 Qiuji Wu1,2,3,4,5,6
Qiuji Wu1,2,3,4,5,6 Awatef Allouch1,2,3,4
Awatef Allouch1,2,3,4 Isabelle Martins1,2,3,4
Isabelle Martins1,2,3,4 Catherine Brenner7Nazanine Modjtahedi2,3,4
Catherine Brenner7Nazanine Modjtahedi2,3,4 Eric Deutsch2,3,4
Eric Deutsch2,3,4 Jean-Luc Perfettini1,2,3,4*
Jean-Luc Perfettini1,2,3,4*
- 1Cell Death and Aging Team, Gustave Roussy Cancer Campus, Villejuif, France
- 2Laboratory of Molecular Radiotherapy, INSERM U1030, Gustave Roussy Cancer Campus, Villejuif, France
- 3Gustave Roussy Cancer Campus, Villejuif, France
- 4Université Paris Saclay, Villejuif, France
- 5Department of Radiation and Medical Oncology, Zhongnan Hospital, Wuhan University, Wuhan, China
- 6Hubei Key Laboratory of Tumor Biological Behaviors, Zhongnan Hospital, Wuhan University, Wuhan, China
- 7Laboratory of Signaling and Cardiovascular Pathophysiology, INSERM UMR-S 1180, Université Paris-Sud, Faculté de Pharmacie, Châtenay-Malabry, France
Radiation therapy is one of the cornerstones of cancer treatment. In tumor cells, exposure to ionizing radiation (IR) provokes DNA damages that trigger various forms of cell death such as apoptosis, necrosis, autophagic cell death, and mitotic catastrophe. IR can also induce cellular senescence that could serve as an additional antitumor barrier in a context-dependent manner. Moreover, accumulating evidence has demonstrated that IR interacts profoundly with tumor-infiltrating immune cells, which cooperatively drive treatment outcomes. Recent preclinical and clinical successes due to the combination of radiation therapy and immune checkpoint blockade have underscored the need for a better understanding of the interplay between radiation therapy and the immune system. In this review, we will present an overview of cell death modalities induced by IR, summarize the immunogenic properties of irradiated cancer cells, and discuss the biological consequences of IR on innate immune cell functions, with a particular attention on dendritic cells, macrophages, and NK cells. Finally, we will discuss their potential applications in cancer treatment.
Introduction
Radiation therapy has been used in cancer treatment for over a century and represents one of the most efficient treatment modalities in the oncology field. Over 50% of all cancer patients receive radiation therapy during the course of their disease. Radiation therapy is widely used in many localized solid tumors, ranging from brain tumors, head and neck cancer, lung cancer, esophageal cancer, breast cancer, rectal cancer, and cervical cancer to prostate cancer among others. Radiation therapy is also used for the management of metastatic diseases such as brain or bone metastasis (1). Despite the fact that radiation therapy contributes to approximately 40% of all cancer cures (2), treatment failure is frequently observed due to local recurrence and distal metastasis (3).
Antitumor effects of radiation therapy are mainly due to the induction of an important cellular stress that triggers cell cycle arrest and leads eventually to either cellular senescence or cell death depending on the doses and the irradiation schedules used. Today it is also established that these local biological effects stimulate both innate and adaptive immune cells present in the tumor microenvironment and elicit an antitumor response at distance of the irradiated tumor sites. This biological process is also known as “abscopal” effect. The antitumor response elicited by radiation therapy can be enhanced by unleashing immune resistance mechanisms through the use of immune checkpoint blockers [such as anti-cytotoxic T-lymphocyte-associated protein-4 (anti-CTLA-4) or anti-PD-L1 antibodies], revealing that the modulation of the cross-talk between the biological effects of radiation therapy and the immune system is central for optimal tumor growth inhibition (4). The identification of rational approaches to design therapeutic strategies for the combination of radiation therapy with immunotherapy is still an unmet need. A better understanding of the molecular and cellular components of the emerging field of radio-oncoimmunology is central for the development of novel therapeutic approaches aiming at improving the effectiveness of radiotherapy.
In this review, we first highlight the diversity of cell death modalities elicited by ionizing radiation (IR) and focus on their immunogenic potentials. Next, we will briefly describe the roles of main innate immune cells in tumor microenvironment and then discuss the impacts of IR on various innate cells functions. We will also discuss how the modulation of innate immune cell functions by IR impacts on cancer treatment. A particular attention will be paid to dendritic cells (DCs), macrophages, natural killer (NK) cells, and myeloid-derived suppressor cells (MDSCs), since currently much more is known about these specific cell types.
Ionizing Radiation Dictates the Death and the Immunogenicity of Cancer Cells
Despite the fact that radiation therapy plays a central role in cancer treatment, the biological processes that are involved in the effectiveness of radiotherapy are poorly understood. Even though various forms of cell death, including apoptosis, autophagic cell death, mitotic catastrophe, and cellular senescence, have been detected after IR (5, 6), the precise contribution of these lethal events to the biological effects of IR remains elusive.
Ionizing Radiation Can Eliminate Cancer Cells through Distinct Cell Death Modalities
After exposure to IR, cancer cells may die through distinct modalities (Figure 1). Apoptosis, autophagic cell death, necrosis, and necroptosis are cell death modalities that have been extensively studied and characterized. A nomenclature mainly based on morphological, biochemical, and enzymatic criteria has been proposed and ordered lethal processes in three types, with apoptosis as the type I cell death modality, the autophagic cell death as the type II cell death, and necrosis or necroptosis as type III cell modalities (7).
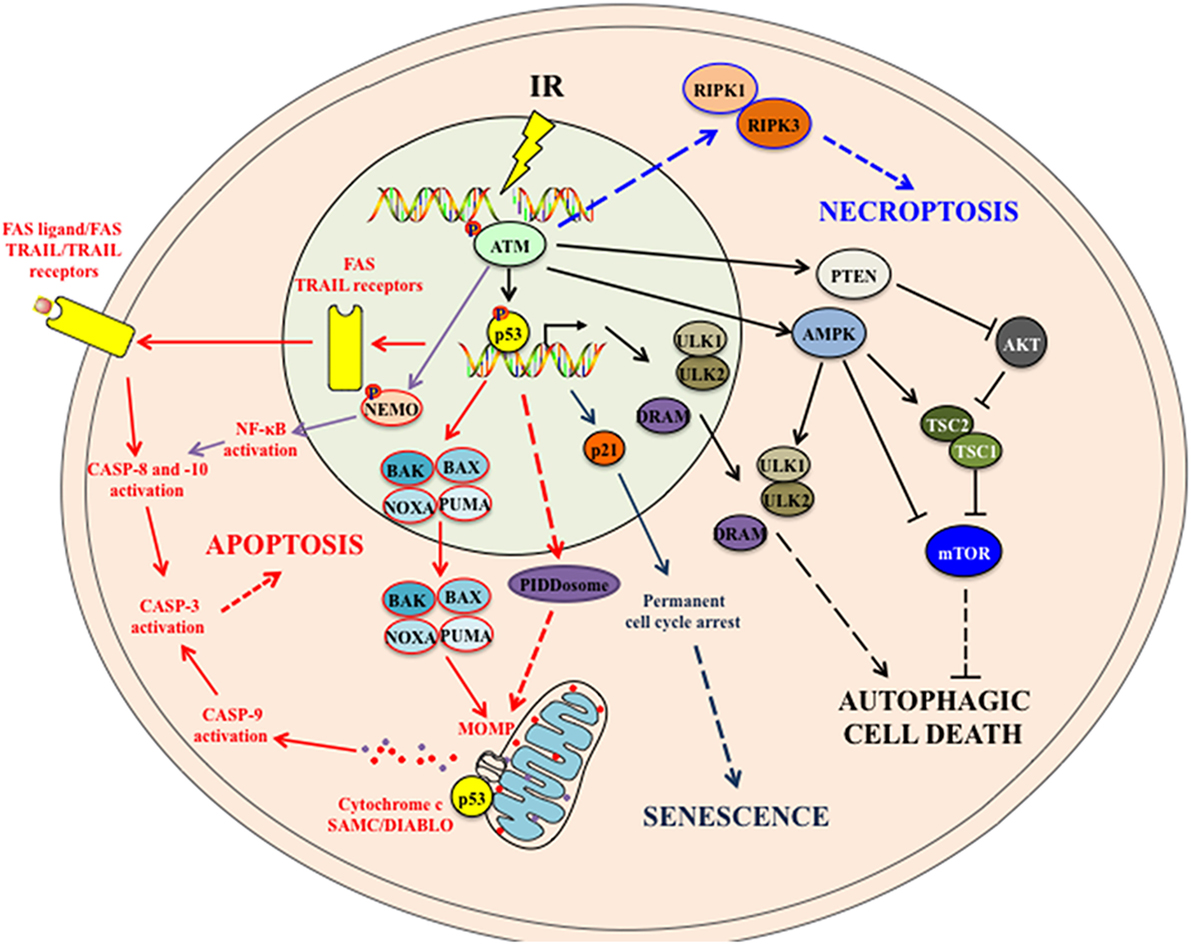
Figure 1. The intracellular signaling pathways associated with IR-induced cell death modalities. IR induces cellular apoptosis by activating both the intrinsic apoptotic pathway (through proapoptotic proteins-mitochondrial outer membrane permeabilization (MOMP)-Cytochrome c/SMAC/DIABLO release-caspase activation) and the extrinsic apoptotic pathway (through the upregulation of death receptors and the activation of downstream caspases). p53 also induces the expression of p53-inducible death domain (PIDD) protein in response to ionizing radiation, which acts as an effector of p53-dependent apoptosis. In addition, activated ATM following IR may activate the NF-κB pathway that in turn induces apoptosis. IR also leads to persistent DNA damages, which induce to p53 activation and p21 upregulation. p21 mediates cell cycle arrest and cellular senescence. Following IR, both activated ATM and p53 may trigger autophagic cell death to cells. ATM can activate AMPK and PTEN that suppress mTOR complex and induce autophagy. In addition, p53 upregulates the expression of autophagy-initiating kinase ULK1 and ULK2 and the damage-regulated autophagy modulator (DRAM) that subsequently induce autophagy. Note that it is still not certain whether this IR-induced autophagy would systematically lead to autophagic cell death. How IR induces necroptosis is still not fully understood. Some studies suggest that, in absence of caspase 8 activation, activated ATM following DNA damages (such as those induced by alkylating agent treatment) might mediate necroptosis by activating RIPK1 and RIPK3. See the main text for details. Abbreviations: AMPK, adenosine 5′-monophosphate (AMP)-activated protein kinase; ATM, ataxia-telangiectasia mutated; BAK, BCL-2 homologous antagonist/killer; BAX, BCL-2-associated X protein; IR, ionizing radiation; mTOR, mammalian target of rapamycin; NEMO, NF-kappa-B essential modulator; NF-κB, nuclear factor kappa B; PTEN, phosphatase and tensin homolog; PUMA, p53 upregulated modulator of apoptosis; RIPK, receptor-interacting protein kinase; TRAIL, TNF-related apoptosis-inducing ligand; TSC, tuberous sclerosis complex; ULK, UNC-51-like kinase.
Apoptosis, which is the principal death modality detected after IR, is described as a programmed cell death (PCD) with specific morphological alterations such as the chromatin condensation (also known as pyknosis), the nuclear fragmentation (also known as karyorhexis), the plasma membrane blebbing, and the formation of apoptotic bodies that could be engulfed by phagocytes (7). Apoptosis can be triggered by two distinct interlinked signaling pathways, namely the intrinsic pathway driven by intracellular cues (such as DNA damage or metabolic alterations) and the extrinsic pathway driven by extracellular signals such as death ligands. In both pathways, apoptotic signals lead to the activation of initiator caspases (CASP) (such as CASP-9 for the intrinsic pathway and CASP-8 and -10 for the extrinsic pathway), through proteolytic cleavages. Once activated, these initiator proteases trigger a cascade of CASP activation by cleaving and activating downstream effector CASP (including CASP-3, -6, and -7). Consequently, the proteolytic processing of numerous cytoplasmic or nuclear substrates of CASP triggers the typical morphology of apoptotic cells. Initially associated with the induction of apoptosis, the biological activities of CASP may also participate to cellular processes that are independent of cell death modalities (such as macrophage activation or differentiation of skeletal myoblasts and keratinocytes) (8), indicating that the detection of the enzymatic activity of caspases in response to IR may not always be indicative of the execution of an apoptotic death.
Irradiated cells may also die through type a II cell death modality that is known as autophagic cell death (9). Initially, misnamed as autophagy (10), the autophagic cell death is a biological process distinct from autophagy. Autophagy is an evolutionarily conserved lysosomal pathway that participates in the maintenance of the cellular homeostasis by preventing the accumulation of misfolded and aggregated proteins as well as damaged organelles (11). This process, which starts with the nucleation of phagophore forms, produces, through lipid incorporation, the autophagosomes that will fuse with lysosomes to become autolysosomes that orchestrate the degradation of the sequestered content. This autophagic flux that is tightly regulated by autophagy-related (ATG) proteins (12) may either favor tumor growth by favoring the survival of cancer cells under unfavorable conditions (such as hypoxia and nutriment deprivation) or contribute to tumor suppression by triggering the death of cancer cells when they are resistant to apoptosis (13). The autophagic cell death is defined as a cell death process that occurs after the induction of autophagy and is blocked by inhibitors of autophagy function and/or genetic inactivation of autophagic modulators (14). The autophagic protein ATG5 was recently implicated in the induction of IR-induced autophagic cell death (15). This process is distinct from the induction of autophagy after IR where the inhibition of the mammalian target of Rapamycin (mTOR) or the kinase AKT increases cytotoxicity of IR (13), confirming that autophagy may also contribute to the resistance of cancer cells to IR. We recently revealed that autophagy may also be involved in the enhancement of radiation therapy effects in immune-competent mice (16), highlighting the fact that the autophagic machinery can contribute to the regulation of cancer cell fate during cancer treatment.
Necrosis and necroptosis are stereotypical forms of type III cell death modalities that are also detected after IR. Necrosis was initially described as an unordered cell death mode associated with an organelle swelling, the rupture of their plasma membrane and the cell lysis. This “accidental” death leads to the passive release of intracellular components such as adenosine triphosphate (ATP) or high mobility group box 1 (HMGB1) protein and causes an intense inflammatory response. Low doses of IR generally eliminate cancer cells through apoptosis, whereas high doses of IR can lead to necrosis (17). The characterization of the molecular mechanisms of necroptosis (18) revealed the ability of IR to induce a programmed necrosis in anaplastic thyroid and adrenocortical cancer cells (19). Necroptosis and necrosis share morphological characteristics (such as plasma membrane rupture, cell swelling, and the release of intracellular components to extracellular milieu), but in contrast to necrotic process, necroptosis is a regulated process that can be induced in response to death receptor activation or after apoptosis inhibition and regulated by receptor-interacting protein kinases 1 and 3 (RIPK-1 and -3) or mixed lineage kinase domain-like (18).
Ionizing radiation has also been associated with cell death modalities that do not or partially exhibit the morphological features, the biochemical alterations and the enzymatic activities above described. These less studied cell death processes have been defined as atypical cell death modalities. The mitotic catastrophe is one of these processes that can be induced after radiotherapy. In response to IR, tumor cells carrying mutated or inactivated p53 cannot efficiently activate cell cycle checkpoints (in particular G2/M checkpoint) to initiate cell cycle arrest and carry out DNA repair. Consequently, cancer cells containing unrepaired DNA enter prematurely into mitosis and undergo mitotic catastrophe (5). In addition, the irradiation of human keratinocytes with doses ranging from 0.005 to 0.5 Gy induces early apoptosis and necrosis with a substantial population of cells that undergo G2/M arrest and ultimately die through mitotic cell death (20), indicating that non-tumoral cells may also undergo a mitotic death after IR. Alternatively, mitotic catastrophe may result from the hyper-amplification of centrosomes as a result of failure to repair the DNA damages induced by IR, and lead to multipolar mitotic spindles and abnormal chromosomal segregation (21).
In addition to canonical cell death modalities, cellular senescence can also be induced in dose-dependent and cell type-dependent manners and contribute to the elimination of cancer cells after IR (22, 23). Cellular senescence is a state during which cells undergo irreversible growth arrest in response to various stimuli including oncogene or tumor suppressor gene activation, epigenetic disruption, oxidative stress, as well as DNA damage elicited by IR or several chemotherapeutic agents (24). This cellular process, which is activated and maintained by p53/p21- or p16INK4a/RB-dependent pathways, is considered as an antitumor barrier that halts the proliferation of cancer cells (24, 25). Senescent cells remain metabolically active and can secrete numerous proinflammatory cytokines, chemokines, growth factors, and proteases that collectively are known as senescence-associated secretory phenotype (SASP). Once released, SASP can act in an autocrine and/or paracrine manner to induce numerous either beneficial or noxious activities including induction of angiogenesis, modulation of cell proliferation and stem cell activity, stimulation of epithelial–mesenchymal transition, promotion of chronic inflammation, depending on the specific pathophysiological context (24). Thus, while cellular senescence represents a cell-autonomous tumor suppressor mechanism, radiation-induced senescence could impact on the neighboring cancer cells and favor tumor survival and growth.
The Central Role of the Kinase Ataxia-Telangiectasia Mutated and the Tumor Suppressive Protein p53 in IR-Mediated Cell Killing
The kinase ataxia-telangiectasia mutated (ATM) and the tumor suppressive protein p53 play critical roles in coordinating DNA repair and cell fate determination when DNA damages are not repaired. Following sublethal doses of IR, DNA double-strand breaks are sensed by the MRE11-RAD50-NSB1 (MRN) complex, which in turn recruits and activates the apical kinases ATM mainly by favoring its autophosphorylation at serine 1981. ATM phosphorylates MRN complex, and other substrates including checkpoint kinase 2 (CHK2), p53-binding protein 1, and breast cancer gene 1 protein, which participate in sustaining DNA damage response signaling and in inducing S and G2/M arrest. ATM and CHK2 further phosphorylate p53, leading to its stabilization and activation of its transcription factor function. P53 upregulates the expression of p21 that induces the cell cycle arrest in G1. The initiation of DNA damage response by ATM and the induction of cell cycle arrest by p53 allow an efficient DNA repair process to restore genome integrity (26). However, when damages are not repaired efficiently, cell death programs are initiated.
The Kinase ATM Regulates Cell Death Modalities Elicited by IR
Upon IR-induced DNA DSBs, the kinase ATM and its downstream effector CHK2 kinase are phosphorylated and activate the tumor suppressive protein p53. The tumor suppressive protein p53 regulates through transcription-dependent or independent mechanisms the activation of both intrinsic and extrinsic apoptotic signaling pathways (27). Furthermore, the kinase ATM may also phosphorylate the NF-κB essential modulator (NEMO/IKK-γ) thus, leading to NF-κB activation and subsequent proapoptotic CASP-8 activation (28). The kinase ATM may also regulate autophagy and control the induction of cell death.
Although in some cases, the induction of autophagy via ATM-adenosine monophosphate-activated protein kinase (AMPK)-UNC-51-like kinase (ULK1) pathways was described to confer cytoprotective effect in Temozolomide-treated glioma cells (29), the regulation of autophagy through ATM-AMPK- tuberous sclerosis complex 2 (TSC2)–mediated suppression of mTORC1 by reactive nitrogen species lead to the loss of cell viability in breast cancer cells (30). In response to DNA damage induced by Topotecan, ATM phosphorylates phosphatase and tensin homolog and promotes its nuclear translocation and induces autophagy (31). Whether IR induces autophagy via similar signaling pathways should be further clarified. Instead, it is shown that ATM mediated IR-induced autophagy through activation of p38 mitogen-activated protein kinase (MAPK) and inhibition of mTOR pathway in human cervical cancer Hela cells. Pharmacological and genetic inactivation of ATM lead to decreased autophagy and hypersensitivity of Hela cells to IR (32). The role of ATM in IR-induced necroptosis has not been clearly demonstrated. ATM regulates alkylating DNA-damage agent-induced necroptosis through phosphorylation of histone protein H2AX (33). It is suggested that in response to DNA DSBs and in absence of CASP-8 activation, ATM might activate RIPK1 and RIPK3, which form necrosome and trigger necroptosis. However, this remains yet to be verified and clarified (28).
The Tumor Suppressive Protein p53 Contributes to IR-Induced Cell Death
The tumor suppressor p53 plays a center role in the regulation of numerous IR-induced cell death pathways. Following IR and DNA damages, the tumor suppressive protein 53 is phosphorylated at serine 15 and serine 20 by the kinases ATM and ATR and their downstream mediators CHK2 and CHK1. Once phosphorylated, p53 is dissociated from its negative regulator, the E3 ubiquitin ligase MDM2 and stabilized (34). Radiation can induce cell apoptosis via both intrinsic and extrinsic pathways. In the IR-induced intrinsic pathway, p53 induces the transcription of a number of proapoptotic proteins, including members of B-cell leukemia 2 (BCL-2) family such as the proapoptotic BCL-2-associated X protein (BAX) (35). Apart from its prominent role as a transcription factor, p53 also functions in the cytoplasm to induce apoptosis by directly activating the proapoptotic BAX and BAK (36). BH3-only proteins including p53 upregulated modulator of apoptosis (PUMA), NOXA and Bcl-2 interacting mediator of cell death (BIM) are also key initiators of apoptosis induced by IR (37–40). The protein p53 also induces the expression of p53-inducible death domain protein in response to IR, which acts as an effector of p53-dependent apoptosis (41). In addition, a number of antiapoptotic proteins are repressed, which further enhances IR-induced apoptosis. For instance, p53 negatively regulates Bcl-2 gene expression (42). P53 also transcriptionally represses the expression of antiapoptotic survivin gene (43). Both activation of proapoptotic proteins and repression of ant-apoptotic proteins by IR subsequently lead to the formation of BAX-BAK pores in the mitochondrial outer membrane, triggering mitochondrial outer membrane permeabilization (MOMP). MOMP facilitates the release of toxic proteins such as cytochrome c and the proapoptotic SMAC/DIABLO into the cytosol, leading to the activation of the intrinsic apoptotic pathway by activating the initiator CASP-9 (28). IR triggers also extrinsic apoptotic pathways by upregulating death receptors. IR upregulates Fas expression in tumor cells in a wild type p53-dependent manner (44, 45). IR also induces the expression of the TNF-related apoptosis-inducing ligand (TRAIL) receptors Killer/DR5 (46, 47). Other TRAIL receptors including DCR1, DCR2 and DR4 can also be induced by IR and are regulated by the wild-type p53 (48). The upregulation of these death receptors by IR may facilitate extrinsic apoptosis. The death receptors assemble into a multiprotein complex called death-inducing signaling complex (DISC) which in turn serves as a scaffold for the recruitment and activation of the initiator CASP-8 and CASP-10, leading to the activation of extrinsic apoptosis pathway. In addition to the upregulation of death receptors, IR also generated ceramides via acid sphingomyelinase, which in turn acts on the mitochondrion or activates the proapoptotic stress-activated protein kinase/c-Jun N-terminal kinase pathway and initiates apoptosis (49, 50).
Like its pleiotropic roles in regulating IR-induced apoptosis, p53 also modulates autophagy at multiple levels in IR-exposed cells. The transcription factor p53 upregulates the expression of human autophagy-initiating kinase ULK1 and ULK2 and induces autophagy in response to DNA damage. This p53-regulated autophagy ultimately leads to DNA-damage-induced cell death. Interestingly, p53 also induces the expression of the damage-regulated autophagy modulator (DRAM), a lysosomal protein that induces autophagy, leading to p53-dependent apoptosis, linking autophagy to p53 and damage-induced apoptosis (51).
The cellular senescence induced by IR is mainly mediated by p53. Persistent DNA damage activates p53 that induces p21 expression and cell cycle arrest (24). It is also shown that reactive oxygen species (ROS) are essential for P53-mediated cellular senescence after IR (52). Alteration of p53-dependent activity affects IR-induced cellular senescence. For example, activation of P53 with Nutlin-3a sensitized lung cancer cells to IR through induction of premature senescence (53). The nerve injury-induced protein 1 (Ninjurin1, Ninj1) is a P53 target following IR that in turn suppresses the expression of P53. Accordingly, inactivation of Ninj1 suppresses cell proliferation but enhances P53-mediated apoptosis and cellular senescence (54).
Ionizing Radiation of Tumor Cells also Favors the Development of Anticancer Immune Response
Apart from its direct genotoxic activity and tumor cell killing capacity, IR also enhances immune response via immunogenic properties of IR-induced cell death, upregulation of major histocompatibility complex (MHC) class I molecules and de novo tumor antigen production that collectively and coordinately prime and activate innate and adaptive immune systems to generate tumor-specific immune response.
Ionizing Radiation Induces Immunogenic Cell Death
Immunogenic cell death (ICD) consists of a functionally peculiar type of apoptotic demise triggered by various specific stimuli that is able to activate an adaptive immune response against dead cell-associated antigens. ICD involves the emission of a series of immunostimulatory damage-associated molecular patterns (DAMPs) including cell surface exposure of endoplasmic reticulum chaperone calreticulin (CRT), secretion of ATP, and release of HMGB1 protein, occurring in a defined spatiotemporal sequence. These ICD-associated DAMPs bind to specific receptors, recruits antigen-presenting cells (APCs) that process and present the dead cell-associated antigens to CD8+ cytotoxic T cells. Activated adaptive immune responses mediate direct antitumor effects and may acquire a memory phenotype that contributes to long-term tumor control (55).
Ionizing radiation is shown to effectively promote tumor ICD (56). For example, in a mouse B16F10 melanoma model, irradiation of cutaneous tumor prior to resection is shown to induce a specific antitumor immune response and significantly reduces lung metastasis after systemic challenge with untreated melanoma cells. Radiation induces CRT exposure on melanoma cell surface leading to increased DC phagocytosis of tumor cells (57). Radiation also induces the secretion of ATP and HMBG1 in both dying and live tumor cells, leading to increased antigen-specific cytotoxic T lymphocytes (CTL)-mediated tumor cell lysis (58). The combination of IR and hyperthermia treatment on colorectal cells induces cell surface expression as well as extracellular release of the chaperon molecule heat shock protein 70 (HSP70). HSP70 is able to promote DC maturation as revealed by an upregulation of the co-stimulatory molecule CD80 and the chemokine receptor CCR7. In addition, this combined treatment enhances phagocytic activities of macrophages and DCs along with an augmentation of proinflammatory cytokines [such as interleukin (IL)-8 and IL-12] secretion (59). Importantly, radiation-induced ICD has also been observed in clinical settings. In patients with esophageal squamous cell carcinoma receiving chemo-radiation therapy, tumor antigen-specific T cell response and elevated serum HMGB1 are detected in 38% of patients. HMGB1, which is significantly upregulated in the chemoradiation-treated tumors, is associated with better survival (60).
Ionizing Radiation Induces Tumor Antigen Expression
In addition, IR upregulates tumor associated-antigens and MHC class I complex that increase the recruitment of tumor antigen-specific T cells and activate T cell-mediated tumor killing (61–63). Early studies indicate that high-dose (from 25 to 100 Gy) gamma-irradiation induces the upregulation of the tumor rejection antigen (HSP gp96) on human cervical cancer cells that may increase immunogenicity of tumor cells (64). Other tumor-associated antigens such as carcinoembryonic antigen, colon-specific antigen, mucin-1 and MHC class I are upregulated by irradiation, which enhances antigen-specific T cell response (62, 65). Moreover, irradiation may also enhance FAS expression in tumor cells and sensitizes tumor cells to antigen-specific CTL killing via FAS/FAS ligand pathway. The combination of irradiation and CTL yields enhanced antitumor response (66). Therefore, irradiation may induce an “in situ vaccination” to improve antitumor immune response and also immunotherapy efficacy (61). These properties of IR are important as they contribute to the increased immunotherapy effects even in poorly immunogenic tumors (67).
Ionizing Radiation Modulates Mutational Burden during Anticancer Treatment
In tumor cells, IR provokes massive DNA damages. However, a small part of tumor cells eventually develop resistance to IR-mediated killing and accumulate incorrectly repaired/unrepaired DNA damages. This adds to tumor mutational burden and might enhance tumor aggressiveness. On the other hand, IR-induced mutations might provide a pool of tumor neoantigens that can be recognized and targeted by immune system (68). Indeed, it is shown that IR induces novel peptide synthesis in tumor cells and enhances antigen presentation by MHC class I molecules (63). Consequently, the specific expression of tumor neoantigens driven by tumor-specific mutations could be used as biomarkers of radiation therapy efficacy and could contribute to the development of novel therapeutic approaches (69).
The Tumor Microenvironment Irradiation Dictates Antitumor Innate Immune Response
Tumors are composed of tumor cells and tumor stroma. Tumor stroma contains cellular components (such as fibroblasts, endothelial cells, myeloid-derived cells, and lymphocytes), vascular and lymphatic vessels, non-cellular supporting structures, cytokine, and chemokine milieu. Innate immune cells such as DCs (DCs), macrophages, natural killer (NK) cells, neutrophils, and other myeloid-derived cells such as MDSCs have been found in various tumors (70).
Tumor-infiltrating DCs are found in many different types of cancers and are reported to be associated with both good and poor prognosis depending on the types of studied tumor. Although DCs represent the most important APCs to cross-present tumor antigens to effector T cells and to activate antitumor T cell response, these essential capacities are paralyzed by tumor-derived inhibitory factors including IL-10, TGF-β, vascular endothelial growth factor A (VEGF-A), and arginase (71). In many cases, tumor-infiltrating DCs gradually develop an immunosuppressive phenotype characterized by lower expression of co-stimulatory molecules, decreased antigen-presenting activity and upregulation of regulatory molecules and receptors such as PD-1 and TIM-3 within tumor-microenvironment, as the tumor grow from early stages to advanced diseases (71, 72). Thus, restoring immunostimulatory capacities of tumor-infiltrating DCs and administration of antigen-loaded autologous DC vaccines may have important implications in the development of more efficient antitumor therapies (73, 74).
Tumor-infiltrating macrophages or tumor-associated macrophages (TAMs) are the major myeloid cells found in the tumor area. TAMs are derived from peripheral blood monocytes and are recruited to the tumor area by various tumor-derived chemokines and cytokines such as colony stimulating factor-1 (CSF-1), C-C motif chemokine ligand 2 (CCL2), stromal cell-derived factor-1 (SDF-1), and VEGF-A. Other factors such as hypoxia and tumor cell metabolites also contribute to TAMs infiltration. TAMs are differentiated and skewed toward protumorigenic phenotype within distinct tumor microenvironment such as hypoxia, acidity, and immunosuppressive cytokine milieu (75). TAMs contribute to tumor growth, angiogenesis, invasiveness, and metastasis. TAMs also express high level of ligands for PD-1 and CTLA-4 that exert immunosuppressive functions on T cells. In addition TAMs interfere with T cells activation by depleting L-arginine in the milieu that is important for T cell receptor ζ chain expression. Other inhibitory mechanisms include induction of T cell apoptosis and production of anti-inflammatory cytokines such as IL-10 and TGF-β. In addition, TAMs induce the recruitment of immunosuppressive regulatory T cells through the expression of chemokines such CCL5, CCL20, and CCL22 (76). Thus, TAMs infiltration was associated with poor clinical outcomes in the majority of cancers (77). Reversing these adversary roles of TAMs will be important in improving anticancer therapy efficacies.
NK cells also play important roles in antitumor immunity. This is not only due to their direct tumor cell-killing function via granzyme B/perforin pathway and other death-receptor pathways, but also due to their ability to secrete a plethora of proinflammatory cytokines and chemokines that regulate and promote innate and adaptive immune response (78). However, as in the cases of DCs and macrophages, cytotoxic functions of NK cells are often impaired within tumor microenvironment. Various factors including cytokines and tumor metabolites directly inhibit maturation, proliferation, and functions of NK cells. In addition, other tumor-infiltrating cells such as MDSCs, TAMs, and regulatory T cells also inhibit the functions of NK cells (78). Accordingly, several NK cell-based in vivo approaches including the activation of NK cells with stimulatory cytokines, the induction of antibody-dependent cell-mediated cytotoxicity and IFN-γ production with tumor antigen-specific monoclonal antibodies, and the enhancement of the cytolytic activity of NK cells with blocking antibodies against inhibitory signals, may increase the chances for successful cancer treatment (79).
Myeloid-derived suppressor cells are a group of heterogeneous immature myeloid cells with suppressive activities on both innate and adaptive immunity. MDSCs differentiate from common myeloid progenitors and are often composed of cells at varied differentiation stages. MDSCs may be grouped into monocytic MDSCs and granulocytic MDSCs. Tumor-derived cytokines and growth factors such as VEGF, IL-6, granulocyte CSF, granulocyte-macrophage CSF, and other proinflammatory mediators such as IL-1β, IL-17, HMGB1, cyclooxygenase 2 (COX2), and prostaglandin E2 (PGE2) induce MDSCs accumulation, differentiation, proliferation, and acquisition of immunosuppressive functions (80, 81). MDSCs exert their immunosuppressive roles on T cells through multiple mechanisms, including secretion of anti-inflammatory IL-10 and transforming growth factor-β (TGF-β) that inhibit functions of T cells and NK cells, generation of ROS and nitric oxide (NO) that interfere with T cell proliferation and activation, and interaction with other immune cells such as TAMs that together create a protumorigenic microenvironment (80). Like TAMs, MDSCs express high levels of PD-L1 that induces T cell exhaustion and arginase I that depletes l-arginine that is essential for T cell activation. MDSCs induce also regulatory T cell accumulation and impair NK cell cytotoxicity (80). Therefore, MDSCs are prominent players that can support tumor growth and inhibit antitumor immunity and thus represent another major obstacle to overcome for effective antitumor therapies.
Other tumor-infiltrating innate immune cell such as neutrophils, Langerhans cells, and eosinophils that have emerged as potential players in tumor development are also promising targets to improve the efficacy of cancer treatment (82–85). For example, tumor-associated eosinophils have been revealed to play essential roles in orchestrating effective antitumor response. Eosinophils were shown to produce chemo-attractants that recruit effector T cells into the tumor. Eosinophils induce also macrophage activation and tumor vascular normalization that together contribute to tumor suppression (85). Currently, the role of eosinophils in tumor immunity is under more in depth investigation and the impact of radiation therapy on the functions of tumor-associated eosinophils remains largely unknown.
Ionizing Radiation Modifies Innate Immune Cell Migration and Homing
Tumor irradiation facilitates tumor antigen capturing and enhances tumor antigen presentation by DCs (86). Irradiation down regulates DC chemoattractant CCL21 expression in tumor tissue, which reduces the retention of DCs in tumor area after irradiation (86). On the other hand, irradiation also upregulates the expression of CCL21 on lymphatic vessels (87). These together may facilitate DCs homing to lymph nodes. These effects promote the ability of DCs to cross-prime and activate T cells (86). In contrast, another study demonstrated that gamma-irradiation (2Gy-8Gy) inhibited the migration murine DCs both in vitro and in vivo, in part due to a decreased expression of CCR7 and an increased apoptosis induced by irradiation in DCs (88).
Similarly, IR impacts profoundly on macrophage migration. A total of 10 Gy cranial γ-irradiation induces the expression of inflammatory mediators that serve as chemoattractant to promote the influx of peripheral blood-derived CCR2+ macrophages into the mouse brain (89). In the context of tumors, IR also induces macrophage recruitment. Tumor hypoxia due to a radiation-induced disruption of tumor vessels creates a transient hypoxic microenvironment and increases the expression of tumor-derived CSF-1, SDF-1 that together induces recruitment as well as anti-inflammatory activation of TAMs after radiation therapy (90–92). In addition, IR upregulates M-CSF expression by pancreatic ductal adenocarcinoma cells, which induces macrophage recruitment and differentiation toward M2-like phenotype (93). Of note, clinical studies also revealed that radiation therapy induced CSF-1 augmentation as well as the protumoral activation of macrophages, which were both associated with an impaired radiation therapy efficacy in prostate cancer (94). Combined radiation therapy with a anti-CSF-1 antibody or CSF-1R inhibitor treatment showed an improved antitumor effect (95) and will be significant to be further evaluated in clinical trials. Another important monocyte-chemoattractant CCL2 is also upregulated by IR and mediates macrophage recruitment into non-small cell lung cancer (96).
Irradiation-induced apoptosis increased neutrophils infiltration to the thymus (97). These recruited neutrophils were important in thymus regeneration after whole-body X-irradiation through their expression of SDF-1 (98, 99). Further characterization of the neutrophil infiltrating the tumors and the functional impact of irradiation on tumor-associated neutrophils should help for the development of novel therapeutic strategies.
Single high-dose (30 Gy) irradiation of the skin induced significant accumulation of eosinophils and the production of eosinophil-related cytokines such as IL4, IL-5, IL-13, IL-33, and CCL11 (100). A recent study showed that although synchroton microbean radiation treatment did not induce a significant difference in eosinophils infiltration pattern in murine mammary tumors as compared to synchroton broad-beam treatment, they did differentially regulate a subset of genes (Ear11, Ccl24, Ccl6, Ccl9) that were related to eosinophil functions and recruitment (101).
Phagocytosis and Antigen Presentation Are Altered after IR
The effect of in vitro direct irradiation on DCs depends on irradiation doses and DCs maturation states. For example, 5 Gy gamma-irradiation downregulated the expression of costimulatory receptors CD80/CD86 on immature monocyte-derived DCs but did not affect these receptors on mature DCs or their ability to stimulate autologous T cells (102). Another study showed that when irradiated at 30 Gy, CD86 expression was increased on immature DCs and decreased on mature DCs, while other markers remained unaffected by irradiation. However, in this study, irradiation impaired the stimulatory effects of both immature and mature DCs on the proliferation of allogenic T cells (103). Irradiation also affected DCs functions differentially in that it inhibited DCs response to endogenous antigens but enhanced DCs response to exogenous antigens (104). The divergent effects of irradiation on DCs were not due to defect in maturation or in presenting endogenous antigens, but were rather a result of the inhibition of proteasome function by irradiation. This in part accounted for the decreased endogenous antigen processing and possibly enhanced MHC class I molecules recycling and exogenous antigen presentation. Accordingly, irradiation abrogated DCs-induced endogenous antigen-specific T cell response and tumor suppression. On the contrary, irradiation enhanced the ability of DCs to activate T cell response to exogenous antigens and inhibited the growth of exogenous antigen-expressing tumors (104). Therefore, different irradiation doses, DCs maturation states and different types of antigens influence the outcomes of DCs activation following direct irradiation.
Like DCs, Langerhans cells residing in the skin and mucosa are endowed with potent antigen-presenting capacities at the first line of immune defense (105). An early study examining the prognostic role of Langerhans cell infiltration in uterine cervical squamous cell carcinoma patients treated with radiation therapy, showed that Langerhans cell infiltration was significantly associated with higher 5-year overall survival, suggesting that Langerhans cell infiltration after radiation therapy might mediate the immune response through their antigen presenting capacity and enhance the antitumor effect (106). Indeed, it was demonstrated that Langerhans cell infiltration after radiation therapy was associated with increased T cell infiltration and with improved local tumor control in cervical cancer (107, 108). However, in other settings, Langerhans cells may also limit the effect of radiation therapy. Epidermal Langerhans cells are more radioresistant than dermal DCs due to an overexpression of p21 and the capacity of the rapid repair of DNA damages induced by irradiation. Following radiation, Langerhans cells migrate to skin-draining lymph nodes in a CCR7-dependent manner. It is shown that Langerhans cell induced immunosuppressive regulatory T cell accumulation in the tumor is in part due to an upregulation of MHC class II expression on migratory Langerhans cells after irradiation. Consequently, Treg cells accumulation mediates immune suppression and tumor resistance to radiation therapy (105). Therefore, it appears that the in vivo impacts of IR on Langerhans cells might depend on the tumor types as well as the induction of different types of T cell infiltration (effector T cells or regulatory T cells).
The Differentiation and the Activation of Innate Immune Cells Is Modulated by IR
Radiation induces tumor cells death that leads to the release of tumor antigens, HSPs and other danger signals. These products then stimulate DC maturation. Although some in vitro studies arguing that IR compromises the stimulatory activities of DCs, in vivo models demonstrate that IR enhances the ability of DCs to capture tumor antigens (86) and promotes DC migration to draining lymph nodes in a way that is dependent on toll-like receptor signaling pathway, where they present tumor antigens to T cells and induce antigen-specific T cell response (109).
Various factors determine the impacts of IR on macrophage functions. One prominent factor is irradiation doses. For example, it was reported in many studies that low-dose (≤1 Gy) irradiation inhibited the proinflammatory activation of macrophages (110). Low-dose irradiation also inhibited oxidative burst in activated macrophages (111). On the contrary, high-dose (≥1 Gy) irradiation tends to induce a proinflammatory phenotype on macrophages with increased production of proinflammatory cytokines such as IL-1β and expression of induced nitric oxide synthase (iNOS) (112–114). Another important factor lies in macrophages. Macrophages from different mouse strains show variant intrinsic radiosensitivity. For example, irradiation enhanced anti-inflammatory characteristics of macrophages from C57BL/6 mice that are supposed to be more radioresistant, whereas macrophages from CBA/Ca mice that are more radiosensitive retain a proinflammatory feature after irradiation (115). Irradiation also differentially affected functions of macrophages from BALB/c and C57BL/6 mice (116).
In the tumor context, to date IR has been shown to either enhance the protumorigenic properties of TAMs or reprogram them toward antitumoral phenotypes in different experimental settings. For examples, IR induces M2-like protumorigenic TAMs that contribute to tumor recurrence and treatment failure. This is due to CSF-1 expression in murine prostate tumor cells that induced the recruitment of TAMs and MDSCs. Combined treatment with irradiation and CSF-1R inhibitor markedly improved antitumor efficacy (94). Macrophages from irradiated tumors show increased expression of arginase 1 (Arg1), COX2, and iNOS that promote tumor growth (117, 118). Macrophages also increased the expression of VEGF that led to tumor neovasculogenesis (119). However, there were also studies showing that radiation therapy could redirect TAMs from protumorigenic to antitumoral cells. For example, low-dose (2 Gy) whole-body irradiation induced iNOS expression and the production of proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), IL-12 (p70), and IFN-γ in peritoneal macrophages and TAMs (120). A recent study on murine insulinoma demonstrated that low-dose (2 Gy) irradiation induced iNOS expression in macrophages both in vitro and in vivo. This reprograming of proinflammatory macrophages by irradiation led to tumor vascular normalization and increased the effect of T cell immunotherapy (121). Furthermore, irradiation combined with 2-deoxy-d-glucose or hyperthermia also activated macrophages toward proinflammatory phenotype (122). These results suggest that depending on studied tumor models and the specificity of the used treatment regimen, irradiation may have different effects on TAMs functions that can in turn impact on tumor response and treatment outcomes.
The roles of neutrophils in tumor immunobiology are just emerging and little is known at the moment about the impact of IR on tumor-associated neutrophils. For instance, low-dose (0.512 Gy) irradiation suppressed myeloperoxidase activity and reactive nitrogen species generation in neutrophils from guinea pig (123). On the other hand, high-dose (20 Gy) irradiation induced oxygen free radicals in rat neutrophils (124). However, the effects of irradiation on human neutrophils are less known.
Ionizing Radiation Changes Cytokine Secretion Profiles
Different doses of irradiation yield different functional modulations to DCs. Low-dose irradiation seems to have divergent effects on DCs in many reports, possibly due to different experiment designs. For instance, low-dose at 0.05 Gy of gamma-irradiation of murine DCs significantly induced IL-2, IL-12, and interferon-γ (IFN-γ) production in DCs that promote T cells proliferation (125). At a dose of 0.2 Gy, gamma irradiation increases the surface expression of CD80, CD86, MHC class I and II molecules in murine DCs but inhibits their capacity of antigen uptake. In addition, this low-dose irradiation suppresses IL-12 production in DCs, but increases IL-10 production, implying a shift to immune tolerance (126). However, low-dose irradiation (from 0.05 to 1.0 Gy) did not affect surface markers or cytokine production in neither immature nor mature human DCs, and had no influence on the capacity of DCs to stimulate T cell proliferation (127), suggesting that the impact of low-dose irradiation on DCs function might be different from mouse to human.
High dose of irradiation also impacts on DCs differently. Irradiation at 30 Gy did not impact on DCs endocytic, phagocytic and migratory capacity but significantly inhibited IL-12 production by mature DCs while IL-10 production was unaffected (103). Inhibition of IL-12 expression in DCs by irradiation was in part mediated by an increase of IL-6 and activation of down stream signal transducer and activator of transcription 3, which led to inhibition of c-REL transcription factor (128). In addition, irradiated peptide-pulsed mature DCs showed impaired ability to prime naïve CTL (103). Likewise, gamma-irradiated (30 Gy) DCs derived from peripheral blood mononuclear cell of multiple sclerosis patients showed significantly reduced surface expression of costimulatory CD86 and had lower capacity to promote T cell proliferation as compared to non-irradiated DCs. These irradiated DCs also upregulated IL-2 and IL-4 secretion by T cells (129). Although high-dose irradiation might directly inhibit functions of DCs, another study showed that irradiation (3 × 5 Gy) induced tumor cell death that triggers DC maturation and production of proinflammatory cytokines such as IL-6, IL-8, IL-12p70, and TNF-α (130). Irradiation from 10 to 60 Gy also upregulates CD70 expression on mature DCs, an event that is correlated with the ability of these cells to stimulate T cell proliferation and IFN-γ production (131).
Although, in many in vitro studies, irradiation was shown to inhibit the antigen presentation capacity and the production of proinflammatory cytokines in DCs, in vivo studies seems to reflect opposite effects, possibly due to the complexity of the microenvironment that cooperatively influences the maturation and the activation of DCs. It might also be possible that combined direct and indirect effects of in vivo irradiation promote distinct DC functions in a context that significantly differed from in vitro irradiations. For example, although X-ray irradiation at 6 Gy significantly suppressed IL-23 secretion and slightly inhibited IL-12p70 production in DCs, irradiated fibroblast still interacted with and stimulated DCs to maintain IL-23/Th17 response (132). Thus, direct and indirect impacts of high-dose irradiation on DC activation could be quite different even opposite. This may explain why in many preclinical models, additive or synergic effects of DCs administration and radiation therapy were often documented.
As mentioned above, IR can directly modulate macrophage activation phenotype and their cytokine expression profiles. In addition, IR impacts on macrophage functions indirectly through the interaction of IR-induced cell death with macrophages. Irradiation-induced tumor cell death, in particular apoptosis, has previously been regarded as non-immunogenic (133). Apoptotic cells induced the secretion of anti-inflammatory cytokine IL-10 in macrophages (134). However, accumulating studies have also pointed out that apoptosis triggered by a subset of antitumor treatments may have immunogenic effects (133, 135). In addition, while the engulfment of apoptotic cells by non-stimulated or M2 macrophages induced the expression of anti-inflammatory macrophage markers such as TGF-β, such engulfment by M1 macrophages enhanced proinflammatory properties as indicated by an increased production of iNOS, superoxide, IL-6, and TNF-α (136). ICD induced by irradiation leads to the release of HMGB1 and the secretion of ATP (56). Upon ligation with TLR4, HMGB1 triggers NF-κB activation (137). ATP binds to P2X7 purinergic receptor and activates the NLRP3 inflammasome (138). NF-κB and NLRP3 inflammasome activation are both involved in the expression and maturation of proinflammatory cytokines such as IL-1β (139).
Innate Immune Cell-Mediated Cytotoxicity Is Affected by IR
Interestingly, apart from the enhancement of antigen-presenting capacity of DCs, irradiated tumor cells can induce the expression of granzyme B and perforin in DCs and directly stimulate DCs cytotoxicity to kill tumor cells (140). Although gamma-irradiation induces DCs accumulation in the tumor area that further activates tumor-specific T cell (141), it is noteworthy that radiation therapy induced upregulation of tumor antigens may also confer suppressive effects on DCs. For example, radiation-induced breast tumor-derived gamma-synuclein was shown to inhibit the expression of costimulatory molecules CD40 and CD86, and decrease the expression of proinflammatory cytokines in DCs. Gamma-synuclein-treated DCs also inhibit T cell proliferation but induce TGF-β production in T cells and increase the population of immunosuppressive regulatory T cells (142).
It was also demonstrated that in irradiated tumors, while the expression of costimulatory molecules is upregulated, the expression of PD-L1 and PD-L2 on DCs (140), which are known to inhibit antitumor immunity (143), are significantly reduced. Contradictorily, some other studies show that IR upregulate the expression of PD-L1 on tumor cells, DCs and TAMs that limit the antitumor effect of radiotherapy. The combined therapy of irradiation and anti-PD-L1 treatment resulted in activation of cytotoxic T cells and synergistic elimination of MDSCs by T cell-generated TNF, which is associated with delayed tumor growth (4, 144).
Irradiation can directly affect NK cell functions. In vitro studies showed that X-ray irradiation at 5 to 15 Gy could transiently increase human NK cell activity to lyse tumor cells that could be maintained in the presence of interferon (145). It was reported that the cytotoxic activity of human peripheral blood NK cells augmented following an irradiation dose at 1 Gy that peaked at 6 Gy and then decreased gradually when irradiation dose reached 16 Gy. Similarly other studies showed that human NK cells activity was enhanced following irradiation at 5–20 Gy (146, 147). In addition, low-dose gamma irradiation at ≤0.2 Gy induced expansion of NK cells, augmented NK cell cytotoxicity (148) and the expression of Fas ligands and perforin, and significantly increased the expression of IFN-γ and TNF-α in NK cells in a p38MAPK-dependent manner (149). Irradiation can also affect NK cell functions through the modulation of interaction between tumor cells and NK cells. For example, irradiation upregulated the expression of natural-killer group 2, member D (NKG2D) ligand and HSP70 in tumor cells that may increase susceptibilities of tumor cells to NK cell-mediated cytolytic attack (150, 151). Combined treatment of radiation therapy and histone deacetylase inhibitor was shown to increase the expression of NKG2D ligand expression and enhance the susceptibilities of lung cancer cells to NK cell cytotoxic activities (152). IR also triggers the release of second mitochondria-derived activator of caspase (Smac) from mitochondria that competes with X-linked inhibitor of apoptosis protein and enhances NK cell-mediated apoptosis of tumor cells (153).
Ionizing Radiation May Also Trigger the Elimination of Innate Immune Cells
Radiation therapy is a prominent source of myelosuppression during cancer treatment, especially when combined with chemotherapy. This is in particular the case when radiation therapy is delivered to pelvis such as for cervical cancer, rectal cancer and prostate cancer, during which a large proportion of bone marrow is affected (154). Neutrophils are the major innate immune cells that are decreased by radiation therapy. Up to 90 and 80% of cervical cancer patients underwent a grade II or worse neutropenia during 3D conformal radiotherapy and intense-modulated radiation therapy, respectively (155).
Myeloid-derived suppressor cells have been shown to accumulate in many cancer patients. In hepatocellular carcinoma, the basal level of CD14+HLA−DR−/low MDSCs is higher than that in healthy controls. Radiotherapy significantly reduced the frequency of CD14+HLA−DR−/low MDSCs that was negatively correlated to patient overall survival, indicating that a reduction of MDSCs after radiotherapy could be used as a prognostic factor in hepatocellular carcinoma patients (156). Radiation therapy of tumors also leads to a decrease of peripheral MDSCs that re-expand upon tumor recurrence. Declined MDSCs population was associated with increased T cells proliferation and T cells response to tumor-associated antigens (157). In patients with oligometastases, stereotactic body radiotherapy (SBRT) when combined with the multitargeted tyrosine kinase inhibitor Sunitinib, induced a decrease of peripheral blood CD33+CD14+CD16+ monocytic MDSCs as well as Tregs and B cells, along with an increase of Tbet expression in primary CD4+ and CD8+ T cells, which was associated with improved progression-free survival. A reduction of monocytic MDSC in this setting thus may be considered a valuable biomarker for predicting clinical outcomes (158).
Early studies have shown that gamma-ray or X-ray irradiation also decreases the number of epidermal Langerhans cell in human skin (159, 160). Similarly, in a dose-dependent manner, irradiation depleted mouse epidermal Langerhans cells population that was recovered after the stop of irradiation (161–163).
Effective DNA damage sensing followed by efficient and faithful DNA repair to restore genome integrity is vital for cell functions and cell survival, as reflected by the fact that germline mutation of ATM and TP53 caused hereditary defects in DNA damage signaling and repair pathway lead to predisposition of cancer and many other diseases such as immune deficiency (164).
Dysfunction in ATM (murine analog of human ATM) results in the accumulation of unrepaired DNA in the cytoplasm upon DNA damage. These free DNA fragments are sensed by STING (stimulator of interferon genes)-mediated pathway, which activates the expression of Toll-like receptors (TLRs), RIG-I-like receptors and promotes induction of type I interferons, leading to enhanced antiviral and antibacterial response in Atm−/− mice (165). DNA DSBs also activate the transcription factor interferon regulatory factor 3 (IRF-3) in a manner dependent on ATM-IKKα/β, leading to cell-autonomous production of interferon β (166). Further, persistent ROS are shown to induce chronic activation of ATM that triggers a continuous activation of NF-κB pathways, contributing to aggressive phenotype of cancer cells (167). Indeed, ATM has been shown to regulate NF-κB activity by mediating nuclear NEMO SUMOylation and subsequent ubiquitination, an event that leads to NEMO relocation to the cytoplasm and NF-κB activation through the canonical pathway (168).
P53 was recently demonstrated to participate in the regulation of macrophages functions. P53 is involved in the proinflammatory macrophage activation and in addition, P53 suppresses the anti-inflammation phenotype of macrophages (15). P53 cooperates with NF-κB to induce proinflammatory genes expression in macrophages (169). P53 may directly activate IRF-5 (170), a dominant transcription factor in proinflammatory macrophage activation (171).
Concluding Remarks
While interventions aiming at improving the efficacy of IR by the combination T cell directed approaches (such as PD-1/PD-L1 blockades) and IR are growing in the clinic, there is mounting evidence that IR also primes and induces the activation of an adaptive antitumor immunity through the induction of ICD, the release of tumor antigen, the stimulation of inflammatory response, and the modulation of immune cell functions, which can facilitate and enhance immunotherapy effects and potentially reduce immunotherapy-related adverse events (Figure 2). However, the impact of radiation on innate immune cells may be tumor type dependent and vary in relation with the specificity of the used treatment protocol. On the other hand, many reports indicate that in certain cases radiation therapy creates a more immunosuppressive microenvironment due to the upregulation of PD-L1, a transient potentiation of tumor hypoxia, or an alternative activation of TAMs, indicating that the addition of immunotherapy to the treatment protocol can overcome these obstacles, increase radiosensitivity and may lead to an enhanced systemic effect of radiation therapy. For these reasons, there is a strong rationality for combining radiation with immunotherapy for cancer treatment. A deeper understanding of the molecular mechanisms that are involved in the modulation of innate immune cell functions, particularly in the context of tumor microenvironment, is thus fundamental for the development of new therapeutic strategies targeting the inhibitory effects of tumor-infiltrating cells and for the restoration of their antitumor activities.
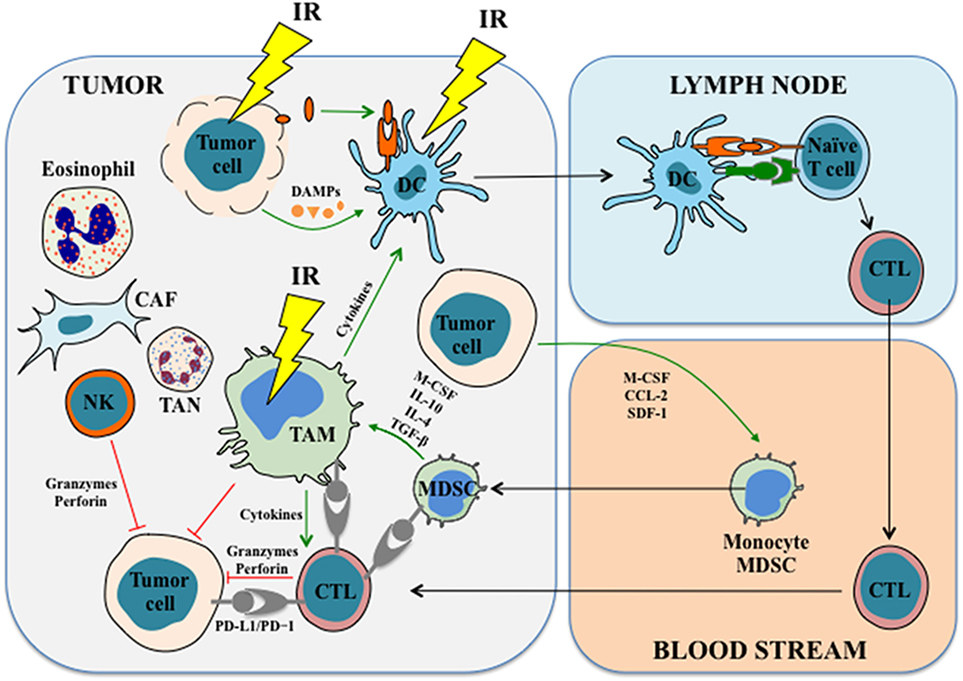
Figure 2. The effects of IR on immune cells. IR induces immunogenic cell death in tumor cells, leading to the release of tumor antigens and damage-associated molecular patterns (DAMPs), which in turn prime and activate antigen-presenting cells (APCs) such as dendritic cells. APCs stimulate and activate T cells in the lymph nodes and lead to the generation as well as the proliferation of tumor antigen-specific cytotoxic T cells (CTLs), which then migrate into the tumor to exert antitumor functions and mount the antitumor immune response. IR also has profound impacts on tumor-associated macrophages (TAMs). For example, IR induces macrophage infiltration and differentiation in the tumor. In some cases, IR promotes proinflammatory macrophage activation and enhances their immunostimulatory and tumoricidal activities. In addition, accumulating studies revealed that IR might modulate functions of other innate immune cells, such as myeloid-derived suppressor cells, NK cells, tumor-associated neutrophils, and probably other types of cells. See the main text for details. Abbreviations: CAF, cancer-associated fibroblast; CCL-2, chemokine (C–C motif) ligand-2; CTL, cytotoxic T lymphocyte; DC, dendritic cell; IL, interleukin; IR, ionizing radiation; M-CSF, macrophage colony-stimulating factor; MDSC, myeloid-derived suppressor cell; NK cell, natural killer cell; PD-1, programmed cell death protein 1; PD-L1, programmed cell death ligand 1; SDF-1, stromal cell-derived factor-1; TAN, tumor-associated neutrophil; TGF-β, transforming growth factor β.
Author Contributions
QW, AA, IM, CB, NM, ED, and JLP provided advices and wrote the article. QW designed and produced the figures. JLP edited the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hoegler D. Radiotherapy for palliation of symptoms in incurable cancer. Curr Probl Cancer (1997) 21:129–83. doi: 10.1016/S0147-0272(97)80004-9
2. Baskar R, Lee KA, Yeo R, Yeoh KW. Cancer and radiation therapy: current advances and future directions. Int J Med Sci (2012) 9:193–9. doi:10.7150/ijms.3635
3. Vatner RE, Formenti SC. Myeloid-derived cells in tumors: effects of radiation. Semin Radiat Oncol (2015) 25:18–27. doi:10.1016/j.semradonc.2014.07.008
4. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature (2015) 520:373–7. doi:10.1038/nature14292
5. Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumour Biol (2010) 31:363–72. doi:10.1007/s13277-010-0042-8
6. Yang Y, Yang Y, Yang X, Zhu H, Guo Q, Chen X, et al. Autophagy and its function in radiosensitivity. Tumour Biol (2015) 36:4079–87. doi:10.1007/s13277-015-3496-x
7. Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ (2007) 14:1237–43. doi:10.1038/sj.cdd.4402148
8. Perfettini JL, Kroemer G. Caspase activation is not death. Nat Immunol (2003) 4:308–10. doi:10.1038/ni0403-308
9. Cui L, Song Z, Liang B, Jia L, Ma S, Liu X. Radiation induces autophagic cell death via the p53/DRAM signaling pathway in breast cancer cells. Oncol Rep (2016) 35:3639–47. doi:10.3892/or.2016.4752
10. Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol (2008) 9:1004–10. doi:10.1038/nrm2529
11. Dunn WA Jr. Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J Cell Biol (1990) 110:1935–45. doi:10.1083/jcb.110.6.1935
12. Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ (2015) 22:367–76. doi:10.1038/cdd.2014.143
13. Zeng X, Kinsella TJ. Impact of autophagy on chemotherapy and radiotherapy mediated tumor cytotoxicity: “To Live or not to Live”. Front Oncol (2011) 1:30. doi:10.3389/fonc.2011.00030
14. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell (2008) 132:27–42. doi:10.1016/j.cell.2007.12.018
15. Jo GH, Bogler O, Chwae YJ, Yoo H, Lee SH, Park JB, et al. Radiation-induced autophagy contributes to cell death and induces apoptosis partly in malignant glioma cells. Cancer Res Treat (2015) 47:221–41. doi:10.4143/crt.2013.159
16. Ko A, Kanehisa A, Martins I, Senovilla L, Chargari C, Dugue D, et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ (2014) 21:92–9. doi:10.1038/cdd.2013.124
17. Rainaldi G, Ferrante A, Indovina PL, Santini MT. Induction of apoptosis or necrosis by ionizing radiation is dose-dependent in MG-63 osteosarcoma multicellular spheroids. Anticancer Res (2003) 23:2505–18.
18. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol (2010) 11:700–14. doi:10.1038/nrm2970
19. Nehs MA, Lin CI, Kozono DE, Whang EE, Cho NL, Zhu K, et al. Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery (2011) 150:1032–9. doi:10.1016/j.surg.2011.09.012
20. Jella KK, Garcia A, Mcclean B, Byrne HJ, Lyng FM. Cell death pathways in directly irradiated cells and cells exposed to medium from irradiated cells. Int J Radiat Biol (2013) 89:182–90. doi:10.3109/09553002.2013.734942
21. Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol (2011) 12:385–92. doi:10.1038/nrm3115
22. Sabin RJ, Anderson RM. Cellular senescence – its role in cancer and the response to ionizing radiation. Genome Integr (2011) 2:7. doi:10.1186/2041-9414-2-7
23. Suzuki M, Boothman DA. Stress-induced premature senescence (SIPS) – influence of SIPS on radiotherapy. J Radiat Res (2008) 49:105–12. doi:10.1269/jrr.07081
24. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol (2013) 75:685–705. doi:10.1146/annurev-physiol-030212-183653
25. Campisi J, d’adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol (2007) 8:729–40. doi:10.1038/nrm2233
26. Mckinnon PJ. DNA repair deficiency and neurological disease. Nat Rev Neurosci (2009) 10:100–12. doi:10.1038/nrn2559
27. Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene (2003) 22:9030–40. doi:10.1038/sj.onc.1207116
28. Matt S, Hofmann TG. The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell Mol Life Sci (2016) 73:2829–50. doi:10.1007/s00018-016-2130-4
29. Zou Y, Wang Q, Li B, Xie B, Wang W. Temozolomide induces autophagy via ATMAMPKULK1 pathways in glioma. Mol Med Rep (2014) 10:411–6. doi:10.3892/mmr.2014.2151
30. Tripathi DN, Chowdhury R, Trudel LJ, Tee AR, Slack RS, Walker CL, et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci U S A (2013) 110:E2950–7. doi:10.1073/pnas.1307736110
31. Chen JH, Zhang P, Chen WD, Li DD, Wu XQ, Deng R, et al. ATM-mediated PTEN phosphorylation promotes PTEN nuclear translocation and autophagy in response to DNA-damaging agents in cancer cells. Autophagy (2015) 11:239–52. doi:10.1080/15548627.2015.1009767
32. Liang N, Jia L, Liu Y, Liang B, Kong D, Yan M, et al. ATM pathway is essential for ionizing radiation-induced autophagy. Cell Signal (2013) 25:2530–9. doi:10.1016/j.cellsig.2013.08.010
33. Baritaud M, Cabon L, Delavallee L, Galan-Malo P, Gilles ME, Brunelle-Navas MN, et al. AIF-mediated caspase-independent necroptosis requires ATM and DNA-PK-induced histone H2AX Ser139 phosphorylation. Cell Death Dis (2012) 3:e390. doi:10.1038/cddis.2012.120
34. Shi D, Gu W. Dual roles of MDM2 in the regulation of p53: ubiquitination dependent and ubiquitination independent mechanisms of MDM2 repression of p53 activity. Genes Cancer (2012) 3:240–8. doi:10.1177/1947601912455199
35. Zhan Q, Fan S, Bae I, Guillouf C, Liebermann DA, O’connor PM, et al. Induction of bax by genotoxic stress in human cells correlates with normal p53 status and apoptosis. Oncogene (1994) 9:3743–51.
36. Perfettini JL, Kroemer RT, Kroemer G. Fatal liaisons of p53 with Bax and Bak. Nat Cell Biol (2004) 6:386–8. doi:10.1038/ncb0504-386
37. Erlacher M, Michalak EM, Kelly PN, Labi V, Niederegger H, Coultas L, et al. BH3-only proteins Puma and Bim are rate-limiting for gamma-radiation- and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood (2005) 106:4131–8. doi:10.1182/blood-2005-04-1595
38. Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell (2003) 4:321–8. doi:10.1016/S1535-6108(03)00244-7
39. Michalak EM, Villunger A, Adams JM, Strasser A. In several cell types tumour suppressor p53 induces apoptosis largely via Puma but Noxa can contribute. Cell Death Differ (2008) 15:1019–29. doi:10.1038/cdd.2008.16
40. Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science (2003) 302:1036–8. doi:10.1126/science.1090072
41. Lin Y, Ma W, Benchimol S. Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat Genet (2000) 26:122–7. doi:10.1038/79102
42. Miyashita T, Harigai M, Hanada M, Reed JC. Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer Res (1994) 54:3131–5.
43. Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem (2002) 277:3247–57. doi:10.1074/jbc.M106643200
44. Sheard MA, Vojtesek B, Janakova L, Kovarik J, Zaloudik J. Up-regulation of Fas (CD95) in human p53wild-type cancer cells treated with ionizing radiation. Int J Cancer (1997) 73:757–62. doi:10.1002/(SICI)1097-0215(19971127)73:5<757::AID-IJC24>3.0.CO;2-1
45. Sheard MA, Uldrijan S, Vojtesek B. Role of p53 in regulating constitutive and X-radiation-inducible CD95 expression and function in carcinoma cells. Cancer Res (2003) 63:7176–84.
46. Wu GS, Burns TF, Mcdonald ER III, Jiang W, Meng R, Krantz ID, et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet (1997) 17:141–3. doi:10.1038/ng1097-141
47. Wu GS, Kim K, El-Deiry WS. KILLER/DR5, a novel DNA-damage inducible death receptor gene, links the p53-tumor suppressor to caspase activation and apoptotic death. Adv Exp Med Biol (2000) 465:143–51. doi:10.1007/0-306-46817-4_13
48. Guan B, Yue P, Clayman GL, Sun SY. Evidence that the death receptor DR4 is a DNA damage-inducible, p53-regulated gene. J Cell Physiol (2001) 188:98–105. doi:10.1002/jcp.1101
49. Bonnaud S, Niaudet C, Pottier G, Gaugler MH, Millour J, Barbet J, et al. Sphingosine-1-phosphate protects proliferating endothelial cells from ceramide-induced apoptosis but not from DNA damage-induced mitotic death. Cancer Res (2007) 67:1803–11. doi:10.1158/0008-5472.CAN-06-2802
50. Kolesnick R, Fuks Z. Radiation and ceramide-induced apoptosis. Oncogene (2003) 22:5897–906. doi:10.1038/sj.onc.1206702
51. Crighton D, Wilkinson S, O’prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell (2006) 126:121–34. doi:10.1016/j.cell.2006.05.034
52. Fitzgerald AL, Osman AA, Xie TX, Patel A, Skinner H, Sandulache V, et al. Reactive oxygen species and p21Waf1/Cip1 are both essential for p53-mediated senescence of head and neck cancer cells. Cell Death Dis (2015) 6:e1678. doi:10.1038/cddis.2015.44
53. Luo H, Yount C, Lang H, Yang A, Riemer EC, Lyons K, et al. Activation of p53 with Nutlin-3a radiosensitizes lung cancer cells via enhancing radiation-induced premature senescence. Lung Cancer (2013) 81:167–73. doi:10.1016/j.lungcan.2013.04.017
54. Cho SJ, Rossi A, Jung YS, Yan W, Liu G, Zhang J, et al. Ninjurin1, a target of p53, regulates p53 expression and p53-dependent cell survival, senescence, and radiation-induced mortality. Proc Natl Acad Sci U S A (2013) 110:9362–7. doi:10.1073/pnas.1221242110
55. Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology (2014) 3:e955691. doi:10.4161/21624011.2014.955691
56. Galluzzi L, Kepp O, Kroemer G. Immunogenic cell death in radiation therapy. Oncoimmunology (2013) 2:e26536. doi:10.4161/onci.26536
57. Perez CA, Fu A, Onishko H, Hallahan DE, Geng L. Radiation induces an antitumour immune response to mouse melanoma. Int J Radiat Biol (2009) 85:1126–36. doi:10.3109/09553000903242099
58. Gameiro SR, Jammeh ML, Wattenberg MM, Tsang KY, Ferrone S, Hodge JW. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget (2014) 5:403–16. doi:10.18632/oncotarget.1719
59. Schildkopf P, Frey B, Ott OJ, Rubner Y, Multhoff G, Sauer R, et al. Radiation combined with hyperthermia induces HSP70-dependent maturation of dendritic cells and release of pro-inflammatory cytokines by dendritic cells and macrophages. Radiother Oncol (2011) 101:109–15. doi:10.1016/j.radonc.2011.05.056
60. Suzuki Y, Mimura K, Yoshimoto Y, Watanabe M, Ohkubo Y, Izawa S, et al. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res (2012) 72:3967–76. doi:10.1158/0008-5472.CAN-12-0851
61. Vanpouille-Box C, Pilones KA, Wennerberg E, Formenti SC, Demaria S. In situ vaccination by radiotherapy to improve responses to anti-CTLA-4 treatment. Vaccine (2015) 33:7415–22. doi:10.1016/j.vaccine.2015.05.105
62. Garnett CT, Palena C, Chakraborty M, Tsang KY, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res (2004) 64:7985–94. doi:10.1158/0008-5472.CAN-04-1525
63. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med (2006) 203:1259–71. doi:10.1084/jem.20052494
64. Santin AD, Hermonat PL, Ravaggi A, Chiriva-Internati M, Hiserodt JC, Batchu RB, et al. The effects of irradiation on the expression of a tumour rejection antigen (heat shock protein gp96) in human cervical cancer. Int J Radiat Biol (1998) 73:699–704. doi:10.1080/095530098141951
65. Modrak DE, Gold DV, Goldenberg DM, Blumenthal RD. Colonic tumor CEA, CSAp and MUC-1 expression following radioimmunotherapy or chemotherapy. Tumour Biol (2003) 24:32–9. doi:10.1159/000070658
66. Chakraborty M, Abrams SI, Camphausen K, Liu K, Scott T, Coleman CN, et al. Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J Immunol (2003) 170:6338–47. doi:10.4049/jimmunol.170.12.6338
67. Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res (2005) 11:728–34.
68. Kalbasi A, June CH, Haas N, Vapiwala N. Radiation and immunotherapy: a synergistic combination. J Clin Invest (2013) 123:2756–63. doi:10.1172/JCI69219
69. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi:10.1126/science.aaa4971
70. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer (2012) 12:298–306. doi:10.1038/nrc3245
71. Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL. Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol (2015) 194:2985–91. doi:10.4049/jimmunol.1403134
72. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol (2012) 13:832–42. doi:10.1038/ni.2376
73. Anguille S, Smits EL, Lion E, Van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol (2014) 15:e257–67. doi:10.1016/S1470-2045(13)70585-0
74. Krempski J, Karyampudi L, Behrens MD, Erskine CL, Hartmann L, Dong H, et al. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol (2011) 186:6905–13. doi:10.4049/jimmunol.1100274
75. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23:549–55. doi:10.1016/S1471-4906(02)02302-5
76. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity (2014) 41:49–61. doi:10.1016/j.immuni.2014.06.010
77. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One (2012) 7:e50946. doi:10.1371/journal.pone.0050946
78. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol (2016) 17:1025–36. doi:10.1038/ni.3518
79. Pietra G, Vitale C, Pende D, Bertaina A, Moretta F, Falco M, et al. Human natural killer cells: news in the therapy of solid tumors and high-risk leukemias. Cancer Immunol Immunother (2016) 65:465–76. doi:10.1007/s00262-015-1744-y
80. Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res (2015) 128:95–139. doi:10.1016/bs.acr.2015.04.002
81. De Sanctis F, Solito S, Ugel S, Molon B, Bronte V, Marigo I. MDSCs in cancer: conceiving new prognostic and therapeutic targets. Biochim Biophys Acta (2016) 1865:35–48. doi:10.1016/j.bbcan.2015.08.001
82. Stoitzner P, Sparber F, Tripp CH. Langerhans cells as targets for immunotherapy against skin cancer. Immunol Cell Biol (2010) 88:431–7. doi:10.1038/icb.2010.31
83. Kim J, Bae JS. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediators Inflamm (2016) 2016:6058147. doi:10.1155/2016/6058147
84. Powell DR, Huttenlocher A. Neutrophils in the tumor microenvironment. Trends Immunol (2016) 37:41–52. doi:10.1016/j.it.2015.11.008
85. Carretero R, Sektioglu IM, Garbi N, Salgado OC, Beckhove P, Hammerling GJ. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8(+) T cells. Nat Immunol (2015) 16:609–17. doi:10.1038/ni.3159
86. Teitz-Tennenbaum S, Li Q, Okuyama R, Davis MA, Sun R, Whitfield J, et al. Mechanisms involved in radiation enhancement of intratumoral dendritic cell therapy. J Immunother (2008) 31:345–58. doi:10.1097/CJI.0b013e318163628c
87. Cummings RJ, Gerber SA, Judge JL, Ryan JL, Pentland AP, Lord EM. Exposure to ionizing radiation induces the migration of cutaneous dendritic cells by a CCR7-dependent mechanism. J Immunol (2012) 189:4247–57. doi:10.4049/jimmunol.1201371
88. Liu C, Lin J, Zhao L, Yang Y, Gao F, Li B, et al. Gamma-ray irradiation impairs dendritic cell migration to CCL19 by down-regulation of CCR7 and induction of cell apoptosis. Int J Biol Sci (2011) 7:168–79. doi:10.7150/ijbs.7.168
89. Morganti JM, Jopson TD, Liu S, Gupta N, Rosi S. Cranial irradiation alters the brain’s microenvironment and permits CCR2+ macrophage infiltration. PLoS One (2014) 9:e93650. doi:10.1371/journal.pone.0093650
90. Russell JS, Brown JM. The irradiated tumor microenvironment: role of tumor-associated macrophages in vascular recovery. Front Physiol (2013) 4:157. doi:10.3389/fphys.2013.00157
91. Shiao SL, Ruffell B, Denardo DG, Faddegon BA, Park CC, Coussens LM. TH2-polarized CD4(+) T cells and macrophages limit efficacy of radiotherapy. Cancer Immunol Res (2015) 3:518–25. doi:10.1158/2326-6066.CIR-14-0232
92. Chiang CS, Fu SY, Wang SC, Yu CF, Chen FH, Lin CM, et al. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front Oncol (2012) 2:89. doi:10.3389/fonc.2012.00089
93. Seifert L, Werba G, Tiwari S, Giao Ly NN, Nguy S, Alothman S, et al. Radiation therapy induces macrophages to suppress T-cell responses against pancreatic tumors in mice. Gastroenterology (2016) 150:1659.e–72.e. doi:10.1053/j.gastro.2016.02.070
94. Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res (2013) 73:2782–94. doi:10.1158/0008-5472.CAN-12-3981
95. Stafford JH, Hirai T, Deng L, Chernikova SB, Urata K, West BL, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol (2016) 18:797–806. doi:10.1093/neuonc/nov272
96. Wang X, Yang X, Tsai Y, Yang L, Chuang KH, Keng PC, et al. IL-6 mediates macrophage infiltration after irradiation via up-regulation of CCL2/CCL5 in non-small cell lung cancer. Radiat Res (2017) 187:50–9. doi:10.1667/RR14503.1
97. Uchimura E, Watanabe N, Niwa O, Muto M, Kobayashi Y. Transient infiltration of neutrophils into the thymus in association with apoptosis induced by whole-body X-irradiation. J Leukoc Biol (2000) 67:780–4.
98. Fujiwara H, Yamazaki T, Uzawa A, Nagata K, Kobayashi Y. Transient infiltration of neutrophils into the thymus following whole-body X-ray irradiation in IL-10 knockout mice. Biochem Biophys Res Commun (2008) 369:432–6. doi:10.1016/j.bbrc.2008.02.043
99. Nakayama E, Shiratsuchi Y, Kobayashi Y, Nagata K. The importance of infiltrating neutrophils in SDF-1 production leading to regeneration of the thymus after whole-body X-irradiation. Cell Immunol (2011) 268:24–8. doi:10.1016/j.cellimm.2011.01.006
100. Lee EJ, Kim JW, Yoo H, Kwak W, Choi WH, Cho S, et al. Single high-dose irradiation aggravates eosinophil-mediated fibrosis through IL-33 secreted from impaired vessels in the skin compared to fractionated irradiation. Biochem Biophys Res Commun (2015) 464:20–6. doi:10.1016/j.bbrc.2015.05.081
101. Ibahim MJ, Yang Y, Crosbie JC, Stevenson A, Cann L, Paiva P, et al. Eosinophil-associated gene pathways but not eosinophil numbers are differentially regulated between synchrotron microbeam radiation treatment and synchrotron broad-beam treatment by 48 hours postirradiation. Radiat Res (2016) 185:60–8. doi:10.1667/RR14115.1
102. Reuben JM, Korbling M, Gao H, Lee BN. The effect of low dose gamma irradiation on the differentiation and maturation of monocyte derived dendritic cells. J Gravit Physiol (2004) 11:49–52.
103. Merrick A, Errington F, Milward K, O’donnell D, Harrington K, Bateman A, et al. Immunosuppressive effects of radiation on human dendritic cells: reduced IL-12 production on activation and impairment of naive T-cell priming. Br J Cancer (2005) 92:1450–8. doi:10.1038/sj.bjc.6602518
104. Liao YP, Wang CC, Butterfield LH, Economou JS, Ribas A, Meng WS, et al. Ionizing radiation affects human MART-1 melanoma antigen processing and presentation by dendritic cells. J Immunol (2004) 173:2462–9. doi:10.4049/jimmunol.173.4.2462
105. Price JG, Idoyaga J, Salmon H, Hogstad B, Bigarella CL, Ghaffari S, et al. CDKN1A regulates Langerhans cell survival and promotes Treg cell generation upon exposure to ionizing irradiation. Nat Immunol (2015) 16:1060–8. doi:10.1038/ni.3270
106. Nakano T, Oka K, Arai T, Morita S, Tsunemoto H. Prognostic significance of Langerhans’ cell infiltration in radiation therapy for squamous cell carcinoma of the uterine cervix. Arch Pathol Lab Med (1989) 113:507–11.
107. Nakano T, Oka K, Takahashi T, Morita S, Arai T. Roles of Langerhans’ cells and T-lymphocytes infiltrating cancer tissues in patients treated by radiation therapy for cervical cancer. Cancer (1992) 70:2839–44. doi:10.1002/1097-0142(19921215)70:12<2839::AID-CNCR2820701220>3.0.CO;2-7
108. Nakano T, Oka K, Sugita T, Tsunemoto H. Antitumor activity of Langerhans cells in radiation therapy for cervical cancer and its modulation with SPG administration. In Vivo (1993) 7:257–63.
109. Chen Z, Xia D, Bi X, Saxena A, Sidhu N, El-Gayed A, et al. Combined radiation therapy and dendritic cell vaccine for treating solid tumors with liver micro-metastasis. J Gene Med (2005) 7:506–17. doi:10.1002/jgm.692
110. Lodermann B, Wunderlich R, Frey S, Schorn C, Stangl S, Rodel F, et al. Low dose ionising radiation leads to a NF-kappaB dependent decreased secretion of active IL-1beta by activated macrophages with a discontinuous dose-dependency. Int J Radiat Biol (2012) 88:727–34. doi:10.3109/09553002.2012.689464
111. Schaue D, Marples B, Trott KR. The effects of low-dose X-irradiation on the oxidative burst in stimulated macrophages. Int J Radiat Biol (2002) 78:567–76. doi:10.1080/09553000210126457
112. Mckinney LC, Aquilla EM, Coffin D, Wink DA, Vodovotz Y. Ionizing radiation potentiates the induction of nitric oxide synthase by IFN-gamma and/or LPS in murine macrophage cell lines: role of TNF-alpha. J Leukoc Biol (1998) 64:459–66.
113. Mckinney LC, Aquilla EM, Coffin D, Wink DA, Vodovotz Y. Ionizing radiation potentiates the induction of nitric oxide synthase by interferon-gamma and/or lipopolysaccharide in murine macrophage cell lines. Role of tumor necrosis factor-alpha. Ann N Y Acad Sci (2000) 899:61–8. doi:10.1111/j.1749-6632.2000.tb06176.x
114. Lee H, Ahn YT, Park SH, Park DY, Jin YW, Kim CS, et al. Lactobacillus plantarum HY7712 protects against the impairment of NK-cell activity caused by whole-body gamma-irradiation in mice. J Microbiol Biotechnol (2014) 24:127–31. doi:10.4014/jmb.1307.07001
115. Coates PJ, Rundle JK, Lorimore SA, Wright EG. Indirect macrophage responses to ionizing radiation: implications for genotype-dependent bystander signaling. Cancer Res (2008) 68:450–6. doi:10.1158/0008-5472.CAN-07-3050
116. Frischholz B, Wunderlich R, Ruhle PF, Schorn C, Rodel F, Keilholz L, et al. Reduced secretion of the inflammatory cytokine IL-1beta by stimulated peritoneal macrophages of radiosensitive Balb/c mice after exposure to 0.5 or 0.7 Gy of ionizing radiation. Autoimmunity (2013) 46:323–8. doi:10.3109/08916934.2012.747522
117. Tsai CS, Chen FH, Wang CC, Huang HL, Jung SM, Wu CJ, et al. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int J Radiat Oncol Biol Phys (2007) 68:499–507. doi:10.1016/j.ijrobp.2007.01.041
118. Okubo M, Kioi M, Nakashima H, Sugiura K, Mitsudo K, Aoki I, et al. M2-polarized macrophages contribute to neovasculogenesis, leading to relapse of oral cancer following radiation. Sci Rep (2016) 6:27548. doi:10.1038/srep27548
119. Meng Y, Beckett MA, Liang H, Mauceri HJ, Van Rooijen N, Cohen KS, et al. Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res (2010) 70:1534–43. doi:10.1158/0008-5472.CAN-09-2995
120. Prakash H, Klug F, Nadella V, Mazumdar V, Schmitz-Winnenthal H, Umansky L. Low doses of gamma irradiation potentially modifies immunosuppressive tumor microenvironment by retuning tumor-associated macrophages: lesson from insulinoma. Carcinogenesis (2016) 37:301–13. doi:10.1093/carcin/bgw007
121. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell (2013) 24:589–602. doi:10.1016/j.ccr.2013.09.014
122. Farooque A, Afrin F, Adhikari JS, Dwarakanath BS. Polarization of macrophages towards M1 phenotype by a combination of 2-deoxy-d-glucose and radiation: implications for tumor therapy. Immunobiology (2016) 221:269–81. doi:10.1016/j.imbio.2015.10.009
123. Balabanli B, Turkozkan N, Balabanli S, Erdamar H, Akmansu M. The effect of vitamin A pretreatment on radiation induced alteration in neutrophil functions. Mol Cell Biochem (2006) 286:103–5. doi:10.1007/s11010-005-9099-x
124. Panes J, Granger DN. Neutrophils generate oxygen free radicals in rat mesenteric microcirculation after abdominal irradiation. Gastroenterology (1996) 111:981–9. doi:10.1016/S0016-5085(96)70065-3
125. Shigematsu A, Adachi Y, Koike-Kiriyama N, Suzuki Y, Iwasaki M, Koike Y, et al. Effects of low-dose irradiation on enhancement of immunity by dendritic cells. J Radiat Res (2007) 48:51–5. doi:10.1269/jrr.06048
126. Chun SH, Park GY, Han YK, Kim SD, Kim JS, Lee CG, et al. Effect of low dose radiation on differentiation of bone marrow cells into dendritic cells. Dose Response (2012) 11:374–84. doi:10.2203/dose-response.12-041.Lee
127. Jahns J, Anderegg U, Saalbach A, Rosin B, Patties I, Glasow A, et al. Influence of low dose irradiation on differentiation, maturation and T-cell activation of human dendritic cells. Mutat Res (2011) 709–710:32–9. doi:10.1016/j.mrfmmm.2011.02.007
128. Lee EJ, Lee SJ, Kim JH, Kim KJ, Yang SH, Jeong KY, et al. Radiation inhibits interleukin-12 production via inhibition of C-Rel through the interleukin-6/signal transducer and activator of transcription 3 signaling pathway in dendritic cells. PLoS One (2016) 11:e0146463. doi:10.1371/journal.pone.0146463
129. Cao MD, Chen ZD, Xing Y. Gamma irradiation of human dendritic cells influences proliferation and cytokine profile of T cells in autologous mixed lymphocyte reaction. Cell Biol Int (2004) 28:223–8. doi:10.1016/j.cellbi.2003.12.006
130. Kulzer L, Rubner Y, Deloch L, Allgauer A, Frey B, Fietkau R, et al. Norm- and hypo-fractionated radiotherapy is capable of activating human dendritic cells. J Immunotoxicol (2014) 11:328–36. doi:10.3109/1547691X.2014.880533
131. Huang J, Wang QJ, Yang S, Li YF, El-Gamil M, Rosenberg SA, et al. Irradiation enhances human T-cell function by upregulating CD70 expression on antigen-presenting cells in vitro. J Immunother (2011) 34:327–35. doi:10.1097/CJI.0b013e318216983d
132. Malecka A, Wang Q, Shah S, Sutavani RV, Spendlove I, Ramage JM, et al. Stromal fibroblasts support dendritic cells to maintain IL-23/Th17 responses after exposure to ionizing radiation. J Leukoc Biol (2016) 100:381–9. doi:10.1189/jlb.3A1015-474R
133. Kepp O, Tesniere A, Schlemmer F, Michaud M, Senovilla L, Zitvogel L, et al. Immunogenic cell death modalities and their impact on cancer treatment. Apoptosis (2009) 14:364–75. doi:10.1007/s10495-008-0303-9
134. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature (1997) 390:350–1. doi:10.1038/37022
135. Garrido G, Rabasa A, Sanchez B, Lopez MV, Blanco R, Lopez A, et al. Induction of immunogenic apoptosis by blockade of epidermal growth factor receptor activation with a specific antibody. J Immunol (2011) 187:4954–66. doi:10.4049/jimmunol.1003477
136. Rastogi S, Boylan M, Wright EG, Coates PJ. Interactions of apoptotic cells with macrophages in radiation-induced bystander signaling. Radiat Res (2013) 179:135–45. doi:10.1667/RR2969.1
137. El-Saghire H, Michaux A, Thierens H, Baatout S. Low doses of ionizing radiation induce immune-stimulatory responses in isolated human primary monocytes. Int J Mol Med (2013) 32:1407–14. doi:10.3892/ijmm.2013.1514
138. Luna-Gomes T, Santana PT, Coutinho-Silva R. Silica-induced inflammasome activation in macrophages: role of ATP and P2X7 receptor. Immunobiology (2015) 220:1101–6. doi:10.1016/j.imbio.2015.05.004
139. Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat Immunol (2013) 14:480–8. doi:10.1038/ni.2563
140. Kim SK, Yun CH, Han SH. Enhanced anti-cancer activity of human dendritic cells sensitized with gamma-irradiation-induced apoptotic colon cancer cells. Cancer Lett (2013) 335:278–88. doi:10.1016/j.canlet.2013.02.038
141. Nikitina EY, Gabrilovich DI. Combination of gamma-irradiation and dendritic cell administration induces a potent antitumor response in tumor-bearing mice: approach to treatment of advanced stage cancer. Int J Cancer (2001) 94:825–33. doi:10.1002/1097-0215(20011215)94:6<825::AID-IJC1545>3.0.CO;2-5
142. Kang SM, Kim MH, Song KH, Jung SY, Ahn J, Hwang SG, et al. Modulation of dendritic cell function by the radiation-mediated secretory protein gamma-synuclein. Cell Death Discov (2015) 1:15011. doi:10.1038/cddiscovery.2015.11
143. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi:10.1038/nrc3239
144. Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest (2014) 124:687–95. doi:10.1172/JCI67313
145. Uchida A, Mizutani Y, Nagamuta M, Ikenaga M. Effects of X-ray irradiation on natural killer (NK) cell system. I. Elevation of sensitivity of tumor cells and lytic function of NK cells. Immunopharmacol Immunotoxicol (1989) 11:507–19. doi:10.3109/08923978909005381
146. Brovall C, Schacter B. Radiation sensitivity of human natural killer cell activity: control by X-linked genes. J Immunol (1981) 126:2236–9.
147. Zarcone D, Tilden AB, Lane VG, Grossi CE. Radiation sensitivity of resting and activated nonspecific cytotoxic cells of T lineage and NK lineage. Blood (1989) 73:1615–21.
148. Sonn CH, Choi JR, Kim TJ, Yu YB, Kim K, Shin SC, et al. Augmentation of natural cytotoxicity by chronic low-dose ionizing radiation in murine natural killer cells primed by IL-2. J Radiat Res (2012) 53:823–9. doi:10.1093/jrr/rrs037
149. Yang G, Kong Q, Wang G, Jin H, Zhou L, Yu D, et al. Low-dose ionizing radiation induces direct activation of natural killer cells and provides a novel approach for adoptive cellular immunotherapy. Cancer Biother Radiopharm (2014) 29:428–34. doi:10.1089/cbr.2014.1702
150. Gehrmann M, Marienhagen J, Eichholtz-Wirth H, Fritz E, Ellwart J, Jaattela M, et al. Dual function of membrane-bound heat shock protein 70 (Hsp70), Bag-4, and Hsp40: protection against radiation-induced effects and target structure for natural killer cells. Cell Death Differ (2005) 12:38–51. doi:10.1038/sj.cdd.4401510
151. Kim JY, Son YO, Park SW, Bae JH, Chung JS, Kim HH, et al. Increase of NKG2D ligands and sensitivity to NK cell-mediated cytotoxicity of tumor cells by heat shock and ionizing radiation. Exp Mol Med (2006) 38:474–84. doi:10.1038/emm.2006.56
152. Son CH, Keum JH, Yang K, Nam J, Kim MJ, Kim SH, et al. Synergistic enhancement of NK cell-mediated cytotoxicity by combination of histone deacetylase inhibitor and ionizing radiation. Radiat Oncol (2014) 9:49. doi:10.1186/1748-717X-9-49
153. Yang KL, Wang YS, Chang CC, Huang SC, Huang YC, Chi MS, et al. Reciprocal complementation of the tumoricidal effects of radiation and natural killer cells. PLoS One (2013) 8:e61797. doi:10.1371/journal.pone.0061797
154. Chang Y, Yang ZY, Li GL, Li Q, Yang Q, Fan JQ, et al. Correlations between radiation dose in bone marrow and hematological toxicity in patients with cervical cancer: a comparison of 3DCRT, IMRT, and RapidARC. Int J Gynecol Cancer (2016) 26:770–6. doi:10.1097/IGC.0000000000000660
155. Hui B, Zhang Y, Shi F, Wang J, Wang T, Wang J, et al. Association between bone marrow dosimetric parameters and acute hematologic toxicity in cervical cancer patients undergoing concurrent chemoradiotherapy: comparison of three-dimensional conformal radiotherapy and intensity-modulated radiation therapy. Int J Gynecol Cancer (2014) 24:1648–52. doi:10.1097/IGC.0000000000000292
156. Wang D, An G, Xie S, Yao Y, Feng G. The clinical and prognostic significance of CD14+HLA-DR-/low myeloid-derived suppressor cells in hepatocellular carcinoma patients receiving radiotherapy. Tumour Biol (2016) 37:10427–33. doi:10.1007/s13277-016-4916-2
157. Crittenden MR, Savage T, Cottam B, Bahjat KS, Redmond WL, Bambina S, et al. The peripheral myeloid expansion driven by murine cancer progression is reversed by radiation therapy of the tumor. PLoS One (2013) 8:e69527. doi:10.1371/journal.pone.0069527
158. Chen HM, Ma G, Gildener-Leapman N, Eisenstein S, Coakley BA, Ozao J, et al. Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clin Cancer Res (2015) 21:4073–85. doi:10.1158/1078-0432.CCR-14-2742
159. Kawase Y, Naito S, Ito M, Sekine I, Fujii H. The effect of ionizing radiation on epidermal Langerhans cells – a quantitative analysis of autopsy cases with radiation therapy. J Radiat Res (1990) 31:246–55. doi:10.1269/jrr.31.246
160. Edwards EK Jr, Edwards EK Sr. The effect of superficial x-radiation on epidermal Langerhans cells in human skin. Int J Dermatol (1990) 29:731–2. doi:10.1111/j.1365-4362.1990.tb03782.x
161. Cole S, Lewkowicz SJ, Townsend KM. Langerhans cell number and morphology in mouse footpad epidermis after X irradiation. Radiat Res (1984) 100:594–606. doi:10.2307/3576422
162. Cole S, Townsend KM. Dose response of Langerhans cells in mouse footpad epidermis after X irradiation. Radiat Res (1985) 103:251–9. doi:10.2307/3576579
163. Cole S. Long-term effects of local ionizing radiation treatment on Langerhans cells in mouse footpad epidermis. J Invest Dermatol (1986) 87:608–12. doi:10.1111/1523-1747.ep12455853
164. Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med (2015) 66:129–43. doi:10.1146/annurev-med-081313-121208
165. Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity (2015) 42:332–43. doi:10.1016/j.immuni.2015.01.012
166. Yu Q, Katlinskaya YV, Carbone CJ, Zhao B, Katlinski KV, Zheng H, et al. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep (2015) 11:785–97. doi:10.1016/j.celrep.2015.03.069
167. Alpay M, Backman LR, Cheng X, Dukel M, Kim WJ, Ai L, et al. Oxidative stress shapes breast cancer phenotype through chronic activation of ATM-dependent signaling. Breast Cancer Res Treat (2015) 151:75–87. doi:10.1007/s10549-015-3368-5
168. Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol (2007) 8:49–62. doi:10.1038/nrm2083
169. Lowe JM, Menendez D, Bushel PR, Shatz M, Kirk EL, Troester MA, et al. p53 and NF-kappaB coregulate proinflammatory gene responses in human macrophages. Cancer Res (2014) 74:2182–92. doi:10.1158/0008-5472.CAN-13-1070
170. Mori T, Anazawa Y, Iiizumi M, Fukuda S, Nakamura Y, Arakawa H. Identification of the interferon regulatory factor 5 gene (IRF-5) as a direct target for p53. Oncogene (2002) 21:2914–8. doi:10.1038/sj.onc.1205459
Keywords: ionizing radiation, tumor cell death, innate immunity, immunotherapy, cancer treatment
Citation: Wu Q, Allouch A, Martins I, Brenner C, Modjtahedi N, Deutsch E and Perfettini JL (2017) Modulating Both Tumor Cell Death and Innate Immunity Is Essential for Improving Radiation Therapy Effectiveness. Front. Immunol. 8:613. doi: 10.3389/fimmu.2017.00613
Received: 26 December 2016; Accepted: 09 May 2017;
Published: 26 May 2017
Edited by:
Serge M. Candéias, CEA, FranceReviewed by:
Sandra Demaria, New York University, United StatesAbhishek D. Garg, University of Leuven, Belgium
Copyright: © 2017 Wu, Allouch, Martins, Brenner, Modjtahedi, Deutsch and Perfettini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Luc Perfettini, perfettini@orange.fr