 Clément Da Silva
Clément Da Silva Camille Wagner
Camille Wagner Johnny Bonnardel
Johnny Bonnardel Jean-Pierre Gorvel
Jean-Pierre Gorvel Hugues Lelouard
Hugues Lelouard- 1Aix-Marseille University, CNRS, INSERM, CIML, Marseille, France
- 2Laboratory of Myeloid Cell Ontogeny and Functional Specialisation, VIB Inflammation Research Center, Ghent, Belgium
The gut represents a potential entry site for a wide range of pathogens including protozoa, bacteria, viruses, or fungi. Consequently, it is protected by one of the largest and most diversified population of immune cells of the body. Its surveillance requires the constant sampling of its encounters by dedicated sentinels composed of follicles and their associated epithelium located in specialized area. In the small intestine, Peyer’s patches (PPs) are the most important of these mucosal immune response inductive sites. Through several mechanisms including transcytosis by specialized epithelial cells called M-cells, access to the gut lumen is facilitated in PPs. Although antigen sampling is critical to the initiation of the mucosal immune response, pathogens have evolved strategies to take advantage of this permissive gateway to enter the host and disseminate. It is, therefore, critical to decipher the mechanisms that underlie both host defense and pathogen subversive strategies in order to develop new mucosal-based therapeutic approaches. Whereas penetration of pathogens through M cells has been well described, their fate once they have reached the subepithelial dome (SED) remains less well understood. Nevertheless, it is clear that the mononuclear phagocyte system (MPS) plays a critical role in handling these pathogens. MPS members, including both dendritic cells and macrophages, are indeed strongly enriched in the SED, interact with M cells, and are necessary for antigen presentation to immune effector cells. This review focuses on recent advances, which have allowed distinguishing the different PP mononuclear phagocyte subsets. It gives an overview of their diversity, specificity, location, and functions. Interaction of PP phagocytes with the microbiota and the follicle-associated epithelium as well as PP infection studies are described in the light of these new criteria of PP phagocyte identification. Finally, known alterations affecting the different phagocyte subsets during PP stimulation or infection are discussed.
Introduction
In mammals, the gastrointestinal mucosa is the largest surface of interaction with the external environment. This ensures an efficient absorption of nutrients, electrolytes, and water but concomitantly it exposes the body to environmental threats through the ingestion of contaminated food or drinks. Thus, the gut represents a privileged site of entry for various pathogen agents, such as protozoa, bacteria, viruses, toxins, or prion. Different mechanisms of defense exist to protect the body integrity against these threats. The efficient and size-selective shield provided by the mucus layer and the glycocalyx above the villous epithelium favors the uptake of small diffusible molecules while preventing microorganisms from reaching the epithelium. Protection is also ensured through secretion of antimicrobial compounds and innate polyreactive and antigen-specific secretory immunoglobulin A (sIgA) in the intestinal lumen. Finally, the intestinal epithelium forms a physical barrier between the lumen and the lamina propria. However, pathogens, such as Salmonella and Shigella, can survive challenging environmental conditions, disrupt the mucus and the continuity of the epithelial barrier, and penetrate the epithelium to reach interstitial tissues (1). It is, therefore, important for the mucosal immune system to be aware of the presence of pathogens as soon as possible. A simple way to achieve this objective is to provide a facilitated access to the gut luminal content toward the mucosal surface at restricted areas distributed regularly along the gastrointestinal tract. The mammal small intestine possesses such specific sentinel sites marked by the presence of lymphoid follicles. Peyer’s patches (PPs) are the most important of these monitoring sites since they are constituted of several clustered B-cell follicles forming domes interspersed with T-cell zones termed interfollicular regions (IFR). While villi are specialized for absorption of nutrients, PPs are dedicated to the sampling of foreign material and to the induction of mucosal immune responses (2–4). Due to the low number of mucus-secreting goblet cells and lack of polymeric immunoglobulin receptor expression in the follicle-associated epithelium (FAE), PP have a reduced mucus layer and no IgA secretion, respectively, which may favor interaction with pathogens (5, 6). Moreover, the FAE is characterized by the presence of specialized epithelial cells termed M cells, which lack a typical brush border and possess a thin glycocalyx that give a better accessibility to large particulate antigens (7–12). The underlying stromal cell network ensures at least in part this specialization of the FAE. Thus, subepithelial stromal cells express high amounts of the cytokine RANKL, which is necessary to both the production of the chemokine CCL20 by the FAE and the development of M cells (13, 14). The latter display specific carbohydrates and receptors that are used as binding sites by pathogens (15–22). Following their adherence to M cells, particulate antigens are rapidly transported from the lumen to the subepithelial dome (SED) or to an invagination of the basolateral membrane of M cells forming a pocket in which phagocytes, T and B cells reside. Importantly, the presence of M cells is critical for the sampling of both commensals and pathogens (17, 23–25). Once delivered into the basolateral pocket or in the SED, uptake, degradation, and presentation of antigens by the mononuclear phagocyte system (MPS), i.e., macrophages (MF) and dendritic cells (DCs), are key steps to induce a mucosal immune response. During infection, subepithelial phagocytes are, therefore, involved both in PP innate defense and in the initiation of the mucosal immune response (11). However, the role of each phagocyte subpopulation in infection has remained elusive due to an absence of consensual phenotype markers for each subset. Studies have indeed pointed out the substantial overlap in several key surface markers between MF and DC (e.g., CD11c, CD11b, SIRPα, and the major histocompatibility complex class II, MHCII) (26). Thus, until very recently, the characterization of MF in PP has been hampered by the lack of reliable markers. Finally, each dome of a given PP is surrounded by villi, thus preventing an easy discrimination of phagocytes from dome and dome-associated villus (DAV). Although IFRs are located on the sides of each dome, we, hereafter, refer FAE, SED, follicle, and IFR-located phagocytes jointly as dome phagocytes by opposition with DAV phagocytes.
In this review, we focus on recent advances, which have allowed distinguishing the different dome mononuclear phagocyte subsets. We provide an overview of their phenotype, distribution, ontogeny, lifespan, transcriptional profile, and function. We then consider some PP functional studies in the context of these new criteria to propose an identification of implicated dome phagocytes. Finally, we discuss alterations affecting the different phagocyte subsets upon PP stimulation or infection.
Diversity and Specificity of the PP MPS
Recent progresses in the characterization of PP MPS have demonstrated that dome DC and MF display unique characteristics very distinct from their DAV counterparts (Table 1).
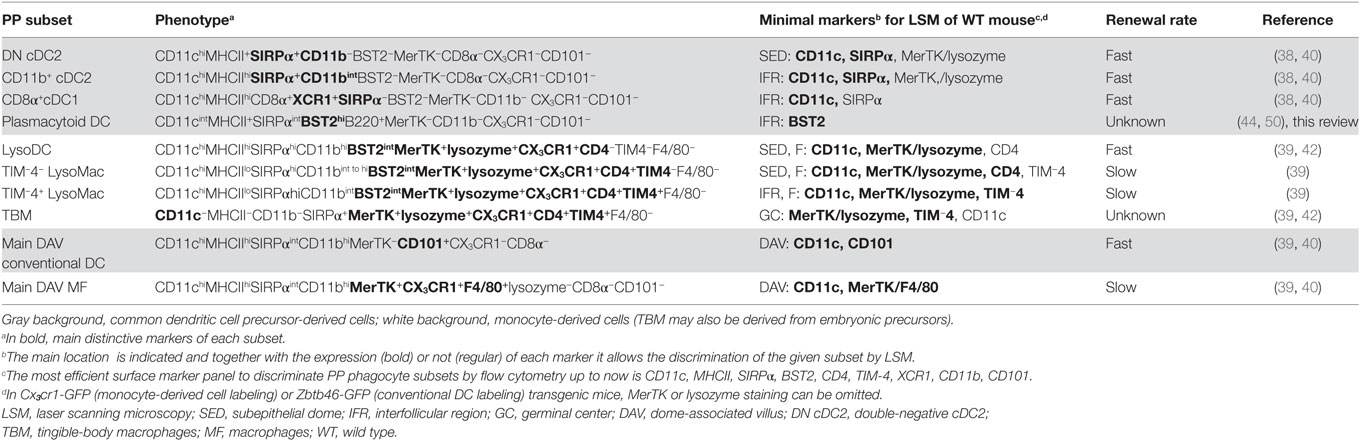
Table 1. Peyer’s patch (PP) phagocyte subsets at steady state.
Dome Conventional DC
Mouse common DC precursor (CDP)-derived DC, also called conventional DC (cDC), comprise two major subsets, which have been first identified through the expression of either CD8α (cDC1) or CD11b (cDC2) in addition to CD11c and MHCII (27, 28). Recently, more reliable, specific, and cross-species conserved markers for cDC1, such as XCR1 and Clec9a, have been identified (29–34). Similarly, SIRPα is a more widely distributed marker of cDC2 than CD11b, although shared with MF (35). Both cDC1 and cDC2 are present in domes (Figure 1) (36, 37). In addition, a third cDC subset, termed double negative cDC (DN cDC), which neither expresses CD11b nor CD8α, has been described in PP (37, 38). However, DN cDC have been recently identified as belonging to the cDC2 subset (Figure 1). They indeed share key surface markers with cDC2, such as SIRPα and Clec4a4, and, unlike cDC1, do not depend on Batf3 for their differentiation (39, 40). In addition, the transcriptional programs of CD11b+ and DN cDC are very close from each other. Notably, CD11b+ cDC express more MHCII at their surface and higher levels of key maturation marker genes such as Stat4, Ccr7, Ccl22, Socs2, and Il6 than DN cDC (40). Moreover, the latter are able to express CD11b upon in vitro culture and are recruited in PP before CD11b+ cDC (40). Therefore, it is assumed that DN and CD11b+ dome cDC represent immature and mature homeostatic differentiation stages of cDC2, respectively. Dome cDC2 encompass actually a developmental continuum of cells with gradual surface acquisition of CCR7, CD11b, EpCAM, JAM-A, and MHCII and decrease of CD24 expression (40). Importantly, dome cDC2 are distinct from DAV cDC2 (Table 1). Thus, the latter display more CD11b and less SIRPα at their surface than dome cDC2. Moreover, most of them express CD101 whereas dome cDC2 do not (40).
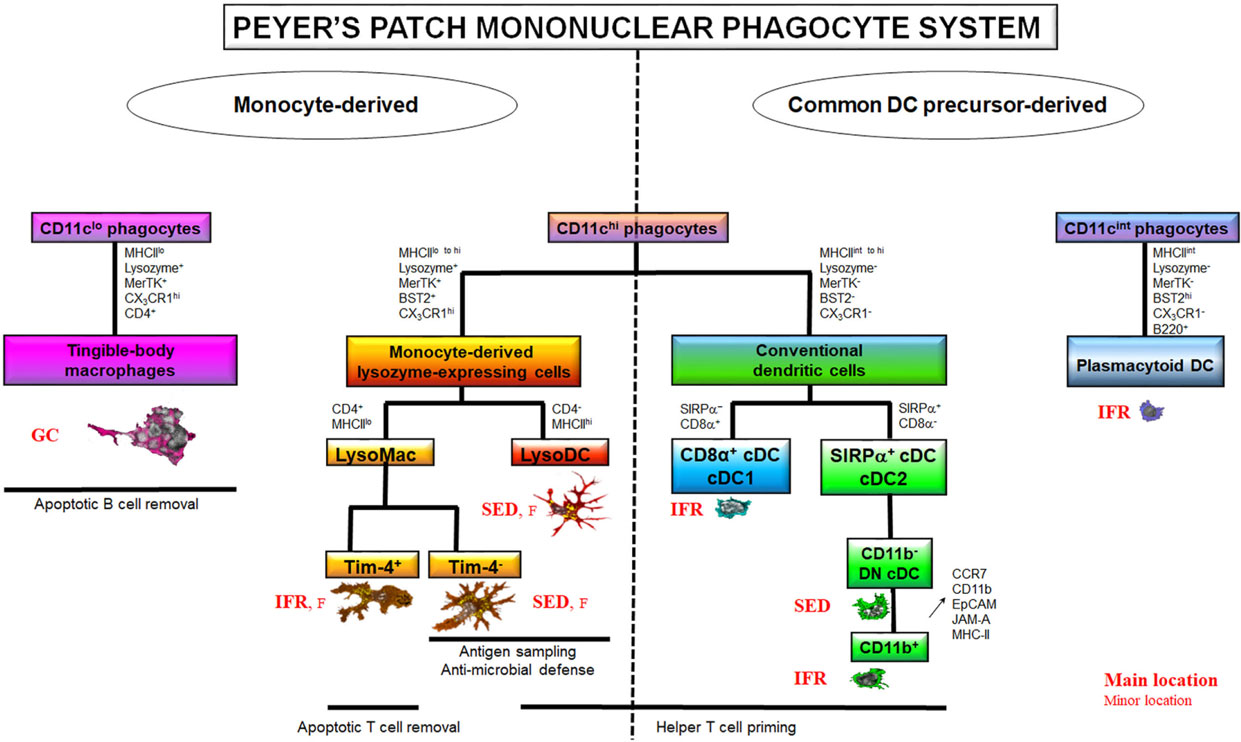
Figure 1. The Peyer’s patch (PP) mononuclear phagocyte system (MPS). The PP MPS encompasses two large families of cells based on their origin, the common DC precursor (CDP)-derived and the monocyte-derived phagocytes. The CDP-derived cells comprise CD11chi conventional DC (cDC) and CD11cint plasmacytoid DC. Among cDC, cDC1 are CD8α+ but SIRPα− whereas cDC2 are SIRPα+ but CD8α−. cDC2 exist in several stages of differentiation among which the two extremes are the so-called double negative (DN) cDC2, which do not express CD11b, and the CD11b+ cDC2. CD11b+ cDC2 derive from DN cDC2 through the upregulation of CCR7, CD11b, EpCAM, JAM-A, and MHCII. CDP-derived cells are mainly located in the T cell zones, i.e., interfollicular regions (IFR), at the exception of DN cDC2, which transit through the subepithelial dome (SED). cDC excel in helper T cell priming but are poorly phagocytic. On the contrary, CD11chi monocyte-derived cells are strongly phagocytic. They also display a broad range of antimicrobial defense mechanisms. CD11chi monocyte-derived cells encompass two main subsets based on their phenotype, lifespan, and ability to prime T cells: macrophages (MF) and the monocyte-derived dendritic cell (DC) termed LysoDC. LysoDC are CD4−MHCIIhi short-lived SED-located DC with helper T cell priming ability. CD11chi MF, also called LysoMac, are CD4+MHCIIlo long-lived cells without any helper T cell priming ability. TIM-4− LysoMac are mainly located in the SED whereas TIM-4+ LysoMac are mainly located in the IFR. A third type of MF, termed tingible-body macrophages, reside in the germinal center (GC) of the follicle (F) where they are involved in apoptotic B cell removal. Unlike other PP MF, they do not express CD11c. Although shown on the monocyte-derived cell part of the diagram, it is currently unknown whether they truly derive from monocytes or whether they self-renew from embryonic precursors. Adapted from Ref. (39).
Dome MF
Unlike villous MF, identification of dome MF has remained unsolved for decades due to the lack of expression of classic macrophage markers such as F4/80 (EMR1), sialoadhesin (Siglec1/CD169), Mannose Macrophage Receptor (MMR/CD206), or Fc Gamma Receptor I (FcGRI/CD64) (39). Moreover, a substantial overlap of key surface markers, such as CD11c, CD11b, MHCII, and SIRPα, exists between MF and cDC (26). By the past, this has led to a great confusion concerning location and functions of both dome phagocyte populations. However, recent works managed distinguishing dome cDC from monocyte-derived cells (Table 1) (39–41). The latter encompass dome MF and the monocyte-derived DC termed LysoDC (Figure 1). Most dome monocyte-derived cells express CD11c, CD11b, SIRPα, BST2, CX3CR1, MerTK, and lysozyme (Table 1). BST2 and lysozyme expression are hallmarks of dome monocyte-derived cells since DAV MF express little or none of these molecules (39, 42). Thus, CD11c+ dome MF have been termed LysoMac by analogy with LysoDC and by opposition to villous MF, which do not express lysozyme. LysoMac are strongly autofluorescent large long-lived cells, which depend on the growth factor M-CSF to develop (39). They express CD4 but only low levels of MHCII. They encompass two main subsets based on the expression of the phosphatidylserine receptor TIM-4 (Figure 1) (39).
Tingible-body macrophages (TBM), which also display TIM-4 at their surface, form a third dome macrophage subset (39). Like LysoMac, TBM express MerTK, CX3CR1, SIRPα, lysozyme, and CD4 but typically lack CD11c, CD11b, and MHCII expression (Table 1) (39, 42). BST2 expression in TBM has not been investigated so far. Although easily detectable in situ through the presence of many internalized apoptotic bodies, they have been poorly characterized and their origin, either circulating monocytes, embryonic precursors or both, is unknown. In addition to these subsets, PP contain a layer of poorly described serosal/muscularis MF, which, depending on their location, express or not CD169 (see below) (39).
LysoDC
LysoDC are short-lived monocyte-derived DC (Figure 1; Table 1) (39). Unlike LysoMac, they are weakly autofluorescent, express very high levels of MHCII, but no CD4, and are strongly dependent on CCR2, the chemokine receptor that allows monocyte egress from the bone marrow (39). Morphologically, they are large stellate motile cells (42, 43). Upon stimulation with the TLR7 agonist R848, they secrete IL-6 and TNF but no IL-10 (Table 2) (39). So far, no equivalent of LysoDC has been described in villous lamina propria. LysoDC are present in mouse, rat, and human PP (42). Thus, these phagocytes seem to be specific of PP and maybe of isolated lymphoid follicles in several species including humans.
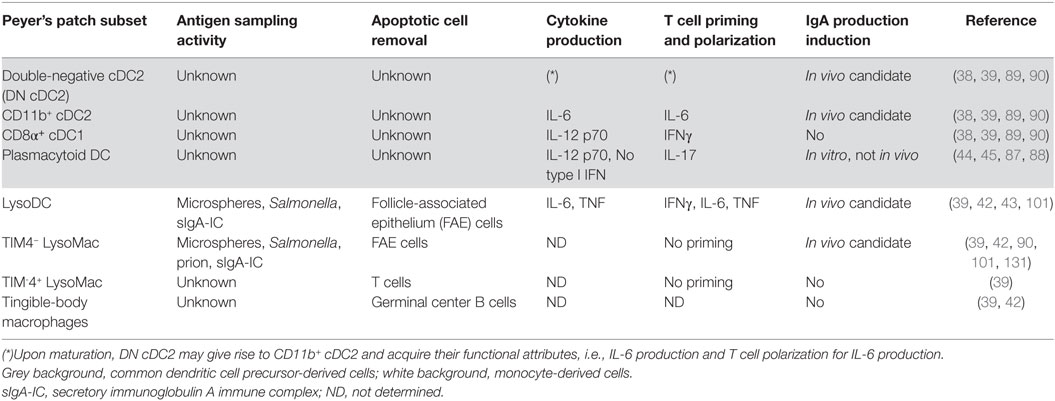
Table 2. Functions of dome phagocyte subsets.
Plasmacytoid DC
Although PP plasmacytoid DCs (pDCs) share BST2 expression with monocyte-derived cells, they constantly express higher levels of BST2 and lower levels of CD11c and SIRPα than LysoDC and LysoMac (Table 1) (39, 40). One can also distinguish PP pDC from monocyte-derived cells, thanks to their B220 expression. PP pDCs are distinct from pDCs isolated from other tissues by their inability to secrete abundant type I IFN in response to the TLR9 agonist CpG (Table 2) (44). Expression of the mucosal migratory receptor CCR9 and of the specific transcriptional regulator of the pDC lineage E2-2 is also reduced in PP pDCs as compared to other pDCs (45). Like other pDCs, PP pDCs are derived from the CDP and are induced by Flt3L, but their recruitment also requires type I IFN/STAT1 signaling (45). This type I IFN conditioning of PP pDC could favor the production of the inflammatory cytokines IL-6, IL-23, and TNF instead of type I IFNs (45).
Anatomic Localization of PP Phagocyte Subsets at Steady State
Each region of the dome, i.e., FAE, SED, follicle, germinal center (GC), and IFR, is populated with specific subsets of phagocytes (Figure 2).
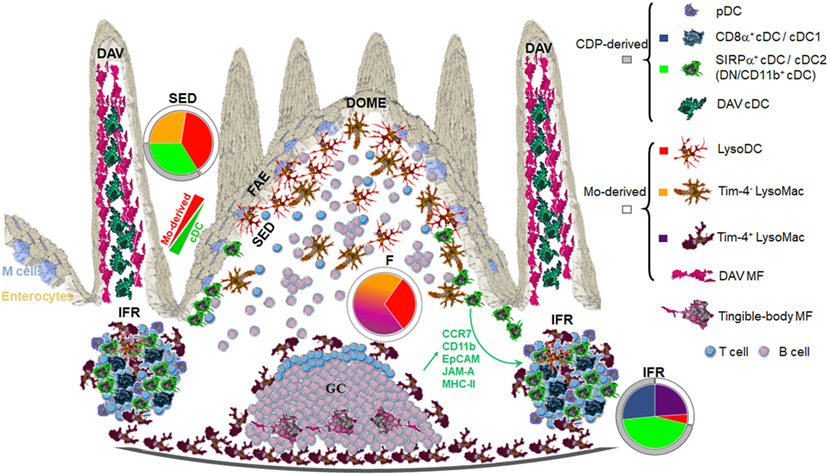
Figure 2. Anatomic localization of Peyer’s patch (PP) phagocyte subpopulations. Origin and shape of each PP phagocyte subset is displayed on the right. Color codes correspond to colors displayed in pie charts. The latter show in each region of the dome the distribution of PP phagocyte subsets at the exception of plasmacytoid DC (pDC). FAE, follicle-associated epithelium; SED, subepithelial dome; F, follicle; IFR, interfollicular region; DAV, dome-associated villus; MF, macrophages. Adapted from Ref. (40).
FAE and SED
Subepithelial phagocytes are mainly composed of CD11c+ CD11b+ cells (37, 40, 42). Due to the expression of both surface markers, these cells were initially thought to be cDC2. However, these subepithelial CD11c+CD11b+ cells also express CX3CR1 and lysozyme and belong to the monocyte-derived family of phagocytes, i.e., LysoDC and LysoMac (40). Both represent actually two-third of subepithelial phagocytes with increasing ratio while reaching the upper part of the dome (Figure 2). By contrast, the SED contains few cDC, mainly DN cDC2 (JAM-A−CCR7−CD11b−SIRPα+ cDC), which are rather located in the lower part of the dome (Figure 3) (40). Both subepithelial LysoDC and DN cDC2 can penetrate the FAE and strongly interact with M cells, whereas LysoMac remain mainly located in the SED (40). Heterogeneity in dome macrophage-associated phenotype is tightly linked to their different anatomic localization within PP (Figure 2). This suggests that these phenotype differences reflect an important regional specialization of macrophage functions. Thus, subepithelial LysoMac do not express TIM-4 (39).
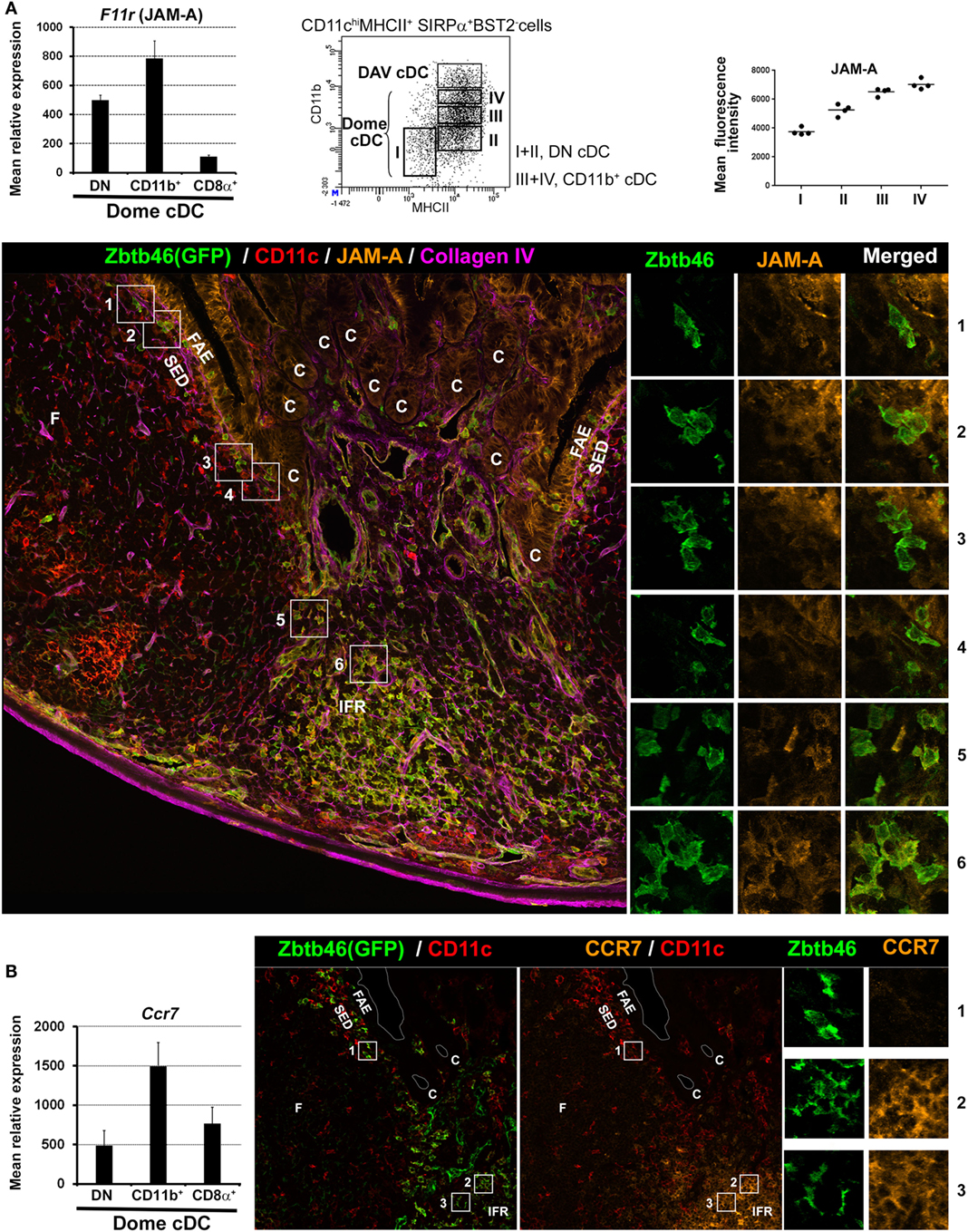
Figure 3. Location of dome double negative (DN) and CD11b+ cDC2 based on JAM-A and CCR7 expression. (A) Left panel: normalized mean relative expression ± SD of F11r (JAM-A) in dome conventional DC (cDC) subsets. Mid-panel: identification of four developmental stages of dome cDC2 based on CD11b and MHCII surface expression. Stage I, CD11b−MHCIIlo; Stage II, CD11b−MHCIIint; Stage III, CD11bloMHCIIhi; Stage IV, CD11bintMHCIIhi. Right panel: mean fluorescence intensity of JAM-A in the four developmental stages of dome cDC2. JAM-A expression increases from stage I (DN cDC2) to stage IV (CD11b+ cDC2). Lower panel: confocal microscopy projection of a Zbtb46-GFP−/+ mouse Peyer’s patch (PP) section stained for EGFP (green), CD11c (red), JAM-A (orange), and collagen IV (magenta). Higher magnifications of the numbered boxed area are shown on the right. cDC (CD11c+GFP+ cells) are mainly located in the IFR. However, some of them are located in the SED with a progressive decrease in numbers while reaching the upper part of the dome. Like LysoDC, they can penetrate into the follicle-associated epithelium (FAE). Subepithelial cDC2 (boxed area 1–4) express no or faint levels of JAM-A (stage I or II of dome cDC2; DN cDC2) whereas interfollicular cDC2 (boxed area 5 and 6) express it (stage III or IV of dome cDC2; CD11b+ cDC2). (B) Left panel: normalized mean relative expression ± SD of Ccr7 in dome cDC subsets. Right panel: confocal microscopy projection of a Zbtb46-GFP−/+ mouse PP section stained for EGFP (green), CD11c (red), and CCR7 (orange). Higher magnifications of the numbered boxed area are shown on the right. Subepithelial cDC2 (boxed area 1) do not express CCR7 (DN cDC2) whereas interfollicular cDC2 (boxed area 2 and 3) do (CD11b+ cDC2). Parts of (A,B) are adapted from Ref. (40).
Follicle and GC
Conventional DCs are generally absent from the follicle and from the GC. The upper part of the follicle comprises exclusively scattered LysoDC and TIM-4− LysoMac, whereas in its lower part, TIM-4+ LysoMac replace TIM-4− LysoMac (39). Finally, TBM are the only phagocytes of the GC.
Interfollicular Regions
Interfollicular regions contain mainly cDC1 (SIRPα− cDC), CD11b+ cDC2 (JAM-A+CCR7+CD11b+SIRPα+ cDC), and scattered TIM-4+ LysoMac (Figures 2 and 3) (39, 40). Of note, by microscopy, CD11b staining is not detectable in interfollicular CD11b+ cDC2 and TIM-4+ LysoMac due to its low levels of expression in these cells (40). Only LysoDC and subepithelial TIM-4− LysoMac actually stain for CD11b in the dome. Since interfollicular cDC (cDC1 and CD11b+ cDC2) express CCR7 whereas subepithelial cDC (DN cDC2) do not (Figure 3B), the former are likely recruited through the specific expression of CCL19 and CCL21 in the IFR whereas the latter are likely recruited in the SED through their expression of CCR6 and secretion of CCL20 by the FAE (37, 40, 46–49). Surprisingly, interfollicular TIM-4+ LysoMac do not express CCR7, indicating that another factor may be involved in their addressing to the IFR (40). Interestingly, a layer of these MF surrounds the IFR, thus forming border guards of the T cell zone. Finally, CD169 is only expressed by MF located at the base of the IFR toward the serosa, including serosal/muscularis MF (39).
BST2 has been extensively used to identify pDC in different mouse organs including PP (44, 50). Based on this marker, pDCs were first supposed to be located in the SED and in the IFR (44, 50). However, PP monocyte-derived cells express BST2 at steady state (Figure 4A) (39). Moreover, stimuli that trigger interferon responses induce BST2 expression in several cell types (51). We, therefore, decided to re-investigate pDC location. We found that pDCs are mainly located in the IFR but not in the SED where BST2 is weakly displayed by monocyte-derived cells (Figure 4B).
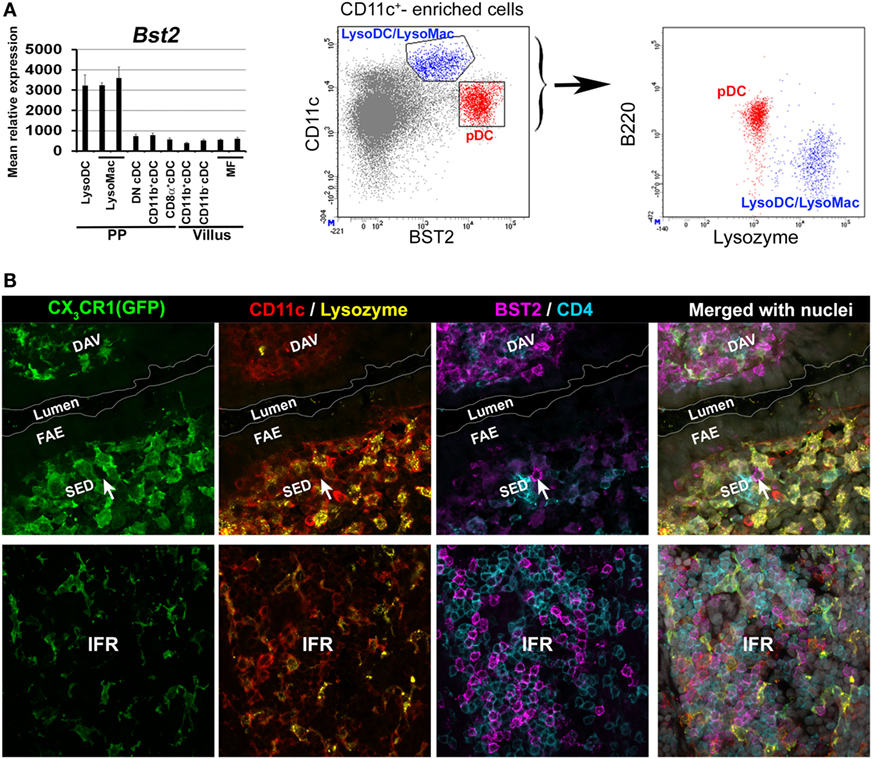
Figure 4. Location of plasmacytoid DC (pDC) in Peyer’s patch (PP). (A) Left panel: normalized mean relative expression ± SD of Bst2 in intestinal phagocytes based on Immgen database (139) and on the PP phagocyte microarray data deposited to NCBI GEO under accession numbers GSE94380 and GSE65514 (39, 40). Expression of Bst2 by LysoDC and LysoMac is shown. Right panel: in PP, monocyte-derived cells (CD11chiB220−lysozyme+ in blue), i.e., LysoDC and LysoMac, express lower levels of BST2 than pDC (CD11cintB220+lysozyme− in red). (B) Confocal microscopy projection of CX3CR1-EGFP−/+ mouse PP sections stained for EGFP (green), CD11c (red), lysozyme (yellow), CD4 (blue), and BST2 (magenta). Upper panel: BST2 is strongly expressed by cells of the dome-associated villus (DAV) but only weakly by LysoDC and LysoMac (CX3CR1+CD11c+lysozyme+ cells) in the subepithelial dome (SED). A single putative pDC (arrow) strongly stained for BST2 is located in the SED. Lower panel: unlike the SED, the IFR is enriched in pDC. (A) is adapted from Ref. (39).
Functions of PP Phagocyte Subsets
Interaction with the FAE and Antigen Sampling Activity
The preferential uptake of luminal particulate antigens in PP as compared to villi first relies on the specific characteristics of the FAE (5–10, 52). Some of these properties, such as low levels of mucin expression, altered surface glycosylation, and lack of secretion of antimicrobial proteins, depend on IL-22 signaling inhibition through the production of IL-22 binding protein (IL-22BP) by CD11c+CD11b+MHCII+ cells of the SED (53). Thereby, IL-22BP promotes microbial uptake into PP by influencing the FAE transcriptional program. Unfortunately, the markers used to identify IL-22BP-secreting cells do not allow distinguishing cDC from monocyte-derived cells (53). In order to better characterize these IL-22BP-secreting phagocytes, we decided to interrogate the gene expression database of dome CD11chi phagocytes (NCBI GEO accession numbers GSE94380 and GSE65514) for IL-22BP transcripts (Il22ra2). Il22ra2 was enriched in LysoDC and TIM-4− LysoMac as compared to cDC (Figure 5A). These results, together with the preferential location of LysoDC and TIM-4− LysoMac in the SED, support their role in the secretion of IL-22BP, which in turn inhibits IL-22 signaling, alters the FAE transcriptional program, and favors the internalization of both commensal and pathogenic bacteria (53).
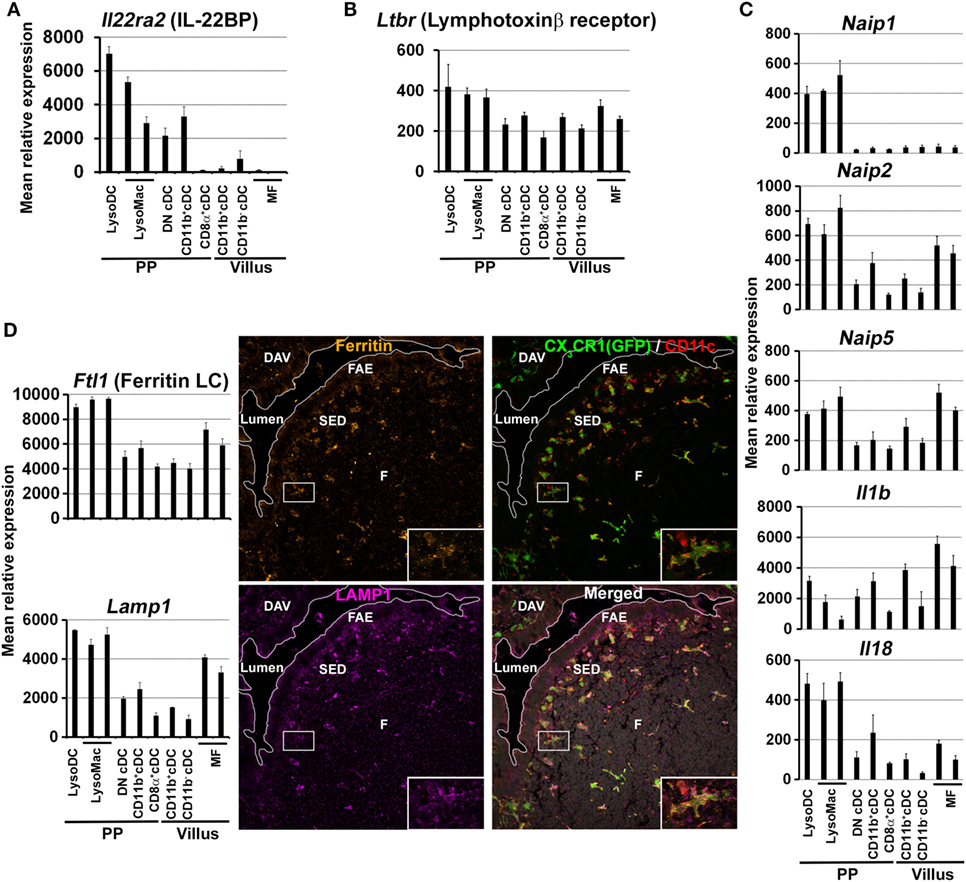
Figure 5. Expression of defined markers by Peyer’s patch (PP) phagocyte subpopulations. (A–D) Normalized mean relative expression ± SD of Il22ra2, Ltbr, Naip1, Naip2, Naip5, Il1b, Il18, Ftl1, and Lamp1 in intestinal phagocytes based on Immgen database (139) and on the PP phagocyte microarray data deposited to NCBI GEO under accession numbers GSE94380 and GSE65514 (39, 40). (A) In the gut, Il22ra2 [IL-22 binding protein (IL-22BP)] is mainly expressed by LysoDC and TIM-4− LysoMac. (B) In PP, Ltbr (lymphotoxin β receptor) is mainly expressed by LysoDC, LysoMac and, to a lesser extent, conventional DC (cDC). (C) Enrichment of some members of the NAIP/NLRC4 inflammasome pathway (Naip1, Naip2, Naip5, Il1b, and Il18) in LysoDC and LysoMac. Note that Naip1 is only expressed by dome monocyte-derived cells. (D) Left panel: in the gut, LysoDC and LysoMac express higher levels of Ftl1 (ferritin light chain) and Lamp1 than other phagocytes. Right panel: labeling of a PP section shows enrichment of ferritin and LAMP1 expression in LysoDC and LysoMac of the subepithelial dome (SED) and of the follicle (F). Inserts: higher magnification of the boxed area showing one LysoDC strongly stained for ferritin and LAMP1.
In addition to this strong influence on FAE global characteristics, monocyte-derived cells and especially LysoDC maintain privileged interaction with M cells. Thus, LysoDC are able to extend dendrites through M cell specific transcellular pores to gain access to the lumen (Figure 6) (43). The cell adhesion molecules EpCAM and JAM-A are recruited at the M cell pore-forming membrane but neither the tight junction proteins ZO-1 and occludin nor the adherens junction proteins E-cadherin and β-catenin. Therefore, the formation of these M cell transcellular pores does not alter the integrity of the epithelial barrier. JAM-A is also enriched at the trans-M cell dendrite (TMD) membrane, which may favor homotypic interaction. In addition, there is a strong recruitment of filamentous actin in TMD in agreement with their high level of motility. These TMD scan indeed rapidly the surface of M cells and attract particulate antigens and bacteria from the lumen to capture them (43). Since blockade of the M cell-specific chemokine CCL9 drastically reduces the number of CD11c+CD11b+ cells in the SED and since LysoDC strongly express its receptor CCR1, it is tempting to speculate that the degree of interaction between M cell and LysoDC is regulated through the release control of this chemokine by M cells (39, 49).
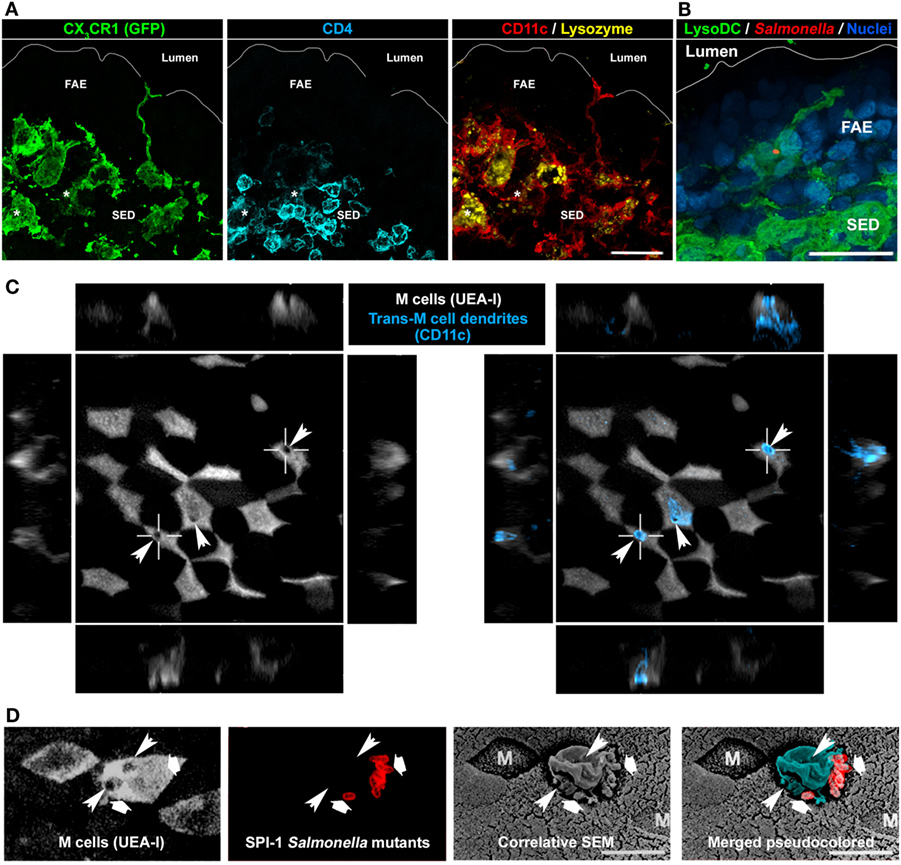
Figure 6. Involvement of LysoDC trans-M cell dendrites in the sampling of Salmonella typhimurium. (A) A CX3CR1-deficient LysoDC (GFP in place of CX3CR1 in green; CD11c in red; lysozyme in yellow) extends a dendrite through the follicle-associated epithelium (FAE) to reach the lumen. In addition to CX3CR1, CD11c, and lysozyme, LysoMac (*) stain for CD4 (blue). (B) 2 h postoral infection, a Salmonella (red) is taken up by a LysoDC (green) extending a dendrite into an uninfected FAE. (C) M cell transcellular pores (arrowheads), through which trans-M cell dendrites (CD11c in blue) cross the FAE, are highlighted by circular holes in the UEA-I cell surface staining. (D) By correlative scanning electron microscopy (SEM), Salmonella (second and last panels in red, large arrows) are located at the periphery of a protrusion (pseudocolored in blue) arising from an M cell. Circular holes (thin arrowheads) in the UEA-I cell surface staining indicate the presence of M cell transcellular pores. (B,C) are adapted from Ref. (43), (D) is adapted from Ref. (110).
Although LysoDC are the main TMD-forming phagocytes, subepithelial LysoDC and TIM-4− LysoMac equally internalize particulate antigens (Table 2) (39). This suggests that, at least at steady state, the main route of particulate antigen sampling across the FAE is mediated through M cell transcytosis rather than by TMD. The lack of particulate antigen uptake by subepithelial cDC (DN cDC2) in vivo is in agreement first with their low number in this region and second with in vitro microparticle uptake assays showing a much more efficient phagocytic activity of LysoDC and LysoMac as compared to dome cDC (40, 42). Interestingly, in addition to transporting luminal antigens in their basolateral pocket or in the SED, M cells constitutively release on their basal side microvesicles, which are taken up by subepithelial CD11c+CD11b+CX3CR1+ cells, i.e., LysoDC and TIM-4− LysoMac (54). Finally, monocyte-derived cells and M cell cooperation extend beyond cell death since the former engulf dying M cells (Table 2) (42). In summary, particulate antigen uptake in the SED is mainly performed by subepithelial monocyte-derived cells and occurs through at least four distinct mechanisms, which all involve M cells: (i) M cell-mediated transcytosis; (ii) M cell microvesicle shedding; (iii) formation of TMD; (iv) dying M cell phagocytosis.
Innate Defense Functions
LysoDC and LysoMac have been first identified through their strong expression of the antibacterial compound lysozyme (42). Then, they have been distinguished from dome cDC by their surface expression of the host antiviral restriction factor BST2 (39). This suggests that, in addition to playing a primary role in antigen sampling, this family of dome phagocytes are strongly involved in the innate defense of PP. Analysis of their transcriptional profile confirmed this assumption (39). Dome monocyte-derived cells display indeed a strong antibacterial and antiviral gene signature as compared to cDC. This includes genes encoding for viral and bacterial-associated molecular pattern recognition molecules such as toll-like receptors (TLRs), NAIPs, STING, DAI, and RIG-I. Several pathways of antiviral and antibacterial defense such as replication inhibition, metal sequestration, NLRC4 inflammasome formation, and detoxification mechanisms are also upregulated in monocyte-derived cells as compared to cDC. Therefore, LysoDC and LysoMac, but not cDCs, display strong innate defense mechanisms against both viral and bacterial infections (Figure 1).
Priming of T Cells
Conventional DC have been long recognized as the most efficient professional antigen-presenting cells to initiate an antigen-specific immune response through the priming of both naïve CD4+ and CD8+ T cells (55). Upon activation, cDC initiate a process of differentiation, also termed maturation, which involves an important genetic reprogramming (56, 57). This induces profound phenotypic, morphological, and functional changes, which allow their migration to lymph node T cell zones and their antigen presentation and naïve T cell priming ability. Depletion of CD11c+ phagocytes showed that in PP they are involved both in the retention of interfollicular naive helper T cells and in their priming following antigen feeding (58, 59). Most dome cDC reside in the IFR and, therefore, do not require to migrate to encounter naïve T cells (40). Although it may be convenient and secure to rapidly prime naïve T cells, it raises indubitably the question of antigen acquisition. It could be, however, hypothesized that, after acquisition of soluble antigens or transfer of particulate antigens from monocyte-derived cells, DN cDC2 upregulate CCR7 and downregulate CCR6 to rapidly shuttle from the SED to the IFR. Their constant migratory activity would thus lead to the apparent underrepresentation of cDC in the SED where most of the luminal antigen sampling activity is performed (39, 40). By contrast, LysoDC, which are highly efficient in particulate antigen sampling, are mainly located in the SED and, to some extent, in the follicle but their migration to the T cell zone or their ability to prime T cells in vivo in the SED remain pending issues (39, 42, 43).
In vitro, unlike LysoMac, both dome cDC and LysoDC are able to induce naïve antigen-specific T helper cell proliferation (Table 2) (39). This is in line with the fact that, upon TLR7 stimulation, LysoDC upregulate MHCII and the co-stimulatory molecules CD40 and CD86 whereas LysoMac do not. Both cDC1 and LysoDC prime naïve antigen-specific T helper cells for IFNγ production. LysoDC also induce the production of IL-6, a property shared with cDC2 (Table 2).
Interaction with the Microbiota and Induction of the Mucosal Humoral Immune Response
Peyer’s patches are the primary site of antigen-specific sIgA-secreting cell induction (3, 4, 60–64). Production of sIgA is rapidly induced upon microbial colonization and strongly reduced in germ-free animals (65). Moreover, sIgA coating of the microbiota plays a critical role in its diversification (66–68). Interestingly, sIgA predominantly target specific members of the microbiota, especially those residing in the small intestine and those considered as pathobionts (69–71). Therefore, microbiota influences the mucosal immune system, which in turn regulates symbiont diversity and stability (72, 73). Among commensal bacteria that profoundly influence the mucosal immune system in mouse, segmented filamentous bacteria (SFB) play a privileged role, through induction of Th17 cells and IgA-secreting cells (74–76). This SFB-induced immune response largely depends on the stimulation of PPs and isolated lymphoid follicles (77). Interestingly, specific members of the microbiota, including SFB, colonize PPs (77–80). Moreover, M cells are able to transport different defined commensal bacteria, which induce distinct M cell transcriptional programs (81). This sampling of gut microbiota through M cell-mediated pathways is crucial to initiate mucosal sIgA production (25). Thus, it is tempting to speculate that microbiota members capable of inducing strong humoral immune responses are those that strongly interact with the FAE. Once translocated in the SED, SFBs are internalized by CD11cintCD11b+ and CD11chiCD11b+ phagocytes (82). Other IgA-inducer commensals, especially Alcaligenes species, reside inside CD11c+ cells within isolated lymphoid follicles, PP, and mesenteric lymph nodes (MLN) of mice and humans (79, 80, 83). PP CD11c+ phagocytes are known to carry commensal bacteria through a CCR7-dependent mechanism to the MLN, which are required to restrict commensal-loaded CD11c+ phagocytes to the mucosal compartment but are dispensable for IgA induction (84). However, the accurate identification of the CD11c+ phagocyte subset(s), which sample(s) and carry(ies) commensal bacteria, remains currently unknown.
In vitro, the role of PP phagocytes in IgA class switching has long been recognized (85, 86). More recently, it has been shown that PP and MLN pDC efficiently promote IgA class switch recombination through their expression of membrane-bound BAFF (B-cell activating factor) and APRIL (a proliferation-inducing ligand) independently of any T-cell or microbial stimulus (Table 2) (87). However, a recent pDC depletion study showed that pDCs are dispensable for intestinal IgA production in vivo (88). CD11chiCD11b+B220− phagocytes have also been implicated in the differentiation of naïve B cells into sIgA-secreting cells in vitro (89). It remains, however, to establish whether these CD11chiCD11b+B220− phagocytes are LysoDC, LysoMac, or dome CD11b+ cDC2 and to confirm their function in vivo. The role of each dome phagocyte subset in sIgA-secreting cell commitment in vivo is indeed not well-established (Table 2). However, efficient IgA class switching requires interaction of CCR6+ B cells with lymphotoxin-dependent CD11chiMHCII+CD11b+ phagocytes in the SED (90). Since lymphotoxin β receptor transcripts (Ltbr) are expressed by monocyte-derived cells and, to a lesser extent, by cDC (Figure 5B), it remains to establish whether these CD11chiMHCII+CD11b+ phagocytes correspond to CD11b+ cDC2, LysoDC, or LysoMac. Nevertheless, the sampling activity and the anatomic localization of the different phagocyte subsets as described above would argue for LysoDC/LysoMac rather than for CD11b+ cDC2 involvement. The lymphotoxin required to maintain these phagocytes in the SED could be mainly provided by subepithelial innate lymphoid cells type 3 (ILC3) (90). However, a recent report indicates that microbiota-derived butyrate suppresses ILC3 in terminal ileal PP, rendering their role in subepithelial phagocyte maintenance unlikely at least in this part of the gut (91). Lack of CCR6 expression by B cells or RANKL expression deficiency in subepithelial stromal cells, which results in inhibition of CCL20 production by the FAE, prevents B cells migration into the SED, precludes their interaction with CD11chiMHCII+CD11b+ phagocytes and finally inhibits IgA class switching and bacteria-specific sIgA production (14, 90). The integrin complex αvβ8 expressed by CD11chiMHCII+CD11b+ phagocytes could directly activate TGFβ during the interaction of these phagocytes with CCR6+ B cells and promote IgA class switching (90). In summary, induction of commensal bacteria-specific sIgA-secreting cells is a complex process involving many different cell types: (i) subepithelial stromal cells producing RANKL for the formation of M cells and for the production of CCL20, which recruits CCR6+ B cell and DN cDC2 into the SED; (ii) M cells for antigen sampling; (iii) lymphotoxin-producing cells for the maintenance of CD11chiMHCII+CD11b+ phagocytes; (iv) CD11chiMHCII+CD11b+ phagocytes for activation of CCR6+ B cells.
Regulation of the Adaptive Immune Response
Tingible-body macrophages are critical in the removal of apoptotic B cells during the selection process that occurs in the GC (Table 2) (92). Defect in this scavenging function leads to secondary necrosis, release of noxious molecules and pro-inflammatory signals, and is linked to autoantibody production and autoimmune disease development. This scavenging function requires the expression of the apoptotic receptor MerTK by TBM and of the soluble bridging molecule MFG-E8 by follicular DC, the GC stromal cells involved in the shaping of the B cell response (93–96). A number of other factors and receptors, such as TIM-4, have also been implicated in this process and their deficiency leads to autoimmunity, too (92). Therefore, although some of these molecules may have redundant roles, they may also function together to be more efficient in apoptotic cell removal through several mechanisms, thus preventing the arising of autoimmunity (97).
Like TBM, interfollicular MF express the apoptotic cell receptor TIM-4 and are located in a region of effector cell priming (39). Although the function of these TIM-4+ LysoMac remains elusive, they are thus likely to participate to the clearance of dying T cells like TBM contribute to the removal of dying B cells. Interestingly, TIM-4 deficiency leads to not only B cell but also T cells hyperactivity (98). Moreover, TIM-4 functions have been linked to the control of the adaptive immune response and tolerance through the removal of antigen-specific T cells (99, 100). Concerning PP phagocytes, while LysoDC interact with and prime naïve helper T cells in vitro, in the same conditions LysoMac phagocytize them, supporting their role in the removal of T cells (Table 2) (39). Thus, the location of TIM-4+ LysoMac in the T cell zone correlates well with a possible function in the removal of some naïve T cells during their priming process. Therefore, TBM and TIM-4+ LysoMac could perform a crucial role in PP adaptive immune response regulation at the level of GC and IFR, respectively.
PP Phagocytes in Infection
Sensing and Uptake of Immune Complexes
Innate polyreactive and antigen-specific sIgA are secreted by lamina propria plasma cells and transported through the epithelium by the polymeric immunoglobulin receptor to be finally released in the lumen. During infection, sIgA recognize and bind pathogens, thus participating to their clearance through a process called immune exclusion. Interestingly, M cells express on their surface dectin-1 and Siglec-F, which can serve as sIgA receptors allowing the uptake of luminal sIgA immune complexes (101). Since uptake of sIgA-coated bacteria persists in PP of dectin-1-deficient mice, Siglec-F expression may be sufficient to mediate sIgA binding to M cells (102). In the SED, sIgA immune complexes are associated with CD11c+CD11b+CX3CR1+MHCII+ cells, i.e., LysoDC and/or TIM-4− LysoMac (101). Entry of IgA-coated bacteria into PP does not require CX3CR1 expression (102). However, this does not preclude a potential role of TMD in sIgA immune complex uptake since CX3CR1 is not involved in TMD formation (Figure 6A) (39). Finally, this mechanism of immune complex sampling ensures the constant monitoring of sIgA-coated antigens present in the gut, including both pathogens and symbionts (103, 104). Through this process, new antigen-specific IgA-secreting cells could be produced to allow a better exclusion of pathogens.
Bacterial Infection: The Case of Salmonella
Salmonella enterica is an enteroinvasive bacterium typically acquired by ingestion of contaminated water or food. In the absence of dysbiosis, the primary invasion sites of Salmonella enterica serovar Typhimurium, the murine model of systemic salmonellosis, are PP of the distal ileum and cecal patches (105, 106). Through their fimbrial FimH adhesin, Salmonella Typhimurium are able to bind to the GP2 molecules expressed at the surface of M cells (17). Then, they use their Salmonella pathogenicity island 1 (SPI-1) type III secretion system to deliver effector proteins, which reorganize the cytoskeleton and allow the translocation of bacteria through M cells by inducing membrane ruffling (17, 107, 108). However, even SPI-1 and FimH Salmonella mutants or GP2-deficient mice show, to some extent, bacterial translocation into PP (17, 102, 109–111). One possible mechanism for this residual penetration could be transcytosis mediated by M cells, as observed with inert particles (13, 112, 113). Alternatively, these bacteria could be directly sampled by TMD (Figure 6) (43). Salmonella Typhimurium indeed induce TMD that rapidly internalize bacteria before retracting back to the SED (43). As soon as 2 h after oral infection, bacteria are present in LysoDC extending dendrites into the FAE in absence of any bacterial invasion of epithelial cells (Figure 6B). Interestingly, similar trans-M cell passages of uncharacterized leukocytes have been observed by electron microscopy using rabbit intestinal loop models of Streptococcus pneumoniae and Vibrio cholerae infection (114, 115). TMD are, therefore, infection-inducible and transient processes, which allow a fast M cell-regulated uptake of luminal material by immunocompetent cells. Such mechanism of sampling may notably avoid risks of massive penetration by pathogens. A typical feature of TMD formation is the appearance of a circular hole at the M cell apical membrane (Figure 6C). Interestingly, correlative scanning electron microscopy studies of PP in mouse intestinal loop infected with SPI-1 Salmonella mutants, which are unable to induce epithelial cell apical membrane ruffling, have highlighted the formation of membrane protrusions above M cell surface circular holes (Figure 6D) (109, 110). Therefore, these protrusions, which do not express the M cell marker UEA-I but bind bacteria, are likely TMD.
Once translocated by M cells or internalized by TMD, Salmonella Typhimurium are predominantly found in subepithelial lysozyme-expressing cells, i.e., TIM-4− LysoMac and/or LysoDC (Table 2) (42). Importantly, these phagocytes express genes involved in innate defense against Salmonella (39). These genes notably include Naip1, Naip2, and Naip5, which encode cytosolic receptors for the needle and inner rod proteins of the type III secretion system and for flagellin, respectively (Figure 5C) (116). Upon recognition of their ligands, NAIP proteins co-oligomerize with the adaptor NLRC4 to form an inflammasome complex and to recruit and activate caspase-1, which in turn process IL-1β and IL-18 into their active form. Interestingly, monocyte-derived cells express high levels of Il1b and Il18, indicating that, upon inflammasome activation, they may secrete large amounts of these pro-inflammatory cytokines (Figure 5C). Despite the expression of all these defense genes, it is currently unknown whether TIM-4− LysoMac and/or LysoDC are able to kill internalized Salmonella and die from pyroptosis upon inflammasome activation or whether bacteria have evolved strategies to survive, replicate inside, and kill these phagocytes.
Invasion of PP by Salmonella also induces the CCR6-dependent recruitment of CD11c+ cells in the SED and the FAE, probably through the release of CCL20 by the latter (117). As mentioned above, CCL20 is indeed specifically expressed by the FAE, thanks to its contact with RANKL-producing stromal cells (14). Among PP CD11c+ phagocytes, CCR6 expression is restricted to cDC2, and more specifically, DN cDC2 (40, 118). Thus, Salmonella induce the recruitment of DN cDC2 in the SED and the FAE. CCR6 expression also promotes the activation of Salmonella-specific T cells upon infection (117). Whether this activation relies on the cooperation between monocyte-derived cells that internalize bacteria and DN cDC2, which are recruited to the SED, remains to establish. Interestingly, upon inflammasome activation, pyroptosis of monocyte-derived cells could lead to the release of bacterial antigens and presentation of the latter to T cells by cDC2 as demonstrated in in vitro models using mouse bone marrow-derived DC and MF (119). However, since CCR6 is expressed by many other PP immune cell types and is involved in many cellular processes such as B cell migration in the SED and M cell differentiation, it remains to establish whether CCR6+ DN cDC2 are directly involved in activation of Salmonella-specific T cells upon infection (90, 118, 120–122).
Viral Infection: Reovirus and Norovirus
Reovirus enters the host through intestinal M cells and lack of M cells prevents from productive infection (18, 23, 123). Similarly, norovirus infection is reduced in M cell-deficient mice (23). Thus, M cells represent preferential entry sites for viruses, in addition to enteropathogenic bacteria. Although noroviruses have a tropism for DC and MF, their precise target following transport through M cells is currently unknown (124, 125). Unlike norovirus, reovirus preferentially replicates within the FAE (126). Interestingly, CD11c+ cells of the SED internalize reovirus-infected apoptotic FAE cells (127). As mentioned above, lysozyme-expressing CD11c+ cells, i.e., LysoDC and TIM-4− LysoMac, internalize apoptotic FAE cells (Table 2), suggesting their involvement in apoptotic epithelial cell-derived viral antigen handling (42). Importantly, PP CD11c+ cells from infected mice are able to process and present viral antigens from apoptotic cells to activate reovirus-primed T cells (127). Finally, initiation of an anti-reovirus immune response characterized by the production of virus-specific sIgA and cytotoxic T cells occurs in PP.
Prion Infection
Infectious prions are proteins with an abnormal conformation, which, upon conversion of the normally folded endogenous cellular prion protein and spreading to the central nervous system, lead to neurodegenerative diseases. Natural infection occurs mainly by oral consumption of prion-contaminated food. After oral exposure, uptake of infectious prions by M cells and their accumulation and replication upon follicular DCs in small intestine PP are essential for the efficient spread of disease to the brain (128, 129). CD11c+ cells are also required for the early stage of PP infection (130). In the SED, infectious prions are located in cells enriched for ferritin and LAMP1 but not MHCII (131). To better characterize these subepithelial phagocytes, we examined the expression of ferritin and LAMP1 transcripts in the gene expression database of dome phagocytes. We found that these transcripts are enriched in LysoDC and LysoMac as compared to cDC (Figure 5D). We also confirmed by immunostaining of PP sections the increased expression of ferritin and LAMP1 inside subepithelial LysoDC and LysoMac (CD11c+CX3CR1+ cells) as compared to other cells (Figure 5D). Since LysoDC express high levels of MHCII, this rather supports a role of TIM-4− LysoMac in the transmission of infectious prion (Table 2). Another population of infectious prion-loaded CD11b+ phagocytes is located in the subfollicular area of PP (132). These subfollicular phagocytes are absent from uninfected animals, which suggests that TIM-4− LysoMac could migrate from the SED to this subfollicular area upon infection. Interestingly, CXCR5 expression deletion in CD11c+ cells delays accumulation of infectious prion upon follicular DC and impedes oral prion disease pathogenesis (133). This suggests that CXCR5 could allow migration of CD11c+ phagocytes from the SED to the follicle or subfollicular area, which would promote spreading of infectious prion to follicular DC. However, when we interrogated the gene expression database of dome phagocytes, we did not find significant CXCR5 expression in any CD11c+ phagocytes. Therefore, identity of CD11c+CXCR5+ cells in PP as well as the mechanism of transfer of infectious prion from TIM-4− LysoMac to follicular DC remain pending issues.
Behavior of PP Phagocytes upon Infection
Our knowledge on the alteration induced by pathogens on PP phagocyte populations is scarce. What we know relies mainly on stimulation of PP with pathogen-derived compounds or mimetics. Thus, the cholera toxin induces the migration of CD11c+ cells into the FAE. Several TLR ligands induce similar CD11c+ cell relocation (112, 134–136). This is in line with the recruitment of DN cDC2 and formation of TMD observed shortly after Salmonella infection (43, 117). Thus, the first event that occurs upon pathogen detection is an increase of the sampling activity by recruitment in the FAE of both DN cDC2 and LysoDC. Then, SED-located CD11c+ cells are thought to migrate from the SED to the IFR in order to prime naïve T cells. Microsphere-loaded CD11c+ cells usually located in the SED are indeed observed in the IFR after cholera toxin or Salmonella Typhimurium-induced stimulation (137). Moreover, systemic injection of soluble Toxoplasma gondii tachyzoite antigen leads to a loss of CD11c+CD11b+ cells in the SED combined with the recruitment of CD11c+CD11b+ cells in the IFR (37). Actually, all activated dome cDC are located in the IFR as exemplified by their specific expression of CD83, CD86, CD205, CCL22, and CCR7 (40). Finally, the number of interfollicular cDC increase in the IFR of R848-fed animals (40, 138). However, this is at least in part due to interfollicular cDC1 number increase and to DAV cDC2 recruitment through a TNF-dependent pathway (40). The respective contribution of DAV and SED cDC2 to the migratory pool of interfollicular cDC is currently unknown, as well as their role in the induction of the mucosal immune response. Nevertheless, DAV and SED cDC recruitment in the IFR upon stimulation may allow in a single region the presentation of antigens sampled both in DAV and in SED. Since uptake of pathogens is facilitated in the FAE as compared to villous epithelium (11, 25, 42, 43), such mechanism of antigen sorting could help the mucosal immune system to discriminate between innocuous and harmful matters. Whether other stimuli than R848 induce similar recruitment of DAV cDC in the IFR is, however, currently unknown. If so, the current model of PP phagocyte activation will have to be modified to include DAV cDC as an integral part of the process.
Concluding Remarks
Although PP phagocytes are now well characterized, many efforts have to be done in order to understand the role of each phagocyte population in the mucosal immune response initiation during enteric infection. Importantly, to assess carefully these functions, a convenient and well-established panel of markers should be used in the different research laboratories in order to clearly identify each subset and avoid confusion between them. Here, we propose two panels of markers, one for microscopy and one for flow cytometry, which allow distinguishing each PP subset including DAV cDC and DAV MF (Table 1). These panels undoubtedly identify each subset of PP phagocytes and in the future should help clarify their functions in the initiation of the mucosal immune response.
Author Contributions
HL wrote the manuscript. CDS, CW, JB, and J-PG gave feedback and revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mark A. Jepson for sharing data and opinion with us. We acknowledge the CIML histology, cytometry, and mouse house facilities. We thank the PICSL imaging facility of the CIML (ImagImm), member of the national infrastructure France-BioImaging supported by the French National Research Agency (ANR-10-INBS-04). The project leading to this publication has received funding from Excellence Initiative of Aix-Marseille University—A*MIDEX, a French “Investissements d’Avenir” programme. The studies performed by the authors were supported by institutional grants from INSERM, CNRS, and Aix-Marseille University to the CIML. CDS is supported by the FRM fellowship FDT20160434982.
References
1. Sperandio B, Fischer N, Sansonetti PJ. Mucosal physical and chemical innate barriers: lessons from microbial evasion strategies. Semin Immunol (2015) 27(2):111–8. doi:10.1016/j.smim.2015.03.011
2. Jung C, Hugot JP, Barreau F. Peyer’s patches: the immune sensors of the intestine. Int J Inflam (2010) 2010:823710. doi:10.4061/2010/823710
3. Suzuki K, Kawamoto S, Maruya M, Fagarasan S. GALT: organization and dynamics leading to IgA synthesis. Adv Immunol (2010) 107:153–85. doi:10.1016/b978-0-12-381300-8.00006-x
4. Reboldi A, Cyster JG. Peyer’s patches: organizing B-cell responses at the intestinal frontier. Immunol Rev (2016) 271(1):230–45. doi:10.1111/imr.12400
5. Bhalla DK, Owen RL. Cell renewal and migration in lymphoid follicles of Peyer’s patches and cecum – an autoradiographic study in mice. Gastroenterology (1982) 82(2):232–42.
6. Pappo J, Owen RL. Absence of secretory component expression by epithelial cells overlying rabbit gut-associated lymphoid tissue. Gastroenterology (1988) 95(5):1173–7. doi:10.1016/0016-5085(88)90347-2
7. Frey A, Giannasca KT, Weltzin R, Giannasca PJ, Reggio H, Lencer WI, et al. Role of the glycocalyx in regulating access of microparticles to apical plasma membranes of intestinal epithelial cells: implications for microbial attachment and oral vaccine targeting. J Exp Med (1996) 184(3):1045–59. doi:10.1084/jem.184.3.1045
8. Mabbott NA, Donaldson DS, Ohno H, Williams IR, Mahajan A. Microfold (M) cells: important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol (2013) 6(4):666–77. doi:10.1038/mi.2013.30
10. Owen RL, Jones AL. Epithelial cell specialization within human Peyer’s patches: an ultrastructural study of intestinal lymphoid follicles. Gastroenterology (1974) 66(2):189–203.
11. Schulz O, Pabst O. Antigen sampling in the small intestine. Trends Immunol (2013) 34(4):155–61. doi:10.1016/j.it.2012.09.006
12. Bockman DE, Cooper MD. Pinocytosis by epithelium associated with lymphoid follicles in the bursa of Fabricius, appendix, and Peyer’s patches. An electron microscopic study. Am J Anat (1973) 136(4):455–77. doi:10.1002/aja.1001360406
13. Knoop KA, Kumar N, Butler BR, Sakthivel SK, Taylor RT, Nochi T, et al. RANKL is necessary and sufficient to initiate development of antigen-sampling M cells in the intestinal epithelium. J Immunol (2009) 183(9):5738–47. doi:10.4049/jimmunol.0901563
14. Nagashima K, Sawa S, Nitta T, Tsutsumi M, Okamura T, Penninger JM, et al. Identification of subepithelial mesenchymal cells that induce IgA and diversify gut microbiota. Nat Immunol (2017) 18(6):675–82. doi:10.1038/ni.3732
15. Clark MA, Jepson MA, Simmons NL, Booth TA, Hirst BH. Differential expression of lectin-binding sites defines mouse intestinal M-cells. J Histochem Cytochem (1993) 41(11):1679–87. doi:10.1177/41.11.7691933
16. Giannasca PJ, Giannasca KT, Falk P, Gordon JI, Neutra MR. Regional differences in glycoconjugates of intestinal M cells in mice: potential targets for mucosal vaccines. Am J Physiol (1994) 267(6 Pt 1):G1108–21.
17. Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, et al. Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature (2009) 462(7270):226–30. doi:10.1038/nature08529
18. Helander A, Silvey KJ, Mantis NJ, Hutchings AB, Chandran K, Lucas WT, et al. The viral sigma1 protein and glycoconjugates containing alpha2-3-linked sialic acid are involved in type 1 reovirus adherence to M cell apical surfaces. J Virol (2003) 77(14):7964–77. doi:10.1128/JVI.77.14.7964-7977.2003
19. Lelouard H, Reggio H, Mangeat P, Neutra M, Montcourrier P. Mucin-related epitopes distinguish M cells and enterocytes in rabbit appendix and Peyer’s patches. Infect Immun (1999) 67(1):357–67.
20. Lelouard H, Reggio H, Roy C, Sahuquet A, Mangeat P, Montcourrier P. Glycocalyx on rabbit intestinal M cells displays carbohydrate epitopes from Muc2. Infect Immun (2001) 69(2):1061–71. doi:10.1128/IAI.69.2.1061-1071.2001
21. Clark MA, Hirst BH, Jepson MA. M-cell surface beta1 integrin expression and invasin-mediated targeting of Yersinia pseudotuberculosis to mouse Peyer’s patch M cells. Infect Immun (1998) 66(3):1237–43.
22. Nakato G, Hase K, Suzuki M, Kimura M, Ato M, Hanazato M, et al. Cutting edge: Brucella abortus exploits a cellular prion protein on intestinal M cells as an invasive receptor. J Immunol (2012) 189(4):1540–4. doi:10.4049/jimmunol.1103332
23. Gonzalez-Hernandez MB, Liu T, Payne HC, Stencel-Baerenwald JE, Ikizler M, Yagita H, et al. Efficient norovirus and reovirus replication in the mouse intestine requires microfold (M) cells. J Virol (2014) 88(12):6934–43. doi:10.1128/jvi.00204-14
24. Kanaya T, Hase K, Takahashi D, Fukuda S, Hoshino K, Sasaki I, et al. The Ets transcription factor Spi-B is essential for the differentiation of intestinal microfold cells. Nat Immunol (2012) 13(8):729–36. doi:10.1038/ni.2352
25. Rios D, Wood MB, Li J, Chassaing B, Gewirtz AT, Williams IR. Antigen sampling by intestinal M cells is the principal pathway initiating mucosal IgA production to commensal enteric bacteria. Mucosal Immunol (2016) 9(4):907–16. doi:10.1038/mi.2015.121
26. Cerovic V, Bain CC, Mowat AM, Milling SW. Intestinal macrophages and dendritic cells: what’s the difference? Trends Immunol (2014) 35(6):270–7. doi:10.1016/j.it.2014.04.003
27. Vremec D, Shortman K. Dendritic cell subtypes in mouse lymphoid organs: cross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J Immunol (1997) 159(2):565–73.
28. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol (2014) 14(8):571–8. doi:10.1038/nri3712
29. Sancho D, Mourao-Sa D, Joffre OP, Schulz O, Rogers NC, Pennington DJ, et al. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J Clin Invest (2008) 118(6):2098–110. doi:10.1172/jci34584
30. Huysamen C, Willment JA, Dennehy KM, Brown GD. CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J Biol Chem (2008) 283(24):16693–701. doi:10.1074/jbc.M709923200
31. Caminschi I, Proietto AI, Ahmet F, Kitsoulis S, Shin Teh J, Lo JC, et al. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood (2008) 112(8):3264–73. doi:10.1182/blood-2008-05-155176
32. Dorner BG, Dorner MB, Zhou X, Opitz C, Mora A, Guttler S, et al. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity (2009) 31(5):823–33. doi:10.1016/j.immuni.2009.08.027
33. Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, et al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med (2010) 207(6):1283–92. doi:10.1084/jem.20100223
34. Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med (2010) 207(6):1273–81. doi:10.1084/jem.20100348
35. Gurka S, Hartung E, Becker M, Kroczek RA. Mouse conventional dendritic cells can be universally classified based on the mutually exclusive expression of XCR1 and SIRPalpha. Front Immunol (2015) 6:35. doi:10.3389/fimmu.2015.00035
36. Anjuere F, Martin P, Ferrero I, Fraga ML, del Hoyo GM, Wright N, et al. Definition of dendritic cell subpopulations present in the spleen, Peyer’s patches, lymph nodes, and skin of the mouse. Blood (1999) 93(2):590–8.
37. Iwasaki A, Kelsall BL. Localization of distinct Peyer’s patch dendritic cell subsets and their recruitment by chemokines macrophage inflammatory protein (MIP)-3alpha, MIP-3beta, and secondary lymphoid organ chemokine. J Exp Med (2000) 191(8):1381–94. doi:10.1084/jem.191.8.1381
38. Iwasaki A, Kelsall BL. Unique functions of CD11b+, CD8 alpha+, and double-negative Peyer’s patch dendritic cells. J Immunol (2001) 166(8):4884–90. doi:10.4049/jimmunol.166.8.4884
39. Bonnardel J, Da Silva C, Henri S, Tamoutounour S, Chasson L, Montanana-Sanchis F, et al. Innate and adaptive immune functions of Peyer’s patch monocyte-derived cells. Cell Rep (2015) 11(5):770–84. doi:10.1016/j.celrep.2015.03.067
40. Bonnardel J, Da Silva C, Wagner C, Bonifay R, Chasson L, Masse M, et al. Distribution, location, and transcriptional profile of Peyer’s patch conventional DC subsets at steady state and under TLR7 ligand stimulation. Mucosal Immunol (2017). doi:10.1038/mi.2017.30
41. Bonnardel J, Da Silva C, Masse M, Montanana-Sanchis F, Gorvel JP, Lelouard H. Gene expression profiling of the Peyer’s patch mononuclear phagocyte system. Genom Data (2015) 5:21–4. doi:10.1016/j.gdata.2015.05.002
42. Lelouard H, Henri S, De Bovis B, Mugnier B, Chollat-Namy A, Malissen B, et al. Pathogenic bacteria and dead cells are internalized by a unique subset of Peyer’s patch dendritic cells that express lysozyme. Gastroenterology (2010) 138(1):173–84.e1–3. doi:10.1053/j.gastro.2009.09.051
43. Lelouard H, Fallet M, de Bovis B, Meresse S, Gorvel JP. Peyer’s patch dendritic cells sample antigens by extending dendrites through M cell-specific transcellular pores. Gastroenterology (2012) 142(3):592–601.e3. doi:10.1053/j.gastro.2011.11.039
44. Contractor N, Louten J, Kim L, Biron CA, Kelsall BL. Cutting edge: Peyer’s patch plasmacytoid dendritic cells (pDCs) produce low levels of type I interferons: possible role for IL-10, TGFbeta, and prostaglandin E2 in conditioning a unique mucosal pDC phenotype. J Immunol (2007) 179(5):2690–4. doi:10.4049/jimmunol.179.5.2690
45. Li HS, Gelbard A, Martinez GJ, Esashi E, Zhang H, Nguyen-Jackson H, et al. Cell-intrinsic role for IFN-alpha-STAT1 signals in regulating murine Peyer patch plasmacytoid dendritic cells and conditioning an inflammatory response. Blood (2011) 118(14):3879–89. doi:10.1182/blood-2011-04-349761
46. Gunn MD, Tangemann K, Tam C, Cyster JG, Rosen SD, Williams LT. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc Natl Acad Sci U S A (1998) 95(1):258–63. doi:10.1073/pnas.95.1.258
47. Ngo VN, Tang HL, Cyster JG. Epstein-Barr virus-induced molecule 1 ligand chemokine is expressed by dendritic cells in lymphoid tissues and strongly attracts naive T cells and activated B cells. J Exp Med (1998) 188(1):181–91. doi:10.1084/jem.188.1.181
48. Willimann K, Legler DF, Loetscher M, Roos RS, Delgado MB, Clark-Lewis I, et al. The chemokine SLC is expressed in T cell areas of lymph nodes and mucosal lymphoid tissues and attracts activated T cells via CCR7. Eur J Immunol (1998) 28(6):2025–34. doi:10.1002/(SICI)1521-4141(199806)28:06<2025::AID-IMMU2025>3.0.CO;2-C
49. Zhao X, Sato A, Dela Cruz CS, Linehan M, Luegering A, Kucharzik T, et al. CCL9 is secreted by the follicle-associated epithelium and recruits dome region Peyer’s patch CD11b+ dendritic cells. J Immunol (2003) 171(6):2797–803. doi:10.4049/jimmunol.171.6.2797
50. Asselin-Paturel C, Brizard G, Pin JJ, Briere F, Trinchieri G. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J Immunol (2003) 171(12):6466–77. doi:10.4049/jimmunol.171.12.6466
51. Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, Colonna M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J Immunol (2006) 177(5):3260–5. doi:10.4049/jimmunol.177.5.3260
52. Owen RL, Pierce NF, Apple RT, Cray WC Jr. M cell transport of Vibrio cholerae from the intestinal lumen into Peyer’s patches: a mechanism for antigen sampling and for microbial transepithelial migration. J Infect Dis (1986) 153(6):1108–18. doi:10.1093/infdis/153.6.1108
53. Jinnohara T, Kanaya T, Hase K, Sakakibara S, Kato T, Tachibana N, et al. IL-22BP dictates characteristics of Peyer’s patch follicle-associated epithelium for antigen uptake. J Exp Med (2017) 214(6):1607–18. doi:10.1084/jem.20160770
54. Sakhon OS, Ross B, Gusti V, Pham AJ, Vu K, Lo DD. M cell-derived vesicles suggest a unique pathway for trans-epithelial antigen delivery. Tissue Barriers (2015) 3(1–2):e1004975. doi:10.1080/21688370.2015.1004975
55. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392(6673):245–52. doi:10.1038/32588
56. Dalod M, Chelbi R, Malissen B, Lawrence T. Dendritic cell maturation: functional specialization through signaling specificity and transcriptional programming. EMBO J (2014) 33(10):1104–16. doi:10.1002/embj.201488027
57. Manh TP, Alexandre Y, Baranek T, Crozat K, Dalod M. Plasmacytoid, conventional, and monocyte-derived dendritic cells undergo a profound and convergent genetic reprogramming during their maturation. Eur J Immunol (2013) 43(7):1706–15. doi:10.1002/eji.201243106
58. Fahlen-Yrlid L, Gustafsson T, Westlund J, Holmberg A, Strombeck A, Blomquist M, et al. CD11c(high)dendritic cells are essential for activation of CD4+ T cells and generation of specific antibodies following mucosal immunization. J Immunol (2009) 183(8):5032–41. doi:10.4049/jimmunol.0803992
59. Obata T, Shibata N, Goto Y, Ishikawa I, Sato S, Kunisawa J, et al. Critical role of dendritic cells in T cell retention in the interfollicular region of Peyer’s patches. J Immunol (2013) 191(2):942–8. doi:10.4049/jimmunol.1200636
60. Craig SW, Cebra JJ. Peyer’s patches: an enriched source of precursors for IgA-producing immunocytes in the rabbit. J Exp Med (1971) 134(1):188–200. doi:10.1084/jem.134.1.188
61. Gutzeit C, Magri G, Cerutti A. Intestinal IgA production and its role in host-microbe interaction. Immunol Rev (2014) 260(1):76–85. doi:10.1111/imr.12189
62. Husband AJ, Gowans JL. The origin and antigen-dependent distribution of IgA-containing cells in the intestine. J Exp Med (1978) 148(5):1146–60. doi:10.1084/jem.148.5.1146
63. Lycke NY, Bemark M. The role of Peyer’s patches in synchronizing gut IgA responses. Front Immunol (2012) 3:329. doi:10.3389/fimmu.2012.00329
64. Macpherson AJ, Geuking MB, Slack E, Hapfelmeier S, McCoy KD. The habitat, double life, citizenship, and forgetfulness of IgA. Immunol Rev (2012) 245(1):132–46. doi:10.1111/j.1600-065X.2011.01072.x
65. Benveniste J, Lespinats G, Salomon J. Serum and secretory IgA in axenic and holoxenic mice. J Immunol (1971) 107(6):1656–62.
66. Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y, et al. Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity (2014) 41(1):152–65. doi:10.1016/j.immuni.2014.05.016
67. Reikvam DH, Derrien M, Islam R, Erofeev A, Grcic V, Sandvik A, et al. Epithelial-microbial crosstalk in polymeric Ig receptor deficient mice. Eur J Immunol (2012) 42(11):2959–70. doi:10.1002/eji.201242543
68. Wei M, Shinkura R, Doi Y, Maruya M, Fagarasan S, Honjo T. Mice carrying a knock-in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense. Nat Immunol (2011) 12(3):264–70. doi:10.1038/ni.1991
69. Bunker JJ, Flynn TM, Koval JC, Shaw DG, Meisel M, McDonald BD, et al. Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity (2015) 43(3):541–53. doi:10.1016/j.immuni.2015.08.007
70. Kau AL, Planer JD, Liu J, Rao S, Yatsunenko T, Trehan I, et al. Functional characterization of IgA-targeted bacterial taxa from undernourished Malawian children that produce diet-dependent enteropathy. Sci Transl Med (2015) 7(276):276ra24. doi:10.1126/scitranslmed.aaa4877
71. Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell (2014) 158(5):1000–10. doi:10.1016/j.cell.2014.08.006
72. Macpherson AJ, Koller Y, McCoy KD. The bilateral responsiveness between intestinal microbes and IgA. Trends Immunol (2015) 36(8):460–70. doi:10.1016/j.it.2015.06.006
73. Pabst O, Cerovic V, Hornef M. Secretory IgA in the coordination of establishment and maintenance of the microbiota. Trends Immunol (2016) 37(5):287–96. doi:10.1016/j.it.2016.03.002
74. Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity (2009) 31(4):677–89. doi:10.1016/j.immuni.2009.08.020
75. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139(3):485–98. doi:10.1016/j.cell.2009.09.033
76. Klaasen HL, Van der Heijden PJ, Stok W, Poelma FG, Koopman JP, Van den Brink ME, et al. Apathogenic, intestinal, segmented, filamentous bacteria stimulate the mucosal immune system of mice. Infect Immun (1993) 61(1):303–6.
77. Lecuyer E, Rakotobe S, Lengline-Garnier H, Lebreton C, Picard M, Juste C, et al. Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity (2014) 40(4):608–20. doi:10.1016/j.immuni.2014.03.009
78. Jepson MA, Clark MA, Simmons NL, Hirst BH. Actin accumulation at sites of attachment of indigenous apathogenic segmented filamentous bacteria to mouse ileal epithelial cells. Infect Immun (1993) 61(9):4001–4.
79. Obata T, Goto Y, Kunisawa J, Sato S, Sakamoto M, Setoyama H, et al. Indigenous opportunistic bacteria inhabit mammalian gut-associated lymphoid tissues and share a mucosal antibody-mediated symbiosis. Proc Natl Acad Sci U S A (2010) 107(16):7419–24. doi:10.1073/pnas.1001061107
80. Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science (2012) 336(6086):1321–5. doi:10.1126/science.1222551
81. Lapthorne S, Macsharry J, Scully P, Nally K, Shanahan F. Differential intestinal M-cell gene expression response to gut commensals. Immunology (2012) 136(3):312–24. doi:10.1111/j.1365-2567.2012.03581.x
82. Morikawa M, Tsujibe S, Kiyoshima-Shibata J, Watanabe Y, Kato-Nagaoka N, Shida K, et al. Microbiota of the small intestine is selectively engulfed by phagocytes of the lamina propria and Peyer’s patches. PLoS One (2016) 11(10):e0163607. doi:10.1371/journal.pone.0163607
83. Fung TC, Bessman NJ, Hepworth MR, Kumar N, Shibata N, Kobuley D, et al. Lymphoid-tissue-resident commensal bacteria promote members of the IL-10 cytokine family to establish mutualism. Immunity (2016) 44(3):634–46. doi:10.1016/j.immuni.2016.02.019
84. Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science (2004) 303(5664):1662–5. doi:10.1126/science.1091334
85. Spalding DM, Griffin JA. Different pathways of differentiation of pre-B cell lines are induced by dendritic cells and T cells from different lymphoid tissues. Cell (1986) 44(3):507–15. doi:10.1016/0092-8674(86)90472-1
86. Spalding DM, Williamson SI, Koopman WJ, McGhee JR. Preferential induction of polyclonal IgA secretion by murine Peyer’s patch dendritic cell-T cell mixtures. J Exp Med (1984) 160(3):941–6. doi:10.1084/jem.160.3.941
87. Tezuka H, Abe Y, Asano J, Sato T, Liu J, Iwata M, et al. Prominent role for plasmacytoid dendritic cells in mucosal T cell-independent IgA induction. Immunity (2011) 34(2):247–57. doi:10.1016/j.immuni.2011.02.002
88. Moro-Sibilot L, This S, Blanc P, Sanlaville A, Sisirak V, Bardel E, et al. Plasmacytoid dendritic cells are dispensable for noninfectious intestinal IgA responses in vivo. Eur J Immunol (2016) 46(2):354–9. doi:10.1002/eji.201545977
89. Sato A, Hashiguchi M, Toda E, Iwasaki A, Hachimura S, Kaminogawa S. CD11b+ Peyer’s patch dendritic cells secrete IL-6 and induce IgA secretion from naive B cells. J Immunol (2003) 171(7):3684–90. doi:10.4049/jimmunol.171.7.3684
90. Reboldi A, Arnon TI, Rodda LB, Atakilit A, Sheppard D, Cyster JG. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science (2016) 352(6287):aaf4822. doi:10.1126/science.aaf4822
91. Kim SH, Cho BH, Kiyono H, Jang YS. Microbiota-derived butyrate suppresses group 3 innate lymphoid cells in terminal ileal Peyer’s patches. Sci Rep (2017) 7(1):3980. doi:10.1038/s41598-017-02729-6
92. Rahman ZS. Impaired clearance of apoptotic cells in germinal centers: implications for loss of B cell tolerance and induction of autoimmunity. Immunol Res (2011) 51(2–3):125–33. doi:10.1007/s12026-011-8248-4
93. Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science (2004) 304(5674):1147–50. doi:10.1126/science.1094359
94. Khan TN, Wong EB, Soni C, Rahman ZS. Prolonged apoptotic cell accumulation in germinal centers of Mer-deficient mice causes elevated B cell and CD4+ Th cell responses leading to autoantibody production. J Immunol (2013) 190(4):1433–46. doi:10.4049/jimmunol.1200824
95. Kranich J, Krautler NJ, Heinen E, Polymenidou M, Bridel C, Schildknecht A, et al. Follicular dendritic cells control engulfment of apoptotic bodies by secreting Mfge8. J Exp Med (2008) 205(6):1293–302. doi:10.1084/jem.20071019
96. Rahman ZS, Shao WH, Khan TN, Zhen Y, Cohen PL. Impaired apoptotic cell clearance in the germinal center by Mer-deficient tingible body macrophages leads to enhanced antibody-forming cell and germinal center responses. J Immunol (2010) 185(10):5859–68. doi:10.4049/jimmunol.1001187
97. Nishi C, Toda S, Segawa K, Nagata S. Tim4- and MerTK-mediated engulfment of apoptotic cells by mouse resident peritoneal macrophages. Mol Cell Biol (2014) 34(8):1512–20. doi:10.1128/mcb.01394-13
98. Rodriguez-Manzanet R, Sanjuan MA, Wu HY, Quintana FJ, Xiao S, Anderson AC, et al. T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc Natl Acad Sci U S A (2010) 107(19):8706–11. doi:10.1073/pnas.0910359107
99. Albacker LA, Karisola P, Chang YJ, Umetsu SE, Zhou M, Akbari O, et al. TIM-4, a receptor for phosphatidylserine, controls adaptive immunity by regulating the removal of antigen-specific T cells. J Immunol (2010) 185(11):6839–49. doi:10.4049/jimmunol.1001360
100. Albacker LA, Yu S, Bedoret D, Lee WL, Umetsu SE, Monahan S, et al. TIM-4, expressed by medullary macrophages, regulates respiratory tolerance by mediating phagocytosis of antigen-specific T cells. Mucosal Immunol (2013) 6(3):580–90. doi:10.1038/mi.2012.100
101. Rochereau N, Drocourt D, Perouzel E, Pavot V, Redelinghuys P, Brown GD, et al. Dectin-1 is essential for reverse transcytosis of glycosylated SIgA-antigen complexes by intestinal M cells. PLoS Biol (2013) 11(9):e1001658. doi:10.1371/journal.pbio.1001658
102. Fransen F, Zagato E, Mazzini E, Fosso B, Manzari C, El Aidy S, et al. BALB/c and C57BL/6 mice differ in polyreactive IgA abundance, which impacts the generation of antigen-specific IgA and microbiota diversity. Immunity (2015) 43(3):527–40. doi:10.1016/j.immuni.2015.08.011
103. Kadaoui KA, Corthesy B. Secretory IgA mediates bacterial translocation to dendritic cells in mouse Peyer’s patches with restriction to mucosal compartment. J Immunol (2007) 179(11):7751–7. doi:10.4049/jimmunol.179.11.7751
104. Rol N, Favre L, Benyacoub J, Corthesy B. The role of secretory immunoglobulin A in the natural sensing of commensal bacteria by mouse Peyer’s patch dendritic cells. J Biol Chem (2012) 287(47):40074–82. doi:10.1074/jbc.M112.405001
105. Carter PB, Collins FM. The route of enteric infection in normal mice. J Exp Med (1974) 139(5):1189–203. doi:10.1084/jem.139.5.1189
106. Hohmann AW, Schmidt G, Rowley D. Intestinal colonization and virulence of Salmonella in mice. Infect Immun (1978) 22(3):763–70.
107. Clark MA, Jepson MA, Simmons NL, Hirst BH. Preferential interaction of Salmonella typhimurium with mouse Peyer’s patch M cells. Res Microbiol (1994) 145(7):543–52. doi:10.1016/0923-2508(94)90031-0
108. Jones BD, Ghori N, Falkow S. Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer’s patches. J Exp Med (1994) 180(1):15–23. doi:10.1084/jem.180.1.15
109. Clark MA, Hirst BH, Jepson MA. Inoculum composition and Salmonella pathogenicity island 1 regulate M-cell invasion and epithelial destruction by Salmonella typhimurium. Infect Immun (1998) 66(2):724–31.
110. Clark MA, Reed KA, Lodge J, Stephen J, Hirst BH, Jepson MA. Invasion of murine intestinal M cells by Salmonella typhimurium inv mutants severely deficient for invasion of cultured cells. Infect Immun (1996) 64(10):4363–8.
111. Martinoli C, Chiavelli A, Rescigno M. Entry route of Salmonella typhimurium directs the type of induced immune response. Immunity (2007) 27(6):975–84. doi:10.1016/j.immuni.2007.10.011
112. Chabot S, Wagner JS, Farrant S, Neutra MR. TLRs regulate the gatekeeping functions of the intestinal follicle-associated epithelium. J Immunol (2006) 176(7):4275–83. doi:10.4049/jimmunol.176.7.4275
113. Pappo J, Ermak TH. Uptake and translocation of fluorescent latex particles by rabbit Peyer’s patch follicle epithelium: a quantitative model for M cell uptake. Clin Exp Immunol (1989) 76(1):144–8.
114. Owen RL, Heyworth MF. Lymphocyte migration from Peyer’s patches by diapedesis through M cells into the intestinal lumen. Adv Exp Med Biol (1985) 186:647–54.
115. Regoli M, Borghesi C, Bertelli E, Nicoletti C. A morphological study of the lymphocyte traffic in Peyer’s patches after an in vivo antigenic stimulation. Anat Rec (1994) 239(1):47–54. doi:10.1002/ar.1092390106
116. Vance RE. The NAIP/NLRC4 inflammasomes. Curr Opin Immunol (2015) 32:84–9. doi:10.1016/j.coi.2015.01.010
117. Salazar-Gonzalez RM, Niess JH, Zammit DJ, Ravindran R, Srinivasan A, Maxwell JR, et al. CCR6-mediated dendritic cell activation of pathogen-specific T cells in Peyer’s patches. Immunity (2006) 24(5):623–32. doi:10.1016/j.immuni.2006.02.015
118. Kucharzik T, Hudson JT III, Waikel RL, Martin WD, Williams IR. CCR6 expression distinguishes mouse myeloid and lymphoid dendritic cell subsets: demonstration using a CCR6 EGFP knock-in mouse. Eur J Immunol (2002) 32(1):104–12. doi:10.1002/1521-4141(200201)32:1<104::AID-IMMU104>3.0.CO;2-C
119. Yrlid U, Wick MJ. Salmonella-induced apoptosis of infected macrophages results in presentation of a bacteria-encoded antigen after uptake by bystander dendritic cells. J Exp Med (2000) 191(4):613–24. doi:10.1084/jem.191.4.613
120. Ebisawa M, Hase K, Takahashi D, Kitamura H, Knoop KA, Williams IR, et al. CCR6hiCD11c(int) B cells promote M-cell differentiation in Peyer’s patch. Int Immunol (2011) 23(4):261–9. doi:10.1093/intimm/dxq478
121. Lugering A, Floer M, Westphal S, Maaser C, Spahn TW, Schmidt MA, et al. Absence of CCR6 inhibits CD4+ regulatory T-cell development and M-cell formation inside Peyer’s patches. Am J Pathol (2005) 166(6):1647–54. doi:10.1016/S0002-9440(10)62475-3
122. Westphal S, Lugering A, von Wedel J, von Eiff C, Maaser C, Spahn T, et al. Resistance of chemokine receptor 6-deficient mice to Yersinia enterocolitica infection: evidence of defective M-cell formation in vivo. Am J Pathol (2008) 172(3):671–80. doi:10.2353/ajpath.2008.070393
123. Wolf JL, Rubin DH, Finberg R, Kauffman RS, Sharpe AH, Trier JS, et al. Intestinal M cells: a pathway for entry of reovirus into the host. Science (1981) 212(4493):471–2. doi:10.1126/science.6259737
124. Karst SM, Wobus CE. A working model of how noroviruses infect the intestine. PLoS Pathog (2015) 11(2):e1004626. doi:10.1371/journal.ppat.1004626
125. Wobus CE, Karst SM, Thackray LB, Chang KO, Sosnovtsev SV, Belliot G, et al. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol (2004) 2(12):e432. doi:10.1371/journal.pbio.0020432
126. Bass DM, Trier JS, Dambrauskas R, Wolf JL. Reovirus type I infection of small intestinal epithelium in suckling mice and its effect on M cells. Lab Invest (1988) 58(2):226–35.
127. Fleeton MN, Contractor N, Leon F, Wetzel JD, Dermody TS, Kelsall BL. Peyer’s patch dendritic cells process viral antigen from apoptotic epithelial cells in the intestine of reovirus-infected mice. J Exp Med (2004) 200(2):235–45. doi:10.1084/jem.20041132
128. Donaldson DS, Kobayashi A, Ohno H, Yagita H, Williams IR, Mabbott NA. M cell-depletion blocks oral prion disease pathogenesis. Mucosal Immunol (2012) 5(2):216–25. doi:10.1038/mi.2011.68
129. Donaldson DS, Sehgal A, Rios D, Williams IR, Mabbott NA. Increased abundance of M cells in the gut epithelium dramatically enhances oral prion disease susceptibility. PLoS Pathog (2016) 12(12):e1006075. doi:10.1371/journal.ppat.1006075
130. Raymond CR, Aucouturier P, Mabbott NA. In vivo depletion of CD11c+ cells impairs scrapie agent neuroinvasion from the intestine. J Immunol (2007) 179(11):7758–66. doi:10.4049/jimmunol.179.11.7758
131. Kujala P, Raymond CR, Romeijn M, Godsave SF, van Kasteren SI, Wille H, et al. Prion uptake in the gut: identification of the first uptake and replication sites. PLoS Pathog (2011) 7(12):e1002449. doi:10.1371/journal.ppat.1002449
132. Takakura I, Miyazawa K, Kanaya T, Itani W, Watanabe K, Ohwada S, et al. Orally administered prion protein is incorporated by m cells and spreads into lymphoid tissues with macrophages in prion protein knockout mice. Am J Pathol (2011) 179(3):1301–9. doi:10.1016/j.ajpath.2011.05.058
133. Bradford BM, Reizis B, Mabbott NA. Oral prion disease pathogenesis is impeded in the specific absence of CXCR5-expressing dendritic cells. J Virol (2017) 91(10). doi:10.1128/jvi.00124-17
134. Anosova NG, Chabot S, Shreedhar V, Borawski JA, Dickinson BL, Neutra MR. Cholera toxin, E. coli heat-labile toxin, and non-toxic derivatives induce dendritic cell migration into the follicle-associated epithelium of Peyer’s patches. Mucosal Immunol (2008) 1(1):59–67. doi:10.1038/mi.2007.7
135. Chabot SM, Chernin TS, Shawi M, Wagner J, Farrant S, Burt DS, et al. TLR2 activation by proteosomes promotes uptake of particulate vaccines at mucosal surfaces. Vaccine (2007) 25(29):5348–58. doi:10.1016/j.vaccine.2007.05.029
136. Chabot SM, Shawi M, Eaves-Pyles T, Neutra MR. Effects of flagellin on the functions of follicle-associated epithelium. J Infect Dis (2008) 198(6):907–10. doi:10.1086/591056
137. Shreedhar VK, Kelsall BL, Neutra MR. Cholera toxin induces migration of dendritic cells from the subepithelial dome region to T- and B-cell areas of Peyer’s patches. Infect Immun (2003) 71(1):504–9. doi:10.1128/IAI.71.1.504-509.2003
138. Yrlid U, Milling SW, Miller JL, Cartland S, Jenkins CD, MacPherson GG. Regulation of intestinal dendritic cell migration and activation by plasmacytoid dendritic cells, TNF-alpha and type 1 IFNs after feeding a TLR7/8 ligand. J Immunol (2006) 176(9):5205–12. doi:10.4049/jimmunol.176.9.5205
Keywords: mucosal immunity, Peyer’s patch, dendritic cells, macrophages, M cells, microbiota, IgA, bacterial and viral infections
Citation: Da Silva C, Wagner C, Bonnardel J, Gorvel J-P and Lelouard H (2017) The Peyer’s Patch Mononuclear Phagocyte System at Steady State and during Infection. Front. Immunol. 8:1254. doi: 10.3389/fimmu.2017.01254
Received: 27 July 2017; Accepted: 20 September 2017;
Published: 02 October 2017
Edited by:
Christel Vérollet, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceReviewed by:
Dhanansayan Shanmuganayagam, University of Wisconsin-Madison, United StatesSunil Joshi, Old Dominion University, United States
Copyright: © 2017 Da Silva, Wagner, Bonnardel, Gorvel and Lelouard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hugues Lelouard, lelouard@ciml.univ-mrs.fr