle to the Stroke Division of the Department of Neurology at Columbia University Medical Center.
1. van Coevorden-Hameete MH, de Graaff E, Titulaer MJ, Hoogenraad CC, Sillevis Smitt PA. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun Rev (2014) 13(3):299–312. doi: 10.1016/j.autrev.2013.10.016
2. Leypoldt F, Armangue T, Dalmau J. Autoimmune encephalopathies. Ann N Y Acad Sci (2015) 1338:94–114. doi:10.1111/nyas.12553
3. Sinmaz N, Amatoury M, Merheb V, Ramanathan S, Dale RC, Brilot F. Autoantibodies in movement and psychiatric disorders: updated concepts in detection methods, pathogenicity, and CNS entry. Ann N Y Acad Sci (2015) 1351(1):22–38. doi:10.1111/nyas.12764
4. Brimberg L, Mader S, Fujieda Y, Arinuma Y, Kowal C, Volpe BT, et al. Antibodies as mediators of brain pathology. Trends Immunol (2015) 36(11):709–24. doi:10.1016/j.it.2015.09.008
5. Dubey D, Sawhney A, Greenberg B, Lowden A, Warnack W, Khemani P, et al. The spectrum of autoimmune encephalopathies. J Neuroimmunol (2015) 287:93–7. doi:10.1016/j.jneuroim.2015.08.014
6. Swedo SE. Sydenham’s chorea. A model for childhood autoimmune neuropsychiatric disorders. JAMA (1994) 272(22):1788–91. doi:10.1001/jama.272.22.1788
7. Church AJ, Cardoso F, Dale RC, Lees AJ, Thompson EJ, Giovannoni G. Anti-basal ganglia antibodies in acute and persistent Sydenham’s chorea. Neurology (2002) 59(2):227–31. doi:10.1212/WNL.59.2.227
8. Cunningham MW. Streptococcus and rheumatic fever. Curr Opin Rheumatol (2012) 24(4):408–16. doi:10.1097/BOR.0b013e32835461d3
9. Venkatesan A, Benavides DR. Autoimmune encephalitis and its relation to infection. Curr Neurol Neurosci Rep (2015) 15(3):3. doi:10.1007/s11910-015-0529-1
10. Balint B, Bhatia KP. Stiff person syndrome and other immune-mediated movement disorders – new insights. Curr Opin Neurol (2016) 29(4):496–506. doi:10.1097/WCO.0000000000000351
11. Kothur K, Wienholt L, Brilot F, Dale RC. CSF cytokines/chemokines as biomarkers in neuroinflammatory CNS disorders: a systematic review. Cytokine (2016) 77:227–37. doi:10.1016/j.cyto.2015.10.001
12. Selmi C, Barin JG, Rose NR. Current trends in autoimmunity and the nervous system. J Autoimmun (2016) 75:20–9. doi:10.1016/j.jaut.2016.08.005
13. Vezzani A, Fujinami RS, White HS, Preux PM, Blumcke I, Sander JW, et al. Infections, inflammation and epilepsy. Acta Neuropathol (2016) 131(2):211–34. doi:10.1007/s00401-015-1481-5
14. Becher B, Spath S, Goverman J. Cytokine networks in neuroinflammation. Nat Rev Immunol (2017) 17(1):49–59. doi:10.1038/nri.2016.123
15. Prinz M, Priller J. The role of peripheral immune cells in the CNS in steady state and disease. Nat Neurosci (2017) 20(2):136–44. doi:10.1038/nn.4475
16. Dale RC, Brilot F. Autoimmune basal ganglia disorders. J Child Neurol (2012) 27(11):1470–81. doi:10.1177/0883073812451327
17. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol (2012) 12(9):623–35. doi:10.1038/nri3265
18. Jessen NA, Munk AS, Lundgaard I, Nedergaard M. The glymphatic system: a beginner’s guide. Neurochem Res (2015) 40(12):2583–99. doi:10.1007/s11064-015-1581-6
19. Louveau A, Harris TH, Kipnis J. Revisiting the mechanisms of CNS immune privilege. Trends Immunol (2015) 36(10):569–77. doi:10.1016/j.it.2015.08.006
20. Cunningham MW. Rheumatic fever, autoimmunity, and molecular mimicry: the streptococcal connection. Int Rev Immunol (2014) 33(4):314–29. doi:10.3109/08830185.2014.917411
21. Swedo SE. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS). Mol Psychiatry (2002) 7(Suppl 2):S24–5. doi:10.1038/sj.mp.4001170
22. Williams KA, Swedo SE. Post-infectious autoimmune disorders: Sydenham’s chorea, PANDAS and beyond. Brain Res (2015) 1617:144–54. doi:10.1016/j.brainres.2014.09.071
23. Murphy TK, Snider LA, Mutch PJ, Harden E, Zaytoun A, Edge PJ, et al. Relationship of movements and behaviors to Group A Streptococcus infections in elementary school children. Biol Psychiatry (2007) 61(3):279–84. doi:10.1016/j.biopsych.2006.08.031
24. Kirvan CA, Swedo SE, Heuser JS, Cunningham MW. Mimicry and autoantibody-mediated neuronal cell signaling in Sydenham chorea. Nat Med (2003) 9(7):914–20. doi:10.1038/nm892
25. Kirvan CA, Swedo SE, Snider LA, Cunningham MW. Antibody-mediated neuronal cell signaling in behavior and movement disorders. J Neuroimmunol (2006) 179(1–2):173–9. doi:10.1016/j.jneuroim.2006.06.017
26. Murphy TK, Kurlan R, Leckman J. The immunobiology of Tourette’s disorder, pediatric autoimmune neuropsychiatric disorders associated with Streptococcus, and related disorders: a way forward. J Child Adolesc Psychopharmacol (2010) 20(4):317–31. doi:10.1089/cap.2010.0043
27. Ben-Pazi H, Stoner JA, Cunningham MW. Dopamine receptor autoantibodies correlate with symptoms in Sydenham’s chorea. PLoS One (2013) 8(9):e73516. doi:10.1371/journal.pone.0073516
28. Cox CJ, Zuccolo AJ, Edwards EV, Mascaro-Blanco A, Alvarez K, Stoner J, et al. Antineuronal antibodies in a heterogeneous group of youth and young adults with tics and obsessive-compulsive disorder. J Child Adolesc Psychopharmacol (2015) 25(1):76–85. doi:10.1089/cap.2014.0048
29. Kovacevic M, Grant P, Swedo SE. Use of intravenous immunoglobulin in the treatment of twelve youths with pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. J Child Adolesc Psychopharmacol (2015) 25(1):65–9. doi:10.1089/cap.2014.0067
30. Latimer ME, L’Etoile N, Seidlitz J, Swedo SE. Therapeutic plasma apheresis as a treatment for 35 severely ill children and adolescents with pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. J Child Adolesc Psychopharmacol (2015) 25(1):70–5. doi:10.1089/cap.2014.0080
31. Diamond B, Honig G, Mader S, Brimberg L, Volpe BT. Brain-reactive antibodies and disease. Annu Rev Immunol (2013) 31:345–85. doi:10.1146/annurev-immunol-020711-075041
32. Sauteur PM, Hackenberg A, Tio-Gillen AP, van Rossum AM, Berger C, Jacobs BC. Intrathecal anti-GalC antibodies in bickerstaff brain stem encephalitis. Neuropediatrics (2015) 46(6):428–30. doi:10.1055/s-0035-1566730
33. Vergori A, Masi G, Donati D, Ginanneschi F, Annunziata P, Cerase A, et al. Listeria meningoencephalitis and anti-GQ1b antibody syndrome. Infection (2016) 44(4):543–6. doi:10.1007/s15010-015-0862-y
34. Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). N Engl J Med (1956) 255(2):57–65. doi:10.1056/NEJM195607122550201
35. Bickerstaff ER. Brain-stem encephalitis; further observations on a grave syndrome with benign prognosis. Br Med J (1957) 1(5032):1384–7. doi:10.1136/bmj.1.5032.1384
36. Al-Din AN, Anderson M, Bickerstaff ER, Harvey I. Brainstem encephalitis and the syndrome of Miller Fisher: a clinical study. Brain (1982) 105(Pt 3):481–95. doi:10.1093/brain/105.3.481
37. Chiba A, Kusunoki S, Shimizu T, Kanazawa I. Serum IgG antibody to ganglioside GQ1b is a possible marker of Miller Fisher syndrome. Ann Neurol (1992) 31(6):677–9. doi:10.1002/ana.410310619
38. Yuki N, Yamada M, Sato S, Ohama E, Kawase Y, Ikuta F, et al. Association of IgG anti-GD1a antibody with severe Guillain-Barre syndrome. Muscle Nerve (1993) 16(6):642–7. doi:10.1002/mus.880160610
39. Sano Y, Kanda T. Isolation and properties of endothelial cells forming the blood-nerve barrier. Methods Mol Biol (2011) 686:417–25. doi:10.1007/978-1-60761-938-3_21
40. Kanda T. Biology of the blood-nerve barrier and its alteration in immune mediated neuropathies. J Neurol Neurosurg Psychiatry (2013) 84(2):208–12. doi:10.1136/jnnp-2012-302312
41. Peltonen S, Alanne M, Peltonen J. Barriers of the peripheral nerve. Tissue Barriers (2013) 1(3):e24956. doi:10.4161/tisb.24956
42. Shimizu F, Omoto M, Sano Y, Mastui N, Miyashiro A, Tasaki A, et al. Sera from patients with multifocal motor neuropathy disrupt the blood-nerve barrier. J Neurol Neurosurg Psychiatry (2014) 85(5):526–37. doi:10.1136/jnnp-2013-305405
43. Arias I, Sorlozano A, Villegas E, de Dios Luna J, McKenney K, Cervilla J, et al. Infectious agents associated with schizophrenia: a meta-analysis. Schizophr Res (2012) 136(1–3):128–36. doi:10.1016/j.schres.2011.10.026
44. Hornig M. The role of microbes and autoimmunity in the pathogenesis of neuropsychiatric illness. Curr Opin Rheumatol (2013) 25(4):488–795. doi:10.1097/BOR.0b013e32836208de
45. Goldmann T, Prinz M. Role of microglia in CNS autoimmunity. Clin Dev Immunol (2013) 2013:208093. doi:10.1155/2013/208093
46. Graus F, Dalmau J. Paraneoplastic neurological syndromes: diagnosis and treatment. Curr Opin Neurol (2007) 20(6):732–7. doi:10.1097/WCO.0b013e3282f189dc
47. Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol (2011) 10(1):63–74. doi:10.1016/S1474-4422(10)70253-2
48. Bataller L, Valero C, Diaz R, Froufe A, Garcia-Zarza A, Ribalta T, et al. Cerebellar ataxia associated with neuroendocrine thymic carcinoma and GAD antibodies. J Neurol Neurosurg Psychiatry (2009) 80(6):696–7. doi:10.1136/jnnp.2008.161042
49. Arino H, Hoftberger R, Gresa-Arribas N, Martinez-Hernandez E, Armangue T, Kruer MC, et al. Paraneoplastic neurological syndromes and glutamic acid decarboxylase antibodies. JAMA Neurol (2015) 72(8):874–81. doi:10.1001/jamaneurol.2015.0749
50. Vincent A, Buckley C, Schott JM, Baker I, Dewar BK, Detert N, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain (2004) 127(Pt 3):701–12. doi:10.1093/brain/awh077
51. Pinatel D, Hivert B, Boucraut J, Saint-Martin M, Rogemond V, Zoupi L, et al. Inhibitory axons are targeted in hippocampal cell culture by anti-Caspr2 autoantibodies associated with limbic encephalitis. Front Cell Neurosci (2015) 9:265. doi:10.3389/fncel.2015.00265
52. Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology (2008) 70(20):1883–90. doi:10.1212/01.wnl.0000312275.04260.a0
53. Irani SR, Pettingill P, Kleopa KA, Schiza N, Waters P, Mazia C, et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol (2012) 72(2):241–55. doi:10.1002/ana.23577
54. Vernino S, Lennon VA. Autoantibody profiles and neurological correlations of thymoma. Clin Cancer Res (2004) 10(21):7270–5. doi:10.1158/1078-0432.CCR-04-0735
55. Dileepan T, Smith ED, Knowland D, Hsu M, Platt M, Bittner-Eddy P, et al. Group A Streptococcus intranasal infection promotes CNS infiltration by streptococcal-specific Th17 cells. J Clin Invest (2016) 126(1):303–17. doi:10.1172/JCI80792
56. Brimberg L, Benhar I, Mascaro-Blanco A, Alvarez K, Lotan D, Winter C, et al. Behavioral, pharmacological, and immunological abnormalities after streptococcal exposure: a novel rat model of Sydenham chorea and related neuropsychiatric disorders. Neuropsychopharmacology (2012) 37(9):2076–87. doi:10.1038/npp.2012.56
57. Yaddanapudi K, Hornig M, Serge R, De Miranda J, Baghban A, Villar G, et al. Passive transfer of streptococcus-induced antibodies reproduces behavioral disturbances in a mouse model of pediatric autoimmune neuropsychiatric disorders associated with streptococcal infection. Mol Psychiatry (2010) 15(7):712–26. doi:10.1038/mp.2009.77
58. Lotan D, Benhar I, Alvarez K, Mascaro-Blanco A, Brimberg L, Frenkel D, et al. Behavioral and neural effects of intra-striatal infusion of anti-streptococcal antibodies in rats. Brain Behav Immun (2014) 38:249–62. doi:10.1016/j.bbi.2014.02.009
59. Macri S, Ceci C, Onori MP, Invernizzi RW, Bartolini E, Altabella L, et al. Mice repeatedly exposed to Group-A beta-Haemolytic Streptococcus show perseverative behaviors, impaired sensorimotor gating, and immune activation in rostral diencephalon. Sci Rep (2015) 5:13257. doi:10.1038/srep13257
60. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci (2010) 30(17):5866–75. doi:10.1523/JNEUROSCI.0167-10.2010
61. Wright S, Hashemi K, Stasiak L, Bartram J, Lang B, Vincent A, et al. Epileptogenic effects of NMDAR antibodies in a passive transfer mouse model. Brain (2015) 138(Pt 11):3159–67. doi:10.1093/brain/awv257
62. Hammer C, Stepniak B, Schneider A, Papiol S, Tantra M, Begemann M, et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol Psychiatry (2014) 19(10):1143–9. doi:10.1038/mp.2013.110
63. Planaguma J, Leypoldt F, Mannara F, Gutierrez-Cuesta J, Martin-Garcia E, Aguilar E, et al. Human N-methyl d-aspartate receptor antibodies alter memory and behaviour in mice. Brain (2015) 138(Pt 1):94–109. doi:10.1093/brain/awu310
64. Manto M-U, Laute M-A, Aguera M, Rogemond V, Pandolfo M, Honnorat J. Effects of anti-glutamic acid decarboxylase antibodies associated with neurological diseases.: GAD-Ab and cerebellar function. Ann Neurol (2007) 61(6):544–51. doi:10.1002/ana.21123
65. Geis C, Weishaupt A, Hallermann S, Grunewald B, Wessig C, Wultsch T, et al. Stiff person syndrome-associated autoantibodies to amphiphysin mediate reduced GABAergic inhibition. Brain (2010) 133(11):3166–80. doi:10.1093/brain/awq253
66. Hackert JK, Muller L, Rohde M, Bien CG, Kohling R, Kirschstein T. Anti-GAD65 containing cerebrospinal fluid does not alter GABAergic transmission. Front Cell Neurosci (2016) 10:130. doi:10.3389/fncel.2016.00130
67. Stemmler N, Rohleder K, Malter MP, Widman G, Elger CE, Beck H, et al. Serum from a patient with GAD65 antibody-associated limbic encephalitis did not alter GABAergic neurotransmission in cultured hippocampal networks. Front Neurol (2015) 6:189. doi:10.3389/fneur.2015.00189
68. Wang B, Dileepan T, Briscoe S, Hyland KA, Kang J, Khoruts A, et al. Induction of TGF-beta1 and TGF-beta1-dependent predominant Th17 differentiation by group A streptococcal infection. Proc Natl Acad Sci U S A (2010) 107(13):5937–42. doi:10.1073/pnas.0904831107
69. Dileepan T, Linehan JL, Moon JJ, Pepper M, Jenkins MK, Cleary PP. Robust antigen specific th17 T cell response to group A Streptococcus is dependent on IL-6 and intranasal route of infection. PLoS Pathog (2011) 7(9):e1002252. doi:10.1371/journal.ppat.1002252
70. Hoffman KL, Hornig M, Yaddanapudi K, Jabado O, Lipkin WI. A murine model for neuropsychiatric disorders associated with group A beta-hemolytic streptococcal infection. J Neurosci (2004) 24(7):1780–91. doi:10.1523/JNEUROSCI.0887-03.2004
71. Cutforth T, DeMille MM, Agalliu I, Agalliu D. CNS autoimmune disease after Streptococcus pyogenes infections: animal models, cellular mechanisms and genetic factors. Future Neurol (2016) 11(1):63–76. doi:10.2217/fnl.16.4
72. Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol (2016) 15(4):391–404. doi:10.1016/S1474-4422(15)00401-9
73. Wurdemann T, Kersten M, Tokay T, Guli X, Kober M, Rohde M, et al. Stereotactic injection of cerebrospinal fluid from anti-NMDA receptor encephalitis into rat dentate gyrus impairs NMDA receptor function. Brain Res (2016) 1633:10–8. doi:10.1016/j.brainres.2015.12.027
74. Guasp M, Sola-Valls N, Martinez-Hernandez E, Gil MP, Gonzalez C, Brieva L, et al. Cerebellar ataxia and autoantibodies restricted to glutamic acid decarboxylase 67 (GAD67). J Neuroimmunol (2016) 300:15–7. doi:10.1016/j.jneuroim.2016.09.019
75. Haselmann H, Ropke L, Werner C, Kunze A, Geis C. Interactions of human autoantibodies with hippocampal GABAergic synaptic transmission – analyzing antibody-induced effects ex vivo. Front Neurol (2015) 6:136. doi:10.3389/fneur.2015.00136
76. Rodella U, Scorzeto M, Duregotti E, Negro S, Dickinson BC, Chang CJ, et al. An animal model of Miller Fisher syndrome: mitochondrial hydrogen peroxide is produced by the autoimmune attack of nerve terminals and activates Schwann cells. Neurobiol Dis (2016) 96:95–104. doi:10.1016/j.nbd.2016.09.005
77. Maier H, Schmidbauer M, Pfausler B, Schmutzhard E, Budka H. Central nervous system pathology in patients with the Guillain-Barre syndrome. Brain (1997) 120(Pt 3):451–64. doi:10.1093/brain/120.3.451
78. Okumura A, Ushida H, Maruyama K, Itomi K, Ishiguro Y, Takahashi M, et al. Guillain-Barre syndrome associated with central nervous system lesions. Arch Dis Child (2002) 86(4):304–6. doi:10.1136/adc.86.4.304
79. Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol (2010) 9(8):776–85. doi:10.1016/S1474-4422(10)70137-X
80. Kleopa KA, Elman LB, Lang B, Vincent A, Scherer SS. Neuromyotonia and limbic encephalitis sera target mature shaker-type K+ channels: subunit specificity correlates with clinical manifestations. Brain (2006) 129(Pt 6):1570–84. doi:10.1093/brain/awl084
81. Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology (2011) 77(2):179–89. doi:10.1212/WNL.0b013e318224afde
82. Shillito P, Molenaar PC, Vincent A, Leys K, Zheng W, van den Berg RJ, et al. Acquired neuromyotonia: evidence for autoantibodies directed against K+ channels of peripheral nerves. Ann Neurol (1995) 38(5):714–22. doi:10.1002/ana.410380505
83. Kaur C, Rathnasamy G, Ling EA. The choroid plexus in healthy and diseased brain. J Neuropathol Exp Neurol (2016) 75(3):198–213. doi:10.1093/jnen/nlv030
84. Liddelow SA, Temple S, Mollgard K, Gehwolf R, Wagner A, Bauer H, et al. Molecular characterisation of transport mechanisms at the developing mouse blood-CSF interface: a transcriptome approach. PLoS One (2012) 7(3):e33554. doi:10.1371/journal.pone.0033554
85. Liddelow SA. Development of the choroid plexus and blood-CSF barrier. Front Neurosci (2015) 9:32. doi:10.3389/fnins.2015.00032
86. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, et al. C-C chemokine receptor 6-regulated entry of Th-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol (2009) 10(5):514–8. doi:10.1038/ni.1716
87. Rudick RA, Eskin TA. Neuropathological features of a lupus-like disorder in autoimmune mice. Ann Neurol (1983) 14(3):325–32. doi:10.1002/ana.410140311
88. Zameer A, Hoffman SA. Increased ICAM-1 and VCAM-1 expression in the brains of autoimmune mice. J Neuroimmunol (2003) 142(1–2):67–74. doi:10.1016/S0165-5728(03)00262-5
89. Chan OT, Madaio MP, Shlomchik MJ. The central and multiple roles of B cells in lupus pathogenesis. Immunol Rev (1999) 169:107–21. doi:10.1111/j.1600-065X.1999.tb01310.x
90. Wen J, Stock AD, Chalmers SA, Putterman C. The role of B cells and autoantibodies in neuropsychiatric lupus. Autoimmun Rev (2016) 15(9):890–5. doi:10.1016/j.autrev.2016.07.009
91. Wolak DJ, Thorne RG. Diffusion of macromolecules in the brain: implications for drug delivery. Mol Pharm (2013) 10(5):1492–504. doi:10.1021/mp300495e
92. Zhao H, Aoshi T, Kawai S, Mori Y, Konishi A, Ozkan M, et al. Olfactory plays a key role in spatiotemporal pathogenesis of cerebral malaria. Cell Host Microbe (2014) 15(5):551–63. doi:10.1016/j.chom.2014.04.008
93. Meredith ME, Salameh TS, Banks WA. Intranasal delivery of proteins and peptides in the treatment of neurodegenerative diseases. AAPS J (2015) 17(4):780–7. doi:10.1208/s12248-015-9719-7
94. van Riel D, Verdijk R, Kuiken T. The olfactory nerve: a shortcut for influenza and other viral diseases into the central nervous system. J Pathol (2015) 235(2):277–87. doi:10.1002/path.4461
95. Schafer A, Brooke CB, Whitmore AC, Johnston RE. The role of the blood-brain barrier during Venezuelan equine encephalitis virus infection. J Virol (2011) 85(20):10682–90. doi:10.1128/JVI.05032-11
96. Munster VJ, Prescott JB, Bushmaker T, Long D, Rosenke R, Thomas T, et al. Rapid Nipah virus entry into the central nervous system of hamsters via the olfactory route. Sci Rep (2012) 2:736. doi:10.1038/srep00736
97. Herbert RP, Harris J, Chong KP, Chapman J, West AK, Chuah MI. Cytokines and olfactory bulb microglia in response to bacterial challenge in the compromised primary olfactory pathway. J Neuroinflammation (2012) 9:109. doi:10.1186/1742-2094-9-109
98. Lochhead JJ, Wolak DJ, Pizzo ME, Thorne RG. Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J Cereb Blood Flow Metab (2015) 35(3):371–81. doi:10.1038/jcbfm.2014.215
99. Kumar NN, Gautam M, Lochhead JJ, Wolak DJ, Ithapu V, Singh V, et al. Relative vascular permeability and vascularity across different regions of the rat nasal mucosa: implications for nasal physiology and drug delivery. Sci Rep (2016) 6:31732. doi:10.1038/srep31732
100. Kaminski M, Bechmann I, Kiwit J, Glumm J. Migration of monocytes after intracerebral injection. Cell Adh Migr (2012) 6(3):164–7. doi:10.4161/cam.20281
101. Mohammad MG, Tsai VW, Ruitenberg MJ, Hassanpour M, Li H, Hart PH, et al. Immune cell trafficking from the brain maintains CNS immune tolerance. J Clin Invest (2014) 124(3):1228–41. doi:10.1172/JCI71544
102. Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell (2015) 163(5):1064–78. doi:10.1016/j.cell.2015.10.067
103. Knowland D, Arac A, Sekiguchi KJ, Hsu M, Lutz SE, Perrino J, et al. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron (2014) 82(3):603–17. doi:10.1016/j.neuron.2014.03.003
104. Vajkoczy P, Laschinger M, Engelhardt B. Alpha4-integrin-VCAM-1 binding mediates G protein-independent capture of encephalitogenic T cell blasts to CNS white matter microvessels. J Clin Invest (2001) 108(4):557–65. doi:10.1172/jci12440
105. Lengfeld JE, Lutz SE, Smith JR, Diaconu C, Scott C, Kofman SB, et al. Endothelial Wnt/beta-catenin signaling reduces immune cell infiltration in multiple sclerosis. Proc Natl Acad Sci U S A (2017) 114(7):E1168–77. doi:10.1073/pnas.1609905114
106. Bartanusz V, Jezova D, Alajajian B, Digicaylioglu M. The blood-spinal cord barrier: morphology and clinical implications. Ann Neurol (2011) 70(2):194–206. doi:10.1002/ana.22421
107. Cruz-Orengo L, Daniels BP, Dorsey D, Basak SA, Grajales-Reyes JG, McCandless EE, et al. Enhanced sphingosine-1-phosphate receptor 2 expression underlies female CNS autoimmunity susceptibility. J Clin Invest (2014) 124(6):2571–84. doi:10.1172/jci73408
108. Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B, et al. A specialized vascular niche for adult neural stem cells. Cell Stem Cell (2008) 3(3):279–88. doi:10.1016/j.stem.2008.07.025
109. Winkler CW, Race B, Phillips K, Peterson KE. Capillaries in the olfactory bulb but not the cortex are highly susceptible to virus-induced vascular leak and promote viral neuroinvasion. Acta Neuropathol (2015) 130(2):233–45. doi:10.1007/s00401-015-1433-0
110. Colin-Castelan D, Ramirez-Santos J, Gutierrez-Ospina G. Differential vascular permeability along the forebrain ventricular neurogenic niche in the adult murine brain. J Neurosci Res (2016) 94(2):161–9. doi:10.1002/jnr.23682
111. Sajja RK, Rahman S, Cucullo L. Drugs of abuse and blood-brain barrier endothelial dysfunction: a focus on the role of oxidative stress. J Cereb Blood Flow Metab (2016) 36(3):539–54. doi:10.1177/0271678X15616978
112. Argaw AT, Zhang Y, Snyder BJ, Zhao M-L, Kopp N, Lee SC, et al. IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol (2006) 177(8):5574–84. doi:10.4049/jimmunol.177.8.5574
113. Lakhan SE, Kirchgessner A, Tepper D, Leonard A. Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front Neurol (2013) 4:32. doi:10.3389/fneur.2013.00032
114. Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR II, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol (2010) 189(1):111–26. doi:10.1083/jcb.200902153
115. Alvarez JI, Cayrol R, Prat A. Disruption of central nervous system barriers in multiple sclerosis. Biochim Biophys Acta (2011) 1812(2):252–64. doi:10.1016/j.bbadis.2010.06.017
116. Stamatovic SM, Keep RF, Wang MM, Jankovic I, Andjelkovic AV. Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J Biol Chem (2009) 284(28):19053–66. doi:10.1074/jbc.M109.000521
117. Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med (2007) 13(10):1173–5. doi:10.1038/nm1651
118. Alter A, Duddy M, Hebert S, Biernacki K, Prat A, Antel JP, et al. Determinants of human B cell migration across brain endothelial cells. J Immunol (2003) 170(9):4497–505. doi:10.4049/jimmunol.170.9.4497
119. Lehmann-Horn K, Sagan SA, Bernard CC, Sobel RA, Zamvil SS. B-cell very late antigen-4 deficiency reduces leukocyte recruitment and susceptibility to central nervous system autoimmunity. Ann Neurol (2015) 77(5):902–8. doi:10.1002/ana.24387
120. Michel L, Touil H, Pikor NB, Gommerman JL, Prat A, Bar-Or A. B cells in the multiple sclerosis central nervous system: trafficking and contribution to CNS-compartmentalized inflammation. Front Immunol (2015) 6:636. doi:10.3389/fimmu.2015.00636
121. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol (2004) 14(2):164–74. doi:10.1111/j.1750-3639.2004.tb00049.x
122. Humby F, Bombardieri M, Manzo A, Kelly S, Blades MC, Kirkham B, et al. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med (2009) 6(1):e1. doi:10.1371/journal.pmed.0060001
123. Claes N, Fraussen J, Stinissen P, Hupperts R, Somers V. B cells are multifunctional players in multiple sclerosis pathogenesis: insights from therapeutic interventions. Front Immunol (2015) 6:642. doi:10.3389/fimmu.2015.00642
124. D’Agostino PM, Kwak C, Vecchiarelli HA, Toth JG, Miller JM, Masheeb Z, et al. Viral-induced encephalitis initiates distinct and functional CD103+ CD11b+ brain dendritic cell populations within the olfactory bulb. Proc Natl Acad Sci U S A (2012) 109(16):6175–80. doi:10.1073/pnas.1203941109
125. Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol (2011) 10(8):759–72.
126. Baburamani AA, Ek CJ, Walker DW, Castillo-Melendez M. Vulnerability of the developing brain to hypoxic-ischemic damage: contribution of the cerebral vasculature to injury and repair? Front Physiol (2012) 3:424. doi:10.3389/fphys.2012.00424
127. Hara K, Yoshizuka M, Doi Y, Fujimoto S. Effect of bis (tributyl tin) oxide on permeability of the blood-brain barrier: a transient increase. Occup Environ Med (1994) 51(11):735–8. doi:10.1136/oem.51.11.735
128. Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Toth M, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med (2014) 6(263):263ra158. doi:10.1126/scitranslmed.3009759
129. Yoshio T, Okamoto H, Hirohata S, Minota S. IgG anti-NR2 glutamate receptor autoantibodies from patients with systemic lupus erythematosus activate endothelial cells. Arthritis Rheum (2013) 65(2):457–63. doi:10.1002/art.37745
130. Odaka M, Yuki N, Yamada M, Koga M, Takemi T, Hirata K, et al. Bickerstaff’s brainstem encephalitis: clinical features of 62 cases and a subgroup associated with Guillain-Barre syndrome. Brain (2003) 126(Pt 10):2279–90. doi:10.1093/brain/awg233
131. Snider LA, Lougee L, Slattery M, Grant P, Swedo SE. Antibiotic prophylaxis with azithromycin or penicillin for childhood-onset neuropsychiatric disorders. Biol Psychiatry (2005) 57(7):788–92. doi:10.1016/j.biopsych.2004.12.035
132. Williams KA, Swedo SE, Farmer CA, Grantz H, Grant PJ, D’Souza P, et al. Randomized, controlled trial of intravenous immunoglobulin for pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections. J Am Acad Child Adolesc Psychiatry (2016) 55(10):860e–7e. doi:10.1016/j.jaac.2016.06.017
133. Dalmau J. NMDA receptor encephalitis and other antibody-mediated disorders of the synapse: the 2016 Cotzias lecture. Neurology (2016) 87(23):2471–82. doi:10.1212/WNL.0000000000003414
134. Nathoo N, Jalal H, Natah SS, Zhang Q, Wu Y, Dunn JF. Hypoxia and inflammation-induced disruptions of the blood-brain and blood-cerebrospinal fluid barriers assessed using a novel T1-based MRI method. Acta Neurochir Suppl (2016) 121:23–8. doi:10.1007/978-3-319-18497-5_5
135. Mulkey SB, Glasier CM, El-Nabbout B, Walters WD, Ionita C, McCarthy MH, et al. Nerve root enhancement on spinal MRI in pediatric Guillain-Barre syndrome. Pediatr Neurol (2010) 43(4):263–9. doi:10.1016/j.pediatrneurol.2010.05.011
136. Yikilmaz A, Doganay S, Gumus H, Per H, Kumandas S, Coskun A. Magnetic resonance imaging of childhood Guillain-Barre syndrome. Childs Nerv Syst (2010) 26(8):1103–8. doi:10.1007/s00381-010-1197-8
137. Ashikari Y, Kobayashi S, Tago A, Yoneyama M, Ito M, Fukuda K, et al. A case of Guillain-Barre syndrome with meningeal irritation. Brain Dev (2016) 38(1):163–6. doi:10.1016/j.braindev.2015.06.001
138. Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature (1996) 380(6573):435–9. doi:10.1038/380435a0
139. Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol (2008) 183(3):409–17. doi:10.1083/jcb.200806024
140. Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science (2008) 322(5905):1247–50. doi:10.1126/science.1164594
141. Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A (2009) 106(2):641–6. doi:10.1073/pnas.0805165106
142. Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med (2013) 19(12):1584–96. doi:10.1038/nm.3407
143. Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, et al. The hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science (2011) 334(6063):1727–31. doi:10.1126/science.1206936
144. Beurel E, Kaidanovich-Beilin O, Yeh WI, Song L, Palomo V, Michalek SM, et al. Regulation of Th1 cells and experimental autoimmune encephalomyelitis by glycogen synthase kinase-3. J Immunol (2013) 190(10):5000–11. doi:10.4049/jimmunol.1203057
Keywords: blood–brain barrier, autoimmune encephalitis, basal ganglia encephalitis, NMDA receptor, dopamine receptor, autoantibodies, pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections, Sydenham’s chorea
Citation: Platt MP, Agalliu D and Cutforth T (2017) Hello from the Other Side: How Autoantibodies Circumvent the Blood–Brain Barrier in Autoimmune Encephalitis. Front. Immunol. 8:442. doi: 10.3389/fimmu.2017.00442
Received: 07 February 2017; Accepted: 30 March 2017;
Published: 21 April 2017
Edited by:
Fabienne Brilot, University of Sydney, AustraliaReviewed by:
Anna Fogdell-Hahn, Karolinska Institutet, SwedenCopyright: © 2017 Platt, Agalliu and Cutforth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tyler Cutforth, dGMyNzU2QGN1bWMuY29sdW1iaWEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.