 Nicola L. Diny
Nicola L. Diny Noel R. Rose2
Noel R. Rose2- 1W. Harry Feinstone Department of Molecular Microbiology and Immunology, Johns Hopkins University Bloomberg School of Public Health, Baltimore, MD, USA
- 2Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA
- 3Department of Pathology, Johns Hopkins University School of Medicine, Baltimore, MD, USA
Eosinophils are multifunctional granulocytes that contribute to initiation and modulation of inflammation. Their role in asthma and parasitic infections has long been recognized. Growing evidence now reveals a role for eosinophils in autoimmune diseases. In this review, we summarize the function of eosinophils in inflammatory bowel diseases, neuromyelitis optica, bullous pemphigoid, autoimmune myocarditis, primary biliary cirrhosis, eosinophilic granulomatosis with polyangiitis, and other autoimmune diseases. Clinical studies, eosinophil-targeted therapies, and experimental models have contributed to our understanding of the regulation and function of eosinophils in these diseases. By examining the role of eosinophils in autoimmune diseases of different organs, we can identify common pathogenic mechanisms. These include degranulation of cytotoxic granule proteins, induction of antibody-dependent cell-mediated cytotoxicity, release of proteases degrading extracellular matrix, immune modulation through cytokines, antigen presentation, and prothrombotic functions. The association of eosinophilic diseases with autoimmune diseases is also examined, showing a possible increase in autoimmune diseases in patients with eosinophilic esophagitis, hypereosinophilic syndrome, and non-allergic asthma. Finally, we summarize key future research needs.
Introduction
The cells of the innate immune system can contribute to autoimmune diseases. Activation of innate immune cells by pathogen-associated molecular patterns and antigen presentation by dendritic cells can result in priming of autoreactive T and B cells and set off an adaptive immune response against self-antigens (1–3). Possible roles for innate immune cells exist not only in the initiation stage of autoimmune diseases but also in the modulation and propagation of inflammation and tissue destruction. Such roles have been proposed for neutrophils (4), natural killer cells (5, 6), macrophages (7), dendritic cells (8, 9), innate lymphoid cells (10), and mast cells (11). Eosinophils have been recognized as a part of the inflammatory infiltrate in several organ-specific autoimmune diseases, but their potential role in autoimmune diseases has not been addressed comprehensively. The aim of this review is to synthesize the role of eosinophils in different autoimmune diseases and explore potential unifying effector mechanisms. We also address the association of autoimmune diseases with eosinophil-associated disease like asthma and eosinophilic esophagitis.
Eosinophils
Eosinophil Biology
Eosinophils are granulocytes that develop in the bone marrow in response to IL-5, with a minor role for IL-3, granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-33 (12–15). IL-5 also mediates the release of mature eosinophils into the bloodstream from where they migrate into tissues (16). In healthy individuals, eosinophils are found in the bone marrow, blood, spleen, thymus, gastrointestinal tract, and uterus (17). Under pathological conditions, eosinophils can infiltrate other tissues as well. Eosinophils are usually enumerated in the blood because tissue eosinophils are hard to measure. Eosinophil counts over 450–500 cells/μl blood are considered mild eosinophilia and counts over 1,500 cells/μl are characterized as hypereosinophilia (18).
The main chemotaxins for eosinophils are eotaxins, which homeostatically recruit eosinophils to the gastrointestinal tract, thymus, and uterus (19–21) and to other organs in disease states (22–24). Humans express three functional eotaxins (CCL11, CCL24, and CCL26), whereas mice only express two (CCL11 and CCL24) (25–28). The eotaxin receptor, CCR3, is highly expressed on eosinophils and to a low level on human basophils, mast cells, and Th2 cells (29, 30). Other eosinophil chemoattractants include CCL5 and lipid mediators such as leukotriene B4 and prostaglandin D2, although these factors are not specific for eosinophils (31) (Figure 1).
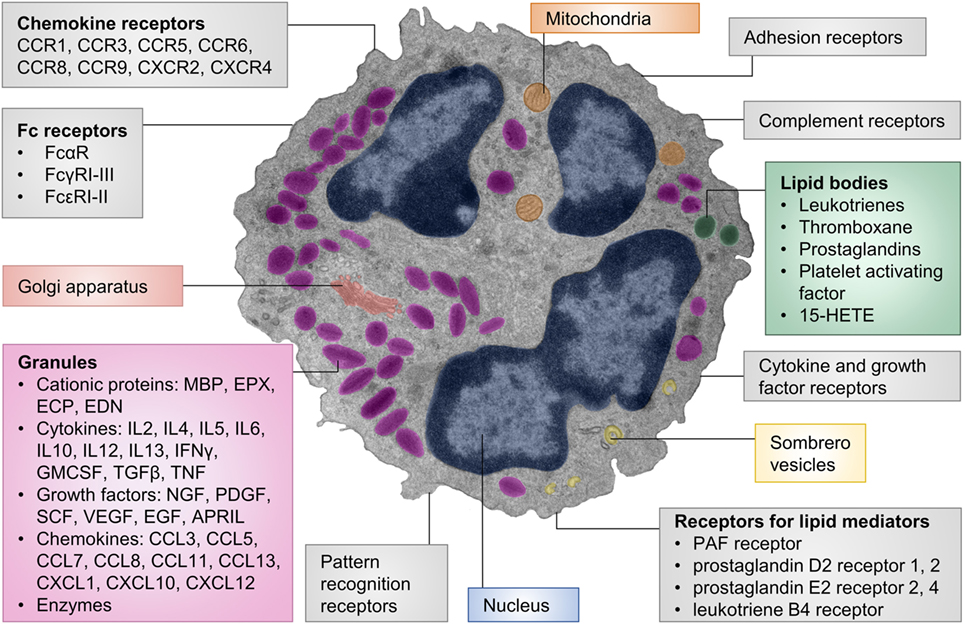
Figure 1. Cellular structure, receptors, and mediators of eosinophils. The pseudocolored composite electron micrograph of an eosinophil highlights cellular structures. Characteristic features of eosinophils include the multilobed nucleus, specific eosinophil granules, lipid bodies, and sombrero vesicles. Eosinophil granules contain cationic proteins, cytokines, growth factors, chemokines, and enzymes. The granule contents can be released upon stimulation. Lipid bodies are the place of synthesis for numerous lipid mediators. Granule contents can be released through sombrero vesicles. Eosinophils carry numerous cell surface receptors including chemokine receptors, Fc receptors, pattern recognition receptors, receptors for lipid mediators, cytokine receptors, complement receptors, and adhesion receptors. Abbreviations: 15-HETE, 15-hydroxyeicosatetraenoic acid; APRIL, a proliferation-inducing ligand; CCL, CC-chemokine ligand; CCR, CC-chemokine receptor; CXCL, CXC-chemokine ligand; CXCR, CXC-chemokine receptor; ECP, eosinophil cationic protein; EDN, eosinophil-derived neurotoxin; EGF, epidermal growth factor; EPX, eosinophil peroxidase; GMCSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; MBP, major basic protein; NGF, nerve growth factor; PDGF, platelet-derived growth factor; PAF, platelet-activating factor; SCF, stem cell factor; TGF, transforming growth factor; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor. The electron micrograph was generously provided by Dr. Isabelle Coppens, Johns Hopkins University, Baltimore, MD, USA.
A unique characteristic of eosinophils are their specific (also termed secondary or secretory) granules. These are secretory vesicles with an electron-dense core and an electron-lucent matrix. Eosinophil granules contain four major granule proteins and numerous cytokines, chemokines, and growth factors (31) (Figure 1). Cytotoxic effects to host tissues and pathogens have been demonstrated for all major granule proteins: eosinophil cationic protein (ECP), eosinophil-derived neurotoxin (EDN), eosinophil peroxidase (EPX), and major basic protein (MBP) (32). MBP can disrupt the cell membrane and is therefore highly cytotoxic to mammalian cells, helminths, and bacteria (33–35). Other effects of MBP include altering smooth muscle contraction, inducing mast cell and basophil degranulation, provoking acetylcholine release from peripheral nerves, and promoting nerve cell survival (36–39). The granule proteins ECP and EDN are ribonucleases (13, 40) with neurotoxic and strong antiviral activities (41, 42) and immune modulatory functions (43). EPX generates reactive oxygen species that are directed extracellularly (44). These products have cytotoxic, prothrombotic, and pro-inflammatory effects (44–46). Granule contents are generally preformed in eosinophils and released upon stimulation. Piecemeal degranulation is the most common process by which eosinophils release their granule contents (47–49). Specific granule factors, rather than the entire granule, are released in response to an activating signal. This leaves the eosinophil intact and able to respond to subsequent stimulation.
Possible Eosinophil Effector Functions in Autoimmune Diseases
Eosinophils are extremely versatile effector cells that damage tissues or modulate the activity of other immune and stromal cells. One could envision many of these effector functions playing a role in the context of autoimmune diseases as well (Box 1). Damage of tissues and cells is a feature of many organ-specific autoimmune diseases. Eosinophils are well known for their strong cytotoxic properties, mediated mostly through granule proteins. This could contribute to organ destruction in autoimmune inflammation.
Box 1. Possible eosinophil effector functions in autoimmune diseases.
• Damage of tissues by cytotoxic granule proteins
• Antibody-dependent cellular cytotoxicity
• Activation of tissue remodeling and fibrosis
• Antigen presentation
• Modulation of the adaptive immune response
• Promotion of B cell responses
• Induction of tissue repair processes.
The ability of eosinophils to bind antibodies and consequently degranulate and kill cells links the adaptive autoimmune response to eosinophil effector functions. Eosinophils express complement receptors (50) and Fc receptors (FcαR, FcγRI–III, and FcεRI–II) either constitutively or under inflammatory conditions (51–54). As a result, they are capable of antibody-dependent cellular cytotoxicity (ADCC) to parasites and mammalian targets (55–57). In autoimmune diseases, eosinophils may kill host cells bound by autoantibodies.
Eosinophils also interact with stromal cells. Actively degranulating eosinophils are frequently found in areas of fibrogenesis, suggesting a potential profibrotic role (58–61). Granule proteins and eosinophil-derived transforming growth factor (TGF)β1 were demonstrated to affect tissue remodeling and fibrosis. Eosinophils can promote fibroblast proliferation (62–64), proteoglycan accumulation (65), matrix metalloproteinase and TGFβ expression, and extracellular matrix protein synthesis (66). These profibrotic functions of eosinophils may add to tissue dysfunction in autoimmune diseases. In chronic inflammatory conditions, eosinophils preferentially locate to nerves (67–70). This interaction results in activation of eosinophils (71–73), nerve damage (74, 75), altered nerve growth (76, 77), and neuropeptide release (78). Contact between eosinophils and nerves has functional consequences. For example, it is one of the causes of airway hypersensitivity in asthma (38, 79).
Eosinophils can form extracellular DNA traps by quickly releasing mitochondrial DNA and granule proteins (80). These structures bind and kill pathogens and contribute to tissue injury in inflammatory conditions (81). DNA extracellular traps have been described in allergic asthma (82), drug hypersensitivity reactions, and allergic contact dermatitis (83). Eosinophils may initiate or perpetuate inflammation by releasing cytokines and chemokines and by interacting with other innate immune cells. For example, eosinophils release MBP, IL-9, stem cell factor, or nerve growth factor, which affect mast cell maturation, survival, and histamine release (84–87).
Eosinophils can also influence the adaptive immune response. They are capable antigen-presenting cells that upregulate MHCII and costimulatory molecules in the context of parasitic infection or allergic asthma (88–90). Moreover, eosinophils migrate to draining lymph nodes (91), and in vitro experiments have demonstrated their ability to present antigen to and activate T cells (92–94). Eosinophils may contribute to the initiation of autoimmune responses by presenting antigen to and activating T cells.
Eosinophil granules contain numerous cytokines such as IL-4, IL-13, IL-25, TGFβ, IL-10, or IDO (31), which suggests an ability to affect T cell differentiation. Eosinophils were shown to suppress Th1/Th17 differentiation (95) or activate Th2 responses in draining lymph nodes (96). In addition, they modulate dendritic cell activity, thereby indirectly affecting polarization of naïve T cells into Th2 cells (97). Eosinophils also shape the humoral immune response. In the bone marrow, eosinophils stimulate plasma cell survival by producing IL-6 and a proliferation-inducing ligand (98), and in the intestine, they promote class-switching to IgA (99, 100). These properties enable eosinophils to shape the adaptive immune response in autoimmune diseases.
Eosinophils may also fulfill immune regulatory and protective functions. Eosinophil-derived mediators like TGFβ and TGFα (101), platelet-derived growth factor (102), vascular endothelial growth factor (103), and fibroblast growth factor (104) can all contribute to tissue repair and angiogenesis. IL-4 released from eosinophils was shown to play a role in liver (105) and muscle (106) regeneration. Whether eosinophils contribute to tissue repair or tissue damage is likely context and disease dependent.
Identification of Eosinophils in Blood and Tissues
Numerous methods exist to identify eosinophils in blood and tissues. Blood eosinophils are routinely counted in clinical settings in differential white blood counts. Human eosinophils (and to a lesser extent mouse eosinophils) are easily identified by hematoxylin and eosin staining of histological sections due to the bright pink staining of the basic granules, which gave them the name eosinophils (107). Mouse blood eosinophils can be detected using modified Giemsa stain or by flow cytometry. Their characteristic forward scatter-side scatter profile in flow cytometry allows for approximation of eosinophils in blood even without specific antibody staining. Antibodies that can be used to stain blood eosinophils target Siglec-F (mouse) or CCR3 (mouse and human) (108, 109). Eosinophils are also positive for unspecific receptors such as CD11b and (at low/intermediate levels) Ly6G. Staining with several antibodies is often required for identification and characterization of eosinophils from tissues, as none of them are absolutely specific for eosinophils (110). Tissue eosinophils can also be detected using immunohistochemistry or immunofluorescence staining with antibodies against MBP or EPX (111). Electron microscopy is another method used to distinguish eosinophils from other cells based on the morphology of eosinophil granules. Moreover, with this technique, the extent and mechanism of degranulation of eosinophils can be determined (112).
Role of Eosinophils in Autoimmune Diseases
Bullous Pemphigoid
Bullous pemphigoid is a blistering disease of the skin with a well-established autoimmune etiology (113). Autoantibodies bind to hemidesmosomal proteins BP180 and BP230 at the dermal–epidermal junction and other extracellular matrix proteins (114–119). Hemidesmosomes are part of the complexes that anchor the cytoskeleton of basal keratinocytes to the dermis (120). Autoantibody binding triggers complement activation, recruitment of immune cells, and release of proteases. This results in tissue damage and blistering (121–124). Neutrophils and eosinophils infiltrate the dermal–epidermal junction and, together with mast cells, are thought to play a key role in bullous pemphigoid (125–127). Increased numbers of peripheral blood eosinophils has long been recognized as a characteristic of bullous pemphigoid patients (128, 129). A positive correlation between blood eosinophil numbers and disease severity has been observed in some reports (130, 131) but not others (132). Eosinophilia in bullous pemphigoid patients is likely caused by increased levels of IL-5, which can be detected at high levels in the serum and blister fluid (133, 134). Keratinocytes in the blisters express eotaxin-1, which directs eosinophil infiltration (133, 135). Eotaxin-1 expression is positively correlated with the number of infiltrating eosinophils in blisters. Eosinophil localization to the basement membrane zone is autoantibody and complement dependent in a human cryosection model of bullous pemphigoid (127). Eosinophils from blisters release IL-6, IL-8, and IL-1β and show an activated phenotype with high CD11b expression (136). Blister eosinophils also underwent apoptosis more readily compared to eosinophils from healthy donors (136).
Several mechanisms by which eosinophils (and other granulocytes) contribute to lesion formation have been identified. Eosinophils in lesional skin were shown to degranulate, and granule proteins are deposited in blisters (125, 137). The eosinophil granule protein ECP can be readily detected in serum and blister fluid of bullous pemphigoid patients (132). However, it is not clear if granule proteins contribute to tissue damage. Eosinophils and neutrophils have been shown to release proteases, matrix metalloproteinase 9 (MMP9), and neutrophil elastase, in lesional biopsies and blister fluid. These proteases can degrade extracellular matrix proteins and BP180, which contributes to dermal–epidermal separation and blister formation (138–140). Blister formation also depends on autoantibodies (141), which are of the IgG1, IgG4, and IgE subtype (142, 143). Recently, eosinophils from bullous pemphigoid patients were shown to express the high-affinity IgE receptor FcεRIα (144), which may trigger eosinophil activation by IgE autoantibodies. Additional evidence for a pathogenic role for eosinophils comes from a case report of a patient with hypereosinophilic syndrome and bullous pemphigoid who was treated with imatinib (a tyrosine kinase inhibitor). In response to imatinib, both conditions resolved and his eosinophil count normalized (145).
Taken together, there is strong evidence from patient studies, in vitro experiments, and animal models for a pathogenic role of eosinophils in bullous pemphigoid. In addition to the mouse model of passive antibody transfer, which reproduces blister formation but not eosinophil infiltration (146), a new model with genetically modified mice has been established (147). Mice with a deletion in the BP180 (collagen XVII) gene spontaneously develop eosinophilia, blister formation, itch, and eosinophil infiltration into the skin lesions. This new model could be used to test for the requirement and pathologic role of eosinophils (and eosinophil products) in future studies. Novel eosinophil-specific drugs may also help to clarify the role of eosinophils in bullous pemphigoid. Trials of bertilimumab, an anti-eotaxin-1 antibody, and mepolizumab, an anti-IL-5 antibody, are currently ongoing (http://ClinicalTrials.gov identifiers: NCT02226146 and NCT01705795).
Inflammatory Bowel Diseases
The etiology of the inflammatory bowel diseases, Crohn’s disease and ulcerative colitis, is not fully understood. Evidence for the involvement of autoimmune processes exists for both (148, 149). Both diseases are associated with other autoimmune diseases, characterized by lymphocytic infiltration, and respond to corticosteroid treatment. Patients with ulcerative colitis carry autoantibodies against colonic epithelial cells and often perinuclear antineutrophilic cytoplasmic (ANCA) antibodies. Specific autoantibodies have not been found in Crohn’s disease patients. The evidence for autoimmunity is stronger in ulcerative colitis than in Crohn’s disease. Here, we will discuss the role of eosinophils in both diseases with a focus on ulcerative colitis.
Eosinophils have long been recognized as a prominent feature of the infiltrate in inflammatory bowel diseases (150–154). Eosinophil numbers in the colon are substantially increased in inflammatory bowel disease patients and display an activated phenotype (154–157). The number of infiltrating eosinophils is positively correlated with disease severity in ulcerative colitis and Crohn’s disease (23, 158–161). In mouse models, the absence of eosinophils dramatically reduces disease severity. In the model of DSS-induced colitis, two different strains of eosinophil-deficient mice were protected compared to controls (23, 162). Depletion of eosinophils in a model of colitis due to Helicobacter hepaticus infection also reduced disease severity (163). Similarly, in a model of TNBS-induced colitis, eosinophil-deficient mice fared better, while hypereosinophilic mice developed more severe disease (164).
Eosinophil migration into the colon mucosa occurs in response to eotaxins. Patients with inflammatory bowel diseases have elevated serum eotaxin-1 levels (158, 165, 166), which correlates positively with disease activity (158, 165). Tissue expression of eotaxin-1, and to a lesser extent eotaxin-2, is increased in ulcerative colitis patients and positively correlated with the number of infiltrating eosinophils and histopathologic disease severity (23, 158). Another study found increased expression of all three eotaxins and IL-5 in ulcerative colitis, but only eotaxin-1 correlated with eosinophil numbers (167). The relative significance of eotaxin-2 and -3 is less clear. Eotaxin-3 was found to be increased in active lesions in ulcerative colitis and to a lesser extent in Crohn’s disease (168). Gene polymorphisms in eotaxin-2 are associated with ulcerative colitis (169). This suggests that all eotaxins may contribute to eosinophil trafficking. The cellular source of eotaxin-1 was identified as CD68+ macrophages and epithelial cells (23) or as CD14+ mononuclear cells (167). Colonic myofibroblasts express eotaxin-3, which is increased in response to IL-4 and IL-13 (168). In the mouse model of DSS-induced colitis, expression of eotaxin-1 and -2 in the colon is increased and deficiency of eotaxin-1, but not eotaxin-2, decreases eosinophil infiltration. This demonstrates that eotaxin-1 is the major chemoattractant for eosinophils in experimental colitis (23, 170). In this mouse model, macrophages are the major eotaxin-1-producing cell type (23, 171). The increased expression of eotaxins, particularly eotaxin-1, in inflammatory bowel diseases shows that eosinophils are specifically recruited to the site of inflammation.
Electron microscopy and immunohistochemistry of colonic biopsies show degranulation of eosinophils (157, 172, 173). Eosinophil granule proteins are also found in the feces (156, 174), gut perfusates (175), and gut lavage fluids (176) of patients with ulcerative colitis and Crohn’s disease. Eosinophil granule proteins in serum or intestine are positively correlated with disease activity in ulcerative colitis (23, 156, 161, 177, 178). Polymorphisms in the genes of ECP and EPX are associated with inflammatory bowel diseases (179). These findings suggest a pathogenic role of eosinophil granule proteins in inflammatory bowel diseases. In one study, however, eosinophil activation was observed during the remission phase (180). EPX is pathogenic in mouse models of DSS- and H. hepaticus-induced colitis. Genetic deficiency or inhibition of EPX reduced disease severity (163, 170).
Several pathogenic functions of eosinophils have been suggested in recent years. Eosinophils were found to increase mucosal barrier permeability in ulcerative colitis by releasing MBP (181) or corticotropin-releasing factor (182). IL-22 is increased in patients with ulcerative colitis or Crohn’s disease (183, 184), and animal studies showed that it is crucial to restore epithelial homeostasis (185). IL-22 induces antimicrobial peptides, mucus production, and epithelial tight junctions (186). Recently, eosinophils were identified as the main source of IL-22-binding protein (IL-22BP), inhibiting the protective actions of IL-22 in DSS-induced experimental colitis and in patients with inflammatory bowel disease (187). In another study, eosinophils were found to localize to nerves in the colonic mucosa in ulcerative colitis and Crohn’s disease (70). Th17 responses have been implicated in inflammatory bowel diseases (188). A possible link between the downstream effector of Th17 responses, GM-CSF, and eosinophils was found recently. GM-CSF enhances eosinophilopoiesis, induces cytokine secretion from eosinophils, and promotes eosinophil survival (163, 189).
In summary, tissue eosinophils are increased in patients with inflammatory bowel diseases, are associated with disease severity, and are specifically recruited through eotaxin-1. Eosinophils likely contribute to the disease process by releasing granule proteins (EPX) or other mediators that affect the intestinal barrier. Thus, there is a strong evidence for a pathogenic role of eosinophils in inflammatory bowel diseases, particularly in ulcerative colitis.
Eosinophilic Granulomatosis with Polyangiitis (EGPA)
Eosinophilic granulomatosis with polyangiitis was first described by Churg and Strauss in 1951 (190). The disease progresses through three overlapping phases: adult-onset asthma, peripheral and tissue eosinophilia, and necrotizing vasculitis with tissue infiltration of eosinophils (191–193). EGPA is an idiopathic type of small vessel vasculitis and is also part of the hypereosinophilic syndromes (194). It is associated with HLA and IL-10 polymorphisms (195). About 40% of EGPA patients have perinuclear ANCA antibodies against myeloperoxidase (MPO), resulting in the classification of EGPA as an ANCA-associated vasculitis (192, 193). The presence or absence of ANCA in EGPA may indicate two clinical subtypes with different organ involvement. ANCA-positive patients have more frequent vasculitis and glomerulonephritis, whereas ANCA-negative patients have more frequent heart and lung involvement (196, 197).
Blood and tissue eosinophilia are diagnostic criteria for EGPA, yet little is known about the pathogenic role of eosinophils in this disease (192). One reason for the absence of mechanistic data is the lack of suitable animal models. The transfer of MPO-positive human serum to mice causes vasculitis, but the eosinophilic component is missing (198). Therefore, all knowledge about the role of eosinophils in EGPA comes from patient studies. An increased eosinophil count during active disease is associated with increased Th2 cytokines IL-5 in serum (199, 200) and increased production of IL-4, IL-5, and IL-13 by T cells (201, 202). CCL17, a chemokine that recruits Th2 cells into tissues, is increased in the serum and biopsies of EGPA patients and is positively correlated with blood eosinophils and IgE (199, 203). This increase in Th2 activity likely contributes to eosinophilia.
Blood eosinophils in EGPA show an activated phenotype expressing high levels of CD69 and CD11b (200, 204). Moreover, they express IL-25, a cytokine that increases release of IL-4, -5, and -13 from T cells. Serum IL-25 is increased in patients with active EGPA compared to inactive disease or healthy controls. It is also detectable in eosinophils from lesional biopsies. T cells in these biopsies and in the blood express the IL-25 receptor IL-17RB (205). This suggests a feed-forward loop between eosinophils and Th2 cells in EGPA.
Neuropathy is a common symptom of EGPA (196). Interestingly, different mechanisms lead to nerve damage depending on the presence or absence of MPO–ANCA. In patients with autoantibodies, MPO–ANCA-induced necrotizing vasculitis results in ischemic damage to the nerves (206–209). In the absence of autoantibodies, massive eosinophil infiltration into the epineurium and occasionally endoneurium is observed. These eosinophils are degranulating and cytotoxic to nerves (209, 210). Sometimes eosinophils form part of the inflammatory infiltrate surrounding necrotizing vessels (209, 211, 212). This may accelerate damage of blood vessels because eosinophils were shown to be directly cytotoxic to endothelial cells in vitro (34, 213). This damage may be mediated by ECP, which is deposited on endothelial surfaces in patients with eosinophilic endomyocarditis (214–216), or by MBP, which is cytotoxic in vitro (34).
Eosinophil chemotaxis into affected tissues in EGPA patients occurs in response to eotaxin-3. Serum levels of eotaxin-3 are substantially higher in EGPA patients with active disease compared to those with inactive disease, healthy controls, or patients with other eosinophil-associated diseases (217, 218). In contrast, there is no increase in serum eotaxin-1 or -2 (217). Eotaxin-3 is also readily detected in biopsies of affected tissues from EGPA patients (217). Eotaxin-3 localizes to endothelial cells of small vessels, smooth muscle cells of small arterioles, the perineurium of the sural nerve, and the respiratory epithelium of the nose. An analysis of single-nucleotide polymorphisms in the eotaxin-3 gene in 161 EGPA patients found no significant associations (218), suggesting that eotaxin-3 polymorphisms may not be causal in EGPA.
The strongest evidence for a pathogenic role of eosinophils in EGPA comes from novel biological treatments that target IL-5 and thereby drastically reduce eosinophil levels. Two open-label trials with the anti-IL-5 antibody mepolizumab demonstrated its efficacy as a steroid-sparing agent and its ability to induce remission over 9 months (219–221). Upon termination of mepolizumab treatment, the majority of patients developed relapses. In one trial, eosinophil count and serum ECP were strongly correlated with disease activity (221). A double-blind randomized placebo-controlled trial of mepolizumab in EGPA is currently ongoing (http://ClinicalTrials.gov identifier: NCT02020889).
Several key findings amount to moderate evidence for a pathogenic role of eosinophils in EGPA. (1) The number of eosinophils and serum ECP correlate with disease severity. (2) Eosinophil infiltration and degranulation in tissues causes organ damage. (3) A potential feed-forward loop between Th2 cells and eosinophils may propagate disease. (4) IL-5-targeted therapies showed beneficial effects.
Eosinophilic Myocarditis
Myocarditis is the inflammation of the heart muscle with or without damage or necrosis of adjacent myocytes in the absence of an ischemic event (222). A wide range of causes from viral, bacterial, and parasitic infections to toxic effects of drugs or hypersensitivity reactions can cause myocarditis, and in many cases, the etiology is unknown (223). Autoimmune processes often play a role either causally or as postinfection autoimmunity: autoantibodies against cardiac antigens are present in the majority of myocarditis patients, myocarditis is associated with other autoimmune diseases, and some patients benefit from immunosuppressive treatment (224, 225). Animal models provide further evidence for autoimmune mechanisms. Cardiac autoantibodies induce disease in rats, and immunization with cardiac myosin peptide in adjuvants induces experimental autoimmune myocarditis (EAM) in mice (224).
Eosinophils form a major part of the inflammatory infiltrate in subtypes of myocarditis, namely in eosinophilic myocarditis and giant cell myocarditis. These subtypes are usually idiopathic. Eosinophilic myocarditis is associated with hypereosinophilic syndrome (HES) and EGPA, but it can also develop in the absence of eosinophilia. About one-third of EGPA patients and 20–50% of HES patients develop cardiovascular manifestations (196, 197, 226–229). Myocarditis is more frequent in ANCA-negative EGPA patients (196, 229). Parasitic infections and hypersensitivity reactions to drugs are other potential causes of eosinophilic myocarditis (230, 231). Giant cell myocarditis and eosinophilic myocarditis are usually treated with strong immunosuppressive agents.
Eosinophils likely play a pathogenic role in the heart (227, 232, 233). Eosinophil granule proteins are deposited in the myocardium during eosinophilic myocarditis and may be cytotoxic to cardiomyocytes (61, 234–236). Eosinophils have also been proposed to activate cardiac mast cells (237) or release prothrombotic tissue factor (238). In HES, eosinophils are thought to damage the endocardium, which results in thrombosis and endocarditis and eventually leads to endomyocardial fibrosis and valvular complications (226, 228). Animal studies further strengthen the evidence that eosinophils contribute to pathology and mortality in eosinophilic myocarditis. Hypereosinophilic mice with transgenic expression of IL-5 (IL-5Tg) spontaneously develop eosinophilic myocarditis at a low frequency (239). We found that induction of EAM in these IL-5Tg mice reliably induces eosinophilic myocarditis with over 60% of the heart-infiltrating cells being eosinophils (240). Induction of EAM in A/J mice causes myocarditis with numerous infiltrating eosinophils (241). Blockade of IL-4 in this model reduces eosinophil infiltration and disease severity (241). Induction of EAM in BALB/c mice that lack interferon (IFN)γ and IL-17A (IFNγ−/−IL-17A−/−) results in severe eosinophilic myocarditis with about 50% fatality by day 21 (242). Ablation of eosinophils in these mice improved survival. In another model, natural killer cell depletion resulted in increased eosinophil infiltration in the heart and aggravated myocarditis. In eosinophil-deficient mice, however, natural killer cell depletion did not increase disease severity (243). These results show that eosinophils are pathogenic in myocarditis.
A major burden of myocarditis lies in the sequela inflammatory dilated cardiomyopathy (DCM), which is the major cause of heart failure in patients younger than 40 years and has a poor 5-year survival rate of less than 50% (244, 245). It is not known at what rate eosinophilic myocarditis patients progress to DCM or how this rate compares to other myocarditis subtypes. By using the EAM model, we found that eosinophil-deficient mice are protected from DCM following myocarditis, while hypereosinophilic IL-5Tg mice developed more severe DCM. This process was dependent on eosinophil-derived IL-4 (240). This suggests that eosinophils drive the chronic disease that ensues myocarditis and impair cardiac function.
Little is known about the mediators that induce eosinophil infiltration into the heart. We found increased expression of eotaxin-1 and eotaxin-3 in endomyocardial biopsies from patients with eosinophilic myocarditis compared to chronic lymphocytic myocarditis (24). In the eosinophilic myocarditis mouse model of EAM in IFNγ−/−IL-17A−/− mice, cardiac expression of eotaxin-1 and -2 is highly increased compared to naïve mice or WT controls (24, 242). In this model, the eotaxin-CCR3 pathway is necessary for eosinophil trafficking to the heart during myocarditis (24).
In summary, there is substantial evidence that eosinophils play a pathogenic role in myocarditis during acute and chronic stages. Several studies in animal models offered mechanistic insight into how eosinophils contribute to myocarditis. It will be interesting to see if eosinophil-targeted therapies in patients with HES or EGPA will reduce the incidence of eosinophilic myocarditis in this high-risk group.
Neuromyelitis Optica
Neuromyelitis optica (NMO) is a demyelinating disease of the central nervous system (CNS) that usually affects the optic nerve and spinal cord. Lesions are necrotic, cavitary, and infiltrated with macrophages and granulocytes. NMO is an autoimmune disease. Anti-aquaporin 4 (AQP4) autoantibodies are present in the majority of patients. These pathogenic antibodies are highly specific for NMO and are one of the features that distinguish it from multiple sclerosis. NMO patients often carry multiple other autoantibodies, and there is a strong association with other autoimmune diseases. Moreover, NMO is much more common in women than men (246).
Neuromyelitis optica has only recently been distinguished from multiple sclerosis with eosinophil infiltration being one of the distinctive features (247). The first description of eosinophil infiltration in NMO lesions was by Lucchinetti and colleagues in 2002. In analyzing lesions from NMO patient autopsies, they found eosinophil infiltration in early active lesions (248). Eosinophils infiltration is located meningeal and perivascular in spinal cord lesions. Both intact and degranulating eosinophils are found (248). Since this original observation, multiple studies have described eosinophil infiltration in the spinal cord, optic nerve (249), brainstem (250, 251), and cerebrospinal fluid (252). Another study found that the cerebrospinal fluid from patients with NMO contains higher levels of eotaxin-2, eotaxin-3, and ECP compared to healthy controls or multiple sclerosis patients. In addition, stimulation of cerebrospinal fluid cells with myelin oligodendrocyte glycoprotein results in increased IL-5 production in NMO compared to controls (253). Together, these studies clearly establish that eosinophils infiltrate and degranulate in NMO lesions, which suggest a pathogenic role for eosinophils.
A recent elegant study used in vitro experiments and a mouse model to determine the role of eosinophils in NMO (254). Bone marrow-derived eosinophils exhibit ADCC when cocultured with a cell line expressing AQP4 in the presence of anti-AQP4. Similar effects of eosinophils are observed on spinal cord slide cultures. Stimulation of eosinophils with platelet-activating factor (PAF), which induces the release of EPX, results in damage to spinal cord slice cultures independent of anti-AQP4 antibody. The authors developed a mouse model of NMO by continuously infusing anti-AQP4 antibodies and human complement intracerebrally for 3 days. In this model, depletion of neutrophils, eosinophils, or both reduces pathology. Likewise, eosinophil-deficient mice have less severe lesions. Induction of disease in hypereosinophilic mice results in more severe lesions with increased eosinophil and neutrophil infiltration (254). This clearly established a pathogenic role for eosinophils in NMO and highlights mechanisms (ADCC and degranulation) by which eosinophils can damage neural tissues.
Primary Biliary Cirrhosis
Primary biliary cirrhosis is a chronic disease of the small intrahepatic bile ducts that eventually leads to cirrhosis. It shows several hallmarks of an autoimmune disease: highly specific antimitochondrial autoantibodies, association with other autoimmune diseases, a female to male ratio of 10:1, and a strong genetic component (255). Histologically, damaged biliary epithelial cells and infiltration of the portal area with plasma cells, T cells, NK cells, macrophages, neutrophils, and eosinophils are visible (255, 256). Cytokine expression in the liver of primary biliary cirrhosis patients is similarly mixed. Compared to other liver diseases, increased hepatic expression of IL-5, IL-6, IFNγ, TGFβ, and IL-2 has been noted (257, 258). Recent studies also identified key Th1 and Th17 cytokines in the liver (259) and on blood cells (260) and a decreased T regulatory to Th17 cell balance in peripheral blood cells (261).
Patients with primary biliary cirrhosis have a higher frequency and increased absolute numbers of eosinophils in peripheral blood and the liver, particularly around damaged bile ducts (262–265). Eosinophil infiltration is higher in the early stages of the disease (stages I–II versus III–IV) (263, 265). Increased eosinophil infiltration was positively associated with liver IL-5 expression (258) and mast cell infiltration (264). Infiltrating eosinophils are degranulating, releasing ECP, MBP, and EDN, which can also be detected in the serum (262, 265). Some patients have autoantibodies to EPX (266), although it is unclear whether these have any pathologic relevance. Two of the established mouse models for primary biliary cirrhosis show eosinophil infiltration in the liver and could be useful for further studies on the role of eosinophils (267–269).
Ursodeoxycholic acid (UDCA) is the only approved drug for primary biliary cirrhosis patients. UDCA can delay disease progression and improve liver biochemistry (255). Of note, a higher frequency of blood eosinophils is associated with better response to UDCA treatment (263). UDCA treatment decreases the frequency and number of eosinophils in the blood (263, 270) and the liver (265) and decreases degranulation of tissue eosinophils and serum MBP and EDN (265). UDCA may suppress tissue eosinophilia by altering dendritic cells and the local cytokine milieu (271).
In some cases, eosinophilia may precede the detection of liver pathology, suggesting that eosinophils are involved early in the disease processes. One study reports on four cases of asymptomatic women with eosinophilia detected during random investigation. All of them had elevated liver enzymes and were diagnosed with primary biliary cirrhosis (272). In another patient eventually diagnosed with primary biliary cirrhosis, eosinophilia was detected 18 months prior to diagnosis, but liver enzymes were still normal 12 months prior to diagnosis, suggesting that eosinophilia can precede overt liver pathology (273). In conclusion, there is some evidence for a role of eosinophils in the early stages of primary biliary cirrhosis.
Other Diseases
Several rare diseases with a possible autoimmune etiology are associated with eosinophils. Usually only case reports or small case series are available for these diseases, making it very difficult to assign a pathologic or protective role to eosinophils.
Eosinophilic Cellulitis
Eosinophilic cellulitis (Wells’ syndrome) is a very rare skin disease characterized by recurrent edematous erythema. Eosinophilic cellulitis is potentially associated with EGPA, HES, UC, or other causes, but the etiology is unknown (274, 275). The typical histopathological sign is flame figures, the focal accumulation of disintegrating eosinophils and collagen fibers. Early stages are characterized by predominantly eosinophilic infiltration (274). Blood eosinophilia is present in 15–67% of patients (275). Blood eosinophils from patients with eosinophilic cellulitis express the high-affinity IL-2 receptor CD25 (276). In vitro, IL-2 treatment of CD25+ eosinophils resulted in priming and increased release of ECP upon subsequent PAF stimulation. This suggests that eosinophils in patients with eosinophilic cellulitis may degranulate more easily. Indeed, extracellular MBP staining is readily observed in flame figures (277, 278) and may be in amyloid form, a sign of large-scale degranulation (279). Eosinophil chemotactic factors CCL17 and CCL24 have been detected in lesions (280). It is possible that eosinophils play a pathogenic role through degranulation.
Eosinophilic Fasciitis
Eosinophilic fasciitis is characterized by thickening and inflammation of the fascia resulting in painful swelling and progressive induration of the skin and soft tissues (281, 282). The etiology of eosinophilic fasciitis is unknown. Autoimmune disease, infections, drugs, physical exertion, and other factors are discussed as potential triggers (283). There is no clear predominance by sex. Antinuclear antibodies are present in 15–20% of patients (284). Blood eosinophilia is present in most cases (60–90%), and eosinophils infiltrate the fascia and sometimes the perimysium (285–287). This increase in eosinophils is not always present and may be a transient feature. In one study, blood or tissue eosinophilia was not associated with the clinical outcome (cure versus residual fibrosis) (286).
IgG4-Related Disease
The fibroinflammatory IgG4-related disease can affect multiple organs including the pancreas, salivary and lacrimal glands, lungs, retroperitoneum, and other tissues (288, 289). While it seems to be an immune-mediated disease, no target antigen (autoimmune or microbial) has been identified (290). The etiology and triggering factors are unknown (289). High serum IgG4 is present in 60–70% of patients (291), but it is unclear whether these IgG4 antibodies are directly pathogenic (289, 290, 292, 293). Key histopathological features are a dense lymphoplasmacytic infiltrate, storiform fibrosis, and obliterative phlebitis (288). Peripheral blood eosinophilia is found in about 30% of patients (294). Eosinophils also infiltrate the tissues and are present in the majority of lesions. Eosinophil infiltration is usually mild to moderate but can be predominant in some cases (288). To date, there is no clear evidence for or against a pathogenic role of eosinophils. However, several potential mechanisms have been hypothesized: antigen presentation, release of profibrotic factors, and promotion of plasma cell survival for IgG4 production (290).
Autoimmune Diseases in Patients with Eosinophil-Associated Diseases
Eosinophils are a key feature of asthma, hypereosinophilic syndromes, and eosinophilic gastrointestinal diseases. An increased frequency of autoimmune diseases in patients with these eosinophil-associated diseases would be suggestive of a possible role for eosinophils in autoimmunity.
Hypereosinophilic Syndrome
There are numerous case reports of patients with hypereosinophilic syndrome who also suffer from an autoimmune disease including ulcerative colitis, autoimmune hepatitis, autoimmune thyroiditis, multiple sclerosis, systemic lupus erythematosus, antiphospholipid syndrome, myasthenia gravis, and rheumatoid arthritis (295–310). Some of these patients had more than one autoimmune disease. From these case reports, the overall frequency of autoimmune diseases in HES cannot be determined. It is also not clear if HES precedes autoimmune disease or vice versa. In a trial of mepolizumab therapy for HES, 5 of 78 patients under follow-up developed autoimmune diseases (rheumatoid arthritis, polymyalgia rheumatica, temporal arteritis, lichen planus, and autoimmune thrombocytopenia) (311). These diseases were likely revealed by the tapering of glucocorticoids, but an effect of mepolizumab cannot be excluded. It seems that these are in excess of the expected prevalence of autoimmune diseases; however, future studies are required to determine if this is the case.
Eosinophilic Gastrointestinal Diseases
Autoimmune diseases may be associated with eosinophilic gastrointestinal diseases. A recent literature review summarized case reports of autoimmune connective tissue diseases (SLE, rheumatoid arthritis, systemic sclerosis, and inflammatory myositis) in patients with eosinophilic gastroenteritis (312). These patients were mostly female even though eosinophilic gastroenteritis shows a male predominance. The issue remains that case reports do not allow for any conclusion of association. A recent population-based cohort study on eosinophilic esophagitis found that the risk of several autoimmune diseases was substantially increased in patients compared to controls (313). Eosinophilic esophagitis patients had an increased risk of celiac disease, Crohn’s disease, ulcerative colitis, rheumatoid arthritis, lupus, systemic sclerosis, Hashimoto’s thyroiditis, and multiple sclerosis. No increased risk was found for pernicious anemia or vitiligo. Whether female compared to male eosinophilic esophagitis patients were more likely to also suffer from an autoimmune disease was not assessed. Genome-wide association studies have identified risk loci for eosinophilic esophagitis that were previously associated with autoimmune diseases, suggesting a potential common genetic cause (314). It will be interesting to see whether eosinophilic esophagitis patients with autoimmune comorbidities differ from those without and whether an association of other eosinophilic gastrointestinal diseases with autoimmune diseases can be proven.
Asthma
Eosinophils are a prominent feature of allergic asthma and, to a lesser extent, non-allergic asthma (315). A potential role for autoimmune processes in asthma has been proposed (316). Certain autoantibodies are found more frequently in patients with asthma than in healthy controls (317), and some studies have found a positive association of asthma with autoimmune diseases (318–320). However, others report negative associations between these two conditions (321, 322). It has also been suggested that sex hormones may contribute to asthma severity (323). Non-allergic asthma has a female predominance, while allergic asthma does not (324, 325). Whether autoimmunity plays a role in asthma pathology or is increased in affected individuals remains controversial.
To date, there is no strong evidence for increased autoimmune diseases in patients with HES, asthma, or eosinophilic gastrointestinal disease, and only one study determined an increased risk in EoE patients. Future cross-sectional or cohort studies will be required to determine if the prevalence of autoimmune diseases is truly higher in patients with eosinophil-associated diseases.
Conclusion
There is clear evidence for a pathogenic role of eosinophils in several autoimmune diseases (Table 1). Protective functions of eosinophils have not been identified. Eosinophils contribute to autoimmune diseases in vastly different organs, from the CNS to the skin, gastrointestinal tract, and cardiovascular system. These include tissues where eosinophils reside in healthy individuals, such as the intestine, as well as those where eosinophil are usually absent, such as the heart or CNS. In all of these organs, eotaxins seem to be the main chemokines for eosinophil recruitment. Different eotaxins attract eosinophils to different tissues. Eotaxin-1 is essential for eosinophil trafficking to the intestine, while eotaxin-3 is most important in EGPA. Both eotaxin-1 and -3 attract eosinophils to the heart and eotaxin-2 and -3 recruit eosinophils to the CNS in NMO. Eosinophil infiltration into tissues is usually accompanied by eosinophilia, which may be transient, and is often caused by an increase in serum IL-5 or tissue IL-5. Other cytokines like GM-CSF may also be increased and contribute to eosinophilia. Compared to healthy controls, eosinophils from affected tissues or blood of patients show an activated phenotype, upregulating CD11b and CD69, and releasing cytokines such as IL-25, IL-6, IL-8, and IL-1β.
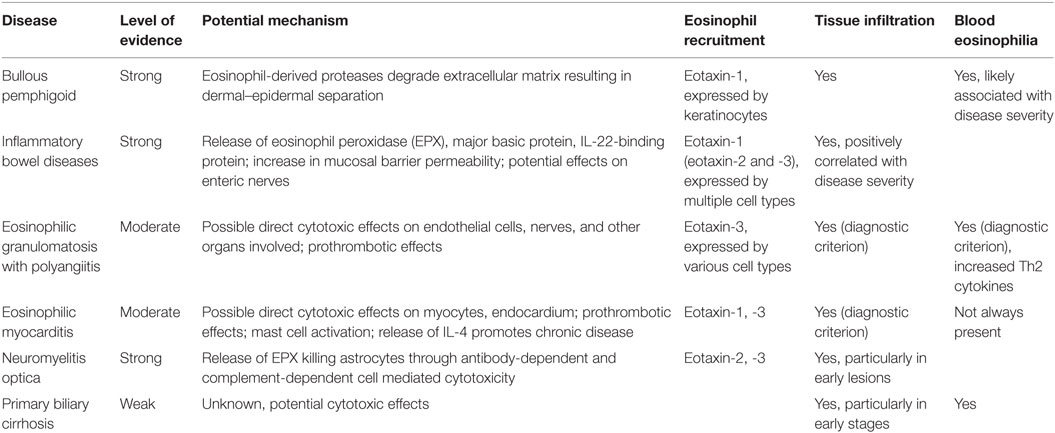
Table 1. Autoimmune diseases with potential eosinophil involvement.
Multiple effector mechanisms have been identified (Figure 2). Degranulation of eosinophils is noted most frequently, perhaps because it is easily visualized by immunofluorescence or histology of biopsies. Degranulating eosinophils are often seen adjacent to dying cells, such as endothelial cells of the vasculature and endocardium, nerve cells, dermis, and intestinal mucosa. As a result, direct cytotoxicity of eosinophils to other cells has been proposed as a mechanism in all autoimmune diseases discussed above. EPX, MBP, and ECP all have strong cytotoxic properties, and often multiple mediators are released. The ability of eosinophils to bind antibodies and subsequently degranulate and kill cells links the adaptive autoimmune response to eosinophil effector functions. Antibody-dependent cell-mediated cytotoxicity by eosinophils was shown for NMO. Antibodies in BP likely cause degranulation of eosinophils and blister formation. Eosinophils are frequently associated with tissue remodeling. In BP, eosinophil-derived MMP9 and neutrophil elastase were shown to degrade extracellular matrix proteins resulting in dermal–epidermal separation. In ulcerative colitis, MBP and corticotrophin-releasing factor from eosinophils downregulate tight-junction proteins on epithelial cells, which decrease their barrier function. Eosinophil-derived cytokines modulating the function of other immune or stromal cells also play a role in autoimmune diseases. Eosinophil-derived IL-4 is important for chronic disease progression in myocarditis, and IL-22BP blocks protective functions of IL-22 in UC. In several diseases, eosinophil infiltration was particularly pronounced in the early stages. This may hint to a role in initiation of the autoimmune response, a hypothesis that is difficult to prove in humans.
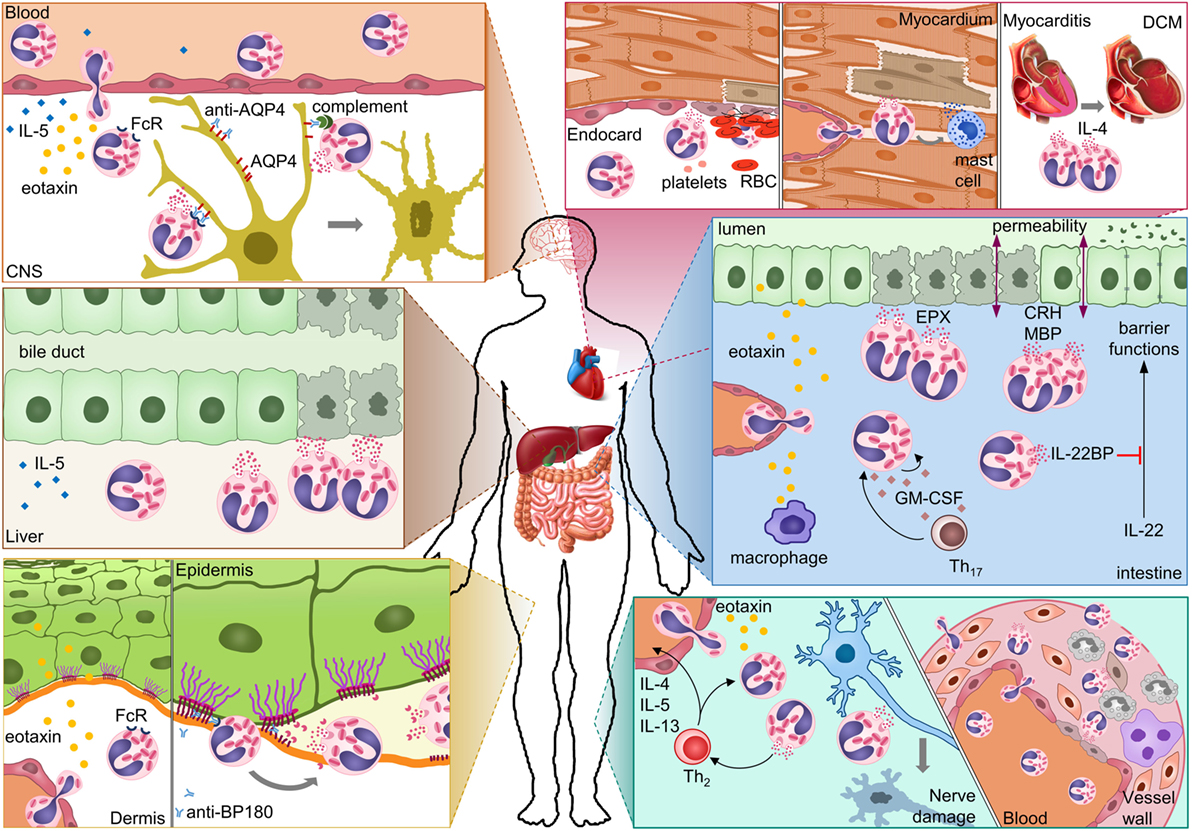
Figure 2. Possible mechanisms of eosinophil-mediated damage in different autoimmune diseases. In neuromyelitis optica, eosinophils damage astrocytes through antibody-dependent and complement-dependent cell-mediated cytotoxicity. Eosinophil degranulation in damaged bile ducts was shown for primary biliary cirrhosis. In bullous pemphigoid, eosinophils release proteases that degrade the dermal–epidermal anchoring complex. Eosinophil infiltration in the heart results in damage to the endocardium and myocardium either directly or indirectly through mast cells. Eosinophil-derived IL-4 can drive progression from autoimmune myocarditis to DCM. In inflammatory bowel diseases, eosinophils can damage the mucosa through multiple mechanisms. Eosinophil granule proteins damage epithelial cells and increase epithelial barrier permeability. Eosinophil-derived IL-22BP blocks the protective effects of IL-22 on epithelial cells. GM-CSF may prolong survival and activation of eosinophils in the intestine. In eosinophilic granulomatosis with polyangiitis, eosinophils damage nerves and blood vessels. Abbreviations: CNS, central nervous system; AQP4, aquaporin 4; FcR, Fc receptor; BP180, bullous pemphigoid 180; RBC, red blood cell; DCM, dilated cardiomyopathy; EPX, eosinophil peroxidase; CRH, corticotropin-releasing hormone; MBP, major basic protein; IL-22BP, IL-22-binding protein; GM-CSF, granulocyte-macrophage colony-stimulating factor; Th17, T-helper 17 cell; Th2, T-helper 2 cell.
Particularly for rare diseases, the evidence for eosinophil involvement is mostly based on case reports, which makes it difficult to exclude associations by chance. Because autoimmune disease patients may receive many drugs, it is worth considering that hypersensitivity reactions to drugs are often accompanied by eosinophilia. On the other hand, it is difficult to ascertain the role of eosinophils in patients treated with glucocorticoids, which are highly effective at reducing eosinophil numbers in blood and organs (326, 327). Eosinophils may be reduced to normal or below normal levels in patients under treatment, and this could mask any associations. Novel targeted therapeutics that affect only specific arms of the immune response and do not dampen eosinophils may reveal new associations.
Future Research Needs
To verify some of the proposed mechanisms and potentially identify new mechanisms of eosinophil-mediated pathology or protection in autoimmune disease, animal models will aid greatly. The lack of in vitro or animal models has hampered research in several autoimmune diseases such as EGPA and primary biliary cirrhosis. In addition, epidemiological studies including larger patient cohorts will be required to determine whether autoimmune diseases are indeed increased in patients with eosinophil-associated diseases such as eosinophilic esophagitis, hypereosinophilic syndrome, or asthma. Finally, viewing and analyzing autoimmune diseases with eosinophil involvement as a group with possible shared mechanisms may advance our understanding and point to common processes.
Author Contributions
ND reviewed the literature and wrote the manuscript. NR conceptualized the idea for the review and edited the manuscript. DČ edited the manuscript and contributed to defining the scope and content of the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all participants of the roundtable on Eosinophils, Type II Immunity and Autoimmune Diseases held in June 2015 in Washington, DC, USA for their insights and discussion on the role of eosinophils in autoimmune diseases. We thank Dr. Isabelle Coppens, Johns Hopkins University, Baltimore, MD, USA, for providing the electron microscopy image and Sean Doughty for language editing.
Funding
This work was supported by NIH/NHLBI grants R01HL113008 and R01HL118183 to DČ, American Heart Association Predoctoral Fellowship 15PRE25400010, and Johns Hopkins Bloomberg School of Public Health Richard J and Margaret Conn Himelfarb Student Support fund to ND. A roundtable on Eosinophils, Type II Immunity and Autoimmune Diseases held in June 2015 was funded by the American Autoimmune Related Diseases Association.
References
1. Ohashi PS, Oehen S, Buerki K, Pircher H, Ohashi CT, Odermatt B, et al. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell (1991) 65(2):305–17. doi:10.1016/0092-8674(91)90164-T
2. Waldner H, Collins M, Kuchroo VK. Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. J Clin Invest (2004) 113:990–7. doi:10.1172/JCI19388
3. Ichikawa HT, Williams LP, Segal BM. Activation of APCs through CD40 or toll-like receptor 9 overcomes tolerance and precipitates autoimmune disease. J Immunol (2002) 169(5):2781–7. doi:10.4049/jimmunol.169.5.2781
4. Huang J, Xiao Y, Xu A, Zhou Z. Neutrophils in type 1 diabetes. J Diabetes Investig (2016) 7(5):652–63. doi:10.1111/jdi.12469
5. Fraker C, Bayer AL. The expanding role of natural killer cells in type 1 diabetes and immunotherapy. Curr Diab Rep (2016) 16(11):109. doi:10.1007/s11892-016-0806-7
6. Jiao G, Wang B. NK cell subtypes as regulators of autoimmune liver disease. Gastroenterol Res Pract (2016) 2016:6903496. doi:10.1155/2016/6903496
7. Zhou Z, Ding M, Huang L, Gilkeson G, Lang R, Jiang W. Toll-like receptor-mediated immune responses in intestinal macrophages; implications for mucosal immunity and autoimmune diseases. Clin Immunol (2016) 173:81–6. doi:10.1016/j.clim.2016.09.005
8. Waldner H. The role of innate immune responses in autoimmune disease development. Autoimmun Rev (2009) 8:400–4. doi:10.1016/j.autrev.2008.12.019
9. Sie C, Korn T. Dendritic cells in central nervous system autoimmunity. Semin Immunopathol (2017) 39(2):99–111. doi:10.1007/s00281-016-0608-7
10. Exley MA, Tsokos GC, Mills KHG, Elewaut D, Mulhearn B. What rheumatologists need to know about innate lymphocytes. Nat Rev Rheumatol (2016) 12:658–68. doi:10.1038/nrrheum.2016.140
11. Conti P, Kempuraj D. Important role of mast cells in multiple sclerosis. Mult Scler Relat Disord (2016) 5:77–80. doi:10.1016/j.msard.2015.11.005
12. Lopez AF, Begley CG, Williamson DJ, Warren DJ, Vadas MA, Sanderson CJ. Murine eosinophil differentiation factor. An eosinophil-specific colony-stimulating factor with activity for human cells. J Exp Med (1986) 163:1085–99. doi:10.1084/jem.163.5.1085
13. Rothenberg ME, Pomerantz JL, Owen WF, Avraham S, Soberman RJ, Austen KF, et al. Characterization of a human eosinophil proteoglycan, and augmentation of its biosynthesis and size by interleukin 3, interleukin 5, and granulocyte/macrophage colony stimulating factor. J Biol Chem (1988) 263:13901–8.
15. Johnston LK, Hsu C-L, Krier-Burris RA, Chhiba KD, Chien KB, McKenzie A, et al. IL-33 precedes IL-5 in regulating eosinophil commitment and is required for eosinophil homeostasis. J Immunol (2016) 197(9):3445–53. doi:10.4049/jimmunol.1600611
16. Collins PD, Marleau S, Griffiths-Johnson DA, Jose PJ, Williams TJ. Cooperation between interleukin-5 and the chemokine eotaxin to induce eosinophil accumulation in vivo. J Exp Med (1995) 182:1169–74. doi:10.1084/jem.182.4.1169
17. Rothenberg ME, Hogan SP. The eosinophil. Annu Rev Immunol (2006) 24:147–74. doi:10.1146/annurev.immunol.24.021605.090720
18. Kovalszki A, Weller PF. Eosinophilia. Prim Care (2016) 43(4):607–17. doi:10.1016/j.pop.2016.07.010
19. Matthews AN, Friend DS, Zimmermann N, Sarafi MN, Luster AD, Pearlman E, et al. Eotaxin is required for the baseline level of tissue eosinophils. Proc Natl Acad Sci U S A (1998) 95:6273–8. doi:10.1073/pnas.95.11.6273
20. Zhang J, Lathbury LJ, Salamonsen LA. Expression of the chemokine eotaxin and its receptor, CCR3, in human endometrium. Biol Reprod (2000) 62:404–11. doi:10.1095/biolreprod62.2.404
21. Gouon-Evans V, Pollard JW. Eotaxin is required for eosinophil homing into the stroma of the pubertal and cycling uterus. Endocrinology (2001) 142:4515–21. doi:10.1210/endo.142.10.8459
22. Pope SM, Zimmermann N, Stringer KF, Karow ML, Rothenberg ME. The eotaxin chemokines and CCR3 are fundamental regulators of allergen-induced pulmonary eosinophilia. J Immunol (2005) 175:5341–50. doi:10.4049/jimmunol.175.8.5341
23. Ahrens R, Waddell A, Seidu L, Blanchard C, Carey R, Forbes E, et al. Intestinal macrophage/epithelial cell-derived CCL11/eotaxin-1 mediates eosinophil recruitment and function in pediatric ulcerative colitis. J Immunol (2008) 181:7390–9. doi:10.4049/jimmunol.181.10.7390
24. Diny NL, Hou X, Barin JG, Chen G, Talor MV, Schaub J, et al. Macrophages and cardiac fibroblasts are the main producers of eotaxins and regulate eosinophil trafficking to the heart. Eur J Immunol (2016) 46:2749–60. doi:10.1002/eji.201646557
25. Kitaura M, Nakajima T, Imai T, Harada S, Combadiere C, Tiffany HL, et al. Molecular cloning of human eotaxin, an eosinophil-selective CC chemokine, and identification of a specific eosinophil eotaxin receptor, CC chemokine receptor 3. J Biol Chem (1996) 271:7725–30. doi:10.1074/jbc.271.13.7725
26. Kitaura M, Suzuki N, Imai T, Takagi S, Suzuki R, Nakajima T, et al. Molecular cloning of a novel human CC chemokine (Eotaxin-3) that is a functional ligand of CC chemokine receptor 3. J Biol Chem (1999) 274:27975–80. doi:10.1074/jbc.274.39.27975
27. Patel VP, Kreider BL, Li Y, Li H, Leung K, Salcedo T, et al. Molecular and functional characterization of two novel human C-C chemokines as inhibitors of two distinct classes of myeloid progenitors. J Exp Med (1997) 185:1163–72. doi:10.1084/jem.185.7.1163
28. Lee JJ, Jacobsen EA, Ochkur SI, McGarry MP, Condjella RM, Doyle AD, et al. Human versus mouse eosinophils: “that which we call an eosinophil, by any other name would stain as red”. J Allergy Clin Immunol (2012) 130:572–84. doi:10.1016/j.jaci.2012.07.025
29. Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science (1997) 277:2005–7. doi:10.1126/science.277.5334.2005
30. Bochner BS. Novel therapies for eosinophilic disorders. Immunol Allergy Clin North Am (2015) 35:577–98. doi:10.1016/j.iac.2015.05.007
31. Rosenberg HF, Dyer KD, Foster PS. Eosinophils: changing perspectives in health and disease. Nat Rev Immunol (2013) 13:9–22. doi:10.1038/nri3341
32. Acharya KR, Ackerman SJ. Eosinophil granule proteins: form and function. J Biol Chem (2014) 289:17406–15. doi:10.1074/jbc.R113.546218
33. Butterworth AE, Wassom DL, Gleich GJ, Loegering DA, David JR. Damage to schistosomula of Schistosoma mansoni induced directly by eosinophil major basic protein. J Immunol (1979) 122:221–9.
34. Gleich GJ, Frigas E, Loegering DA, Wassom DL, Steinmuller D. Cytotoxic properties of the eosinophil major basic protein. J Immunol (1979) 123:2925–7.
35. Frigas E, Loegering DA, Gleich GJ. Cytotoxic effects of the guinea pig eosinophil major basic protein on tracheal epithelium. Lab Invest (1980) 42:35–43.
36. Zheutlin LM, Ackerman SJ, Gleich GJ, Thomas LL. Stimulation of basophil and rat mast cell histamine release by eosinophil granule-derived cationic proteins. J Immunol (1984) 133:2180–5.
37. Piliponsky AM, Pickholtz D, Gleich GJ, Levi-Schaffer F. Human eosinophils induce histamine release from antigen-activated rat peritoneal mast cells: a possible role for mast cells in late-phase allergic reactions. J Allergy ClinImmunol (2001) 107:993–1000. doi:10.1067/mai.2001.114656
38. Jacoby DB, Costello RM, Fryer AD. Eosinophil recruitment to the airway nerves. J Allergy ClinImmunol (2001) 107:211–8. doi:10.1067/mai.2001.112940
39. Morgan RK, Costello RW, Durcan N, Kingham PJ, Gleich GJ, McLean WG, et al. Diverse effects of eosinophil cationic granule proteins on IMR-32 nerve cell signaling and survival. Am J Respir Cell Mol Biol (2005) 33:169–77. doi:10.1165/rcmb.2005-0056OC
40. Rosenberg HF, Ackerman SJ, Tenen DG. Human eosinophil cationic protein. Molecular cloning of a cytotoxin and helminthotoxin with ribonuclease activity. J Exp Med (1989) 170:163–76. doi:10.1084/jem.170.1.163
41. Sorrentino S, Glitz DG, Hamann KJ, Loegering DA, Checkel JL, Gleich GJ. Eosinophil-derived neurotoxin and human liver ribonuclease. Identity of structure and linkage of neurotoxicity to nuclease activity. J Biol Chem (1992) 267:14859–65.
42. Domachowske JB, Dyer KD, Bonville CA, Rosenberg HF. Recombinant human eosinophil-derived neurotoxin/RNase 2 functions as an effective antiviral agent against respiratory syncytial virus. J Infect Dis (1998) 177:1458–64. doi:10.1086/515322
43. Yang D, Chen Q, Rosenberg HF, Rybak SM, Newton DL, Wang ZY, et al. Human ribonuclease A superfamily members, eosinophil-derived neurotoxin and pancreatic ribonuclease, induce dendritic cell maturation and activation. J Immunol (2004) 173:6134–42. doi:10.4049/jimmunol.173.10.6134
44. Lacy P, Latif DA, Steward M, Musat-Marcu S, Man SFP, Moqbel R. Divergence of mechanisms regulating respiratory burst in blood and sputum eosinophils and neutrophils from atopic subjects. J Immunol (2003) 170:2670–9. doi:10.4049/jimmunol.170.5.2670
45. Wang J-G, Mahmud SA, Nguyen J, Slungaard A. Thiocyanate-dependent induction of endothelial cell adhesion molecule expression by phagocyte peroxidases: a novel HOSCN-specific oxidant mechanism to amplify inflammation. J Immunol (2006) 177:8714–22. doi:10.4049/jimmunol.177.12.8714
46. Wang J-G, Mahmud SA, Thompson JA, Geng J-G, Key NS, Slungaard A. The principal eosinophil peroxidase product, HOSCN, is a uniquely potent phagocyte oxidant inducer of endothelial cell tissue factor activity: a potential mechanism for thrombosis in eosinophilic inflammatory states. Blood (2006) 107:558–65. doi:10.1182/blood-2005-05-2152
47. Dvorak AM, Estrella P, Ishizaka T. Vesicular transport of peroxidase in human eosinophilic myelocytes. Clin Exp Allergy (1994) 24(1):10–8. doi:10.1111/j.1365-2222.1994.tb00910.x
48. Melo RCN, Spencer LA, Perez SAC, Ghiran I, Dvorak AM, Weller PF. Human eosinophils secrete preformed, granule-stored interleukin-4 through distinct vesicular compartments. Traffic (2005) 6:1047–57. doi:10.1111/j.1600-0854.2005.00344.x
49. Spencer LA, Melo RCN, Perez SAC, Bafford SP, Dvorak AM, Weller PF. Cytokine receptor-mediated trafficking of preformed IL-4 in eosinophils identifies an innate immune mechanism of cytokine secretion. Proc Natl Acad Sci U S A (2006) 103:3333–8. doi:10.1073/pnas.0508946103
50. Fischer E, Capron M, Prin L, Kusnierz JP, Kazatchkine MD. Human eosinophils express CR1 and CR3 complement receptors for cleavage fragments of C3. Cell Immunol (1986) 97(2):297–306. doi:10.1016/0008-8749(86)90400-4
51. Gounni AS, Lamkhioued B, Ochiai K, Tanaka Y, Delaporte E, Capron A, et al. High-affinity IgE receptor on eosinophils is involved in defence against parasites. Nature (1994) 367(6459):183–6. doi:10.1038/367183a0
52. Grangette C, Gruart V, Ouaissi MA, Rizvi F, Delespesse G, Capron A, et al. IgE receptor on human eosinophils (FcERII). Comparison with B cell CD23 and association with an adhesion molecule. J Immunol (1989) 143(11):3580–8.
53. Hartnell A, Kay AB, Wardlaw AJ. IFN-gamma induces expression of Fc gamma RIII (CD16) on human eosinophils. J Immunol (1992) 148(5):1471–8.
54. Decot V, Woerly G, Loyens M, Loiseau S, Quatannens B, Capron M, et al. Heterogeneity of expression of IgA receptors by human, mouse, and rat eosinophils. J Immunol (2005) 174(2):628–35. doi:10.4049/jimmunol.174.2.628
55. Hallam C, Pritchard DI, Trigg S, Eady RP. Rat eosinophil-mediated antibody-dependent cellular cytotoxicity: investigations of the mechanisms of target cell lysis and inhibition by glucocorticoids. Clin Exp Immunol (1982) 48(3):641–8.
56. Van Epps DE, Bankhurst AD. Human eosinophils: surface receptors and antibody-dependent cytotoxicity. J Lab Clin Med (1978) 91(4):612–7.
57. Parrillo JE, Fauci AS. Human eosinophils. Purification and cytotoxic capability of eosinophils from patients with the hypereosinophilic syndrome. Blood (1978) 51:457–73.
58. Noguchi H, Kephart GM, Colby TV, Gleich GJ. Tissue eosinophilia and eosinophil degranulation in syndromes associated with fibrosis. Am J Pathol (1992) 140(2):521–8.
59. Phipps S, Ying S, Wangoo A, Ong Y-E, Levi-Schaffer F, Kay AB. The relationship between allergen-induced tissue eosinophilia and markers of repair and remodeling in human atopic skin. J Immunol (2002) 169:4604–12. doi:10.4049/jimmunol.169.8.4604
60. Birring SS, Parker D, McKenna S, Hargadon B, Brightling CE, Pavord ID, et al. Sputum eosinophilia in idiopathic pulmonary fibrosis. Inflamm Res (2005) 54:51–6. doi:10.1007/s00011-004-1321-x
61. Corradi D, Vaglio A, Maestri R, Legname V, Leonardi G, Bartoloni G, et al. Eosinophilic myocarditis in a patient with idiopathic hypereosinophilic syndrome: insights into mechanisms of myocardial cell death. Hum Pathol (2004) 35:1160–3. doi:10.1016/j.humpath.2004.05.008
62. Pincus SH, Ramesh KS, Wyler DJ. Eosinophils stimulate fibroblast DNA synthesis. Blood (1987) 70(2):572–4.
63. Kay AB, Phipps S, Robinson DS. A role for eosinophils in airway remodelling in asthma. Trends Immunol (2004) 25:477–82. doi:10.1016/j.it.2004.07.006
64. Minshall EM, Leung DYM, Martin RJ, Song YL, Cameron L, Ernst P, et al. Eosinophil-associated TGF-β1 mRNA expression and airways fibrosis in bronchial asthma. Am J Respir Cell Mol Biol (1997) 17:326–33. doi:10.1165/ajrcmb.17.3.2733
65. Hernnäs J, Särnstrand B, Lindroth P, Peterson CG, Venge P, Malmström A. Eosinophil cationic protein alters proteoglycan metabolism in human lung fibroblast cultures. Eur J Cell Biol (1992) 59:352–63.
66. Pégorier S, Wagner LA, Gleich GJ, Pretolani M. Eosinophil-derived cationic proteins activate the synthesis of remodeling factors by airway epithelial cells. J Immunol (2006) 177:4861–9. doi:10.4049/jimmunol.177.7.4861
67. Milici AJ, Carroll LA, Stukenbrok HA, Shay AK, Gladue RP, Showell HJ. Early eosinophil infiltration into the optic nerve of mice with experimental allergic encephalomyelitis. Lab Invest (1998) 78(10):1239–44.
68. Costello RW, Schofield BH, Kephart GM, Gleich GJ, Jacoby DB, Fryer AD. Localization of eosinophils to airway nerves and effect on neuronal M2 muscarinic receptor function. Am J Physiol (1997) 273(1 Pt 1):L93–103.
69. Fryer AD, Stein LH, Nie Z, Curtis DE, Evans CM, Hodgson ST, et al. Neuronal eotaxin and the effects of CCR3 antagonist on airway hyperreactivity and M2 receptor dysfunction. J Clin Invest (2006) 116(1):228–36. doi:10.1172/JCI25423
70. Smyth CM, Akasheh N, Woods S, Kay E, Morgan RK, Thornton MA, et al. Activated eosinophils in association with enteric nerves in inflammatory bowel disease. PLoS One (2013) 8:e64216. doi:10.1371/journal.pone.0064216
71. Sawatzky DA, Kingham PJ, Court E, Kumaravel B, Fryer AD, Jacoby DB, et al. Eosinophil adhesion to cholinergic nerves via ICAM-1 and VCAM-1 and associated eosinophil degranulation. Am J Physiol Lung Cell Mol Physiol (2002) 282(6):L1279–88. doi:10.1152/ajplung.00279.2001
72. Kingham PJ, McLean WG, Sawatzky DA, Walsh MT, Costello RW. Adhesion-dependent interactions between eosinophils and cholinergic nerves. Am J Physiol Lung Cell Mol Physiol (2002) 282(6):L1229–38. doi:10.1152/ajplung.00278.2001
73. Iwamoto I, Nakagawa N, Yamazaki H, Kimura A, Tomioka H, Yoshida S. Mechanism for substance P-induced activation of human neutrophils and eosinophils. Regul Pept (1993) 46(1–2):228–30. doi:10.1016/0167-0115(93)90042-7
74. Sunohara N, Furukawa S, Nishio T, Mukoyama M, Satoyoshi E. Neurotoxicity of human eosinophils towards peripheral nerves. J Neurol Sci (1989) 92(1):1–7. doi:10.1016/0022-510X(89)90170-6
75. Hogan SP, Mishra A, Brandt EB, Royalty MP, Pope SM, Zimmermann N, et al. A pathological function for eotaxin and eosinophils in eosinophilic gastrointestinal inflammation. Nat Immunol (2001) 2(4):353–60. doi:10.1038/86365
76. Kobayashi H, Gleich GJ, Butterfield JH, Kita H. Human eosinophils produce neurotrophins and secrete nerve growth factor on immunologic stimuli. Blood (2002) 99(6):2214–20. doi:10.1182/blood.V99.6.2214
77. Kingham PJ, McLean WG, Walsh M-T, Fryer AD, Gleich GJ, Costello RW. Effects of eosinophils on nerve cell morphology and development: the role of reactive oxygen species and p38 MAP kinase. Am J Physiol Lung Cell Mol Physiol (2003) 285(4):L915–24. doi:10.1152/ajplung.00094.2003
78. Garland A, Necheles J, White SR, Neeley SP, Leff AR, Carson SS, et al. Activated eosinophils elicit substance P release from cultured dorsal root ganglion neurons. Am J Physiol (1997) 273(5 Pt 1):L1096–102.
79. Elbon CL, Jacoby DB, Fryer AD. Pretreatment with an antibody to interleukin-5 prevents loss of pulmonary M2 muscarinic receptor function in antigen-challenged guinea pigs. Am J Respir Cell Mol Biol (1995) 12(3):320–8. doi:10.1165/ajrcmb.12.3.7873198
80. Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med (2008) 14:949–53. doi:10.1038/nm.1855
81. Simon D, Simon HU, Yousefi S. Extracellular DNA traps in allergic, infectious, and autoimmune diseases. Allergy (2013) 68:409–16. doi:10.1111/all.12111
82. Dworski R, Simon H-U, Hoskins A, Yousefi S. Eosinophil and neutrophil extracellular DNA traps in human allergic asthmatic airways. J Allergy Clin Immunol (2011) 127(5):1260–6. doi:10.1016/j.jaci.2010.12.1103
83. Simon D, Hoesli S, Roth N, Staedler S, Yousefi S, Simon H-U. Eosinophil extracellular DNA traps in skin diseases. J Allergy Clin Immunol (2011) 127(1):194–9. doi:10.1016/j.jaci.2010.11.002
84. Hartman M-L, Piliponsky AM, Temkin V, Levi-Schaffer F. Human peripheral blood eosinophils express stem cell factor. Blood (2001) 97:1086–91. doi:10.1182/blood.V97.4.1086
85. Piliponsky AM, Gleich GJ, Bar I, Levi-Schaffer F. Effects of eosinophils on mast cells: a new pathway for the perpetuation of allergic inflammation. Mol Immunol (2002) 38:1369–72. doi:10.1016/S0161-5890(02)00090-1
86. Solomon A, Aloe L, Pe’er J, Frucht-Pery J, Bonini S, Bonini S, et al. Nerve growth factor is preformed in and activates human peripheral blood eosinophils. J Allergy Clin Immunol (1998) 102(3):454–60. doi:10.1016/S0091-6749(98)70135-6
87. Otani IM, Anilkumar AA, Newbury RO, Bhagat M, Beppu LY, Dohil R, et al. Anti-IL-5 therapy reduces mast cell and IL-9 cell numbers in pediatric patients with eosinophilic esophagitis. J Allergy Clin Immunol (2013) 131:1576–82. doi:10.1016/j.jaci.2013.02.042
88. Lucey DR, Nicholson-Weller A, Weller PF. Mature human eosinophils have the capacity to express HLA-DR. Proc Natl Acad Sci U S A (1989) 86(4):1348–51. doi:10.1073/pnas.86.4.1348
89. MacKenzie JR, Mattes J, Dent LA, Foster PS. Eosinophils promote allergic disease of the lung by regulating CD4(+) Th2 lymphocyte function. J Immunol (2001) 167(6):3146–55. doi:10.4049/jimmunol.167.6.3146
90. Wang H-B, Ghiran I, Matthaei K, Weller PF. Airway eosinophils: allergic inflammation recruited professional antigen-presenting cells. J Immunol (2007) 179(11):7585–92. doi:10.4049/jimmunol.179.11.7585
91. Shi HZ, Humbles A, Gerard C, Jin Z, Weller PF. Lymph node trafficking and antigen presentation by endobronchial eosinophils. J Clin Invest (2000) 105(7):945–53. doi:10.1172/JCI8945
92. Del Pozo V, De Andrés B, Martín E, Cárdaba B, Fernández JC, Gallardo S, et al. Eosinophil as antigen-presenting cell: activation of T cell clones and T cell hybridoma by eosinophils after antigen processing. Eur J Immunol (1992) 22(7):1919–25. doi:10.1002/eji.1830220736
93. Padigel UM, Lee JJ, Nolan TJ, Schad GA, Abraham D. Eosinophils can function as antigen-presenting cells to induce primary and secondary immune responses to Strongyloides stercoralis. Infect Immun (2006) 74(6):3232–8. doi:10.1128/IAI.02067-05
94. Garro AP, Chiapello LS, Baronetti JL, Masih DT. Rat eosinophils stimulate the expansion of Cryptococcus neoformans-specific CD4(+) and CD8(+) T cells with a T-helper 1 profile. Immunology (2011) 132(2):174–87. doi:10.1111/j.1365-2567.2010.03351.x
95. Jacobsen EA, Zellner KR, Colbert D, Lee NA, Lee JJ. Eosinophils regulate dendritic cells and Th2 pulmonary immune responses following allergen provocation. J Immunol (2011) 187:6059–68. doi:10.4049/jimmunol.1102299
96. Jacobsen EA, Doyle AD, Colbert DC, Zellner KR, Protheroe CA, LeSuer WE, et al. Differential activation of airway eosinophils induces IL-13 mediated allergic Th2 pulmonary responses in mice. Allergy (2015) 70(9):1148–59. doi:10.1111/all.12655
97. Chu DK, Jimenez-Saiz R, Verschoor CP, Walker TD, Goncharova S, Llop-Guevara A, et al. Indigenous enteric eosinophils control DCs to initiate a primary Th2 immune response in vivo. J Exp Med (2014) 211:1657–72. doi:10.1084/jem.20131800
98. Chu VT, Fröhlich A, Steinhauser G, Scheel T, Roch T, Fillatreau S, et al. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol (2011) 12:151–9. doi:10.1038/ni.1981
99. Chu VT, Beller A, Rausch S, Strandmark J, Zänker M, Arbach O, et al. Eosinophils promote generation and maintenance of immunoglobulin-A-expressing plasma cells and contribute to gut immune homeostasis. Immunity (2014) 40:582–93. doi:10.1016/j.immuni.2014.02.014
100. Jung Y, Wen T, Mingler MK, Caldwell JM, Wang YH, Chaplin DD, et al. IL-1β in eosinophil-mediated small intestinal homeostasis and IgA production. Mucosal Immunol (2015) 8(4):930–42. doi:10.1038/mi.2014.123
101. Todd R, Donoff BR, Chiang T, Chou MY, Elovic A, Gallagher GT, et al. The eosinophil as a cellular source of transforming growth factor alpha in healing cutaneous wounds. Am J Pathol (1991) 138:1307.
102. Ohno I, Nitta Y, Yamauchi K, Hoshi H, Honma M, Woolley K, et al. Eosinophils as a potential source of platelet-derived growth factor B-chain (PDGF-B) in nasal polyposis and bronchial asthma. Am J Respir Cell Mol Biol (1995) 13:639–47. doi:10.1165/ajrcmb.13.6.7576701
103. Horiuchi T, Weller PF. Expression of vascular endothelial growth factor by human eosinophils: upregulation by granulocyte macrophage colony-stimulating factor and interleukin-5. Am J Respir Cell Mol Biol (1997) 17:70–7. doi:10.1165/ajrcmb.17.1.2796
104. Stenfeldt A-L, Wennerås C. Danger signals derived from stressed and necrotic epithelial cells activate human eosinophils. Immunology (2004) 112:605–14. doi:10.1111/j.1365-2567.2004.01906.x
105. Goh YPS, Henderson NC, Heredia JE, Red Eagle A, Odegaard JI, Lehwald N, et al. Eosinophils secrete IL-4 to facilitate liver regeneration. Proc Natl Acad Sci U S A (2013) 110:9914–9. doi:10.1073/pnas.1304046110
106. Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell (2013) 153:376–88. doi:10.1016/j.cell.2013.02.053
107. Kay AB. The early history of the eosinophil. Clin Exp Allergy (2015) 45(3):575–82. doi:10.1111/cea.12480
108. Aizawa H, Zimmermann N, Carrigan PE, Lee JJ, Rothenberg ME, Bochner BS. Molecular analysis of human Siglec-8 orthologs relevant to mouse eosinophils: identification of mouse orthologs of Siglec-5 (mSiglec-F) and Siglec-10 (mSiglec-G). Genomics (2003) 82(5):521–30. doi:10.1016/S0888-7543(03)00171-X
109. Ethier C, Lacy P, Davoine F. Identification of human eosinophils in whole blood by flow cytometry. Methods Mol Biol (2014) 1178:81–92. doi:10.1007/978-1-4939-1016-8_8
110. Stevens WW, Kim TS, Pujanauski LM, Hao X, Braciale TJ. Detection and quantitation of eosinophils in the murine respiratory tract by flow cytometry. J Immunol Methods (2007) 327(1–2):63–74. doi:10.1016/j.jim.2007.07.011
111. Filley WV, Ackerman SJ, Gleich GJ. An immunofluorescent method for specific staining of eosinophil granule major basic protein. J Immunol Methods (1981) 47(2):227–38. doi:10.1016/0022-1759(81)90123-X
112. Melo RC, Perez SA, Spencer LA, Dvorak AM, Weller PF. Intragranular vesiculotubular compartments are involved in piecemeal degranulation by activated human eosinophils. Traffic (2005) 6(10):866–79. doi:10.1111/j.1600-0854.2005.00322.x
113. Kasperkiewicz M, Zillikens D. The pathophysiology of bullous pemphigoid. Clin Rev Allergy Immunol (2007) 33:67–77. doi:10.1007/s12016-007-0030-y
114. Jordon RE, Beutner EH, Witebsky E, Blumental G, Hale WL, Lever WF. Basement zone antibodies in bullous pemphigoid. JAMA (1967) 200:751–6. doi:10.1001/jama.200.9.751
115. Stanley JR, Hawley-Nelson P, Yuspa SH, Shevach EM, Katz SI. Characterization of bullous pemphigoid antigen: a unique basement membrane protein of stratified squamous epithelia. Cell (1981) 24:897–903. doi:10.1016/0092-8674(81)90115-X
116. Mutasim DF, Takahashi Y, Labib RS, Anhalt GJ, Patel HP, Diaz LA. A pool of bullous pemphigoid antigen(s) is intracellular and associated with the basal cell cytoskeleton-hemidesmosome complex. J Invest Dermatol (1985) 84:47–53. doi:10.1111/1523-1747.ep12274684
117. Labib RS, Anhalt GJ, Patel HP, Mutasim DF, Diaz LA. Molecular heterogeneity of the bullous pemphigoid antigens as detected by immunoblotting. J Immunol (1986) 136:1231–5.
118. Kiss M, Perényi A, Marczinovits I, Molnár J, Dobozy A, Kemény L, et al. Autoantibodies to human alpha6 integrin in patients with bullous pemphigoid. Ann N Y Acad Sci (2005) 1051:104–10. doi:10.1196/annals.1361.051
119. Bekou V, Thoma-Uszynski S, Wendler O, Uter W, Schwietzke S, Hunziker T, et al. Detection of laminin 5-specific auto-antibodies in mucous membrane and bullous pemphigoid sera by ELISA. J Invest Dermatol (2005) 124(4):732–40. doi:10.1111/j.0022-202X.2005.23646.x
120. Borradori L, Sonnenberg A. Structure and function of hemidesmosomes: more than simple adhesion complexes. J Invest Dermatol (1999) 112:411–8. doi:10.1046/j.1523-1747.1999.00546.x
121. Schmidt-Ullrich B, Rule A, Schaumburg-Lever G, Leblanc C. Ultrastructural localization of in vivo-bound complement in bullous pemphigoid. J Invest Dermatol (1975) 65:217–9. doi:10.1111/1523-1747.ep12598218
122. Gammon WR, Merritt CC, Lewis DM, Sams WM Jr, Wheeler CE Jr, Carlo J. Leukocyte chemotaxis to the dermal-epidermal junction of human skin mediated by pemphigoid antibody and complement: mechanism of cell attachment in the in vitro leukocyte attachment method. J Invest Dermatol (1981) 76:514–22. doi:10.1111/1523-1747.ep12521246
123. Liu Z, Giudice GJ, Swartz SJ, Fairley JA, Till GO, Troy JL, et al. The role of complement in experimental bullous pemphigoid. J Clin Invest (1995) 95:1539–44. doi:10.1172/JCI117826
124. Wintroub BU, Mihm MC Jr, Goetzl EJ, Soter NA, Austen KF. Morphologic and functional evidence for release of mast-cell products in bullous pemphigoid. N Engl J Med (1978) 298:417–21. doi:10.1056/NEJM197802232980803
125. Dvorak AM, Mihm MC Jr, Osage JE, Kwan TH, Austen KF, Wintroub BU. Bullous pemphigoid, an ultrastructural study of the inflammatory response: eosinophil, basophil and mast cell granule changes in multiple biopsies from one patient. J Invest Dermatol (1982) 78:91–101. doi:10.1111/1523-1747.ep12505711
126. Iryo K, Tsuda S, Sasai Y. Ultrastructural aspects of infiltrated eosinophils in bullous pemphigoid. J Dermatol (1992) 19:393–9. doi:10.1111/j.1346-8138.1992.tb03247.x
127. Messingham KN, Wang JW, Holahan HM, Srikantha R, Aust SC, Fairley JA. Eosinophil localization to the basement membrane zone is autoantibody- and complement-dependent in a human cryosection model of bullous pemphigoid. Exp Dermatol (2016) 25(1):50–5. doi:10.1111/exd.12883
128. Bushkell LL, Jordon RE. Bullous pemphigoid: a cause of peripheral blood eosinophilia. J Am Acad Dermatol (1983) 8:648–51. doi:10.1016/S0190-9622(83)70073-3
129. Bernard P, Venot J, Constant F, Bonnetblanc JM. Blood eosinophilia as a severity marker for bullous pemphigoid. J Am Acad Dermatol (1987) 16:879–81. doi:10.1016/S0190-9622(87)80227-X
130. Marzano AV, Tedeschi A, Fanoni D, Bonanni E, Venegoni L, Berti E, et al. Activation of blood coagulation in bullous pemphigoid: role of eosinophils, and local and systemic implications. Br J Dermatol (2009) 160:266–72. doi:10.1111/j.1365-2133.2008.08880.x
131. Yu KK, Crew AB, Messingham KAN, Fairley JA, Woodley DT. Omalizumab therapy for bullous pemphigoid. J Am Acad Dermatol (2014) 71:468–74. doi:10.1016/j.jaad.2014.04.053
132. Tedeschi A, Marzano AV, Lorini M, Balice Y, Cugno M. Eosinophil cationic protein levels parallel coagulation activation in the blister fluid of patients with bullous pemphigoid. J Eur Acad Dermatol Venereol (2015) 29:813–7. doi:10.1111/jdv.12464
133. Wakugawa M, Nakamura K, Hino H, Toyama K, Hattori N, Okochi H, et al. Elevated levels of eotaxin and interleukin-5 in blister fluid of bullous pemphigoid: correlation with tissue eosinophilia. Br J Dermatol (2000) 143:112–6. doi:10.1046/j.1365-2133.2000.03599.x
134. D’Auria L, Pietravalle M, Mastroianni A, Ferraro C, Mussi A, Bonifati C, et al. IL-5 levels in the serum and blister fluid of patients with bullous pemphigoid: correlations with eosinophil cationic protein, RANTES, IgE and disease severity. Arch Dermatol Res (1998) 290:25–7. doi:10.1007/s004030050272
135. Frezzolini A, Teofoli P, Cianchini G, Barduagni S, Ruffelli M, Ferranti G, et al. Increased expression of eotaxin and its specific receptor CCR3 in bullous pemphigoid. Eur J Dermatol (2002) 12:27–31.
136. Engmann J, Rüdrich U, Behrens G, Papakonstantinou E, Gehring M, Kapp A, et al. Increased activity and apoptosis of eosinophils in blister fluids, skin and peripheral blood of patients with bullous pemphigoid. Acta Derm Venereol (2017) 97(4):464–71. doi:10.2340/00015555-2581
137. Borrego L, Maynard B, Peterson EA, George T, Iglesias L, Peters MS, et al. Deposition of eosinophil granule proteins precedes blister formation in bullous pemphigoid. Comparison with neutrophil and mast cell granule proteins. Am J Pathol (1996) 148:897–909.
138. Ståhle-Bäckdahl M, Inoue M, Guidice GJ, Parks WC. 92-kD gelatinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kD bullous pemphigoid autoantigen. J Clin Invest (1994) 93:2022–30. doi:10.1172/JCI117196
139. Niimi Y, Pawankar R, Kawana S. Increased expression of matrix metalloproteinase-2, matrix metalloproteinase-9 and matrix metalloproteinase-13 in lesional skin of bullous pemphigoid. Int Arch Allergy Immunol (2006) 139:104–13. doi:10.1159/000090385
140. Verraes S, Hornebeck W, Polette M, Borradori L, Bernard P. Respective contribution of neutrophil elastase and matrix metalloproteinase 9 in the degradation of BP180 (type XVII collagen) in human bullous pemphigoid. J Invest Dermatol (2001) 117:1091–6. doi:10.1046/j.0022-202x.2001.01521.x
141. Sitaru C, Schmidt E, Petermann S, Munteanu LS, Bröcker EB, Zillikens D. Autoantibodies to bullous pemphigoid antigen 180 induce dermal-epidermal separation in cryosections of human skin. J Invest Dermatol (2002) 118:664–71. doi:10.1046/j.1523-1747.2002.01720.x
142. Bernard P, Aucouturier P, Denis F, Bonnetblanc J-M. Immunoblot analysis of IgG subclasses of circulating antibodies in bullous pemphigoid. Clin Immunol Immunopathol (1990) 54:484–94. doi:10.1016/0090-1229(90)90060-4
143. Döpp R, Schmidt E, Chimanovitch I, Leverkus M, Bröcker EB, Zillikens D. IgG4 and IgE are the major immunoglobulins targeting the NC16A domain of BP180 in Bullous pemphigoid: serum levels of these immunoglobulins reflect disease activity. J Am Acad Dermatol (2000) 42:577–83. doi:10.1016/S0190-9622(00)90168-3
144. Messingham KN, Holahan HM, Frydman AS, Fullenkamp C, Srikantha R, Fairley JA. Human eosinophils express the high affinity IgE receptor, FcεRI, in bullous pemphigoid. PLoS One (2014) 9:e107725. doi:10.1371/journal.pone.0107725
145. Hofmann SC, Technau K, Müller AMS, Lübbert M, Bruckner-Tuderman L. Bullous pemphigoid associated with hypereosinophilic syndrome: simultaneous response to imatinib. J Am Acad Dermatol (2007) 56:S68–72. doi:10.1016/j.jaad.2006.02.059
146. Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest (1993) 92(5):2480–8. doi:10.1172/JCI116856
147. Hurskainen T, Kokkonen N, Sormunen R, Jackow J, Löffek S, Soininen R, et al. Deletion of the major bullous pemphigoid epitope region of collagen XVII induces blistering, autoimmunization, and itching in mice. J Invest Dermatol (2015) 135:1303–10. doi:10.1038/jid.2014.443
148. Snook J. Are the inflammatory bowel diseases autoimmune disorders? Gut (1990) 31:961–3. doi:10.1136/gut.31.9.961
149. Das KM, Biancone L. Is IBD an autoimmune disorder? Inflamm Bowel Dis (2008) 14(Suppl 2):S97–101. doi:10.1002/ibd.20723
150. Truelove SC, Richards WC. Biopsy studies in ulcerative colitis. Br Med J (1956) 1:1315–8. doi:10.1136/bmj.1.4979.1315
151. Bercovitz ZT, Sommers SC. Altered inflammatory reaction in nonspecific ulcerative colitis. Arch Intern Med (1966) 117:504–10. doi:10.1001/archinte.117.4.504
152. Rutgeerts P, Geboes K, Vantrappen G, Kerremans R, Coenegrachts JL, Coremans G. Natural history of recurrent Crohn’s disease at the ileocolonic anastomosis after curative surgery. Gut (1984) 25:665–72. doi:10.1136/gut.25.6.665
153. Walsh RE, Gaginella TS. The eosinophil in inflammatory bowel disease. Scand J Gastroenterol (1991) 26:1217–24. doi:10.3109/00365529108998617
154. Dubucquoi S, Janin A, Klein O, Desreumaux P, Quandalle P, Cortot A, et al. Activated eosinophils and interleukin 5 expression in early recurrence of Crohn’s disease. Gut (1995) 37:242–6. doi:10.1136/gut.37.2.242
155. Makiyama K, Kanzaki S, Yamasaki K, Zea-Iriarte W, Tsuji Y. Activation of eosinophils in the pathophysiology of ulcerative colitis. J Gastroenterol (1995) 30(Suppl 8):64–9.
156. Saitoh O, Kojima K, Sugi K, Matsuse R, Uchida K, Tabata K, et al. Fecal eosinophil granule-derived proteins reflect disease activity in inflammatory bowel disease. Am J Gastroenterol (1999) 94:3513–20. doi:10.1111/j.1572-0241.1999.01640.x
157. Jeziorska M, Haboubi N, Schofield P, Woolley DE. Distribution and activation of eosinophils in inflammatory bowel disease using an improved immunohistochemical technique. J Pathol (2001) 194:484–92. doi:10.1002/path.904
158. Coburn LA, Horst SN, Chaturvedi R, Brown CT, Allaman MM, Scull BP, et al. High-throughput multi-analyte Luminex profiling implicates eotaxin-1 in ulcerative colitis. PLoS One (2013) 8:e82300. doi:10.1371/journal.pone.0082300
159. Zezos P, Patsiaoura K, Nakos A, Mpoumponaris A, Vassiliadis T, Giouleme O, et al. Severe eosinophilic infiltration in colonic biopsies predicts patients with ulcerative colitis not responding to medical therapy. Colorectal Dis (2014) 16:O420–30. doi:10.1111/codi.12725
160. Sadi G, Yang Q, Dufault B, Stefanovici C, Stoffman J, El-Matary W. Prevalence of peripheral eosinophilia at diagnosis in children with inflammatory bowel disease. J Pediatr Gastroenterol Nutr (2016) 62:573–6. doi:10.1097/MPG.0000000000000957
161. Park S, Abdi T, Gentry M, Laine L. Histological disease activity as a predictor of clinical relapse among patients with ulcerative colitis: systematic review and meta-analysis. Am J Gastroenterol (2016) 111(12):1692–701. doi:10.1038/ajg.2016.418
162. Vieira AT, Fagundes CT, Alessandri AL, Castor MGM, Guabiraba R, Borges VO, et al. Treatment with a novel chemokine-binding protein or eosinophil lineage-ablation protects mice from experimental colitis. Am J Pathol (2009) 175:2382–91. doi:10.2353/ajpath.2009.090093
163. Griseri T, Arnold IC, Pearson C, Krausgruber T, Schiering C, Franchini F, et al. Granulocyte macrophage colony-stimulating factor-activated eosinophils promote interleukin-23 driven chronic colitis. Immunity (2015) 43:187–99. doi:10.1016/j.immuni.2015.07.008
164. Radnai B, Sturm EM, Stančić A, Jandl K, Labocha S, Ferreirós N, et al. Eosinophils contribute to intestinal inflammation via chemoattractant receptor-homologous molecule expressed on Th2 cells, CRTH2, in experimental Crohn’s disease. J Crohns Colitis (2016) 10:1087–95. doi:10.1093/ecco-jcc/jjw061
165. Chen W, Paulus B, Shu D, Wilson I, Chadwick V. Increased serum levels of eotaxin in patients with inflammatory bowel disease. Scand J Gastroenterol (2001) 36:515–20. doi:10.1080/003655201750153377
166. Mir A, Minguez M, Tatay J, Pascual I, Peña A, Sanchiz V, et al. Elevated serum eotaxin levels in patients with inflammatory bowel disease. Am J Gastroenterol (2002) 97:1452–7. doi:10.1111/j.1572-0241.2002.05687.x
167. Lampinen M, Waddell A, Ahrens R, Carlson M, Hogan SP. CD14+CD33+ myeloid cell-CCL11-eosinophil signature in ulcerative colitis. J Leukoc Biol (2013) 94:1061–70. doi:10.1189/jlb.1212640
168. Takahashi K, Imaeda H, Fujimoto T, Ban H, Bamba S, Tsujikawa T, et al. Regulation of eotaxin-3/CCL26 expression by Th2 cytokines in human colonic myofibroblasts. Clin Exp Immunol (2013) 173(2):323–31. doi:10.1111/cei.12117
169. Park YR, Choi SC, Lee ST, Kim KS, Chae SC, Chung HT. The association of eotaxin-2 and eotaxin-3 gene polymorphisms in a Korean population with ulcerative colitis. Exp Mol Med (2005) 37:553–8. doi:10.1038/emm.2005.68
170. Forbes E, Murase T, Yang M, Matthaei KI, Lee JJ, Lee NA, et al. Immunopathogenesis of experimental ulcerative colitis is mediated by eosinophil peroxidase. J Immunol (2004) 172:5664–75. doi:10.4049/jimmunol.172.9.5664
171. Waddell A, Ahrens R, Steinbrecher K, Donovan B, Rothenberg ME, Munitz A, et al. Colonic eosinophilic inflammation in experimental colitis is mediated by Ly6C(high) CCR2(+) inflammatory monocyte/macrophage-derived CCL11. J Immunol (2011) 186:5993–6003. doi:10.4049/jimmunol.1003844
172. Dvorak AM. Ultrastructural evidence for release of major basic protein-containing crystalline cores of eosinophil granules in vivo: cytotoxic potential in Crohn’s disease. J Immunol (1980) 125:460–2.
173. Nishitani H, Okabayashi M, Satomi M, Shimoyama T, Dohi Y. Infiltration of peroxidase-producing eosinophils into the lamina propria of patients with ulcerative colitis. J Gastroenterol (1998) 33:189–95. doi:10.1007/s005350050068
174. Bischoff SC, Mayer J, Nguyen QT, Stolte M, Manns MP. Immunnohistological assessment of intestinal eosinophil activation in patients with eosinophilic gastroenteritis and inflammatory bowel disease. Am J Gastroenterol (1999) 94:3521–9. doi:10.1111/j.1572-0241.1999.01641.x
175. Carlson M, Raab Y, Peterson C, Hällgren R, Venge P. Increased intraluminal release of eosinophil granule proteins EPO, ECP, EPX, and cytokines in ulcerative colitis and proctitis in segmental perfusion. Am J Gastroenterol (1999) 94:1876–83. doi:10.1111/j.1572-0241.1999.01223.x
176. Levy AM, Gleich GJ, Sandborn WJ, Tremaine WJ, Steiner BL, Phillips SF. Increased eosinophil granule proteins in gut lavage fluid from patients with inflammatory bowel disease. Mayo Clin Proc (1997) 72:117–23. doi:10.4065/72.2.117
177. Pronk-Admiraal CJ, Linskens RK, Van Bodegraven AA, Tuynman HA, Bartels PC. Serum eosinophil cationic protein in active and quiescent ulcerative colitis. Clin Chem Lab Med (2000) 38:619–22. doi:10.1515/CCLM.2000.090
178. Wedrychowicz A, Tomasik P, Pieczarkowski S, Kowalska-Duplaga K, Grzenda-Adamek Z, Fyderek K. Clinical value of serum eosinophilic cationic protein assessment in children with inflammatory bowel disease. Arch Med Sci (2014) 10:1142–6. doi:10.5114/aoms.2013.34415
179. Blom K, Rubin J, Halfvarson J, Törkvist L, Rönnblom A, Sangfelt P, et al. Eosinophil associated genes in the inflammatory bowel disease 4 region: correlation to inflammatory bowel disease revealed. World J Gastroenterol (2012) 18:6409–19; discussion p. 6417–8. doi:10.3748/wjg.v18.i44.6409
180. Lampinen M, Rönnblom A, Amin K, Kristjansson G, Rorsman F, Sangfelt P, et al. Eosinophil granulocytes are activated during the remission phase of ulcerative colitis. Gut (2005) 54:1714–20. doi:10.1136/gut.2005.066423
181. Furuta GT, Nieuwenhuis EES, Karhausen J, Gleich G, Blumberg RS, Lee JJ, et al. Eosinophils alter colonic epithelial barrier function: role for major basic protein. Am J Physiol Gastrointest Liver Physiol (2005) 289:G890–7. doi:10.1152/ajpgi.00015.2005
182. Wallon C, Persborn M, Jönsson M, Wang A, Phan V, Lampinen M, et al. Eosinophils express muscarinic receptors and corticotropin-releasing factor to disrupt the mucosal barrier in ulcerative colitis. Gastroenterology (2011) 140:1597–607. doi:10.1053/j.gastro.2011.01.042
183. Wolk K, Witte E, Hoffmann U, Doecke W-D, Endesfelder S, Asadullah K, et al. IL-22 induces lipopolysaccharide-binding protein in hepatocytes: a potential systemic role of IL-22 in Crohn’s disease. J Immunol (2007) 178:5973–81. doi:10.4049/jimmunol.178.9.5973
184. Andoh A, Zhang Z, Inatomi O, Fujino S, Deguchi Y, Araki Y, et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology (2005) 129:969–84. doi:10.1053/j.gastro.2005.06.071
185. Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest (2008) 118:534–44. doi:10.1172/JCI33194
186. Sonnenberg GF, Fouser LA, Artis D. Functional biology of the IL-22-IL-22R pathway in regulating immunity and inflammation at barrier surfaces. Adv Immunol (2010) 107:1–29. doi:10.1016/B978-0-12-381300-8.00001-0
187. Martin JC, Bériou G, Heslan M, Bossard C, Jarry A, Abidi A, et al. IL-22BP is produced by eosinophils in human gut and blocks IL-22 protective actions during colitis. Mucosal Immunol (2016) 9(2):539–49. doi:10.1038/mi.2015.83
188. Owaga E, Hsieh R-H, Mugendi B, Masuku S, Shih C-K, Chang J-S. Th17 cells as potential probiotic therapeutic targets in inflammatory bowel diseases. Int J Mol Sci (2015) 16:20841–58. doi:10.3390/ijms160920841
189. Willebrand R, Voehringer D. IL-33-induced cytokine secretion and survival of mouse eosinophils is promoted by autocrine GM-CSF. PLoS One (2016) 11:e0163751. doi:10.1371/journal.pone.0163751
190. Churg J, Strauss L. Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am J Pathol (1951) 27:277–301.
191. Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore) (1984) 63:65–81. doi:10.1097/00005792-198403000-00001
192. Khoury P, Grayson PC, Klion AD. Eosinophils in vasculitis: characteristics and roles in pathogenesis. Nat Rev Rheumatol (2014) 10:474–83. doi:10.1038/nrrheum.2014.98
193. Greco A, Rizzo MI, De Virgilio A, Gallo A, Fusconi M, Ruoppolo G, et al. Churg-Strauss syndrome. Autoimmun Rev (2015) 14:341–8. doi:10.1016/j.autrev.2014.12.004
194. Mahr A, Moosig F, Neumann T, Szczeklik W, Taillé C, Vaglio A, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): evolutions in classification, etiopathogenesis, assessment and management. Curr Opin Rheumatol (2014) 26:16–23. doi:10.1097/BOR.0000000000000015
195. Bonatti F, Reina M, Neri TM, Martorana D. Genetic susceptibility to ANCA-associated vasculitis: state of the art. Front Immunol (2014) 5:577. doi:10.3389/fimmu.2014.00577
196. Comarmond C, Pagnoux C, Khellaf M, Cordier JF, Hamidou M, Viallard JF, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum (2013) 65(1):270–81. doi:10.1002/art.37721
197. Moosig F, Bremer JP, Hellmich B, Holle JU, Holl-Ulrich K, Laudien M, et al. A vasculitis centre based management strategy leads to improved outcome in eosinophilic granulomatosis and polyangiitis (Churg-Strauss, EGPA): monocentric experiences in 150 patients. Ann Rheum Dis (2013) 72(6):1011–7. doi:10.1136/annrheumdis-2012-201531
198. Hellmich B, Csernok E, Gross WL. Proinflammatory cytokines and autoimmunity in Churg-Strauss syndrome. Ann N Y Acad Sci (2005) 1051:121–31. doi:10.1196/annals.1361.053
199. Jakiela B, Sanak M, Szczeklik W, Sokolowska B, Plutecka H, Mastalerz L, et al. Both Th2 and Th17 responses are involved in the pathogenesis of Churg-Strauss syndrome. Clin Exp Rheumatol (2011) 29:S23–34.
200. Tsukadaira A, Okubo Y, Kitano K, Horie S, Momose T, Takashi S, et al. Eosinophil active cytokines and surface analysis of eosinophils in Churg-Strauss syndrome. Allergy Asthma Proc (1999) 20:39–44. doi:10.2500/108854199778681486
201. Kiene M, Csernok E, Müller A, Metzler C, Trabandt A, Gross WL. Elevated interleukin-4 and interleukin-13 production by T cell lines from patients with Churg-Strauss syndrome. Arthritis Rheum (2001) 44:469–73. doi:10.1002/1529-0131(200102)44:2<469::AID-ANR66>3.0.CO;2-0
202. Boita M, Guida G, Circosta P, Elia AR, Stella S, Heffler E, et al. The molecular and functional characterization of clonally expanded CD8+ TCR BV T cells in eosinophilic granulomatosis with polyangiitis (EGPA). Clin Immunol (2014) 152:152–63. doi:10.1016/j.clim.2014.03.001
203. Dallos T, Heiland GR, Strehl J, Karonitsch T, Gross WL, Moosig F, et al. CCL17/thymus and activation-related chemokine in Churg-Strauss syndrome. Arthritis Rheum (2010) 62:3496–503. doi:10.1002/art.27678
204. Nakahigashi K, Otsuka A, Miyachi Y, Kabashima K, Tanioka M. A case of Churg-Strauss syndrome: flow cytometric analysis of the surface activation markers of peripheral eosinophils. Acta Derm Venereol (2013) 93:100–1. doi:10.2340/00015555-1384
205. Terrier B, Bièche I, Maisonobe T, Laurendeau I, Rosenzwajg M, Kahn J-E, et al. Interleukin-25: a cytokine linking eosinophils and adaptive immunity in Churg-Strauss syndrome. Blood (2010) 116:4523–31. doi:10.1182/blood-2010-02-267542
206. Jennette JC, Falk RJ. Pathogenesis of the vascular and glomerular damage in ANCA-positive vasculitis. Nephrol Dial Transplant (1998) 13(Suppl 1):16–20. doi:10.1093/ndt/13.suppl_1.16
207. Wiik A. Clinical and pathophysiological significance of anti-neutrophil cytoplasmic autoantibodies in vasculitis syndromes. Mod Rheumatol (2009) 19:590–9. doi:10.1007/s10165-009-0219-4
208. Bajema IM, Hagen EC, De Heer E, Van Der Woude FJ, Bruijn JA. Colocalization of ANCA-antigens and fibrinoid necrosis in ANCA-associated vasculitis. Kidney Int (2001) 60:2025–30. doi:10.1046/j.1523-1755.2001.00019.x
209. Oka N, Kawasaki T, Matsui M, Shigematsu K, Unuma T, Sugiyama H. Two subtypes of Churg-Strauss syndrome with neuropathy: the roles of eosinophils and ANCA. Mod Rheumatol (2011) 21:290–5. doi:10.1007/s10165-010-0400-9
210. Nagashima T, Cao B, Takeuchi N, Chuma T, Mano Y, Fujimoto M, et al. Clinicopathological studies of peripheral neuropathy in Churg-Strauss syndrome. Neuropathology (2002) 22:299–307. doi:10.1046/j.1440-1789.2002.00454.x
211. Vital A, Vital C, Viallard J-F, Ragnaud J-M, Canron M-H, Lagueny A. Neuro-muscular biopsy in Churg-Strauss syndrome: 24 cases. J Neuropathol Exp Neurol (2006) 65:187–92. doi:10.1097/01.jnen.0000200151.60142.25
212. Chao C-C, Hsieh S-T, Shun C-T, Hsieh S-C. Skin denervation and cutaneous vasculitis in eosinophilia-associated neuropathy. Arch Neurol (2007) 64:959–65. doi:10.1001/archneur.64.7.959
213. Shah AM, Brutsaert DL, Meulemans AL, Andries LJ, Capron M. Eosinophils from hypereosinophilic patients damage endocardium of isolated feline heart muscle preparations. Circulation (1990) 81:1081–8. doi:10.1161/01.CIR.81.3.1081
214. Tai P-C, Spry CF, Olsen EJ, Ackerman S, Dunnette S, Gleich G. Deposits of eosinophil granule proteins in cardiac tissues of patients with eosinophilic endomyocardial disease. Lancet (1987) 329:643–7. doi:10.1016/S0140-6736(87)90412-0
215. Slungaard A, Mahoney JR. Bromide-dependent toxicity of eosinophil peroxidase for endothelium and isolated working rat hearts: a model for eosinophilic endocarditis. J Exp Med (1991) 173:117–26. doi:10.1084/jem.173.1.117
216. Slungaard A, Vercellotti GM, Tran T, Gleich GJ, Key NS. Eosinophil cationic granule proteins impair thrombomodulin function. A potential mechanism for thromboembolism in hypereosinophilic heart disease. J Clin Invest (1993) 91:1721. doi:10.1172/JCI116382
217. Polzer K, Karonitsch T, Neumann T, Eger G, Haberler C, Soleiman A, et al. Eotaxin-3 is involved in Churg-Strauss syndrome – a serum marker closely correlating with disease activity. Rheumatology (Oxford) (2008) 47:804–8. doi:10.1093/rheumatology/ken033
218. Zwerina J, Bach C, Martorana D, Jatzwauk M, Hegasy G, Moosig F, et al. Eotaxin-3 in Churg-Strauss syndrome: a clinical and immunogenetic study. Rheumatology (Oxford) (2011) 50:1823–7. doi:10.1093/rheumatology/keq445
219. Kim S, Marigowda G, Oren E, Israel E, Wechsler ME. Mepolizumab as a steroid-sparing treatment option in patients with Churg-Strauss syndrome. J Allergy Clin Immunol (2010) 125:1336–43. doi:10.1016/j.jaci.2010.03.028
220. Moosig F, Gross WL, Herrmann K, Bremer JP, Hellmich B. Targeting interleukin-5 in refractory and relapsing Churg-Strauss syndrome. Ann Intern Med (2011) 155:341–3. doi:10.7326/0003-4819-155-5-201109060-00026
221. Herrmann K, Gross WL, Moosig F. Extended follow-up after stopping mepolizumab in relapsing/refractory Churg-Strauss syndrome. Clin Exp Rheumatol (2012) 30:S62–5.
222. Aretz HT, Billingham ME, Edwards WD, Factor SM, Fallon JT, Fenoglio JJ Jr, et al. Myocarditis. A histopathologic definition and classification. Am J Cardiovasc Pathol (1987) 1:3–14.
224. Cihakova D, Rose NR. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv Immunol (2008) 99:95–114. doi:10.1016/S0065-2776(08)00604-4
225. Frustaci A, Russo MA, Chimenti C. Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: the TIMIC study. Eur Heart J (2009) 30:1995–2002. doi:10.1093/eurheartj/ehp249
227. Parrillo JE, Borer JS, Henry WL, Wolff SM, Fauci AS. The cardiovascular manifestations of the hypereosinophilic syndrome. Prospective study of 26 patients, with review of the literature. Am J Med (1979) 67(4):572–82. doi:10.1016/0002-9343(79)90227-4
228. Ogbogu P, Rosing DR, Horne MK III. Cardiovascular manifestations of hypereosinophilic syndromes. Immunol Allergy Clin North Am (2007) 27(3):457–75. doi:10.1016/j.iac.2007.07.001
229. Cottin V, Bel E, Bottero P, Dalhoff K, Humbert M, Lazor R, et al. Revisiting the systemic vasculitis in eosinophilic granulomatosis with polyangiitis (Churg-Strauss): a study of 157 patients by the Groupe d’Etudes et de Recherche sur les Maladies Orphelines Pulmonaires and the European Respiratory Society Taskforce on eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Autoimmun Rev (2017) 16(1):1–9. doi:10.1016/j.autrev.2016.09.018
230. Ginsberg F, Parrillo JE. Eosinophilic myocarditis. Heart Fail Clin (2005) 1(3):419–29. doi:10.1016/j.hfc.2005.06.013
232. Fauci AS, Harley JB, Roberts WC, Ferrans VJ, Gralnick HR, Bjornson BH. NIH conference. The idiopathic hypereosinophilic syndrome. Clinical, pathophysiologic, and therapeutic considerations. Ann Intern Med (1982) 97:78–92. doi:10.7326/0003-4819-97-1-78
233. Parrillo JE. Heart disease and the eosinophil. N Engl J Med (1990) 323:1560–1. doi:10.1056/NEJM199011293232211
234. Spry CJ, Tai PC, Davies J. The cardiotoxicity of eosinophils. Postgrad Med J (1983) 59(689):147–53. doi:10.1136/pgmj.59.689.147
235. Tai PC, Hayes DJ, Clark JB, Spry CJ. Toxic effects of human eosinophil products on isolated rat heart cells in vitro. Biochem J (1982) 204(1):75–80. doi:10.1042/bj2040075
236. deMello DE, Liapis H, Jureidini S, Nouri S, Kephart GM, Gleich GJ. Cardiac localization of eosinophil-granule major basic protein in acute necrotizing myocarditis. N Engl J Med (1990) 323(22):1542–5. doi:10.1056/NEJM199011293232207
237. Patella V, de Crescenzo G, Marino I, Genovese A, Adt M, Gleich GJ, et al. Eosinophil granule proteins are selective activators of human heart mast cells. Int Arch Allergy Immunol (1997) 113(1–3):200–2. doi:10.1159/000237546
238. Cugno M, Marzano AV, Lorini M, Carbonelli V, Tedeschi A. Enhanced tissue factor expression by blood eosinophils from patients with hypereosinophilia: a possible link with thrombosis. PLoS One (2014) 9(11):e111862. doi:10.1371/journal.pone.0111862
239. Lee NA, McGarry MP, Larson KA, Horton MA, Kristensen AB, Lee JJ. Expression of IL-5 in thymocytes/T cells leads to the development of a massive eosinophilia, extramedullary eosinophilopoiesis, and unique histopathologies. J Immunol (1997) 158:1332–44.
240. Diny NL, Baldeviano GC, Talor MV, Barin JG, Ong S, Bedja D, et al. Eosinophil-derived IL4 drives progression of myocarditis to inflammatory dilated cardiomyopathy. J Exp Med (2017) 214(4):943–57. doi:10.1084/jem.20161702
241. Afanasyeva M, Wang Y, Kaya Z, Park S, Zilliox MJ, Schofield BH, et al. Experimental autoimmune myocarditis in A/J mice is an interleukin-4-dependent disease with a Th2 phenotype. Am J Pathol (2001) 159:193–203. doi:10.1016/S0002-9440(10)61685-9
242. Barin JG, Baldeviano GC, Talor MV, Wu L, Ong S, Fairweather D, et al. Fatal eosinophilic myocarditis develops in the absence of IFN-γ and IL-17A. J Immunol (2013) 191(8):4038–47. doi:10.4049/jimmunol.1301282
243. Ong S, Ligons DL, Barin JG, Wu L, Talor MV, Diny N, et al. Natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration. Am J Pathol (2015) 185(3):847–61. doi:10.1016/j.ajpath.2014.11.023
244. Daubeney PEF, Nugent AW, Chondros P, Carlin JB, Colan SD, Cheung M, et al. Clinical features and outcomes of childhood dilated cardiomyopathy: results from a national population-based study. Circulation (2006) 114(24):2671–8. doi:10.1161/CIRCULATIONAHA.106.635128
245. Magnani JW, Dec GW. Myocarditis: current trends in diagnosis and treatment. Circulation (2006) 113(6):876–90. doi:10.1161/CIRCULATIONAHA.105.584532
246. Pereira WL, Reiche EMV, Kallaur AP, Kaimen-Maciel DR. Epidemiological, clinical, and immunological characteristics of neuromyelitis optica: a review. J Neurol Sci (2015) 355:7–17. doi:10.1016/j.jns.2015.05.034
247. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology (2006) 66:1485–9. doi:10.1212/01.wnl.0000216139.44259.74
248. Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain (2002) 125:1450–61. doi:10.1093/brain/awf151
249. Saadoun S, Bridges LR, Verkman AS, Papadopoulos MC. Paucity of natural killer and cytotoxic T cells in human neuromyelitis optica lesions. Neuroreport (2012) 23:1044–7. doi:10.1097/WNR.0b013e32835ab480
250. Hengstman GJD, Wesseling P, Frenken CWGM, Jongen PJH. Neuromyelitis optica with clinical and histopathological involvement of the brain. Mult Scler (2007) 13:679–82. doi:10.1177/1352458506070145
251. Popescu BFG, Lennon VA, Parisi JE, Howe CL, Weigand SD, Cabrera-Gómez JA, et al. Neuromyelitis optica unique area postrema lesions: nausea, vomiting, and pathogenic implications. Neurology (2011) 76:1229–37. doi:10.1212/WNL.0b013e318214332c
252. Jarius S, Paul F, Franciotta D, Ruprecht K, Ringelstein M, Bergamaschi R, et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci (2011) 306:82–90. doi:10.1016/j.jns.2011.03.038
253. Correale J, Fiol M. Activation of humoral immunity and eosinophils in neuromyelitis optica. Neurology (2004) 63:2363–70. doi:10.1212/01.WNL.0000148481.80152.BF
254. Zhang H, Verkman AS. Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J Clin Invest (2013) 123:2306–16. doi:10.1172/JCI67554
255. Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet (2015) 386:1565–75. doi:10.1016/S0140-6736(15)00154-3
256. Shi T-Y, Zhang F-C. Role of autoimmunity in primary biliary cirrhosis. World J Gastroenterol (2012) 18:7141–8. doi:10.3748/wjg.v18.i48.7141
257. Martinez OM, Villanueva JC, Gershwin ME, Krams SM. Cytokine patterns and cytotoxic mediators in primary biliary cirrhosis. Hepatology (1995) 21:113–9. doi:10.1016/0270-9139(95)90417-4
258. Nagano T, Yamamoto K, Matsumoto S, Okamoto R, Tagashira M, Ibuki N, et al. Cytokine profile in the liver of primary biliary cirrhosis. J Clin Immunol (1999) 19:422–7. doi:10.1023/A:1020511002025
259. Yang C-Y, Ma X, Tsuneyama K, Huang S, Takahashi T, Chalasani NP, et al. IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: implications for therapy. Hepatology (2014) 59:1944–53. doi:10.1002/hep.26979
260. Qian C, Jiang T, Zhang W, Ren C, Wang Q, Qin Q, et al. Increased IL-23 and IL-17 expression by peripheral blood cells of patients with primary biliary cirrhosis. Cytokine (2013) 64:172–80. doi:10.1016/j.cyto.2013.07.005
261. Rong G, Zhou Y, Xiong Y, Zhou L, Geng H, Jiang T, et al. Imbalance between T helper type 17 and T regulatory cells in patients with primary biliary cirrhosis: the serum cytokine profile and peripheral cell population. Clin Exp Immunol (2009) 156:217–25. doi:10.1111/j.1365-2249.2009.03898.x
262. Terasaki S, Nakanuma Y, Yamazaki M, Unoura M. Eosinophilic infiltration of the liver in primary biliary cirrhosis: a morphological study. Hepatology (1993) 17:206–12. doi:10.1002/hep.1840170208
263. Yamazaki K, Nakadate I, Suzuki K, Sato S, Masuda T. Eosinophilia in primary biliary cirrhosis. Am J Gastroenterol (1996) 91:516–22.
264. Nakamura A, Yamazaki K, Suzuki K, Sato S. Increased portal tract infiltration of mast cells and eosinophils in primary biliary cirrhosis. Am J Gastroenterol (1997) 92:2245–9.
265. Yamazaki K, Suzuki K, Nakamura A, Sato S, Lindor KD, Batts KP, et al. Ursodeoxycholic acid inhibits eosinophil degranulation in patients with primary biliary cirrhosis. Hepatology (1999) 30:71–8. doi:10.1002/hep.510300121
266. Takiguchi J, Ohira H, Rai T, Abe K, Takahashi A, Sato Y. Anti-eosinophil peroxidase antibodies detected in patients with primary biliary cirrhosis. Hepatol Res (2005) 32:33–7. doi:10.1016/j.hepres.2005.01.013
267. Irie J, Wu Y, Wicker LS, Rainbow D, Nalesnik MA, Hirsch R, et al. NOD.c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis. J Exp Med (2006) 203:1209–19. doi:10.1084/jem.20051911
268. Ambrosini YM, Yang G-X, Zhang W, Tsuda M, Shu S, Tsuneyama K, et al. The multi-hit hypothesis of primary biliary cirrhosis: polyinosinic-polycytidylic acid (poly I:C) and murine autoimmune cholangitis. Clin Exp Immunol (2011) 166:110–20. doi:10.1111/j.1365-2249.2011.04453.x
269. Concepcion AR, Medina JF. Mouse models of primary biliary cirrhosis. Curr Pharm Des (2015) 21:2401–13. doi:10.2174/1381612821666150316121622
270. Wirth HP, Meyenberger C, Altorfer J, Ammann R, Blum HE. [Eosinophilia in primary biliary cirrhosis: regression under therapy with ursodeoxycholic acid]. Schweiz Med Wochenschr (1994) 124:810–5.
271. Willart MAM, van Nimwegen M, Grefhorst A, Hammad H, Moons L, Hoogsteden HC, et al. Ursodeoxycholic acid suppresses eosinophilic airway inflammation by inhibiting the function of dendritic cells through the nuclear farnesoid X receptor. Allergy (2012) 67:1501–10. doi:10.1111/all.12019
272. Zachou K, Rigopoulou E, Liaskos C, Patsiaoura K, Makri E, Stathakis N, et al. Primary biliary cirrhosis presented as peripheral eosinophilia in asymptomatic women with or without elevated alkaline phosphatase. Eur J Gastroenterol Hepatol (2004) 16:425–8. doi:10.1097/00042737-200404000-00011
273. Gazzaruso C, Giordanetti S, De Cata P, Poggi G, Fratino P. Early diagnosis of primary biliary cirrhosis in type 1 diabetes: the possible role of eosinophilia. Diabetes Care (2003) 26:2963–4. doi:10.2337/diacare.26.10.2963
274. Long H, Zhang G, Wang L, Lu Q. Eosinophilic skin diseases: a comprehensive review. Clin Rev Allergy Immunol (2016) 50(2):189–213. doi:10.1007/s12016-015-8485-8
275. Weins AB, Biedermann T, Weiss T, Weiss JM. Wells syndrome. J Dtsch Dermatol Ges (2016) 14:989–93. doi:10.1111/ddg.13132
276. Simon H-U, Plötz S, Simon D, Seitzer U, Braathen LR, Menz G, et al. Interleukin-2 primes eosinophil degranulation in hypereosinophilia and Wells’ syndrome. Eur J Immunol (2003) 33:834–9. doi:10.1002/eji.200323727
277. Peters MS, Schroeter AL, Gleich GJ. Immunofluorescence identification of eosinophil granule major basic protein in the flame figures of Wells’ syndrome. Br J Dermatol (1983) 109:141–8. doi:10.1111/j.1365-2133.1983.tb07074.x
278. Wouters J, Waelkens E, Vandoninck S, Segaert S, van den Oord JJ. Mass spectrometry of flame figures. Acta Derm Venereol (2015) 95:734–5. doi:10.2340/00015555-2050
279. Soragni A, Yousefi S, Stoeckle C, Soriaga AB, Sawaya MR, Kozlowski E, et al. Toxicity of eosinophil MBP is repressed by intracellular crystallization and promoted by extracellular aggregation. Mol Cell (2015) 57:1011–21. doi:10.1016/j.molcel.2015.01.026
280. Fujimura T, Kambayashi Y, Furudate S, Kakizaki A, Aiba S. A possible mechanism in the recruitment of eosinophils and Th2 cells through CD163+ M2 macrophages in the lesional skin of eosinophilic cellulitis. Eur J Dermatol (2014) 24:180–5. doi:10.1684/ejd.2014.2283
281. Shulman LE. Diffuse fasciitis with eosinophilia: a new syndrome? Trans Assoc Am Physicians (1975) 88:70–86.
282. Shulman LE. Diffuse fasciitis with hypergammaglobulinemia and eosinophilia: a new syndrome? J Rheumatol (1984) 11(5):569–70.
283. Pinal-Fernandez I, Selva-O’ Callaghan A, Grau JM. Diagnosis and classification of eosinophilic fasciitis. Autoimmun Rev (2014) 13:379–82. doi:10.1016/j.autrev.2014.01.019
284. Lebeaux D, Sene D. Eosinophilic fasciitis (Shulman disease). Best Pract Res Clin Rheumatol (2012) 26:449–58. doi:10.1016/j.berh.2012.08.001
285. Lakhanpal S, Ginsburg WW, Michet CJ, Doyle JA, Moore SB. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum (1988) 17(4):221–31. doi:10.1016/0049-0172(88)90008-X
286. Endo Y, Tamura A, Matsushima Y, Iwasaki T, Hasegawa M, Nagai Y, et al. Eosinophilic fasciitis: report of two cases and a systematic review of the literature dealing with clinical variables that predict outcome. Clin Rheumatol (2007) 26(9):1445–51. doi:10.1007/s10067-006-0525-6
287. Lebeaux D, Francès C, Barete S, Wechsler B, Dubourg O, Renoux J, et al. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology (Oxford, England) (2012) 51(3):557–61. doi:10.1093/rheumatology/ker366
288. Deshpande V, Zen Y, Chan JK, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol (2012) 25(9):1181–92. doi:10.1038/modpathol.2012.72
289. Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet (London, England) (2015) 385(9976):1460–71. doi:10.1016/S0140-6736(14)60720-0
290. Della-Torre E, Lanzillotta M, Doglioni C. Immunology of IgG4-related disease. Clin Exp Immunol (2015) 181(2):191–206. doi:10.1111/cei.12641
291. Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol (2012) 22(1):21–30. doi:10.3109/s10165-011-0571-z
292. Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med (2012) 366(6):539–51. doi:10.1056/NEJMra1104650
293. Khosroshahi A, Carruthers MN, Deshpande V, Unizony S, Bloch DB, Stone JH. Rituximab for the treatment of IgG4-related disease: lessons from 10 consecutive patients. Medicine (2012) 91(1):57–66. doi:10.1097/MD.0b013e3182431ef6
294. Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy (2014) 69(2):269–72. doi:10.1111/all.12320
295. Ishida Y, Hayashi M, Higaki A, Matsumoto K, Iikura Y, Ishikawa J, et al. Hypereosinophilic syndrome with generalized myasthenia gravis. J Pediatr (1996) 128:369–72. doi:10.1016/S0022-3476(96)70284-6
296. Markusse HM, Schravenhoff R, Beerman H. Hypereosinophilic syndrome presenting with diarrhoea and anaemia in a patient with systemic lupus erythematosus. Neth J Med (1998) 52:79–81. doi:10.1016/S0300-2977(97)00086-7
297. Thomeer M, Moerman P, Westhovens R, Van den Eeckhout A, Dequeker J, Demedts M. Systemic lupus erythematosus, eosinophilia and Löffler’s endocarditis. An unusual association. Eur Respir J (1999) 13:930–3. doi:10.1034/j.1399-3003.1999.13d38.x
298. Kane SP. Ulcerative colitis with chronic liver disease, eosinophilia and auto-immune thyroid disease. Postgrad Med J (1977) 53:105–8. doi:10.1136/pgmj.53.616.105
299. Chaudhuri K, Dubey S, Zaphiropoulos G. Idiopathic hypereosinophilic syndrome in a patient with long-standing rheumatoid arthritis: a case report. Rheumatology (Oxford) (2002) 41:349–50. doi:10.1093/rheumatology/41.3.349
300. Bonanno D, Fedele R, Minciullo P, Quattrocchi P, Ferlazzo B. [Idiopathic hypereosinophilic syndrome associated with rheumatoid arthritis. A case report]. Reumatismo (2003) 55:181–3.
301. Habibagahi Z, Ali Nazarinia M, Aaflaki E, Ali Ostovan M, Hadi Bagheri M. Systemic lupus erythematosus and hyper-eosinophilic syndrome: an unusual association. West Indian Med J (2009) 58:69–71.
302. Aydoğdu S, Uçar O, Cetin M. A case of systemic lupus erythematosus presenting with hypereosinophilia and Loeffler endocarditis. Acta Cardiol (2010) 65:571–3. doi:10.2143/AC.65.5.2056245
303. Katz HT, Haque SJ, Hsieh FH. Pediatric hypereosinophilic syndrome (HES) differs from adult HES. J Pediatr (2005) 146:134–6. doi:10.1016/j.jpeds.2004.09.014
304. Terrier B, Fontaine H, Schmitz J, Perdu J, Hermine O, Varet B, et al. Coexistence and parallel evolution of hypereosinophilic syndrome, autoimmune hepatitis, and ulcerative colitis suggest common pathogenic features. Am J Gastroenterol (2007) 102:1132–4. doi:10.1111/j.1572-0241.2007.01180_9.x
305. Awano N, Ryu T, Yoshimura N, Takazoe M, Kitamura S, Tanaka M. Successful treatment of ulcerative colitis associated with hypereosinophilic syndrome/chronic eosinophilic leukemia. Intern Med (2011) 50:1741–5. doi:10.2169/internalmedicine.50.5569
306. Darki A, Kodali PP, McPheters JP, Virk H, Patel MR, Jacobs W. Hypereosinophilic syndrome with cardiac involvement in a pregnant patient with multiple sclerosis. Tex Heart Inst J (2011) 38:163–5.
307. Koneru S, Koshy G, Sharp C, Khalafallah AA. Hypereosinophilic syndrome associated with ulcerative colitis presenting with recurrent Loeffler’s endocarditis and left ventricular thrombus treated successfully with immune suppressive therapy and anticoagulation. BMJ Case Rep (2013) 2013. doi:10.1136/bcr-2013-200919
308. Rosenstein RK, Panush RS, Kramer N, Rosenstein ED. Hypereosinophilia and seroconversion of rheumatoid arthritis. Clin Rheumatol (2014) 33:1685–8. doi:10.1007/s10067-014-2566-6
309. Moxey J, Morrisroe K, Romas E. Rheumatoid arthritis heralded by the hypereosinophilic syndrome. Intern Med J (2016) 46:988–9. doi:10.1111/imj.13152
310. Kharabish A, Haroun D. Cardiac MRI findings of endomyocardial fibrosis (Loeffler’s endocarditis) in a patient with rheumatoid arthritis. J Saudi Heart Assoc (2015) 27:127–31. doi:10.1016/j.jsha.2014.11.002
311. Roufosse FE, Kahn J-E, Gleich GJ, Schwartz LB, Singh AD, Rosenwasser LJ, et al. Long-term safety of mepolizumab for the treatment of hypereosinophilic syndromes. J Allergy Clin Immunol (2013) 131:461–7.e1–5. doi:10.1016/j.jaci.2012.07.055
312. Lecouffe-Desprets M, Groh M, Bour B, Le Jeunne C, Puéchal X. Eosinophilic gastrointestinal disorders associated with autoimmune connective tissue disease. Joint Bone Spine (2016) 83(5):479–84. doi:10.1016/j.jbspin.2015.11.006
313. Peterson K, Firszt R, Fang J, Wong J, Smith KR, Brady KA. Risk of autoimmunity in EoE and families: a population-based cohort study. Am J Gastroenterol (2016) 111:926–32. doi:10.1038/ajg.2016.185
314. Sleiman PMA, Wang M-L, Cianferoni A, Aceves S, Gonsalves N, Nadeau K, et al. GWAS identifies four novel eosinophilic esophagitis loci. Nat Commun (2014) 5:5593. doi:10.1038/ncomms6593
315. Amin K, Lúdvíksdóttir D, Janson C, Nettelbladt O, Bjornsson E, Roomans GM, et al. Inflammation and structural changes in the airways of patients with atopic and nonatopic asthma. Am J Respir Crit Care Med (2000) 162:2295–301. doi:10.1164/ajrccm.162.6.9912001
316. Tedeschi A, Asero R. Asthma and autoimmunity: a complex but intriguing relation. Expert Rev Clin Immunol (2008) 4:767–76. doi:10.1586/1744666X.4.6.767
317. Taillé C, Grootenboer-Mignot S, Estellat C, Roy C, So SLK, Pretolani M, et al. Periplakin is a target for autoimmunity in asthma. Respir Res (2016) 17:126. doi:10.1186/s12931-016-0441-5
318. Kero J, Gissler M, Hemminki E, Isolauri E. Could TH1 and TH2 diseases coexist? Evaluation of asthma incidence in children with coeliac disease, type 1 diabetes, or rheumatoid arthritis: a register study. J Allergy Clin Immunol (2001) 108(5):781–3. doi:10.1067/mai.2001.119557
319. Sheikh A, Smeeth L, Hubbard R. There is no evidence of an inverse relationship between TH2-mediated atopy and TH1-mediated autoimmune disorders: lack of support for the hygiene hypothesis. J Allergy Clin Immunol (2003) 111:131–5. doi:10.1067/mai.2003.8
320. Hemminki K, Li X, Sundquist J, Sundquist K. Subsequent autoimmune or related disease in asthma patients: clustering of diseases or medical care? Ann Epidemiol (2010) 20:217–22. doi:10.1016/j.annepidem.2009.11.007
321. Tirosh A, Mandel D, Mimouni FB, Zimlichman E, Shochat T, Kochba I. Autoimmune diseases in asthma. Ann Intern Med (2006) 144(12):877–83. doi:10.7326/0003-4819-144-12-200606200-00004
322. Thormann A, Koch-Henriksen N, Laursen B, Sørensen PS, Magyari M. Inverse comorbidity in multiple sclerosis: findings in a complete nationwide cohort. Mult Scler Relat Disord (2016) 10:181–6. doi:10.1016/j.msard.2016.10.008
323. van den Berge M, Heijink HI, van Oosterhout AJM, Postma DS. The role of female sex hormones in the development and severity of allergic and non-allergic asthma. Clin Exp Allergy (2009) 39:1477–81. doi:10.1111/j.1365-2222.2009.03354.x
324. Knudsen TB, Thomsen SF, Nolte H, Backer V. A population-based clinical study of allergic and non-allergic asthma. J Asthma (2009) 46:91–4. doi:10.1080/02770900802524657
325. Leynaert B, Sunyer J, Garcia-Esteban R, Svanes C, Jarvis D, Cerveri I, et al. Gender differences in prevalence, diagnosis and incidence of allergic and non-allergic asthma: a population-based cohort. Thorax (2012) 67:625–31. doi:10.1136/thoraxjnl-2011-201249
326. Schleimer RP, Bochner BS. The effects of glucocorticoids on human eosinophils. J Allergy Clin Immunol (1994) 94:1202–13. doi:10.1016/0091-6749(94)90333-6
Keywords: innate immune system, autoimmune diseases, eosinophilia, bullous pemphigoid, neuromyelitis optica, eosinophilic granulomatosis with polyangiitis, myocarditis, inflammatory bowel disease
Citation: Diny NL, Rose NR and Čiháková D (2017) Eosinophils in Autoimmune Diseases. Front. Immunol. 8:484. doi: 10.3389/fimmu.2017.00484
Received: 10 February 2017; Accepted: 07 April 2017;
Published: 27 April 2017
Edited by:
Uday Kishore, Brunel University London, UKReviewed by:
David Voehringer, University of Erlangen-Nuremberg, GermanyCordula M. Stover, University of Leicester, UK
Copyright: © 2017 Diny, Rose and Čiháková. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela Čiháková, dcihako1@jhmi.edu