 Peter I. Lobo
Peter I. Lobo Kailo H. Schlegel
Kailo H. Schlegel Amandeep Bajwa
Amandeep Bajwa Mark D. Okusa
Mark D. Okusa- Division of Nephrology, Center for Immunity, Inflammation and Regenerative Medicine, University of Virginia, Charlottesville, VA, United States
Natural IgM anti-leukocyte autoantibodies (IgM-ALAs) inhibit inflammation by several mechanisms. Here, we show that pan-B cells and bone marrow-derived dendritic cells (BMDCs) are switched to regulatory cells when pretreated ex vivo with IgM. B cells are also switched to regulatory cells when pretreated ex vivo with CpG but not with LPS. Pre-emptive infusion of such ex vivo induced regulatory cells protects C57BL/6 mice from ischemia-induced acute kidney injury (AKI) via regulation of in vivo NKT-1 cells, which normally amplify the innate inflammatory response to DAMPS released after reperfusion of the ischemic kidney. Such ex vivo induced regulatory pan-B cells and BMDC express low CD1d and inhibit inflammation by regulating in vivo NKT-1 in the context of low-lipid antigen presentation and by a mechanism that requires costimulatory molecules, CD1d, PDL1/PD1, and IL10. Second, LPS and CpG have opposite effects on induction of regulatory activity in BMDC and B cells. LPS enhances regulatory activity of IgM-pretreated BMDC but negates the IgM-induced regulatory activity in B cells, while CpG, with or without IgM pretreatment, induces regulatory activity in B cells but not in BMDC. Differences in the response of pan-B and dendritic cells to LPS and CpG, especially in the presence of IgM-ALA, may have relevance during infections and inflammatory disorders where there is an increased IgM-ALA and release of TLRs 4 and 9 ligands. Ex vivo induced regulatory pan-B cells could have therapeutic relevance as these easily available cells can be pre-emptively infused to prevent AKI that can occur during open heart surgery or in transplant recipients receiving deceased donor organs.
Introduction
Several observations clearly demonstrate that both innate and adaptive immune inflammatory responses are suppressed by different regulatory mechanisms to protect the host from an over exuberant inflammatory response and from initiating autoimmune diseases. One such newly described regulatory mechanism is natural IgM, which are produced at birth by B1 cells, independently of exposure to foreign antigens and without the need for T helper cells [reviewed in Ref. (1, 2)]. Natural IgM are polyclonal autoantibodies with differing specificities, some of which have been identified, for example, IgM with specificity to leukocytes, erythrocytes, phosphorylcholine (PC) present on oxidized lipids, apoptotic cells, IgG (referred to as IgM rheumatoid factor), intracellular enzymes, DNA, and complement proteins. Several studies have shown that such autoantibodies are produced to prevent autoimmune-mediated inflammation [reviewed in Ref. (2)]. For example, IgM anti-PC, by binding to PC on oxidized lipids and apoptotic cell membranes, prevents immune activation by blocking neoantigen recognition and enhancing their removal by forming immune complexes that are phagocytosed by immature dendritic cell (DC) and macrophages.
Our laboratory has been interested in IgM anti-leukocyte autoantibodies (IgM-ALAs), which is a subset of natural IgM that binds to non-apoptotic leukocyte membrane receptors [reviewed in Ref. (1)]. Prior observations over the last 40 years, particularly in humans, have clearly demonstrated that IgM-ALA levels increase with various infections (viral and parasitic), autoimmune diseases (SLE), and other inflammatory states (sarcoidosis and end stage renal disease), and IgM-ALA levels normalize with control of infection or the inflammatory state [reviewed in Ref. (3)]. We hypothesized that natural IgM-ALA rapidly increases in <72 h to suppress an exuberant inflammatory response that could occur with infective and inflammatory states as they exhibit certain unique characteristics that are suited for such a purpose. First, IgM-ALAs have been shown to bind to specific leukocyte receptors involved in initiating the inflammatory response (e.g., CD3, CD4, CD40, CD86, and PD1) and in leukocyte trafficking (CXCR4 and CCR5) (3–5). Second, unlike pathogenic IgG autoantibodies, IgM-ALAs have low binding affinity to cells, and at body temperature, the cell-bound IgM does not activate the lytic components of complement. Hence, at body temperature, they are particularly suited to modulate cell receptors without inducing complement-mediated cell damage (6–8). Third, these large pentameric autoantibodies predominantly reside in the vascular compartment and not only bind to leukocytes but also bind to receptors on endothelial cells and inhibit activation of endothelial cells (9).
In support of a regulatory role for IgM-ALA are observations in patients with high levels of IgM-ALA. Such patients have significantly less rejections and prolonged kidney and heart survival after a transplant (10–14), and second, these patients develop significantly lower titers of anti-HLA antibodies after alloantigen sensitization while waiting for a transplant (8). In subsequent studies with murine models of inflammation, we showed that administration of intravenous polyclonal IgM, to increase plasma IgM-ALA levels in wild-type C57BL6 mice (referred to as WT-B6), attenuated the inflammatory response induced by innate (renal ischemia), adaptive (cardiac transplants), and autoimmune (diabetes mellitus in NOD mice)-mediated mechanisms (9, 15). Specifically, we showed that protection was mediated by the IgM-ALA subset present in the purified polyclonal IgM preparation as the protective effect on ischemia-induced acute kidney injury (AKI) was abolished when polyclonal IgM was depleted of IgM-ALA with leukocyte adsorption (9).
The several fold higher binding of IgM-ALA to splenic CD11c+ DC when compared to T cells prompted us to investigate whether IgM-ALA inhibited the innate inflammatory response in part, by modulating the function of DC during antigen presentation. In these studies, bone marrow-derived dendritic cells (BMDC) were cultured ex vivo for 48 h with IgM, and such IgM-pretreated BMDC, when pre-emptively infused, protected WT-B6 mice from ischemia-induced AKI by inhibiting the innate inflammatory response that occurs after reperfusion (4). Such protection was not observed with pre-emptive infusion of IgM-pretreated T cells. We showed that IgM pretreatment switched BMDC to a regulatory phenotype, which required PD1 and IL10 to inhibit the inflammatory response. Furthermore, we showed that LPS enhanced the regulatory activity of IgM-pretreated BMDC.
The current studies were aimed at determining the functional impact of IgM-ALA on splenic pan-B cells especially since we also observed threefold to fourfold more IgM binding to B cells when compared to splenic DC (4). As with BMDC, we show that ex vivo pretreatment of splenic pan-B cells with IgM also induced regulatory activity in B cells, which when intravenously infused, protected mice from subsequent ischemia-induced AKI. In addition, we show that CpG, but not LPS, enhanced the IgM-induced regulatory activity of B cells. We also show that ex vivo induced regulatory B cells and BMDC inhibit the inflammatory response in vivo by regulating NKT-1 cells, which normally amplify the renal ischemia-induced innate inflammatory response in WT-B6 mice (16, 17).
Materials and Methods
IgM and IgG Purification from Plasma
IgM and IgG were purified from heat-inactivated (56°C) WT-B6 murine plasma or normal human plasma using size exclusion column chromatography (Sephacryl S-300 HR) as previously described (4) except for modifications detailed below. Care was taken to separate plasma within 4 h of obtaining blood, and then plasma was sterile filtered to remove any contaminating bacteria. Plasma was stored at 4°C prior to column purification. IgM was not isolated by dialyzing sera in water or by ammonium chloride precipitation as both these techniques yielded IgM with impaired functional activity. Column purified IgM was re-passaged through Sephacryl S-300 to remove contaminating proteins that were not effectively removed with the first passage. Residual IgG in the IgM preparation was removed by using agarose beads covalently linked to goat anti-murine IgG. Protein A was not used as any leftover protein A in the IgM preparation also altered lymphocyte function. Purified IgM and IgG was concentrated to 1.3–1.5 mg/ml and then dialyzed against RPMI-1640 and sterile filtered prior to use in cultures and for in vivo use. Purified IgM was stored at 4°C and not frozen as functional activity of IgM was reduced with freezing. The effect of IgM on in vitro cultures was dose dependent, and the maximum effect of IgM was observed using IgM at a physiological dose of 10–30 μg/250 × 103 cells/0.5 ml of culture. We used two control IgM, i.e., our purified mouse IgM (polyclonal) adsorbed with WT-B6 splenic leukocytes to remove IgM-ALA (see Ref. (4) for adsorption details) and a monoclonal mouse IgM anti-KLH (clone C48-6) obtained from BD Pharmingen.
Permission was obtained from our institutional review board to obtain blood from normal human volunteers for this study.
Mice
We obtained C57BL6 mice (WT-B6) from Charles River Laboratories. IL10 ko (B6.129P2-IL10tm1Cgn/J), CD80/86 ko (B6.129S4-Cd80tm1ShrCd86tm2Shr/J), CD40 ko (B6.129P2-CD40tm1Kik/J), and Rag-1 ko (B6.129S7-Rag1tm1Mom/J) on a C57/BL6 background were from Jackson Laboratories.
Surgical Protocol for Kidney Ischemia–Reperfusion Injury (IRI)
All experiments were performed in accordance with NIH and Institutional Animal Care and Use Guidelines. The Animal Research Committee of the University of Virginia approved all procedures and protocols. All experiments were performed on 8- to 10-week-old male mice, weighing approximately 20 g. Kidney IRI was performed under anesthesia with bilateral flank incisions as we have previously described (4). Both kidney pedicles, consisting of renal artery and vein, were exposed and cross-clamped for 26 min, and then clamps were released and kidneys were re-perfused for 24 h. Body temperature was maintained at 34.5–35°C (rectal temp) during surgery with heating pads. Kidney pedicles were exposed, but not clamped in sham-operated mice. Postoperatively, mice were administered analgesia and maintained at 35–37°C. For splenectomy, mice were anesthetized and via a flank incision, the spleen was exposed, and after ligation of splenic vessels, the spleen was removed. Splenectomy was performed 7 days before mice were subjected to renal IRI.
Assessment of Kidney Function and Histology after Renal Ischemia (IRI)
Plasma creatinine was determined 24 h after renal IRI using an enzymatic assay (Diazyme Laboratories, Poway, CA, USA) according to the manufacturer’s protocol. Kidney histology and quantitation of tubular injury were assessed at 24 h using previously described techniques (4).
Isolation of Splenic Pan-B Cells and BMDC and Ex Vivo Pretreatment of These Cells
Pan-B cells, B1a cells, and follicular (FO) B cells were obtained from murine spleens using magnetic microbead isolation columns specific for Pan-B, B1a, and MZ/FO (Miltenyi Biotech Inc., San Diego, CA, USA) and following instructions in the kit. B1a (CD5+) and FO (CD23+) B cell subsets were isolated in a second step using positive selection. Pan-B cells consisting of B1 and B2 cells were obtained by negative selection. Isolated B cell subsets were >92% pure.
BMDCs were generated from bone marrow that was obtained from murine femurs and cultured in RPMI containing 10% FCS and 2 ng/ml of recombinant GM-CSF as previously described (4). Media was changed every 3 days and supplemented with GM-CSF. Non-adherent cells were used, and after 8–10 days, >90% of these non-adherent cells expressed moderate to high levels of CD11c/MHC-Class II and <15% were GR1 positive.
Ex vivo pretreatment of B cells and BMDC (7–10 days old) involved culturing 1 × 106 cells in 1 ml media in 5 ml falcon plastic tubes for 36–48 h in the presence of LPS (Escherichia coli, 400 ng/ml), CpG (ODN 2395 class C. InvivoGen, 5 μg/ml), and polyclonal IgM (100 μg/ml). IgM was added 40–60 min before LPS or CpG. For controls, we used non-CpG ODN and isotype murine IgM [i.e., IgM anti-hemocyanin, IgM anti-KLH (clone C48-6), BD Pharmingen]. In certain experiments, B cells were activated for 48 h with an agonistic anti-CD40 monoclonal antibody (clone ICD10, Jackson Immunoresearch, West Grove, PA, USA, 1 μg/ml), which was cross-linked by goat anti-rat IgG (Affymetrix). After the culture period, cells were washed two times to remove unbound LPS and IgM, and 0.5 × 106 pretreated BMDC or B cells were injected in the tail vein of each mouse. In certain experiments, anti-CD86 (clone 1D10), PD1 (clone J43), PDL1 (clone MIH5), and CD1d (clone 1D1) blocking antibodies (5 μg/ml) or their isotype controls (obtained from eBioscience) were added to cells after the 48-h culture to block these receptors prior to infusing into mice. Viability of 48 h pretreated B cells and BMDC was >70% as assessed by live/dead stain with flow cytometry.
Induction of Apoptosis in B Cells
Splenic pan-B cells pretreated with IgM for 48 h at 37°C were exposed to 150 mJ/cm2 of ultraviolet C irradiation (Stratagene Stratalinker 2400, Fisher Scientific). Irradiated cells were incubated for 6 h at 37°C to allow for IgM binding to apoptotic cells. Cells were washed prior to intravenous injection into mice. More than 80% of cells were apoptotic (Annexin V+) at the time of injection.
Infusion of Mice with Splenic Pan-B Cells or BMDC
In these experiments, ex vivo pretreated isolated pan-B cells or BMDC (0.5 × 106 cells in 0.5 ml) were infused via the tail vein. Bilateral renal ischemia (26 min) was performed 24 h after the infusion. Mice were sacrificed 20–24 h after renal reperfusion.
Antibodies and Other Reagents
The following monoclonal antibodies were used for flow cytometry or immunostaining of tissues and were obtained from eBioscience, San Diego, CA, USA: anti-PD1 (clone J43), anti-PDL1 (clone MIH5), anti-CD40 (clone 1C10), anti-CD80 (clone 16-10A1), anti-CD86 (clone GL1), anti-CD11c (clone N418), anti-CD3 (clone 145-2C11), anti-CD19 (clone eBio 1D3), and anti-IgM (clone II/41). Appropriate fluorochrome-conjugated, isotype-matched irrelevant mAbs were also obtained from eBioscience. Other reagents LPS-E. coli 0111:B4 (Sigma-Aldrich, St. Louis, MO, USA) and Live/Dead fixable Kit were used (Life Technology, Grand Island, NY). TUNEL in situ cell death detection kit was obtained from Roche diagnostics (IN, USA).
Immunofluoresence Staining of Splenic Pan-B Cells and BMDC
9- to 12-day BMDCs were used in these studies. IgM-mediated receptor downregulation was evaluated by adding IgM 40–60 min before LPS at the initiation of the 37°C culture, which was terminated after 48 h. After culture termination, cells were washed and blocked with unlabeled anti-mouse CD16/32 (clone 2.4G2) prior to adding labeled antireceptor antibody. Appropriate fluorochrome-conjugated, isotype-matched, irrelevant mAbs were used as negative controls. Intracytoplasmic staining was performed using methods as described for splenic leukocytes.
Immunofluoresence Staining of Kidney Sections
Kidney tissue was fixed and frozen as we have previously described in detail (4). Frozen sections (5 μm) of kidney were permeabilized with 0.3% Triton X-100, and non-specific binding was blocked with 10% horse serum and anti-mouse CD16/32. Tissue sections were stained with the following conjugated antibodies: rat anti-Ly-6B.2 (anti-neutrophil, clone 7/4, Bio-Rad, Raleigh, NC, USA) and rat anti CD31 (anti-endothelial cell, clone 390, eBioscience). Nuclei were visualized using DAPI.
Quantitation of Cytokines in 48 h Supernatants of Ex Vivo Induced Regulatory B Cells and BMDC
Cytokines were quantitated on supernatants that were immediately frozen at −80°C. Our flow cytometry core Lab at University of Virginia performed these studies using a Luminex bead assay (obtained from Millipore/Sigma, Billerica, MA, USA) that quantitated a panel of 32 proteins consisting of cytokines (including IFNγ, IL1α, IL1β, IL2, IL4, IL3, IL5, IL6, IL7, IL9, L10, IL12p40, IL12p70, LIF, IL13, IL15, IL17, TNFα, and LIX) as well as chemokines (including IP10, KC, MCP-1, MIP-1α, MIP-1β, M-CSF, MIP-2, MIG, and RANTES) and a separate panel of Luminex beads that only quantitated active (but not pro) TGFβ1, TGFβ2, and TGFβ3. B cells produced no or very low levels (<5 pg/ml) of IL1α, IL1β, IL2, IL4, IL3, IL5, IL7, IL9, IL-12, LIF, IL13, LIX, IL15, IL17, TGFβ2, and TGFβ3 even when activated with CpG or LPS. Except for M-CSF, B cells produced all the tested chemokines that significantly increased with TLR activation.
Statistics
Data in figures were analyzed by one-way ANOVA except where indicated. P < 0.05 was used to indicate significance.
Results
IgM and TLR Ligands Utilize Different Mechanisms to Regulate Pan-B Cells and BMDC
In these studies, we wanted to evaluate if ex vivo pretreatment with IgM switches B cells to a regulatory phenotype especially since, in prior studies, we observed that IgM has several fold higher binding to B cells when compared to splenic DCs (4). Column purified splenic pan-B cells were pretreated with IgM ex vivo, and 0.5 × 106 pretreated pan-B cells were intravenously infused into mice 24 h prior to performing renal ischemia. Such ex vivo IgM-pretreated pan-B cells effectively protected mice from ischemic AKI and adding LPS during the IgM pretreatment negated the regulatory effect of IgM on B cells (Figure 1). As in prior studies with BMDC (4), infusion of pan-B cells, pretreated ex vivo with leukocyte adsorbed IgM (control IgM), failed to protect mice from ischemic AKI (data not shown). The opposite effect of LPS on B cells vs BMDC prompted us to evaluate whether other TLRs were involved in the induction of regulatory activity in B cells and BMDC (Figure 1). We therefore tested our hypothesis by using another TLR agonist, i.e., Class C CpG, a TLR9 agonist. CpG, in the absence of IgM, induced regulatory activity in B cells, but not in BMDC, and CpG enhanced the regulatory effect of IgM on pan-B cells, and unlike LPS, CpG negated the protective effect of IgM on BMDC (Figure 1). As depicted in Figure 2, protected kidneys had relatively well-preserved tubular and endothelial cells as well as no or minimal granulocyte infiltration, thus indicating that the infused ex vivo pretreated B cells inhibited the ischemia-induced inflammatory response that causes renal tubular and endothelial cell injury.
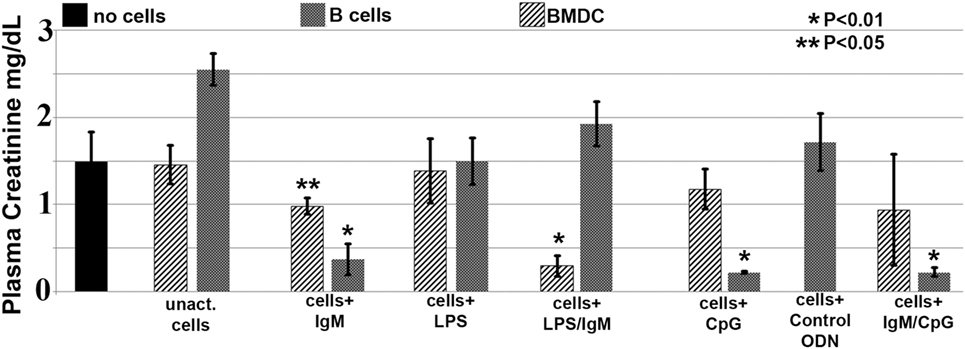
Figure 1. IgM-pretreated B cells are more protective than IgM-pretreated BMDC in preventing ischemia-induced acute kidney injury. Unlike with BMDC, addition of LPS negates the protective effect of IgM-pretreated B cells. CpG activation, unlike LPS, renders B cells, but not BMDC, protective. Use of B cells pretreated with isotype control IgM was not protective (data not shown). Each data point is a mean of at least three separate experiments with at least six mice. * and ** denotes significance (one-way ANOVA) compared to renal ischemia without cell therapy (black bar).
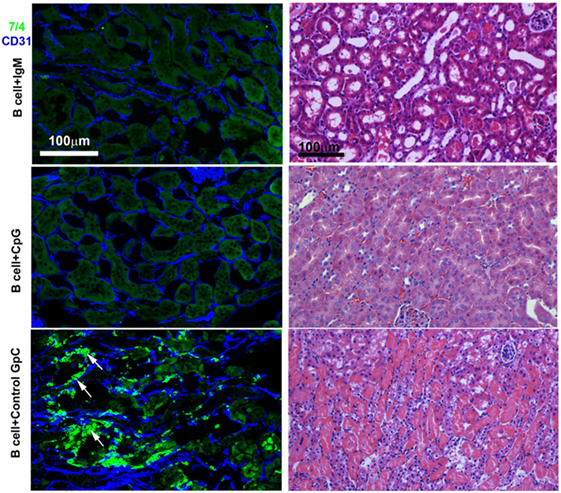
Figure 2. IgM- or CpG ODN-pretreated B cells protect against ischemia-induced renal injury by inhibiting the ischemia-induced inflammatory response (upper two panels). On immunofluorescence staining, note no or minimal 7/4+ inflammatory cell (bright green) infiltration in protected kidneys. CD31+ endothelial cells (blue) are well preserved in protected kidneys. Infusion of B cells pretreated with control GpC ODN failed to protect (bottom panel). Note significant infiltration with 7/4+ inflammatory cells (bright green) many having bilobed nuclei resembling neutrophils (arrows) and damaged CD31+ endothelial cells (blue). The Ly-6B.2 antigen detected by clone 7/4 is expressed on all neutrophils and activated monocytes/macrophages. On H&E-stained histology, note preservation of outer medulla renal tubular histology with minimal interstitial inflammation when mice were infused with IgM- or CpG ODN-pretreated B cells before renal ischemia. There are loss of tubular cells, severe inflammation, and capillary damage with interstitial hemorrhage when mice were infused with B cells pretreated with control GpC ODN. This is a representative example from four separate experiments.
IgM Induces Regulatory Activity in B1a and FO B2 Cells through a Mechanism Not Involving B Cell Apoptosis
Further studies were aimed at defining how IgM induces regulatory activity in pan-B cells. Since others have shown that infusion of apoptotic cells can induce in vivo Bregs (18), and since up to 30% of the infused ex vivo pretreated B cells were apoptotic, we examined this possibility by infusing B cells that were rendered >80% apoptotic with UV irradiation, i.e., after they were pretreated for 48 h with IgM. Data in Figure 3A show that apoptotic B cells, induced by UV irradiation, failed to protect mice from renal ischemia-induced AKI, thus indicating that protection from ischemia-induced AKI was mediated by the live subset of infused IgM-pretreated B cells, which regulate the ischemia-induced inflammatory response. Similarly, ex vivo induced regulatory BMDC failed to be protective in ischemia-induced renal injury when they were rendered apoptotic (4). Hence, these studies would indicate that IgM induces regulatory activity in pan-B cells and BMDC by possibly binding to some undefined inhibitory receptor expressed by these cells.
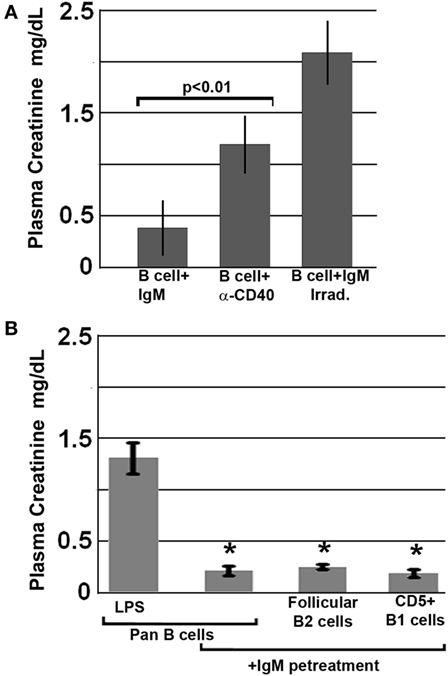
Figure 3. All splenic B cell subsets pretreated ex vivo with IgM are switched to a regulatory phenotype. Bregs lose regulatory activity when irradiated or activated with anti-CD40. (A) IgM-pretreated B cells fail to protect when irradiated just before infusion. Similarly, B cells pretreated with an agonistic anti-CD40 antibody that was also cross-linked are non-protective. (B) IgM-pretreated Pan-B and B cell subsets are equally protective. Data are a mean of three separate experiments with each experimental group having three mice. *Significance when compared to LPS-pretreated pan-B cells.
We next determined whether ex vivo pretreatment of pan-B cells with IgM-induced regulatory activity in B cell subsets arising from certain splenic B cell compartments, i.e., follicles or marginal zone. This question was evaluated by using the Miltenyi Biotec Kit and their MACS columns to isolate pan-B cells, FO B2 cells, or CD5 + B1a cells from murine spleens. Data in Figure 3B show that infusion of 0.5 × 106 pan-B cells, FO B2, or B1a cells, pretreated ex vivo with IgM, protected mice from ischemia-induced renal injury indicating; therefore, that IgM induces regulatory activity in Breg subsets present in the splenic B1a and FO B2 cell compartments.
Human IgM-Pretreated Human B Cells Protect Mice after Renal Ischemia
Since natural IgM is evolutionarily conserved and human IgM can bind to both murine and human B cells, we examined if natural human IgM can regulate human B cells (1, 19). Such human IgM-pretreated human B cells were tested in the murine model of renal ischemia, as in this model, the infused cells exert their functional effects in <48 h, i.e., before these cells are rejected. As shown in Figure 4, infusion of 0.5 × 106 human IgM-pretreated human B cells protected mice from renal ischemia-induced AKI. Therefore, these studies indicate that human IgM and B cells can inhibit the ischemia-induced innate inflammatory response in mice by a mechanism that is also evolutionarily conserved.
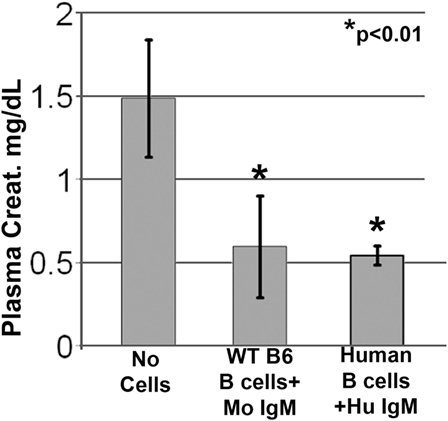
Figure 4. Human IgM pretreatment of human B cells obtained from human blood and murine IgM pretreatment of murine B cells obtained from WT-B6 spleens are protective in renal ischemia. *p value compared to No Cells (one-way ANOVA). Data are a mean of three separate experiments.
Ex Vivo Induced Regulatory B Cells and BMDC Inhibit Renal Ischemia Induced Innate Inflammation by Regulating In Vivo NKT-1 Cells
Acute renal ischemia releases DAMPS and lipid antigen, which normally activate NKT-1 to release large amounts of IFNγ that amplifies the innate inflammatory response (16, 17). We therefore questioned whether infused regulatory B cells or BMDC inhibited in vivo NKT-1 cell function especially since CD1d expression on these regulatory cells was significantly downregulated after ex vivo pretreatment with IgM (Figure 5). Such a possibility seemed plausible as intravenously infused cells have been shown to rapidly enter the splenic marginal zone as well as the liver where they can interact with in vivo NKT-1 cells (20, 21), and second NKT-1 activation by lipid antigen in the context of low CD1d expression has been shown to decrease NKT cytotoxicity and INFγ production (20, 22). Since there was downregulation of CD1d in all ex vivo pretreatment conditions that generated regulatory pan-B or BMDC, we reasoned that infused cells expressing low CD1d could regulate in vivo NKT-1 cells and such NKT-1 cells with a regulatory phenotype may not be able to produce high levels of IFNγ when re-challenged by lipid antigen released after ischemia.
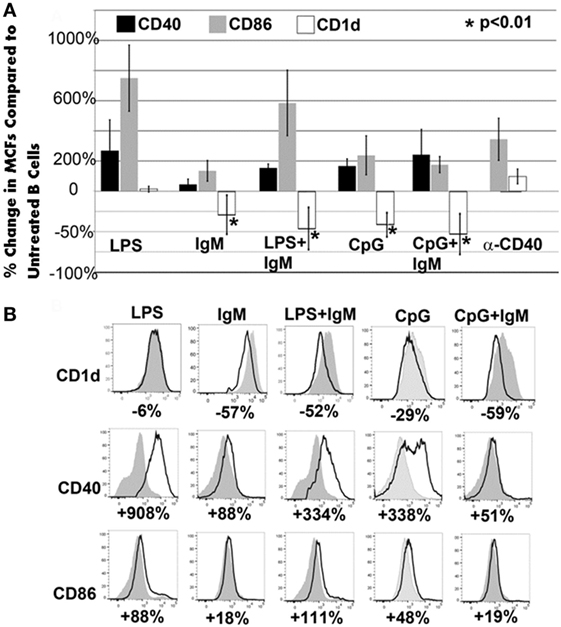
Figure 5. CD1d is downregulated with all ex vivo pretreatments that induce regulatory activity in pan-B and BMDC. (A) Pretreatment of pan-B cells: in all protective pretreatments (i.e., IgM or CpG or CpG/IgM), B cells express decreased CD1d when compared to unactivated cells. Of the three non-protective pretreatments (LPS or LPS + IgM or anti-CD40), only LPS + IgM-pretreated B cells exhibited CD1d downregulation. There is an increased CD86 and CD40 expression when compared to non-activated pan-B cells in both protective and non-protective B cells. Unactivated and pretreated cells were cultured for 48 h with IgM, CpG, or LPS. Change in receptor expression over unactivated cells is expressed as % change. Downregulation is depicted as negative (−) percent change. Asterisks denote significance when the % change is compared to CD1d expression on LPS-pretreated cells (one-way ANOVA). (B) Pretreatment of BMDC: representative example from one of four different experiments is depicted. Note that all protective (IgM or IgM + LPS) BMDCs have significant downregulation of CD1d. However, some non-protective BMDC (i.e., CpG or CpG + IgM) also exhibited downregulation of CD1d. Gray histogram represent unactivated cells. % represents change in receptor expression over unactivated cells. Each data point is a mean of three separate experiments with each experiment having three mice per group.
To test the hypothesis that ex vivo pretreated regulatory B cells and BMDC regulate in vivo NKT-1 cells, we employed four approaches. We initially evaluated if NKT-1 cells had an important role in mediating inflammation in our model of renal ischemia. In this approach, we used WT-B6 mice where NKT-1 cells were rendered anergic. Second, we evaluated if the ex vivo induced regulatory cells required CD1d and costimulatory receptors to interact in vivo with NKT-1 cells to inhibit the ischemia-induced innate inflammatory response. Third, we examined if regulatory B cells, induced ex vivo by either IgM or CpG, require PD1/PDL1 or IL10 to mediate their in vivo regulatory effect on NKT-1 cells. Finally, we evaluated if infused regulatory BMDC and B cells regulated in vivo NKT-1 cells or induced NKT-1 cells to switch to a non-functional or anergic state.
Role of In Vivo NKT-1 in Ischemic AKI
The role of in vivo NKT-1 cells in mediating the innate inflammatory response in our renal ischemia model was tested by performing renal ischemia 72 h after in vivo NKT-1 cells were rendered non-functional or anergic with a large bolus (5 μg) of intraperitoneal α-gal-ceramide as previously described (23). Data in Figure 6 show that 26 min of renal ischemia failed to induce AKI, as determined by an increase in serum creatinine, when in vivo NKT cells were rendered non-functional or anergic with a large bolus of α-gal-ceramide, thus indicating that NKT-1 cells have an important role in mediating the renal ischemia-induced inflammatory response that leads to renal injury in our model of ischemia. In prior studies, using other approaches, including Jα18ko mice, we have also shown that NKT-1 cells by producing high levels of IFNγ have an important role in amplifying the innate inflammatory response in our renal ischemia model (16).
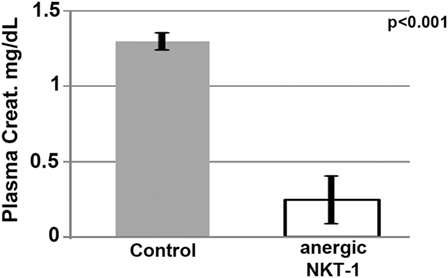
Figure 6. Lack of renal acute kidney injury in mice with anergic NKT-1 cells induced by intraperitoneal α-Gal-Cer (5 μg). Four mice were used in each group.
Interaction of Infused Regulatory Cells with In Vivo NKT-1
Since interaction of infused regulatory cells with in vivo NKT-1 cells requires CD1d to present glycolipids and appropriate costimulatory interaction, we infused ex vivo pretreated B cells and BMDC that were obtained from CD1d and CD 80/86 ko mice. Data in Figure 7A demonstrate that the infused regulatory B cells or IgM + LPS-pretreated regulatory BMDC needed both CD1d and CD80/86 to interact with in vivo NKT-1 cells and protect mice from ischemic AKI (16, 17). Data in Figure 7A also show that CpG but not IgM required CD40 to induce regulatory activity in B cells. This latter finding may be best explained on the role of CD40 in enhancing production of IL10 (24, 25), which is required by CpG-, but not IgM-, induced Bregs (data to be presented in Section “Pan-B Cells and BMDC Regulate In Vivo NKT-1 by Distinct Mechanisms”; Figure 8B).
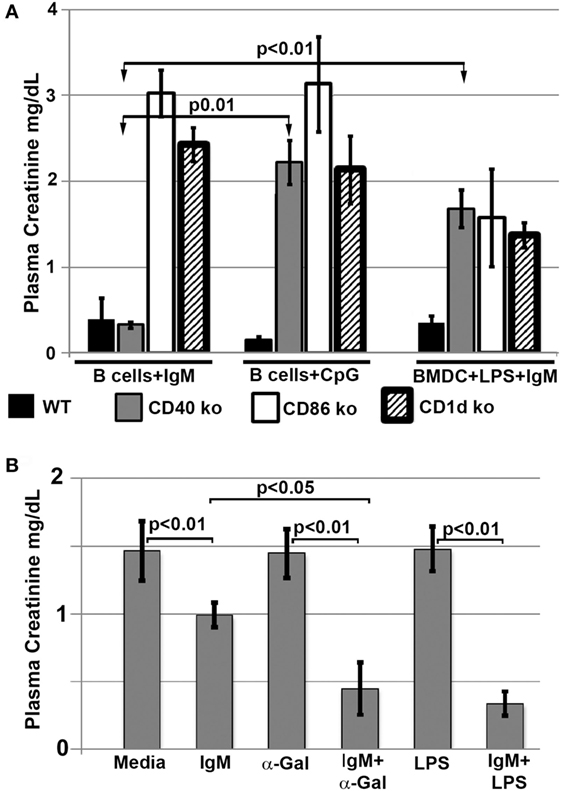
Figure 7. (A) Regulatory B cells and BMDC require CD86, CD40, and CD1d to interact with in vivo NKT. Note that IgM-pretreated regulatory B cells do not need CD40 to interact with NKT-1 cells. (B) α-Gal-Cer or LPS enhance protection mediated by IgM-pretreated BMDC on renal ischemia. BMDCs were cultured with LPS (400 ng/ml) or α-Gal-Cer (100 ng/ml) in the presence of IgM for 48 h at 37°C, washed, and then infused into mice. Data are a mean of at least three separate experiments, and three mice were used for each group in each experiment.
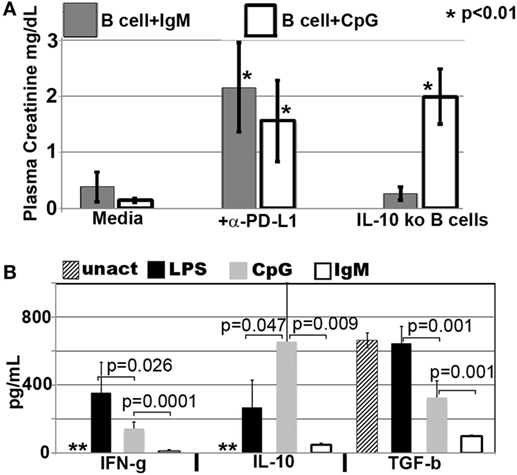
Figure 8. CpG-induced Bregs require PDL1 and IL10 to inhibit inflammation, while IgM-pretreated Bregs require PDL1 but not IL10 or TGFβ1. (A) IgM- and CpG-pretreated B cells lacked protection if PDL1 was blocked just prior to infusion. However, CpG, but not IgM-induced regulatory B cells, required IL10 to mediate in vivo protection. In blocking experiments, pretreated B cells were cultured for 48 h, washed, incubated with anti-PDL1 (5 μg/ml) or isotype control at 4°C for 1 h, re-washed, and then infused (IV) into mice. Isotype control α-PDL1 did not reverse the protective effect of regulatory B cells induced by IgM or CpG (data not shown). *p value compared to media. (B) Levels of IFNγ, IL10, and TGFβ1 present in 48-h culture supernatants of purified B cells that were not pretreated (labeled unact) or pretreated with LPS (400 ng/ml), CpG (5 μg/ml), or IgM (100 μg/ml) during the 48-h culture. 1 × 106 B cells/ml were cultured for 48 h with purified IgM, CpG, or LPS or no additive. Supernatants were obtained before B cells were washed and tested for regulatory activity by intravenous infusion into mice that subsequently underwent renal ischemia. Data are obtained from six to eight different supernatants. **Untreated B cells secreted <1 pg/ml of cytokine. In methods, we provide profile of other cytokines that were also quantitated by the Luminex beads.
In a second, more direct approach, we infused BMDC loaded ex vivo with α-gal-ceramide to directly interact with in vivo NKT-1 cells. As can be seen in Figure 7B, infusion of IgM-pretreated α-gal-ceramide loaded BMDC, expressing low CD1d (Figure 5B), protected mice from ischemic AKI. These studies indicate that protection was mediated by a direct interaction of infused IgM-induced regulatory BMDC (presenting low amounts of α-gal-cer) with in vivo NKT-1. Infusing IgM-pretreated BMDC (i.e., without α-gal-cer loading) was not as effective in protecting against ischemic AKI even though IgM downregulated CD1d (Figures 7B and 5B). However, infusing BMDC pretreated with both LPS and IgM was most effective in protecting against ischemic AKI (Figure 7B) possibly because LPS potentiated the inhibitory effect of IgM on BMDC by upregulating CD40 (Figure 5B), which has an important role in enhancing IL10 production after TLR activation (24, 25).
Pan-B Cells and BMDC Regulate In Vivo NKT-1 by Distinct Mechanisms
In prior studies, we showed that LPS + IgM-pretreated BMDC required PD1 (but not PDL1) and IL10 to regulate the in vivo innate inflammatory response mediated by renal ischemia (4). Therefore, we examined if regulatory B cells, induced ex vivo by either IgM or CpG, require PD1/PDL1 and IL10 to mediate their regulatory or suppressive effect on in vivo NKT-1 cells. This question was examined by using three approaches. First, ex vivo induced regulatory B cells were blocked with either anti-PD1 or anti-PDL1 just before in vivo cell infusion to determine if PD1/PDL1 were required. In these studies, ex vivo induced regulatory B cells were found to require PDL1 (Figure 8A), but not PD1 (data not shown), for their in vivo suppressive effect. Anti-PDL1 was more effective than anti-PD1 in blocking the in vivo inhibitory function of CpG- or IgM-induced regulatory B cells as these pretreated cells expressed high PDL1 and very low PD1 levels. B cells constitutively express high PDL1 and low PD1, and we, like others (26), failed to enhance PD1 expression even after LPS activation (data not shown).
Second, we used IL10 knock-out B cells to test if ex vivo induced regulatory B cells required IL10 to mediate their in vivo inhibitory effect. Rather surprising, IgM-pretreated B cells did not require IL10 to mediate their in vivo regulatory activity while CpG-pretreated B cells required IL10 (Figure 8A). Supernatants from IgM-pretreated B cells had in addition significantly decreased levels of TGFβ1 even when compared to untreated and CpG pretreated B cells (Figure 8B), thus indicating that IgM-pretreated B cells mediate regulatory activity by a mechanism that does not involve TGFβ1 or IL10. Currently, we have not delineated how IgM inhibits IFNγ, IL10, and TGFβ production. However, in the presence of low IFNγ production, B cells pretreated with IgM do not appear to require anti-inflammatory cytokines but require low Cd1d expression and PDL1 to regulate in vivo NKT-1. Such a hypothesis would also explain (i) the lack of regulatory activity in LPS-pretreated B cells where CD1d is not downregulated and production of IFNγ is significantly high when compared to IL10 (Figures 5A and 8B) and (ii) the presence of regulatory activity in CpG-pretreated B cells where CD1d is downregulated and the production of IL10 is significantly higher when compared to IFNγ production (Figures 5A and 8B). The high IL10 requirement for CpG-induced Bregs would also explain why CpG-, but not IgM-, induced Bregs are dependent on CD40 (Figure 7A), which when ligated by CD40L expressed by adjacent B cells in ex vivo culture or by NKT-1 in vivo enhances IL10 production of Bregs (24, 25).
Pretreating B cells with IgM or CpG or LPS did not change the 48-h supernatant levels of IL17, IL4, and IL13, which were produced at very low levels (<5 pg/ml) or TNFα, which was produced at low levels (30–110 pg/ml) (data not shown). Non-pretreated B cells produced no or very low levels (<5 pg/ml) of all cytokines except for high levels of TGFβ1 (Figure 8B). Interestingly, lack of IFNγ and production of high TGFβ1 by untreated B cells was not sufficient to induce regulatory activity in these cells (Figure 1), thus clearly indicating that regulatory activity in B cells requires in addition other factors such as low CD1d levels.
Infused Regulatory Cells Switch NKT-1 to a Regulatory Phenotype
We next determined if intravenously infused regulatory BMDC and B cells protected mice from renal IRI by rendering in vivo NKT-1 non-functional or switching these cells to a regulatory phenotype. Twenty-four hours after infusion of ex vivo pretreated BMDC or B cells, the in vivo function of NKT-1 cells was tested by specifically activating NKT-1 cells with intraperitoneal α-gal-ceramide (5 μg) injection. Under these in vivo activating conditions, only NKT-1 cells are directly activated by α-gal-ceramide to secrete IL4 and IFN-γ, and release of these cytokines can be quantitated in plasma (23). Data in Figure 9 show that NKT-1 cells from mice infused with regulatory BMDC or pan-B cells are switched to a regulatory phenotype, as these NKT-1 cells produce significantly reduced levels of IFNγ but not IL4 when activated with α-gal-ceramide. NKT-1 cells were not rendered anergic as such anergic NKT-1 cells produce extremely low levels of both IFNγ and IL4 when activated with α-gal-ceramide (23). The latter is depicted in Figure 9 where in vivo NKT-1 cells were rendered anergic with a large bolus of α-gal-ceramide and then re-challenged 5 days later with α-gal-ceramide.
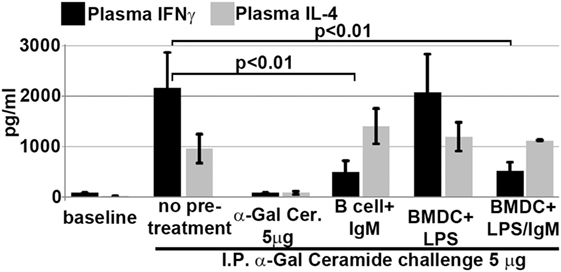
Figure 9. Infusion of IgM + LPS-pretreated BMDC and IgM-pretreated B cells induces in vivo NKT-1 cells to produce less IFNγ but normal or increased IL4 on α-gal-cer challenge. Intraperitoneal (IP) α-Gal-cer (5 μg) was given 24 h after cell infusion, and postchallenge plasma was obtained at 2 h for IL4 and at 18 h for IFN-γ. Mice initially treated with a 5-μg bolus (IP) of α-gal-cer produced very low IFNγ and IL4 upon re-challenge with α-gal-cer. Data are a mean of three separate experiments.
Data in Figures 7A and 8A and our prior studies (4) would indicate that infused regulatory pan-B cells and BMDC require lipid antigen presentation in the context of low CD1d expression, costimulatory interaction, as well as PD1/PDL1 and IL10 to switch in vivo NKT-1 to a regulatory phenotype. However, IgM induces regulatory B cells by a mechanism not involving IL10. In vivo NKT-1 cells with a regulatory phenotype fail to produce high levels of IFNγ (and induce an inflammatory response) when re-challenged with lipid antigens released after ischemic kidney injury.
Low CD1d Expression on Ex Vivo Pretreated Cells Is Not Sufficient to Switch Them to a Regulatory Phenotype
Figure 5 also shows that some non-protective infused cells had downregulation of CD1d expression, thus indicating that low CD1d expression on B cells or BMDC by itself may be insufficient to regulate in vivo NKT-1 as the cross-talk between infused cells and in vivo NKT-1 is also influenced by levels of activating and inhibitory cytokines as well as costimulatory receptors that induce activating or inhibitory signaling pathways. For example, it is possible that the CD1d low LPS + IgM-pretreated pan-B cells did not regulate in vivo NKT-1 as these pretreated B cells expressed high levels of CD86 (Figure 5) and possibly produced high levels of pro-inflammatory cytokines.
Infused Regulatory Cells Do Not Inhibit Ischemia-Induced Innate Inflammation by Regulating In Vivo DC or Innate Effectors or Enhancing Splenic Anti-inflammatory Cells
Further studies were performed in RAG-1 ko mice to determine if the infused IgM-pretreated B cells could ameliorate ischemia-induced inflammation by regulating in vivo DC or innate effectors (e.g., NK or granulocytes) via a mechanism not involving CD1d. To test this question, we infused IgM-pretreated pan-B cells into RAG-1 ko mice. As depicted in Figure 10, IgM-pretreated pan-B cells failed to protect RAG-1 ko mice from renal ischemia-induced renal injury, thus indicating that the infused regulatory B cells cannot directly regulate in vivo DC or innate effectors. In prior studies, we showed that infused IgM + LPS-pretreated regulatory BMDC also failed to protect RAG-1 ko mice from renal ischemia induced injury (4).
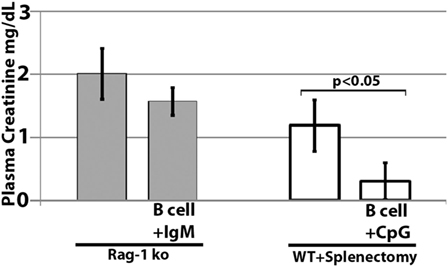
Figure 10. Infused regulatory B cells do not protect Rag-1 ko mice from ischemic acute kidney injury but protect mice that had prior splenectomy. Ex vivo pretreated regulatory B cells were infused in mice splenectomized 7 days before B cell infusion. Four mice were used in each group.
We next determined if there was an increase in splenic CD4+CD25+Foxp3+ T cells and CD4+CD25+IL10+ Treg cells when compared to mice with severe kidney injury. There was no difference in the level of these Treg cells between the two groups (data not shown) when splenic cells from protected mice were examined 24 h after renal ischemia, thus indicating that, in our renal ischemia model, protection was not mediated by an increase in splenic Tregs. However, we did not determine if there was an increase in Tregs several days after infusion of regulatory B cells or BMDC.
In murine models of ischemic AKI, prior splenectomy has been shown to worsen AKI (27) as well as AKI-induced lung injury (28). Potential mechanisms include removal of IL10 producing anti-inflammatory splenocytes as well as removal of anti-inflammatory splenocytes that are dependent on cholinergic stimulation in the spleen (27, 28). In addition, prior splenectomy has been shown to negate the protective effect of regulatory BMDC (i.e., S1P3 deficient), which, when infused, interact in the spleen with CD169+ marginal zone macrophages that induce Foxp3+ Tregs (29). Since significant numbers of infused cells also interact with splenocytes (21), we performed studies on mice splenectomized on day-7, to examine if infused regulatory B cells mediated protection by interacting with anti-inflammatory splenocytes. Data in Figure 10 clearly show that infused regulatory B cells do not require anti-inflammatory splenocytes to mediate protection. Instead, these data on prior splenectomized mice support the concept that infused regulatory cells, induced ex vivo by IgM or CpG, mediate protection by directly interacting and regulating in vivo NKT-1 cells that are also present in other organs, e.g., liver and bone marrow.
Discussion
The current studies were initiated to determine the mechanism by which polyclonal natural IgM inhibits the inflammatory response following renal ischemia. We hypothesized that one potential mechanism involved binding of IgM to receptors on B cells and DCs especially since we noted several fold higher IgM binding to splenic B cells and DC when compared to T cells. To test our hypothesis, we pretreated BMDC or pan B cells with polyclonal IgM and demonstrated that these ex vivo pretreated cells, when intravenously injected 24 h before renal ischemia, protected WT-B6 mice from developing AKI, by inhibiting the innate inflammatory response to DAMPS released by ischemic tubules (4). In this kidney ischemia model, activation of NKT-1 amplifies the inflammatory response which is largely responsible for causing AKI (16). Several observations were made. First, we show that BMDC, pretreated ex vivo with IgM + LPS, or pan-B cells, pretreated ex vivo with IgM or CpG, can be switched to a “regulatory” phenotype with CD1d downregulation. Bregs and regulatory BMDC require CD1d-mediated interaction with NKT-1 to inhibit the innate inflammatory response as CD1d ko B cells or BMDC fail to function as Bregs (Figure 7A). Such ex vivo induced regulatory BMDC or pan-B cells, by presenting lipid antigen in the context of low CD1d, regulate in vivo NKT-1 cells, which when challenged with α-gal-ceramide produce normal or increased IL4 but low IFN-γ (Figure 9). However, our observations indicate that CD1d downregulation by itself is not sufficient for ex vivo induced regulatory pan-B cells or BMDC to switch in vivo NKT-1 to a regulatory phenotype. This is best exemplified by LPS + IgM-pretreated pan-B cells, which failed to regulate in vivo NKT-1 and protect mice from ischemia-induced AKI despite downregulation of CD1d (Figures 1 and 5). These observations indicate that other mechanisms such as inhibitory costimulatory receptors and the balance between pro-inflammatory and anti-inflammatory cytokines are also involved in switching pan-B cells to regulatory cells (Figure 8B). Second, we show that CpG-induced regulatory B cells differ from IgM-induced regulatory B cells. Regulatory B cells induced by CpG require PDL1 (but not PD1) and IL10 to inhibit in vivo NKT-1 cells (Figure 8A) (16), while IgM-induced regulatory B cells require PDL1 but do not require IL10 or TGFβ1 to regulate in vivo NKT-1 (Figure 8A). It is possible that IgM-pretreated pan-B cells, which lack pro-inflammatory cytokines (Figure 8B), regulate in vivo NKT-1 cells by utilizing only PDL1 or in addition by producing other inhibitory molecules such as adenosine or Tim-1 as has been shown with certain other Bregs [reviewed in Ref. (30)]. In the previous study, we show that BMDCs, unlike B cells, require PD1 (but not PDL1) and IL10 to mediate their in vivo regulatory activity (4). Finally, we show that infused cells regulate in vivo NKT-1 cells within 24 h, and this may explain why IgM-pretreated human B cells induced regulatory activity in mice (Figure 4) even though they are most likely rejected within 3 to 4 days after being infused.
The current data and data from our prior studies (4) indicate that the cross-talk between inhibitory signals induced by IgM and signaling induced by TLRs 4 and 9 agonists have different functional effects on B cells and BMDC. First, LPS negates the IgM-induced regulatory activity in B cells while enhancing the IgM-induced regulatory activity in BMDC (Figure 1), and second, CpG induces regulatory activity in B cells but not in BMDC (Figure 1). A potential explanation for these differing effects is that LPS and CpG may bind to other receptors (in addition to TLR) on B cells and BMDC. For example, there is evidence to show that CpG, by binding to the B1 class of scavenger receptors on B cells, regulates B cells (31). This may have clinical relevance as the presence of excess TLR9 ligands released by pathogens or apoptotic cells could induce regulatory activity in B cells and inhibit inflammation mediated by NKT-1.
Currently, studies are in progress to determine how IgM regulates B cells and BMDC. It is possible that IgM binds to different inhibitory receptors on B cells and BMDC and, as a result, activates different inhibitory signaling pathways that differentially affect the cross-talk with pathways induced by TLRs 4 and 9 ligands. IgM can regulate B cells by activating inhibitory receptors that normally inhibit B cell activation. B cells express a number of “inhibitory” receptors namely FcγRIIB, CD22, siglec G, PD1, PDL1, CD5, and CD72. This entire family of “inhibitory” receptors recruits SHP-1 phosphatases to inhibit NFκB activation induced by BcR or TLR activation (32). Siglec receptors (i.e., CD22 and siglec G) are likely candidates as they are expressed on all B cells and IgM binds to these receptors (33, 34). Regulating B cells by IgM binding to FcμR is also a likely possibility as FcμR is expressed on all splenic B cells (35), and there are data to show that FcμR regulates B cell apoptosis, immunoglobulin production (including natural IgM) [reviewed in Ref. (36, 37)], and the density of membrane bound IgM on the BcR receptor (37). In addition, it is possible that B cells, like DC (38), can be regulated by internalization of FcμR bound to natural IgM/autoantigen complexes.
Our IgM-induced regulatory pan-B cells differ from previously described regulatory B cell subsets that regulate autoimmune-mediated inflammation (30) in that (i) they express low levels of CD1d (Figure 5A), (ii) their regulatory activity is negated by TLR4 activation (Figures 1 and 2), (iii) they inhibit innate inflammation by regulating in vivo NKT-1 cells, and (iv) they do not require IL10 for their regulatory activity (Figure 8A). Furthermore, we show that IgM can induce regulatory activity in B cell subsets arising from different splenic compartments, i.e., CD5+ B1 cells, FO B2 cells, and pan-B cells (Figure 3B). In our studies, we did not attempt to evaluate the regulatory effect of infused B cells on in vivo splenic Tregs as mice were sacrificed after 48 h of infusion. Currently, it is unclear how CpG induces regulatory activity in murine splenic pan-B cells but not in BMDC, and second, how LPS negates IgM-induced regulatory activity in B cells but not in BMDC, even though both TLR agonists increase IL10 production. We have not defined the mechanism by which IgM or CpG downregulates CD1d or induces regulatory activity in murine splenic pan-B cell subsets.
We have shown in prior in vitro studies that IgM-ALA (i) inhibits human and murine T cell activation and proliferation in response to anti-CD3 or alloantigens by binding and downregulating CD4 and also by binding to CD3 and inhibiting Zap70 phosphorylation in response to anti-CD3, (ii) inhibits differentiation of naïve murine T cells into TH1 and TH17 effectors, (iii) enhances generation of human and murine Foxp3+ T regs, and (iv) binds to human CCR5 and CXCR4 and inhibits leukocyte chemotaxis in response to chemokines (3, 9). The current studies provide another potential mechanism for IgM-ALA to regulate inflammation, i.e., reprogramming DC and pan-B cells to switch to a CD1dlo regulatory phenotype, which in turn regulates NKT-1 cells.
We propose the following hypothesis based on our findings. Ischemic injury or infectious agents activate DC and B cells with upregulation of costimulatory molecules, cytokine production, and antigen-presenting receptors to induce an inflammatory response. B1 cells are also activated via BCR and TLR, especially with infectious agents, and rapidly increase the production of natural IgM, which binds to certain undefined receptors on DC and B cells and switches these cells to a regulatory phenotype. TLR9 agonists, released by pathogens or apoptotic cells, could also provide another mechanism to regulate pan-B cells in the event of excess LPS or other TLR agonists. IgM-induced regulatory DC and B cells require PD1/PDL1, IL10, and possibly other mediators to mediate suppression and together with other in vivo regulatory mechanisms (e.g., Bregs, Tregs, IL10, FcγRIIB, and IgM) regulate the inflammatory process.
Ethics Statement
All experiments were performed in accordance with the recommendations of the National Institutes of Health and Institutional Animal Care and Use Guidelines. The Animal Research Committee of the University of Virginia approved all procedures and protocols.
Author Contributions
PL conceived the idea, designed the experiments, analyzed data, and wrote the manuscript. MO supervised the kidney ischemia experiments and critically reviewed the data and manuscript. KS performed experiments and prepared the figures. LH performed kidney ischemia experiments and AB performed kidney histology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by National Institutes of Health Grant R01DK083406-01A1 (to PL and MO) and K01DK091444 (AB) and by an American Heart Association career development grant, 11SDG7000007 (AB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health.
References
1. Lobo PI. Role of natural autoantibodies (NAA) and natural IgM anti-leucocyte autoantibodies (IgM-ALA) in health and disease. Front Immunol (2016) 7:198. doi:10.3389/fimmu.2016.00198
2. Gronwall C, Vas J, Silverman GJ. Protective roles of natural IgM antibodies. Front Immunol (2012) 3:66. doi:10.3389/fimmu.2012.00066
3. Lobo PI, Schlegel KH, Spencer CE, Okusa MD, Chisholm C, McHedlishvili N, et al. Naturally occurring IgM anti-leukocyte autoantibodies (IgM-ALA) inhibit T cell activation and chemotaxis. J Immunol (2008) 180(3):1780–91. doi:10.4049/jimmunol.180.3.1780
4. Lobo PI, Schlegel KH, Bajwa A, Huang L, Kurmaeva E, Wang B, et al. Natural IgM switches the function of lipopolysaccharide-activated murine bone marrow-derived dendritic cells to a regulatory dendritic cell that suppresses innate inflammation. J Immunol (2015) 195(11):5215. doi:10.4049/jimmunol.1500052
5. Robey IF, Schluter SF, Akporiaye E, Yocum DE, Marchalonis JJ. Human monoclonal natural autoantibodies against the T-cell receptor inhibit interleukin-2 production in murine T cells. Immunology (2002) 105(4):419–29. doi:10.1046/j.1365-2567.2002.01389.x
6. Terasaki PI, Mottironi VD, Barnett EV. Cytotoxins in disease. N Engl J Med (1970) 283(14):724–8. doi:10.1056/NEJM197010012831403
7. Winfield JB, Winchester RJ, Wernet P, Fu SM, Kunkel HG. Nature of cold-reactive antibodies to lymphocyte surface determinants in systemic lupus erythematosus. Arthritis Rheum (1975) 18(1):1–8. doi:10.1002/art.1780180609
8. Lobo PI. Nature of autolymphocytotoxins present in renal hemodialysis patients: their possible role in controlling alloantibody formation. Transplantation (1981) 32(3):233–7. doi:10.1097/00007890-198109000-00010
9. Lobo PI, Bajwa A, Schlegel KH, Vengal J, Lee SJ, Huang L, et al. Natural IgM anti-leukocyte autoantibodies attenuate excess inflammation mediated by innate and adaptive immune mechanisms involving th-17. J Immunol (2012) 188(4):1675–85. doi:10.4049/jimmunol.1101762
10. Klouda PT, Jeannet M. Cold and warm antibodies and graft survival in kidney allograft recipients. Lancet (1976) 307(7965):876–8. doi:10.1016/S0140-6736(76)92094-8
11. Lobo PI, Rudolf L, Westervelt FB. Enhanced kidney allograft survival across a positive crossmatch (cx) arising from B-cell specific and cold reactive antibodies (ab). Proc Clin Dial Transplant Forum (1977) 7:4–6.
12. Iwaki Y, Park MS, Terasaki PI, Billing R. Enhancement of human kidney allografts by cold B-lymphocyte cytotoxins. Lancet (1978) 311(8076):1228–9. doi:10.1016/S0140-6736(78)92464-9
13. Kerman RH, Susskind B, Buyse I, Pryzbylowski P, Ruth J, Warnell S, et al. Flow cytometry-detected IgG is not a contraindication to renal transplantation: IgM may be beneficial to outcome1. Transplantation (1999) 68(12):1855–8. doi:10.1097/00007890-199912270-00007
14. Przybylowski P, Balogna M, Radovancevic B, Frazier OH, Susskind B, Van Buren C, et al. The role of flow cytometry-detected IgG and IgM anti-donor antibodies in cardiac allograft recipients1. Transplantation (1999) 67(2):258–62. doi:10.1097/00007890-199901270-00012
15. Chhabra P, Schlegel KH, Okusa MD, Lobo PI, Brayman KL. Naturally occurring immunoglobulin M (nIgM) autoantibodies prevent autoimmune diabetes and mitigate inflammation after transplantation. Ann Surg (2012) 256(4):634–41. doi:10.1097/SLA.0b013e31826b4ba9
16. Li L, Huang L, Sung SS, Lobo PI, Brown MG, Gregg RK, et al. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol (2007) 178(9):5899–911. doi:10.4049/jimmunol.178.9.5899
17. Li L, Huang L, Ye H, Song SP, Bajwa A, Lee SJ, et al. Dendritic cells tolerized with adenosine A(2)AR agonist attenuate acute kidney injury. J Clin Invest (2012) 122(11):3931–42. doi:10.1172/JCI63170
18. Gray M, Miles K, Salter D, Gray D, Savill J. Apoptotic cells protect mice from autoimmune inflammation by the induction of regulatory B cells. Proc Natl Acad Sci U S A (2007) 104(35):14080–5. doi:10.1073/pnas.0700326104
19. Dooley H, Flajnik MF. Antibody repertoire development in cartilaginous fish. Dev Comp Immunol (2006) 30(1):43–56. doi:10.1016/j.dci.2005.06.022
20. Wingender G, Krebs P, Beutler B, Kronenberg M. Antigen-specific cytotoxicity by invariant NKT cells in vivo is CD95/CD178-dependent and is correlated with antigenic potency. J Immunol (2010) 185(5):2721–9. doi:10.4049/jimmunol.1001018
21. Barral P, Sanchez-Nino MD, van Rooijen N, Cerundolo V, Batista FD. The location of splenic NKT cells favors their rapid activation by blood-borne antigen. EMBO J (2012) 31(10):2378–90. doi:10.1038/emboj.2012.163
22. Allan LL, Stax AM, Zheng DJ, Chung BK, Kozak FK, Tan R, et al. CD1d and CD1c expression in human B cells is regulated by activation and retinoic acid receptor signaling. J Immunol (2011) 186(9):5261–72. doi:10.4049/jimmunol.1003615
23. Parekh VV, Wilson MT, Olivares-Villagomez D, Singh AK, Wu L, Wang CR, et al. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest (2005) 115(9):2572–83. doi:10.1172/JCI24762
24. Edwards AD, Manickasingham SP, Sporri R, Diebold SS, Schulz O, Sher A, et al. Microbial recognition via toll-like receptor-dependent and -independent pathways determines the cytokine response of murine dendritic cell subsets to CD40 triggering. J Immunol (2002) 169:3652–3600. doi:10.4049/jimmunol.169.7.3652
25. Inoue S, Leitner WW, Golding B, Scott D. Inhibitory effects of B cells on anti-tumor immunity. Cancer Res (2006) 66:7741–7. doi:10.1158/0008-5472.CAN-05-3766
26. Zhong X, Bai C, Gao W, Strom TB, Rothstein TL. Suppression of expression and function of negative immune regulator PD-1 by certain pattern recognition and cytokine receptor signals associated with immune system danger. Int Immunol (2004) 16:1181–8. doi:10.1093/intimm/dxh121
27. Gigliotti JC, Huang L, Bajwa A, Ye H, Mace EH, Hossack JA, et al. Ultrasound modulates the splenic neuroimmune axis in attenuating AKI. J Am Soc Nephrol (2015) 26(10):2470–81. doi:10.1681/ASN.2014080769
28. Andres-Hernando A, Altmann C, Ahuja N, Lanaspa MA, Nemenoff R, He Z, et al. Splenectomy exacerbates lung injury after ischemic acute kidney injury in mice. Am J Physiol Renal Physiol (2011) 301(4):F907–16. doi:10.1152/ajprenal.00107.2011
29. Bajwa A, Huang L, Kurmaeva E, Gigliotti JC, Ye H, Miller J, et al. Sphingosine 1-phosphate receptor 3-deficient dendritic cells modulate splenic responses to ischemia-reperfusion injury. J Am Soc Nephrol (2015) 27(4):1076–90. doi:10.1681/ASN.2015010095
30. Mauri C, Menon M. The expanding family of regulatory B cells. Int Immunol (2015) 27(10):479–86. doi:10.1093/intimm/dxv038
31. Zhu P, Liu X, Treml LS, Cancro MP, Freedman BD. Mechanism and regulatory function of CpG signaling via scavenger receptor B1 in primary B cells. J Biol Chem (2009) 284(34):22878–87. doi:10.1074/jbc.M109.018580
32. Pritchard NR, Smith KG. B cell inhibitory receptors and autoimmunity. Immunology (2003) 108(3):263–73. doi:10.1046/j.1365-2567.2003.01592.x
33. Hanasaki K, Powell LD, Varki A. Binding of human plasma sialoglycoproteins by the B cell-specific lectin CD22. Selective recognition of immunoglobulin M and haptoglobin. J Biol Chem (1995) 270(13):7543–50. doi:10.1074/jbc.270.13.7543
34. Adachi T, Harumiya S, Takematsu H, Kozutsumi Y, Wabl M, Fujimoto M, et al. CD22 serves as a receptor for soluble IgM. Eur J Immunol (2012) 42(1):241–7. doi:10.1002/eji.201141899
35. Shima H, Takatsu H, Fukuda S, Ohmae M, Hase K, Kubagawa H, et al. Identification of TOSO/FAIM3 as an Fc receptor for IgM. Int Immunol (2009) 22(3):149–56. doi:10.1093/intimm/dxp121
36. Wang H, Coligan JE, Morse HC III. Emerging functions of natural IgM and its Fc receptor FCMR in immune homeostasis. Front Immunol (2016) 7:99. doi:10.3389/fimmu.2016.00099
37. Nguyen TT, Kläsener K, Zürn C, Castillo PA, Brust-Mascher I, Imai DM, et al. The IgM receptor Fc [mu] R limits tonic BCR signaling by regulating expression of the IgM BCR. Nat Immunol (2017) 18(3):321–33. doi:10.1038/ni.3677
Keywords: natural IgM, IgM anti-leukocyte antibodies, Bregs, CpG, innate inflammation, ischemia–reperfusion injury, regulatory bone marrow dendritic cells, NKT-1
Citation: Lobo PI, Schlegel KH, Bajwa A, Huang L and Okusa MD (2017) Natural IgM and TLR Agonists Switch Murine Splenic Pan-B to “Regulatory” Cells That Suppress Ischemia-Induced Innate Inflammation via Regulating NKT-1 Cells. Front. Immunol. 8:974. doi: 10.3389/fimmu.2017.00974
Received: 10 May 2017; Accepted: 31 July 2017;
Published: 23 August 2017
Edited by:
Harry W. Schroeder, University of Alabama at Birmingham, United StatesReviewed by:
Raul M. Torres, University of Colorado Denver, United StatesBruce David Mazer, The Research Institute of the McGill University Health Center, Canada
Copyright: © 2017 Lobo, Schlegel, Bajwa, Huang and Okusa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter I. Lobo, pil@virginia.edu