 Yuhai Zhao
Yuhai Zhao Lin Cong1,3
Lin Cong1,3 Walter J. Lukiw
Walter J. Lukiw- 1Neuroscience Center, Louisiana State University School of Medicine, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 2Department of Anatomy and Cell Biology, Louisiana State University School of Medicine, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 3Department of Neurology, Shengjing Hospital, China Medical University, Heping District, Shenyang, China
- 4Department of Neurology, Louisiana State University School of Medicine, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 5Department of Ophthalmology, Louisiana State University School of Medicine, Louisiana State University Health Sciences Center, New Orleans, LA, United States
Abundant clinical, epidemiological, imaging, genetic, molecular, and pathophysiological data together indicate that there occur an unusual inflammatory reaction and a disruption of the innate-immune signaling system in Alzheimer’s disease (AD) brain. Despite many years of intense study, the origin and molecular mechanics of these AD-relevant pathogenic signals are still not well understood. Here, we provide evidence that an intensely pro-inflammatory bacterial lipopolysaccharide (LPS), part of a complex mixture of pro-inflammatory neurotoxins arising from abundant Gram-negative bacilli of the human gastrointestinal (GI) tract, are abundant in AD-affected brain neocortex and hippocampus. For the first time, we provide evidence that LPS immunohistochemical signals appear to aggregate in clumps in the parenchyma in control brains, and in AD, about 75% of anti-LPS signals were clustered around the periphery of DAPI-stained nuclei. As LPS is an abundant secretory product of Gram-negative bacilli resident in the human GI-tract, these observations suggest (i) that a major source of pro-inflammatory signals in AD brain may originate from internally derived noxious exudates of the GI-tract microbiome; (ii) that due to aging, vascular deficits or degenerative disease these neurotoxic molecules may “leak” into the systemic circulation, cerebral vasculature, and on into the brain; and (iii) that this internal source of microbiome-derived neurotoxins may play a particularly strong role in shaping the human immune system and contributing to neural degeneration, particularly in the aging CNS. This “Perspectives” paper will further highlight some very recent developments that implicate GI-tract microbiome-derived LPS as an important contributor to inflammatory-neurodegeneration in the AD brain.
Introduction—Inflammatory Signaling in the Alzheimer’s Disease (AD) Brain
Multiple aspects of increased inflammatory signaling and an altered innate-immune system are consistent features of AD neuropathology; however, it is not well understood where these pathogenic signals originate or how they progressively contribute to the AD process (1–5). AD is characterized by the appearance of complex networks of many different kinds of chemokines and cytokines including, prominently, interleukin 1β (IL-1β) and tumor necrosis factor (TNFα), 40 and 42 amino acid amyloid beta (Aβ40, Aβ42) peptides, and adhesion molecules, in addition to the progressive deposition of these Aβ peptide containing amyloid plaques and neurofibrillary tangles (NFT) in the parenchyma of AD brain (6, 7). Activated microglia, astrocytes, or neurons appear to mediate the release of these pro-inflammatory molecules and cellular immune components (6, 8–12). Indeed, chemokines, cytokines, the insoluble Aβ42-enriched peptide deposits, NFTs, apoptotic, damaged and vanishing neurons, and activated microglia, and other related pro-inflammatory signals are potent neuropathological stimulants that appear to maintain the AD brain in a “chronic state of self-reinforcing inflammation” (2, 7, 10–13). Very recent studies that evaluated the pro-inflammatory potential of several different chemokines, cytokines, Aβ peptides, and lipopolysaccharides (LPS), either alone or in combination, have indicated that when compared, bacterial LPSs exhibit the strongest induction of pro-inflammatory signaling in human neuronal–glial cells in primary coculture of any single inducer, and different LPS extracts from different gastrointestinal (GI)-tract resident Gram-negative bacteria appeared to have different pro-inflammatory potential (12, 14–16). For example, exposure of LPS from the Gram-negative GI-tract abundant Bacteroides fragilis to primary human neuronal–glial cells in coculture was found to be an exceptionally powerful inducer of the NF-κB p50/p65 dimer, a known pro-inflammatory transcription factor complex that triggers the expression of pathogenic pathways involved in neurodegenerative inflammation (15, 16). In both neocortex and hippocampus, LPS has been detected to range from a ~7- to ~21-fold increase abundance in AD brain (Figures 1A–D). Along with an avalanche of very recent work from independent laboratories, these observations prompted us to further examine the presence and anatomical location of LPS in AD brains versus age- and gender-matched controls (12, 17, 18).
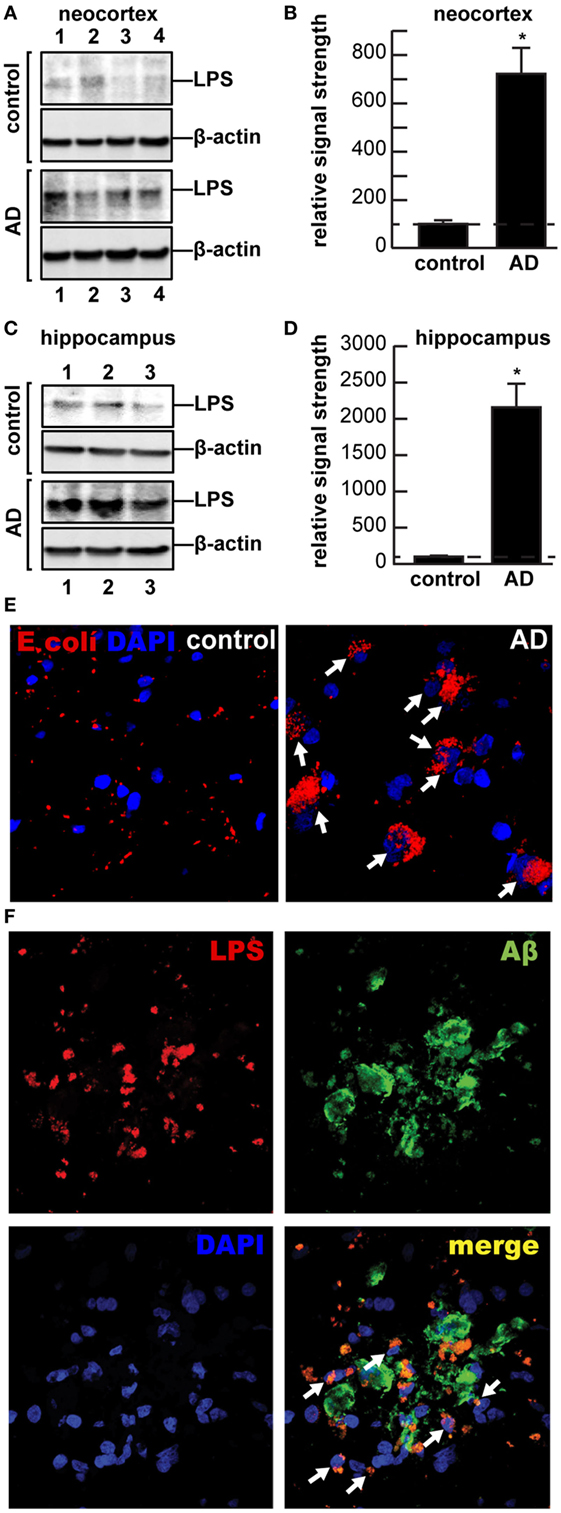
Figure 1. (A–D) Western and (E–F) immunohistochemical analysis of lipopolysaccharide (LPS) (~37 kDa) signals in human brain temporal lobe neocortex [N = 4 control and 4 sporadic Alzheimer’s disease (AD) cases; quantified in (B)]; and (C) hippocampus [N = 3 control and N = 3 sporadic AD cases; quantified in (D)] were compared against β-actin (~42 kDa) abundance in the same sample (using anti-Escherichia coli LPS; cat# ab35654 from Abcam, Cambridge UK and anti-β-actin cat# 3700, Cell Signaling, Danvers, MA, USA). All Western methodologies have been previously described in detail (12, 19). Densitometric readings of immune-reactive bands were obtained using ImageQuantTL [GE Healthcare (12, 19, 20)]; all control and AD tissues were age- and gender-matched; there were no significant differences between the age (control 82.5 ± 8.1 years, AD 81.3 ± 8.8 years), gender (all female), postmortem interval (PMI) (all tissues 3.8 h or less), RNA quality, or RNA yield between each of the two groups; in these samples, LPS abundance was found to be on average greater than sevenfold as abundant in AD when compared to control neocortex; LPS was found to be on average >21-fold as abundant in AD when compared to control hippocampus; in (B,D) a dashed horizontal line at 100 is included for ease of comparison; *p < 0.01 (ANOVA); (E,F) for immunohisto-chemistry control and AD neocortex and/or hippocampal brain tissues were embedded, sectioned (10 μm), fixed, and incubated with primary antibodies (1:1,000; 1× PBS with 2% BSA, 2% goat or donkey serum, and 0.1% TX-100) overnight at 4°C, washed with PBS, and then incubated with Alexa Fluor-conjugated species-specific secondary antibodies (LPS; red fluorescence λmax ~ 650 nm); sections were next counter-stained with DAPI (blue fluorescence; λmax ~ 470 nm) for nuclei (E), and/or Aβ peptide (green fluorescence; λmax ~ 510 nm) (F) and imaged with Zeiss LSM 700 Confocal Laser microscope system (Richmond, VA, USA); note perinuclear staining of LPS in AD; while there appears to be random association of LPS with Aβ deposits in controls, >75% of all LPS signals were found to be associated with brain cell nuclei in AD; the significance of this is not currently known; the association of LPS with the major cellular repository for genetic material suggests that the significance of this association may be genetic; white arrows highlight LPS-nuclear envelope association; a total of 26 control and AD brains (PMI 3.8 h or less) were examined and yielded highly similar results; (E,F) magnification 50×.
Internally Derived Noxious Exudates of the GI-Tract Microbiome
Major Gram-negative bacilli of the human GI-tract, such as the abundant B. fragilis and Escherichia coli (E. coli), are capable of discharging a remarkably complex assortment of pro-inflammatory neurotoxins. These consist of four major components: (i) bacterial amyloids (10, 21); (ii) endotoxins and exotoxins (5, 12); (iii) LPS (12, 18); and (iv) small non-coding RNAs (sncRNAs) [(22–25), unpublished observations]. Either alone or in various combinations, these neurotoxins are intensely pro-inflammatory toward primary human brain cells (12, 15, 16). As integral components of the outer leaflet of the outer membrane of Gram-negative bacteria, LPS shed into the local environment have historically been thought to play some host–pathogen immune-evasion strategy useful to bacterial survival while eliciting strong immune and inflammatory responses within the host. Interestingly, secreted LPS along with proteolytic endotoxins and amyloid monomers are generally soluble as monomers. However, over time, they aggregate into highly insoluble fibrous lipoprotein lesions that associate with the progressive degenerative neuropathology of several common, age-related disorders of the human systemic circulation, and CNS including systemic inflammatory response syndrome, multiple sclerosis, prion disease, and AD (12, 20, 26). LPS, the major molecular component of the outer membrane of Gram-negative bacteria normally serves as a physical barrier providing the bacteria protection from its surroundings. LPS is also recognized by the immune system as a marker for the detection of bacterial pathogen invasion and responsible for the development of inflammatory response is perhaps the most potent stimulator and trigger of inflammation known (27). LPS activates toll-like receptors (TLRs), membrane-spanning protein receptors expressed in microglial cells of the innate-immune system, which recognize common damage- or pathogen-associated molecular-patterns [DAMPS, PAMPs (2, 28)]. Interestingly, of the 13 currently characterized TLRs, the microglial TLR2 and TLR4 are activated by amyloid, LPS, lipoglycans, and/or other microbial triggers that subsequently induce cytokine production, inflammation, phagocytosis, and innate-immune defense responses that directly induce the development of CNS pathology. In addition to the TLR2 and TLR4 receptors, at least one additional microglial transmembrane LPS receptor—CD14 mediates phagocytosis of both bacterial components and Aβ42 peptides, hence expanding roles for microglia and microglial LPS receptors in AD pathophysiology (12, 29).
To cite other recent examples, a secreted, highly pro-inflammatory zinc metalloprotease metalloproteinase B. fragilis endotoxin called fragilysin (BFT) derived from enterotoxigenic strains of B. fragilis have been recently shown to contribute to: (i) anaerobic bacteremia, sepsis and systemic inflammatory distress, diarrheal disease; (ii) systemic inflammation, GI-tract, and colorectal cancers; (iii) inflammatory neurodegeneration in part via the disruption of epithelial cell-based GI-tract barriers via cleavage of the synaptic adhesion zonula adherens protein E-cadherin; and (iv) enterotoxigenic microbes specifically impact microglial-mediated innate-immune responses, detoxifying and phagocytic mechanisms, and amyloidogenesis characteristic of inflammatory aspects of neurodegeneration (12, 15, 16, 30–34). Prokaryotic sncRNAs play essential roles in the regulation of many bacteriological processes including the expression of exotoxins and endotoxins and the regulation of bacterial virulence (22). In eukaryotes, microRNAs (miRNAs) also function as key regulators in many biological processes through posttranscriptional suppression of mRNAs and the downregulation of gene expression. Typical trans-acting microRNA-size sncRNAs are abundant in all prokaryotic cells including bacteria and fungi, but their production, release, and leakage from the confines of a healthy GI-tract into systemic and cerebral circulation and downstream effects along the gut microbiome–brain axis are a highly novel and largely unexplored research area (12, 22, 25). There is considerable speculation that, as for other bacterial exudates, such RNA-based neurotoxins may be pathogenic and highly detrimental to the homeostatic function of the neuronal, glial, endothelial, and other brain cells that comprise the CNS (23, 24).
Leakage of Neurotoxic Molecules into the Systemic Circulation and the CNS
Gram-negative bacterial exudates of the human GI-tract are not only the primary source of a remarkable array of neurotoxic pro-inflammatory amyloids, endo- and exotoxins, LPSs, and sncRNAs but also serve as potent sources of membrane-disrupting agents (12, 15, 16, 35, 36). As aforementioned, BFT can alone induce the disruption of epithelial cell-based GI-tract membrane barriers via presenilin 1-dependent cleavage of the zonula adherens protein E-cadherin, thus leading to progressive functional decline in membrane integrity (12, 15, 16, 30–34). Other recent reports suggest that intestinal dysbiosis and “leaky gut syndrome” constitutes a key pathophysiological link for transport of microbiome-derived toxins across GI-tract and blood–brain biological barriers that result in a progression from systemic to CNS inflammation (12, 21). The progressive failure of major physiological barriers is reminiscent of the activation of the thanatomicrobiome (the “death”-associated microbiome) and the deactivation of protective biological barriers that occurs at the time of death when normal endothelial cell structures and signaling: (i) becomes increasingly inoperative and “leaky” (1, 12, 37); and (ii) progressively unable to support normal homeostatic brain functions that are accompanied by a progressive and insidious functional decline (12, 28, 37). These recent findings indicate that AD-affected brains have remarkably large loads of bacterial-derived toxins compared to controls. The transfer of noxious, pro-inflammatory molecules from the GI-tract microbiome to the CNS may be increasingly important during the course of aging when both the GI-tract and blood–brain barriers become significantly more permeable (12, 28, 38).
Perinuclear Localization of LPS in AD Brains
While other recent studies have reported an LPS-mediated stimulation of chronic inflammation, beta-amyloid accumulation, and episodic memory decline in murine models of AD (39, 40) and a biophysical association of LPS with amyloid deposits and blood vessels in human AD patients (18), here, we provide the first evidence of a perinuclear association of LPS with AD brain cell nuclei (Figures 1E,F). Strong adherence of LPS to the nuclear periphery has recently been shown to inhibit nuclear maturation and function that may impair or block export of mRNA signals from brain cell nuclei, a highly active organelle with extremely high rates of transcription, mRNA processing, and export into the cytoplasm [(41–43), unpublished observations]. This may in part be responsible for the widely observed, generalized downregulation of global gene expression in AD, independently reported by several AD gene expression research laboratories, through the biophysical blockage of mRNA trafficking through nuclear pores (41, 42, 44, 45). LPS may be further injurious to the nuclear membrane just as LPS contributes to cerebrovascular endothelial cell membrane injury (12, 18, 40). Lastly, evidence is accumulating that neurotoxic exudates from other GI-tract microbiota may contribute to dysfunction in additional, ultimately fatal neuropsychiatric illnesses that involve progressive inflammatory neurodegeneration (8, 12). New opportunities to modulate existing gut microbiota and their exudates using probiotics and/or modifications through soluble or insoluble dietary fiber intake could provide novel targets for more effective clinical intervention [Figure 2 (18, 46, 47); unpublished observations]. Interestingly, the high intake of dietary fiber is a strong inhibitor of B. fragilis abundance and proliferation in the intact human GI-tract and as such is a potent inhibitor of the neurotoxic B. fragilis-derived amyloids, LPS, enterotoxins, and sncRNAs. Hence, dietary fiber-mediated suppression of B. fragilis abundance may turn out to be beneficial for both the human GI-tract microbiome and CNS health (34, 38, 46).
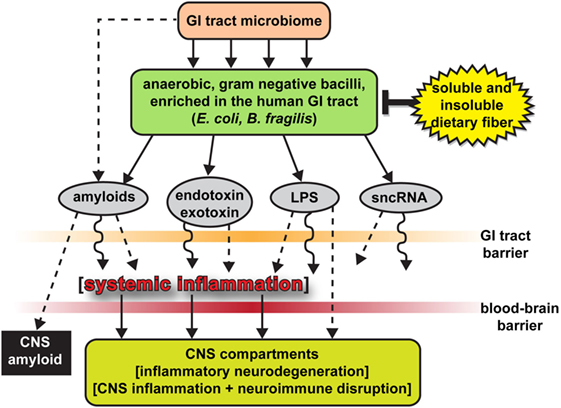
Figure 2. The human gastrointestinal (GI)-tract microbiome as a source of strong pro-inflammatory exudates—highly schematicized depiction of anaerobic, Gram-negative bacilli (such as Escherichia coli and Bacteroides fragilis) of the human GI-tract microbiome and their potentially pathogenic, immunogenic, and pro-inflammatory neurotoxins [amyloids, endotoxins and exotoxins, lipopolysaccharide (LPS), and small non-coding RNAs (sncRNAs)] that may contribute to systemic and CNS inflammation and neuro-immune disruption; two major sources of these complex mixtures are E. coli and B. fragilis; major anaerobic Gram-negative bacilli of the human middle and lower GI-tract, respectively; the B. fragilis toxin (BFT) fragilysin is one of the most potent pro-inflammatory molecules known (12, 15, 16, 30, 37, 38); these intensely pro-inflammatory LPS species may be able to “leak” through at least two major biophysiological barriers—the GI-tract barrier and the blood–brain barrier—to access brain compartments [see Ref. (2, 12, 28, 30, 31, 34)]. Neurotoxic mixtures secreted by multiple GI-tract microbes or other microbial species may have considerable potential to support inflammatory signaling within the CNS (2, 12, 21, 28, 30, 31, 34); B. fragilis proliferation and (BFT) fragilysin levels may be kept in check by increased intake of soluble and insoluble dietary fiber (34, 38, 46); interestingly, BFT-derived fragilysin may exert neurotoxic activities via multiple mechanisms: (i) by increasing the permeability or “leakiness” of the intestinal epithelium via the dissolution of tight junctions in epithelial cells (28, 30); and (ii) by promoting amyloid peptide aggregation and progressive amyloidogenesis (15, 16, 18, 37, 38); Figure 2 modified and updated from Lukiw (15, 16).
Concluding Remarks
It is not generally appreciated that, in the human body, microbial genes outnumber human genes by about 100 to 1, and the impact of bacterial genetics on human health and disease may have been vastly underestimated (8, 12, 15–17, 48). The assumption of the privileged immunological status of the CNS has also been recently questioned in multiple investigations, particularly in terms of inflammatory neurodegenerative diseases such as AD, as both microbial-derived nucleic acid sequences and/or noxious exudates representative of GI-tract Gram-negative bacteria are showing up within CNS compartments, including, prominently, anatomical regions of the CNS involved in inflammatory and pathological signaling and neuro-immune disruptions that characterize the AD process (9, 12, 15, 16, 18, 49). For example, LPS has been recently localized to the same anatomical regions involved in AD-type neuropathology to levels of greater than sevenfold over control in the temporal lobe neocortex and >21-fold over control in the hippocampus. This suggests that GI-tract microbiome-derived LPS may be an important initiator and/or significant contributor to inflammatory degeneration in the AD CNS (Figures 1 and 2). An alternative, yet, highly speculative view is that the human CNS may have its own microbiome, which could also explain the presence of Gram-negative bacterial secretory components in the brain as well as multiple forms of microbial-derived nucleic acid sequences (12, 49).
Author Contributions
YZ, LC, VJ, and WL conceived and discussed the experimental design; YZ, LC, VJ, and WL performed the experiments; YZ and WL performed bioinformatics and contributed to the medical artwork; WL reviewed the results and further researched and wrote this paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The work in this paper was presented in part at the Vavilov Institute Autumn 2016 Seminar Series (Институт Вавилова Осень 2016 Семинар Серии) in Moscow, Russia October 2016, at the Society for Neuroscience (SFN) Annual Meeting, San Diego, CA, USA November 2016, and will be presented in Abstract-Special Symposium format at the Society for Neuroscience (SFN) Annual Meeting, Washington, DC, USA November 2017. Sincere thanks are extended to Drs P. N. Alexandrov, J. G. Cui, F. Culicchia, W. Poon, K. Navel, C. Hebel, C. Eicken, and the late Dr. J. M. Hill for helpful discussions in this research area, for short postmortem interval (PMI) human brain tissues or extracts, for initial bioinformatics and data interpretation, and to D. Guillot and A. I. Pogue for expert technical assistance and medical artwork. Thanks are also extended to the University of California at Irvine Brain Bank and the many neuropathologists, physicians, and researchers of the US and Canada who have provided high quality, short post-mortem interval (PMI) human CNS or extracted tissue fractions for scientific study. Research on the microRNAs, pro-inflammatory, and pathogenic signaling in the Lukiw laboratory involving the innate-immune response, neuroinflammation, and amyloidogenesis in AD, prion, and in other neurological diseases was supported through an unrestricted grant to the LSU Eye Center from Research to Prevent Blindness (RPB); the Louisiana Biotechnology Research Network (LBRN) and NIH grants NEI EY006311, NIA AG18031, and NIA AG038834 (WL).
References
1. Clement C, Hill JM, Dua P, Culicchia F, Lukiw WJ. Analysis of RNA from Alzheimer’s disease post-mortem brain tissues. Mol Neurobiol (2016) 53:1322–8. doi:10.1007/s12035-015-9105-6
2. Richards RI, Robertson SA, O’Keefe LV, Fornarino D, Scott A, Lardelli M, et al. The enemy within: innate surveillance-mediated cell death, the common mechanism of neurodegenerative disease. Front Neurosci (2016) 10:193. doi:10.3389/fnins.2016.00193
3. Franco-Bocanegra DK, Nicoll JAR, Boche D. Innate immunity in Alzheimer’s disease: the relevance of animal models? J Neural Transm (Vienna) (2017). doi:10.1007/s00702-017-1729-4
4. Rojas-Gutierrez E, Muñoz-Arenas G, Treviño S, Espinosa B, Chavez R, Rojas K, et al. Alzheimer’s disease and metabolic syndrome: a link from oxidative stress and inflammation to neurodegeneration. Synapse (2017):e21990. doi:10.1002/syn.21990
5. VanItallie TB. Alzheimer’s disease: innate immunity gone awry? Metabolism (2017) 69S:S41–9. doi:10.1016/j.metabol.2017.01.014
6. Marottoli FM, Katsumata Y, Koster KP, Thomas R, Fardo DW, Tai LM. Peripheral inflammation, apolipoprotein E4, and amyloid-β Interact to induce cognitive and cerebrovascular dysfunction. ASN Neuro (2017) 9. doi:10.1177/1759091417719201
7. McManus RM, Heneka MT. Role of neuroinflammation in neurodegeneration: new insights. Alzheimers Res Ther (2017) 9:14. doi:10.1186/s13195-017-0241-2
8. Foster JA, Lyte M, Meyer E, Cryan JF. Gut microbiota and brain function: an evolving field in neuroscience. Int J Neuropsychopharmacol (2016) 19:yv114. doi:10.1093/ijnp/pyv114
9. Bagyinszky E, Giau VV, Shim K, Suk K, An SSA, Kim S. Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J Neurol Sci (2017) 376:242–54. doi:10.1016/j.jns.2017.03.031
10. Bergman P, Roan NR, Römling U, Bevins CL, Münch J. Amyloid formation: functional friend or fearful foe? J Intern Med (2017) 280:139–52. doi:10.1111/joim.12479
11. Olsen I, Singhrao SK. Inflammasome involvement in Alzheimer’s disease. J Alzheimers Dis (2017) 54:45–53. doi:10.3233/JAD-160197
12. Zhao Y, Jaber V, Lukiw WJ. Secretory products of the human GI-tract microbiome and their potential impact on Alzheimer’s disease (AD): detection of lipopolysaccharide (LPS) in AD hippocampus. Front Cell Infect Microbiol (2017) 7:318. doi:10.3389/fcimb.2017.00318
13. Hill JM, Lukiw WJ. Microbial-generated amyloids and Alzheimer’s disease (AD). Front Aging Neurosci (2015) 7:9. doi:10.3389/fnagi.2015.00009
14. Zhao Y, Lukiw WJ. Microbiome-generated amyloid and potential impact on amyloidogenesis in Alzheimer’s disease (AD). J Nat Sci (2015).
15. Lukiw WJ. The microbiome, microbial-generated pro-inflammatory neurotoxins, and Alzheimer’s disease. J Sport Health Sci (2016) 5(4):393–6. doi:10.1016/j.jshs.2016.08.008
16. Lukiw WJ. Bacteroides fragilis lipopolysaccharide and inflammatory signaling in Alzheimer’s disease. Front Microbiol (2016) 7:1544. doi:10.3389/fmicb.2016.01544
17. Bhattacharjee S, Lukiw WJ. Alzheimer’s disease and the microbiome. Front Cell Neurosci (2013) 7:153. doi:10.3389/fncel.2013.00153
18. Zhan X, Stamova B, Jin LW, DeCarli C, Phinney B, Sharp FR. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology (2016) 87(22):2324–32. doi:10.1212/WNL.0000000000003391
19. Bhattacharjee S, Zhao Y, Dua P, Rogaev EI, Lukiw WJ. microRNA-34a-mediated down-regulation of the microglial-enriched triggering receptor and phagocytosis-sensor TREM2 in age-related macular degeneration. PLoS One (2016) 11(3):e0150211. doi:10.1371/journal.pone.0150211
20. Zhao Y, Dua P, Lukiw WJ. Microbial sources of amyloid and relevance to amyloidogenesis and Alzheimer’s disease (AD). J Alzheimers Dis Parkinsonism (2015) 5(1):177.
21. Köhler CA, Maes M, Slyepchenko A, Berk M, Solmi M, Lanctôt KL, et al. The gut-brain axis, including the microbiome, leaky gut and bacterial translocation: mechanisms and pathophysiological role in Alzheimer’s disease. Curr Pharm Des (2016) 22(40):6152–66. doi:10.2174/1381612822666160907093807
22. Bloch S, Węgrzyn A, Węgrzyn G, Nejman-Faleńczyk B. Small and smaller-sRNAs and microRNAs in the regulation of toxin gene expression in prokaryotic cells: a mini-review. Toxins (Basel) (2017) 9:E181. doi:10.3390/toxins9060181
23. Dendooven T, Luisi BF. RNA search engines empower the bacterial intranet. Biochem Soc Trans (2017):BST20160373. doi:10.1042/BST20160373
24. Nitzan M, Rehani R, Margalit H. Integration of bacterial small RNAs in regulatory networks. Annu Rev Biophys (2017) 46:131–48. doi:10.1146/annurev-biophys-070816-034058
25. Torres-Martínez S, Ruiz-Vázquez RM. The RNAi universe in fungi: a varied landscape of small RNAs and biological functions. Annu Rev Microbiol (2017). doi:10.1146/annurev-micro-090816-093352
26. Clark IA, Vissel B. Amyloid β: one of three danger-associated molecules that are secondary inducers of the proinflammatory cytokines that mediate Alzheimer’s disease. Br J Pharmacol (2015) 172:3714–27. doi:10.1111/bph.13181
27. Li D, Yu F. Peripheral inflammatory biomarkers and cognitive decline in older adults with and without Alzheimer’s disease: a systematic review. J Gerontol Nurs (2017) 1–7. doi:10.3928/00989134-20170519-01
28. Varatharaj A, Galea I. The blood-brain barrier in systemic inflammation. Brain Behav Immun (2017) 60:1–12. doi:10.1016/j.bbi.2016.03.010
29. Jiang Q, Jin S, Jiang Y, Liao M, Feng R, Zhang L, et al. Alzheimer’s disease variants with the genome-wide significance are significantly enriched in immune pathways and active in immune cells. Mol Neurobiol (2017) 54(1):594–600. doi:10.1007/s12035-015-9670-8
30. Sears CL, Geis AL, Housseau F. Bacteroides fragilis subverts mucosal biology: from symbiont to colon carcinogenesis. J Clin Invest (2014) 124:4166–72. doi:10.1172/JCI72334
31. Choi VM, Herrou J, Hecht AL, Teoh WP, Turner JR, Crosson S, et al. Activation of Bacteroides fragilis toxin by a novel bacterial protease contributes to anaerobic sepsis in mice. Nat Med (2016) 22:563–7. doi:10.1038/nm.4077
32. Fathi P, Wu S. Isolation, detection, and characterization of enterotoxigenic Bacteroides fragilis in clinical samples. Open Microbiol J (2016) 10:57–63. doi:10.2174/1874285801610010057
33. Pierce JV, Bernstein HD. Genomic diversity of enterotoxigenic strains of Bacteroides fragilis. PLoS One (2016) 11(6):e0158171. doi:10.1371/journal.pone.0158171
34. Zhan LS, Davies SS. Microbial metabolism of dietary components to bioactive metabolites: opportunities for new therapeutic interventions. Genome Med (2016) 8:46. doi:10.1186/s13073-016-0296-x
35. Negi S, Singh H, Mukhopadhyay A. Gut bacterial peptides with autoimmunity potential as environmental trigger for late onset complex diseases: in-silico study. PLoS One (2017) 12(7):e0180518. doi:10.1371/journal.pone.0180518
36. Zhu X, Han Y, Du J, Liu R, Jin K, Yi W. Microbiota-gut-brain axis and the central nervous system. Oncotarget (2017) 10. doi:10.18632/oncotarget.17754
37. Javan GT, Finley SJ, Can I, Wilkinson JE, Hanson JD, Tarone AM. Human thanatomicrobiome succession and time since death. Sci Rep (2016) 6:29598. doi:10.1038/srep29598
38. Troletti CD, de Goede P, Kamermans A, de Vries HE. Molecular alterations of the blood-brain barrier under inflammatory conditions: the role of endothelial to mesenchymal transition. Biochim Biophys Acta (2016) 1862:452–60. doi:10.1016/j.bbadis.2015.10.010
39. Zhu B, Wang ZG, Ding J, Liu N, Wang DM, Ding LC, et al. Chronic lipopolysaccharide exposure induces cognitive dysfunction without affecting BDNF expression in the rat hippocampus. Exp Ther Med (2014) 7:750–4.
40. Lykhmus O, Mishra N, Koval L, Kalashnyk O, Gergalova G, Uspenska K, et al. Molecular mechanisms regulating LPS-induced inflammation in the brain. Front Mol Neurosci (2016) 9:19. doi:10.3389/fnmol.2016.00019
41. Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res (2002) 70:462–73. doi:10.1002/jnr.10351
42. Lukiw WJ. Gene expression profiling in fetal, aged, and Alzheimer hippocampus: a continuum of stress-related signaling. Neurochem Res (2004) 29:1287–97. doi:10.1023/B:NERE.0000023615.89699.63
43. Magata F, Shimizu T. Effect of lipopolysaccharide on developmental competence of oocytes. Reprod Toxicol (2017) 71:1–7. doi:10.1016/j.reprotox.2017.04.001
44. Mufson EJ, Counts SE, Ginsberg SD. Gene expression profiles of cholinergic nucleus basalis neurons in Alzheimer’s disease. Neurochem Res (2002) 27(10):1035–48. doi:10.1023/A:1020952704398
45. Shi Q, Gibson GE. Oxidative stress and transcriptional regulation in Alzheimer disease. Alzheimer Dis Assoc Disord (2007) 21(4):276–91.
46. Barczynska R, Slizewska K, Litwin M, Szalecki M, Kapusniak J. Effects of dietary fiber preparations made from maize starch on the growth and activity of selected bacteria from the Firmicutes, Bacteroidetes, and Actinobacteria phyla in fecal samples from obese children. Acta Biochim Pol (2016) 63:261–6. doi:10.18388/abp.2015_1068
47. Nguyen PQ. Synthetic biology engineering of biofilms as nanomaterials factories. Biochem Soc Trans (2017) 45:585–97. doi:10.1042/BST20160348
48. Hill JM, Clement C, Pogue AI, Bhattacharjee S, Zhao Y, Lukiw WJ. Pathogenic microbes, the microbiome, and Alzheimer’s disease (AD). Front Aging Neurosci (2014) 6:127. doi:10.3389/fnagi.2014.00127
Keywords: Alzheimer’s disease, inflammatory degeneration, lipopolysaccharide, microbiome, microRNA, small non-coding RNAs
Citation: Zhao Y, Cong L, Jaber V and Lukiw WJ (2017) Microbiome-Derived Lipopolysaccharide Enriched in the Perinuclear Region of Alzheimer’s Disease Brain. Front. Immunol. 8:1064. doi: 10.3389/fimmu.2017.01064
Received: 25 July 2017; Accepted: 16 August 2017;
Published: 04 September 2017
Edited by:
Wesley H. Brooks, University of South Florida, United StatesReviewed by:
Ai-Ling Lin, University of Kentucky, United StatesAbdul Sadiq, University of Malakand, Pakistan
Copyright: © 2017 Zhao, Cong, Jaber and Lukiw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Walter J. Lukiw, wlukiw@lsuhsc.edu