 Rajiv Kumar
Rajiv Kumar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 08 November 2017
Sec. Microbial Immunology
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01492
This article is part of the Research TopicLeishmaniasis: From Innate and Adaptive Immunity to Vaccine DevelopmentView all 15 articles
Leishmaniasis encompasses a group of diseases caused by protozoan parasites belonging to the genus Leishmania. These diseases range from life threatening visceral forms to self-healing cutaneous lesions, and each disease manifestations can progress to complications involving dissemination of parasites to skin or mucosal tissue. A feature of leishmaniasis is the key role host immune responses play in disease outcome. T cells are critical for controlling parasite growth. However, they can also contribute to disease onset and progression. For example, potent regulatory T cell responses can develop that suppress antiparasitic immunity. Alternatively, hyperactivated CD4+ or CD8+ T cells can be generated that cause damage to host tissues. There is no licensed human vaccine and drug treatment options are often limited and problematic. Hence, there is an urgent need for new strategies to improve the efficacy of current vaccine candidates and/or enhance both antiparasitic drug effectiveness and subsequent immunity in treated individuals. Here, we describe our current understanding about host immune responses contributing to disease protection and progression in the various forms of leishmaniasis. We also discuss how this knowledge may be used to develop new strategies for host-directed immune therapy to prevent or treat leishmaniasis. Given the major advances made in immune therapy in the cancer and autoimmune fields in recent years, there are significant opportunities to ride on the back of these successes in the infectious disease domain. Conversely, the rapid progress in our understanding about host immune responses during leishmaniasis is also providing opportunities to develop novel immunotherapy strategies that could have broad applications in diseases characterized by inflammation or immune dysfunction.
Leishmaniasis describes a collection of neglected tropical diseases caused by protozoan parasites of the genus Leishmania that are transmitted by female Phlebotomine sand flies (1). It largely affects the poorest populations living in developing countries and is prevalent throughout the tropical and subtropical regions of Africa, Asia, the Mediterranean, Southern Europe, and South and Central America. Globally, 350 million people are at risk of developing leishmaniasis and 1.5–2 million new cases occur annually (2). The clinical spectrum of leishmaniasis ranges from the life-threatening visceral form to self-healing cutaneous lesions or a more serious mucosal manifestation.
Visceral leishmaniasis (VL), also known as kala-azar, typically involves long-term, low-grade fever, enlarged spleen and liver, weight loss, pancytopenia, and hypergammaglobulinemia [reviewed in Ref. (3)]. Untreated VL cases are almost always fatal, and more than 90% of cases occur in India, Bangladesh, Nepal, Sudan, Ethiopia, and Brazil with an estimated incidence of at least 500,000 new cases and 50,000 deaths each year (2–4). Of note, the state of Bihar in north east India has been the focus of most VL cases for many years (5), but recent efforts toward elimination, and civil unrest in Southern Sudan, have now made the latter region the source of most cases (6). Post kala-azar dermal leishmaniasis (PKDL), which can be a late cutaneous manifestation of VL, either following drug treatment or sometimes independent of VL development, is confined to the Indian subcontinent [India, Nepal and Bangladesh, and East Africa (Sudan)]. It presents as an accumulation of heavily infected macrophages in the skin (7–9), which appear as nodules, papules, or hypopigmented macules. PKDL can appear from 6 months to years after apparent VL cure in the Indian subcontinent, but can also occur earlier (within 6 months) or along with VL in the Sudan, where the incidence of this disease is higher. PKDL heals spontaneously in a proportion of cases in Africa, but rarely in Indian VL patients and requires prolonged treatment. Since PKDL patients harbor increased parasite numbers in their skin, they are thought to act as parasite reservoir and play an important role in disease transmission in endemic regions. As such, they may be an important population to target with effective host-directed immunotherapies.
Cutaneous leishmaniasis (CL) is characterized by the development of ulcerative skin lesions containing parasites and is the most common form of disease occurring mainly in Afghanistan, Algeria, Brazil, Colombia, the Islamic Republic of Iran, Pakistan, Peru, Saudi Arabia, and Syria (2, 10). Cutaneous lesions are generally localized and may persist for months to years or heal spontaneously within weeks. The development of disfiguring scars at the affected skin areas following healing is a major concern.
Mucocutaneous leishmaniasis (MCL) is prevalent in Bolivia, Brazil, Peru, and Ethiopia (WHO, 2014) and is caused by L. baziliensis, L. panamensis, and L. aethiopica. These species metastasize to mucosal tissue in the mouth and upper respiratory tract, leading to localized tissue destruction. MCL can present from several months to years after the development of a cutaneous lesion. Diffused cutaneous leishmaniasis (DCL), which is more common in central and South America, is thought to occur in immunosuppressed individuals, where parasites can readily disseminate to subcutaneous tissue. As both MCL and DCL are associated with strong and weak host inflammatory responses, respectively, which appear to contribute to disease pathology, they also have potential for improved treatment involving host-directed immune therapy.
Studies in experimental VL in mice, caused by infection with the human parasites L. donovani or L. infantum, show the development of antiparasitic IFNγ-producing, Tbet+ CD4+ T (Th1) cells is critical for resistance against infection (11). Many VL patients fail to generate potent cell-mediated immune responses against parasite antigens and this is thought to be an underlying cause of disease. However, enhanced IFNγ mRNA expression in the spleen and bone marrow, as well as increased circulatory plasma IFNγ, TNF and IL-12 in VL patients, suggests that they do not lack a protective Th1 response, but instead, immunosuppressive mechanism are established to prevent parasite killing (12–15). Importantly, antigen-specific responses in whole blood assays indicate that VL patient CD4+ T cells have the capacity to produce IFNγ in response to parasite antigen (16–18). Therefore, attention has now focused on regulatory mechanisms that prevent Th1 cell-mediated control of parasite growth.
CD4+ T cell IL-10 production has emerged as an important mechanism to dampen T cell activation in parasitic infections, including in humans with VL (15, 19, 20). Importantly, most T cell-derived IL-10 is not produced by thymus-derived Foxp3-expressing regulatory T (Treg) cells. Instead, the IL-10 producing CD4+ T cells often co-produce IFNγ and have been designated type 1 regulatory (Tr1) cells (15). They are increasingly recognized as a critical regulatory CD4+ T cell subset that protects tissue from inflammation (21–23). However, Tr1 cells also appear to promote infection by suppressing Th1 cell-mediated immunity. The role of IL-10 in immune suppression and disease progression is well documented in both experimental and human VL (15, 24–28). Human VL is associated with enhanced IL-10 plasma levels, increased IL-10 mRNA expression in lymph nodes, bone marrow, and spleen, and readily detected IL-10 produced by whole blood cells from VL patients following parasite antigen stimulation (15, 28). IL-10 dampens major histocompatibility complex (MHC) class II expression on APC and downregulates TNF and nitric oxide (NO) production, leading to reduced parasite clearance and suppressed activation of Th1 cells (29). Neutralization of IL-10 in VL patient sera can suppress L. donovani replication in macrophages (15, 30), and IL-10 blockade in splenic aspirate cultures from VL patients can limit parasite replication and enhance Th1 cell cytokine production (24). IL-10 can also modulate immune responses by promoting T cell exhaustion (31, 32).
A number of immune checkpoint molecules have also been identified on CD4+ T cells from experimental VL models and VL patients (Table 1). These include CTLA-4 (CD152) and PD-1, which are negative regulators of T cells and are expressed on exhausted or anergic T cells during chronic infection. CTLA-4 binds to the costimulatory ligands B7-1 (CD80) and B7-2 (CD86), with much higher affinity than CD28, while PD-1 interacts with PD-1 ligand 1 (PD-L1; B7.H1) and PD-L2. Activation of CTLA-4 leads to increased levels of TGFβ, as well as apoptosis of CD4+ T cells in murine VL (33). In mice infected with L. donovani, CTLA-4 blockade decreased parasite burden in both liver and spleen, associated with increased frequencies of IFNγ and IL-4 producing cells, and an accelerated hepatic granulomatous response (34, 35). CTLA-4 blockade has also been shown to increase the efficiency of chemotherapy in L. donovani infected mice (36, 37). Similarly, blockade of PD-1 or PD-L1 resulted in enhanced parasite clearance and increased pro-inflammatory cytokine production in experimental VL (38–40). Thus, these studies clearly show the therapeutic potential of targeting immune checkpoint molecules for host-directed immune therapy in VL.
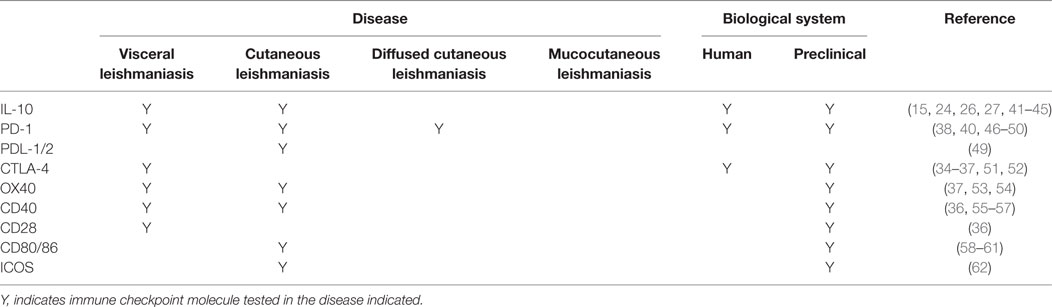
Table 1. Immune checkpoint molecules tested for therapeutic effects in leishmaniasis.
Another important regulatory cytokine involved in VL is IL-27, which is composed of the EBI-3 and p28 sub-units. IL-27 belongs to IL-6/12 cytokine family and was originally described as a co-factor for Th1 cell differentiation, along with IL-12 (63, 64). IL-27 promotes T cell IL-10 production in mice, which is further amplified by autocrine IL-21 production (65, 66). IL-27 receptor-deficient mice infected with L. donovani developed enhanced Th1 responses, but this was associated with severe liver pathology (67). Patients with active VL also presented with enhanced IL-27 plasma levels, as well as increased mRNA transcripts encoding EBI-3 and p28 in splenic aspirates (28). IL-27 produced by CD14+ cells, along with IL-21 from T cell sources, promoted the differentiation and expansion of Ag-specific, IL-10–producing T cells in VL patients. Importantly, pro-inflammatory cytokines, such as IFNγ, act on macrophages and stimulate IL-27 production, suggesting a feedback mechanism to stimulate IL-10 production to control IFNγ levels and protect host tissue. IL-27 has also been associated with suppression of CD4+ T cell IL-17A and IL-22 secretion (68, 69). Since L. donovani antigen-stimulated production of both IL-17A and IL-22 by PBMC in an apparent disease-resistant Sudanese population, these cytokines were proposed to be protective following L. donovani infection and, therefore, elevated IL-27 in VL patients might not only promote disease by increasing IL-10 production, but also by regulating IL-17 production (70). Studies with Indian VL patients showed low levels of IL-17A mRNA transcripts, as well as the IL-17-related transcription factor RORγT during active disease (28). However, there was no direct evidence that this Th17 response was suppressed by IL-27. Furthermore, there is evidence from experimental VL that the impact of IL-17A may depend of the stage of infection, whereby this cytokine impedes antiparasitic immunity early (71), but is protective following establishment of infection (72). Hence, immune dysfunction in VL patients appears to involve multiple immune regulatory pathways that differ both spatially and temporally, and identifying which can be safely and effectively targeted for clinical advantage should be a major research priority.
Other immunosuppressive mechanisms established during VL may be mediated through regulatory T cells. Regulatory T cells can be classified as thymus-derived CD4+CD25+FoxP3+ T (Treg) cells and inducible regulatory T cells that include conventional T cells that convert to FoxP3+ regulatory T cells in peripheral tissues, as well as Tr1 cells (73, 74). To maintain immune homeostatic conditions, Treg cells limit the activity of potentially self-reactive T cell responses and prevent immune-mediated pathology and autoimmunity (75, 76). However, these same mechanisms may also contribute to impaired pathogen clearance during parasitic infection. Treg cells function by secreting regulatory cytokines such IL-10 and TGFβ, as well as expressing inhibitory molecules such as CTLA-4 and IL-35 (77). Treg cells express high levels of CD25 (IL-2R), thereby allowing them to form the high affinity receptor for IL-2, which allows them to deprive conventional T cells of this important growth factor, thus causing apoptosis (78). However, there is little evidence for the involvement of Treg cells in human or experimental VL. Studies from VL patients in Bihar, India showed no accumulation of Treg cells in the spleen or blood, and the frequency of these cells did not change during the course of infection (15, 79). Moreover, FoxP3− T cells were the major source of IL-10 mRNA in VL patient spleens, and this finding was in accordance with murine VL studies where IL-10 secretion by FoxP3−CD4+ T cells correlated with disease severity (19). However, other studies have reported the accumulation of Treg cells at sites of infection and suggested their possible role in disease pathogenesis in both human and experimental VL. One study from India suggested that Treg cells were a major source of IL-10 in the bone marrow of VL patients and that IL-10 secretion from Treg cells suppressed conventional T cells (80). In a different study with Indian VL patients, production of IL-10 and TGFβ by Treg cells was positively correlated with parasite load (81). Similarly, TGFβ-producing Treg cells were shown to accumulate in infected tissues in a murine model of VL (82). Thus, further investigation is needed to establish whether Treg cells are involved in the pathogenesis of VL and whether they can be modulated for therapeutic advantage. Interestingly, TGFβ is also secreted by macrophages and dendritic cells (DCs) during experimental VL. A cathepsin B-like cysteine protease present in L. donovani can activate TGFβ (83), which, in turn, activates arginase-1, leading to enhanced l-ornithine production and reduced NO secretion, thereby promoting parasite survival in infected cells (84, 85). Human VL patients have enhanced TGFβ plasma levels (13) during active disease, suggesting a possible role in pathogenesis. However, more research is needed to better understanding the precise mechanisms of TGFβ-mediated suppression of antiparasitic immunity before it can be considered as an immune therapy target.
The development of regulatory DC subsets following L. donovani infection can also have a major impact on T cell responses during VL. These regulatory DCs are capable of producing anti-inflammatory cytokines, such as IL-10, TGFβ, and IL-27 (86). In experimental VL, it was shown that IL-10+IL-27+ DCs were able to promote IL-10 production by Th1 cells in vivo and identified this cell population as a potential target for immunotherapy (87). Furthermore, CD11cloCD45RB+ DCs in the spleen of L. donovani-infected mice had high levels of IL-10 production, compared to CD11chi populations, and displayed features of immature DCs, including low expression of co-stimulatory molecules and intracellular MHC class II (88). These DCs also produced IL-10 when stimulated with lipopolysaccharide and promoted Treg cell IL-10 production capable of inhibiting mixed lymphocyte reactions driven by conventional DCs (89). The inhibitory effects of these regulatory DCs could be reversed by IL-10 signaling blockade, indicating that IL-10 production was a critical regulatory mechanisms employed by this DC subset (88). Therefore, both cognate and soluble cytokine signals between effector CD4+ T cells, DCs, and infected macrophages have key roles in determining whether parasite growth is controlled and/or disease develops, making these interactions promising targets for immune therapy (Figure 1).
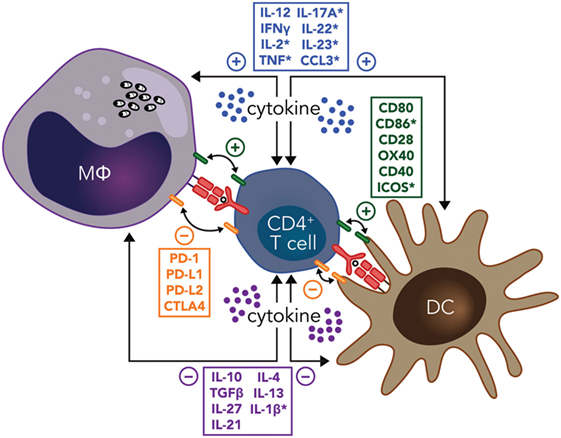
Figure 1. Potential immunotherapy targets to manipulate CD4+ T cell-dendritic cell (DC) and CD4+ T cell-infected macrophage (MΦ) interactions during leishmaniasis include both cognate and soluble cytokine signals. Primary signals between CD4+ T cells and macrophages or DCs through major histocompatibility complex class II antigen presentation of parasite peptide to the T cell receptor are indicated (red), as are both positive (green), and negative (orange) costimulatory signals. Positive (blue) and negative (purple) soluble cytokine signals are also show. Note that many of these signals are bidirectional, as indicated by the double-ended arrows. In addition, molecules highlighted by an asterisk have been reported to have the opposite effects in different type of Leishmania species infections. Amastigotes residing in macrophages are shown in black.
CD8+ T cells can kill Leishmania-infected macrophages by secreting cytolytic enzymes (90, 91). However, studies with human VL blood samples suggest that CD8+ T cells have an anergic or exhausted phenotype, as indicated by high expression of IL-10, CTLA-4, and PD-1, which may hamper the protective efficiency of these cells during active disease (51). A better understanding of role of CD8+ T cells during VL is needed if the antiparasitic potential of these cells through vaccination or immune therapy can be exploited.
Post kala-azar dermal leishmaniasis often develops as a cutaneous complication of VL in apparently cured patients, but can also develop independent of VL. The pathogenesis of PKDL remains poorly understood. It has been postulated that immune suppression may allow multiplication of latent parasites from the viscera or residing in the skin (8). Similar to clinical VL, elevated IFNγ and TNF levels are found in lesions of PKDL patients, with the concurrent presence of the immunosuppressive cytokines IL-10 and TGFβ (92). Despite the presence of high IFN-γ and TNF in these tissues, there is reduced expression of IFNγ and TNF receptors in Indian PKDL patients (92, 93), while genetic polymorphisms in the IFNγ receptor 1 gene promoter region have been reported and found to be associated with susceptibility to PKDL in Sudanese patients (94, 95). Treg cells have also been associated with PKDL in the Indian subcontinent and elevated FoxP3, CD25, and CTLA-4 mRNA expression has been reported in the skin of patients. Furthermore, Foxp3, CD25, and IL-10 mRNA levels directly correlated with parasite load in these PKDL patients (96). Since PKDL either develops soon after VL or independent of VL in the Sudanese population, but takes longer to develop after VL in the Indian subcontinent, the immunopathology of PKDL is likely to differ in these populations. PKDL patients from the Sudan display immune responses similar to cured VL patients and their PBMC proliferate in response to parasite antigens and CD4+ T cells secrete IFNγ and IL-10 (97, 98). However, PKDL patients from the Indian subcontinent have high numbers of CD8+ T cells in their lesions and circulation, along with increased antigen-induced IL-10 production by circulating CD8+ T cells and impaired antigen-induced proliferation (99, 100). Studies with Indian PKDL patients have also demonstrated enhanced Th17 cell responses by analyzing mRNA and protein expression of Th17-related IL-23, IL-17A, and RORγt (101). Stimulation of PKDL patient PBMCs with parasite antigens resulted in IL-17A and IL-23 production, while stimulation with recombinant IL-17A enhanced TNF and NO production. Hence, these data suggest that enhanced Th17 responses may have a role in parasite clearance during PKDL. However, it is still not clear whether regulatory cytokines and/or other mechanisms suppress IL-17-mediated protective responses during active disease. This knowledge is important if we wish to manipulate this immunoregulatory pathway to improve antiparasitic immunity.
Cutaneous leishmaniasis is caused by several Leishmania species, including Leishmania major, L. braziliensis, L. mexicana, and L. amazonensis. Cell-mediated immune responses at the site of cutaneous lesions are of primary importance in determining the outcome of disease. Furthermore, in murine models of CL caused by L. major, the genetic background of mice also determines disease outcome. In C57BL/6 mice, Th1 cell responses promote a self-healing process, while Th2 responses are associated with parasite persistence in the lesions of BALB/c mice [reviewed in Ref. (102, 103)]. In humans infected with L. major, cutaneous lesions have been associated with high IFN-γ, IL-10, and IL-12 mRNA accumulation, indicative of a mixed CD4+ T cell response. Several immune checkpoint molecules have also been identified in experimental CL studies that can modify CD4+ T cell responses to favor parasite clearance (Table 1), again demonstrating immune checkpoint blockade as a potential approach to improve disease treatments.
Following transmission of L. major to mice via sand fly bites or needle, neutrophils rapidly infiltrate the bite site and capture injected parasites (104). Neutrophils rapidly express apoptotic markers following L. major uptake, which attracts monocytes and DCs to the site of infection for removal of apoptotic cells (105, 106). This allows the uptake of parasites into phagocytic cells without triggering inflammation, and thereby enabling establishment of infection. Infected neutrophils also express chemokines, such as CCL-3, to attract DCs to the site of infection (107). This may help to stimulate Th1 cell responses following activation of DCs through interaction of DC-SIGN on DC and specific glycans on neutrophils (108). In addition, CCL3 can induce IL-12 secretion by macrophages in C57BL/6 mice, but not in BALB/c mice (107). Given these latter pro-inflammatory properties of neutrophils, any modifications of neutrophil functions may have to be directed specifically toward their activity as a “Trojan horse” for establishment of infection. However, it is important to remember that the role of neutrophils is critically dependent on the Leishmania species in question, the parasite lifecycle stage and stage of infection, when trying to manipulate neutrophil functions.
Following L. major infection, complement-dependent platelet activation, including the release of platelet-derived growth factors, can stimulate the release of CCL2/MCP-1 by leukocytes and mesenchymal cells, leading to recruitment of Ly6C+ inflammatory monocytes, which can capture and kill parasites via oxidative burst (109). Importantly, these monocytes can migrate to lymph nodes and differentiate into specialized DC subsets during L. major infection. These monocytes-derived DCs secret high levels of IL-12 and stimulate L. major-specific Th1 cell responses, suggesting a contribution to protection against disease (110, 111). Monocytes expressing high levels of CCR2 can also capture L. major at the site of infection in C57BL/6 mice, then migrate to draining lymph nodes and differentiate into inducible nitric oxide (iNOS)-producing DC that also promote Th1 cell-mediated protection (112). Hence, the promotion of these activities in the context of vaccination or drug treatment may be desirable.
NK cells are also recruited to the site of infection in mice infected with L. major and produce IFN-γ, which can amplify DC IL-12 production required for the development of strong Th1 cell responses (113). However, NK cells can also produce IL-10 during L. donovani infection (114), suggesting they can play either antiparasitic or immunoregulatory roles during infection. Depending upon the dose of infection, CD8+ T cells also produce IFN-γ in murine models of CL, which can also help shape early adaptive immune responses associated with protection (115–118). However, resolution of infection following L. major infection is primarily associated with CD4+ T cell-mediated immunity (119–121). Despite healing of cutaneous lesions, parasites continue to persist at the original site of infection, in part due to IL-10-mediated mechanisms, and these persisting parasites are thought to help maintain effector memory CD4+ T cells (TEM) that protect against re-infection (122, 123). This TEM response is lost if parasites are eliminated, as shown by studies in which mice were manipulated to achieve sterile cure (122). Thus, concomitant immunity is compromised and protection against a secondary challenge can be lost in the absence of persisting parasites (124). However, there is also evidence that a pool of long-lasting central memory CD4+ T cells (TCM) can develop in absence of persisting parasites, and that these can acquire effector functions after re-infection leading to protection (125). These TCM cells require additional IL-12 signals to develop into fully functional Th1 cells, and in absence of this signal, they can convert into IL-4-producing Th2 cells (126). TCM cells appear to be generated early during infection, and not only help in controlling secondary infections, but also contribute to clearance of primary infection (127). Hence, these findings suggest both TEM and TCM cells participate in maintaining immunity to L. major infection, but only the TEM require persistent parasite antigen. Therefore, vaccines designed to protect against leishmaniasis should target the expansion of long-lasting TCM cells, rather than short lived TEM cells.
More recently, skin resident memory (TRM) CD8+ T cells have been shown to provide protection against L. major infection, independent of circulatory CD4+ T cells, by recruiting inflammatory monocytes, which rapidly control parasite growth via reactive oxygen species (ROS) and NO generation (128). Thus, these TRM cells also represent a potential target cell population for vaccination. Treg cells appear to play a role in L. major persistence in C57BL/6 mice by suppressing CD4+ T cell effector functions through IL-10-mediated immunosuppressive mechanisms (122). The IL-10 produced by these Treg cells can also promote parasite persistence by modulating APC function and/or inhibiting parasite killing mechanisms in infected macrophages. Thus, the activity of Treg cells at the site of infection can promote concomitant immunity, but also allow parasites to persist. Therefore, although Treg cells could be targeted for immunomodulation, care would have to be taken to ensure that long-term protection was not compromised.
In non-healing CL caused by L. major Seidman strain in C57BL/6 mice, Nlrp3 inflammasome-dependent IL-1β activation plays an important role in determining disease outcome (129). The activation of the Nlrp3 inflammasome enhanced IL-1β activation through caspase-1 cleavage, which caused recruitment of neutrophils to the site of infection, and ultimately resulted in the suppression of immunity, which was confirmed by using neutropenic Genista mice (129). Nlrp3 can promote Th2 cell development in non-healing cutaneous lesions caused by L. major infection in BALB/c mice (130). Thus, this inflammasome and related cell signaling pathways are potential targets for immune therapy to treat and promote healing of cutaneous lesions in human CL (see also below). However, inflammasome- and caspase-1-dependent IL-1-β production has been shown to provide resistance against L. amazonensis infection in mice by triggering NO production (131), thus emphasizing the need for careful consideration in choosing appropriate targets for immune modulation in specific disease settings.
Although self-cure is often the outcome of CL, some patients infected with L. braziliensis, L. panamensis, and L. aethiopica can develop MCL after resolution of their primary lesion, characterized by chronic inflammation of the nasal mucosa and by a hyperactive T-cell response (132, 133), associated with high levels of pro-inflammatory cytokines, such as IFN-γ and TNF-α, and decreased levels of IL-10 and TGF-β (132, 134, 135). Thus, a poorly regulated T cell response is an underlying cause of disease pathogenesis in MCL patients. In patients infected with L. braziliensis, the number of CD8+ T cells recruited to lesions increased as disease progressed, and these cells expressed high levels of granzymes and perforin, indicating they had elevated cytolytic activity (136). In fact, these CD8+ T cells have now been shown to contribute to inflammation and disease pathology via perforin-mediated cytotoxicity (137). In mice co-infected with lymphocytic choriomeningitis virus and L. braziliensis, it was found that perforin-mediated CD8+ T cell cytotoxicity in the lesion resulted in enhanced recruitment of neutrophils and monocytes, which produced IL-1-β that contributed to immunopathology and disease severity (138). Importantly, pharmacological blockade of Nlrp3 reduced inflammation caused by cytotoxic CD8+ T cells in this mouse model of MCL, thus identifying this inflammasome, as well as CD8+ T cell-mediated cytotoxicity, as potential targets for immunotherapy. This was supported by additional data from the same study, using skin biopsies and PBMCs from CL patients infected with L. braziliensis, which IL-1β was highly expressed in skin lesions and blockade of the Nlrp3 inflammasome prevented the IL-1-β secretion from skin biopsies, suggesting a similar pathogenic mechanism might be operating during clinical MCL.
In addition to CD8+ T cell-mediated pathology, Th17 cells have also been associated with pathogenesis in MCL patients (139). MCL lesions were found to have elevated IL-17A mRNA, as well as TGF-β, ROR-γT, and IL-23 mRNA levels, which are associated with Th17 cell differentiation. Interestingly, IL-17 was not only produced by CD4+ T cells but also by CD8+ T cells, CD14+, and CCR6+ Cells. The enhanced production of IL-17 was associated with infiltration and recruitment of neutrophils into the lesion, suggesting that IL-17 may promote inflammatory responses in MCL patients. Thus, IL-17 production could be a therapeutic target in MCL patients to reduce tissue pathology.
Diffused cutaneous leishmaniasis is a severe manifestation of CL characterized by a defective cellular immune response to Leishmania antigens (140). However, this unresponsiveness is restricted to antiparasitic immune responses, as responses to unrelated antigens remain intact (141, 142). DCL patients have high parasite numbers within skin lesions, which has been associated with low levels of IFN-γ and IL-2 mRNA, and concurrent high levels of IL-10, IL-4, and IL-5 mRNA in lesions (135). Therapeutic cure was associated with enhanced IFNγ production, but low IL-10 expression (143), indicating the requirement for a classical Th1 cells response for favorable clinical outcomes. This disease is also associated with high antibody titers and plasma TGF-β [reviewed in Ref. (102)]. IL-10 and TGF-β, along with Treg cells, can antagonize IFN-γ and TNF activities, resulting in impaired microbicidal activities in infected macrophages [reviewed in Ref. (144)]. However, it is not clear in DCL whether high antigen exposure causes T cell unresponsiveness or if impaired T cell responses promote localized parasite growth in the skin [reviewed in Ref. (144)].
Diffused cutaneous leishmaniasis patients respond poorly to conventional drug treatment (145), but some degree of treatment success has been achieved with immune modulation using IFN-γ combined with viable BCG and antimonial drug (146, 147). Since unresponsive T cells often express inhibitory molecules, such as PD-1, CTLA-4, and LAG-3, these may make attractive targets for immune therapy in DCL patients. IL-1-β has also been associated with disease severity in L. mexicana-infected DCL patients (148), making it another potential therapeutic target. Again, care will need to be taken to ensure the promotion of antiparasitic immunity in this context is not at the expense of protection against tissue damage.
The fact that L. major-induced cutaneous lesions often heal spontaneously and protect against future infection is the basis for leishmanization, which involves inoculation with live, virulent parasites in an unexposed part of body to produce a controlled lesion. This strategy has been practiced successfully in the former Soviet Union, Middle East, and Israel, and likely provides protection in humans because it mimics a natural infection, including allowing parasite persistence and development of concomitant immunity. The protection provided by leishmanization is essentially T cell-mediated, whereby IFN-γ-producing CD4+ T cells are recruited to dermal sites of infection where they perform effector functions, including the promotion of microbicidal mechanisms in infected macrophages (123). Importantly, the success of leishmanization depends on the viability and infectivity of injected parasite. Parasites that lost virulence stimulated delayed-type hypersensitive reactions, but did not provide protection from natural re-infection (149). Leishmanization was abandoned in most countries because of logistical problems and safety concerns, due to some immunosuppressed individuals developing non-healing lesions (150). Interestingly, leishmanization can provide cross protection against the visceral form of disease, as leishmanized C57BL/6 mice infected with L. major were protected from heterologous visceral infection with L. infantum, associated with recruitment of IFN-γ-producing Ly6C+CD4+ T cells to both skin and visceral organs (151). Similarly, longitudinal studies in the Sudan indicated that people residing in an L. major endemic area were protected against VL caused by L. donovani (152, 153). In addition, CL caused by a L. donovani strain in Sri-Lanka provided cross protection against visceral disease (154). These findings suggest that leishmanization could be a strategy employed to increase protection against VL. However, a better understanding of the immunoregulatory mechanisms associated with this process is needed to fully exploit the positive aspects of leishmanization with improved safety.
Although different Leishmania species cause a broad range of clinical symptoms, genetic analysis indicates a large degree of genomic conservation between species. Thus, it may be possible to generate broadly effective vaccines against different clinical diseases. However, despite many efforts, there is no effective, licensed vaccine to prevent human leishmaniasis. There is a major need for more efficacious and less toxic adjuvants and immune therapies for better vaccines for patients suffering from leishmaniasis. Studies in VL patients and experimental models (15, 155) indicate the rapid development of immunoregulatory networks following exposure to parasites, which raises questions about how these regulatory networks might influence subsequent immunity, particularly to vaccines. It is noteworthy that many vaccines tested in disease endemic regions have not performed as well as when tested in healthy volunteers. For example, the RTS,S/AS01 vaccine in children and infants affords 36 and 25% efficacy against clinical malaria, respectively (156), while a recent study showed that the efficacy of the same vaccine in healthy volunteers in CHMI studies was 52% (157). Similarly, BCG-mediated protection against pulmonary tuberculosis varies geographically and appears to be much less effective in areas with high incidence of previous infection with M. tuberculosis or sensitization with environmental Mycobacteria (158, 159). Although many reasons could account for the reduced efficacy of vaccines in disease endemic areas, these results suggest that the early establishment of potent, pathogen-specific immunoregulatory networks may be an important factor contributing to this problem (160). Treatment in the field of cancer has been revolutionized by immune checkpoint blockade strategies. These take advantage of the patients own immune system to recognize and kill cancer cells. Although many of the molecules being targeted by this approach were discovered in infectious diseases research, this approach has not been applied to reducing the burden of infection. Therefore, incorporating inhibitors of specific immune checkpoints into vaccine formulations may be one way to transiently reduce immune suppression to allow the generation of robust vaccine-mediated, antiparasitic immunity.
Both CTLA-4 and PD-1 blockade have been successfully used individually and in combination to treat cancer patients (161). Given that leishmaniasis is a chronic infection and shares several key immunoregulatory features with cancer, one strategy could be to “piggy back” on the success of immune checkpoint blockade drugs in cancer to either improve drug treatment protocols by making subsequent immunity more potent and long-lasting or enhance vaccine efficacy. However, it will be important to bear in mind that specific types and combinations of immune checkpoint blockade work best for particular types of cancer, and this is also likely to be the case with the spectrum of diseases caused by Leishmania species. Thus, careful consideration will need to be given to types of immune checkpoint blockade best suited to VL, CL, MCL, PKDL, or DCL because they are likely to differ in their outcomes.
Antimonial chemotherapy was the mainstay for VL treatment for many decades (162). However, parasite resistance against these drugs has developed, especially in the Indian subcontinent (163, 164). Therefore, these drugs are now mainly employed to treat VL in Africa, while drugs such as Amphotericin B, Ambisome, Miltefosine, and paromomycin have been introduced to treat VL in areas of antimonial drug resistance (164, 165). However, these drugs are not without problems, such as toxicity, high cost, potential development of parasite drug resistance, and prolonged treatment regimes [reviewed in Ref. (165)]. Recently, a single dose of Ambisome (lipid formulation of Amphotericin B) was found to be sufficient to successfully treat VL with low toxicity and has now been recommended as a choice of treatment in India subcontinent (166, 167). The oral drug miltefosin has also been used in combination with Amphotericin B. However, based on studies in preclinical models of leishmaniasis, there are concerns that even with combination therapy, drug resistance will develop (168). Further, these drugs do not cause sterile cure and parasites persist in the infected individuals after drug treatment (168–170). This is concerning because these persisting parasites may help promote transmission, with people living in the same household being most at risk of infection (171, 172). Thus, not only should successful cure of disease be a goal of treatment, but lowering the burden of persisting parasites as far as possible is also desired if parasite transmission is to be minimized. However, when considering these goals, it will be important to remember that persistent parasites are also required to maintain concomitant immunity (170), so sterile cure of infected individuals may not necessarily be beneficial. Instead, it may be necessary to establish the level of parasite burden that is low enough to prevent parasite transmission, but at the same time, maintain concomitant immunity, and then try and achieve this through a combination of antiparasitic drug and immunomodulatory strategies.
Drug treatment works most effectively in association with the host immune system, and in particular, cell-mediated immune responses (164). Hence, understanding immunological changes during the course of infection and how these might be modulated to work best with drug is important. The use of biological molecules to stimulate cell-mediated immunity to help achieve therapeutic success has been tested in both preclinical and clinical studies on leishmaniasis (173). In an experimental model of leishmaniasis, treatment with recombinant IL-12 or anti-IL-10 receptor monoclonal antibody, along with pentavalent antimony (Sbv), resulted in improved clearance of L. donovani parasites, compared with animals treated with drug alone (27, 174). In addition, human recombinant IFN-γ has been successfully used to accelerate antiparasitic and clinical responses when used with antimony treatment, and importantly, treat seriously ill VL patients with refractory disease (175–178). Many drugs used to treat VL not only kill parasites, but also promote host immunity. For example, antimonial drugs stimulate the generation of ROS and NO, while miltefosine and Ambisome induce the secretion of IFN-γ, TNF, IL-12, IL-6, and IL-1β from immune cells with a simultaneous decrease in anti-inflammatory cytokine production (179–182). Thus, combining immune-based therapy with conventional antiparasitic drugs is an obvious strategy to improve current treatment protocols.
Rather than supplement or block immune effector molecules, another approach to use host-directed therapy to improve drug treatment is to target intracellular signal transduction pathways. For example, it has been shown that L. infantum infection rapidly induces activation of phosphatidylinositol 3-kinase/Akt and extracellular signal-regulated kinase1/2 in bone marrow-derived dendritic cells (BMDDC), thereby limiting their maturation and pro-inflammatory cytokine secretion. The blockade of this pathway with wortmannin resulted in reduced infection rates of BMDDC (183). Similarly, the rapid activation of protein tyrosine phosphatase, such as SHP-1, by Leishmania is another important parasite evasion strategy, and administration of the SHP-1 inhibitor bpV-phen to mice infected with L. major and L. donovani promoted control of infection via induction of reactive nitrogen intermediates that would otherwise be repressed by parasite-activated SHP-1 (184, 185). Hence, small molecule inhibitors of key cell signaling pathways is another potential approach that could be used with current antiparasitic drug treatment protocols.
Leishmaniasis is generally a disease associated with poverty (186) and as such, diagnosis, treatment, and hospitalization costs are an important consideration in disease control programs. In addition, drug development programs for leishmaniasis are often not a high priority for pharmaceutical companies. Furthermore, even if effective, high-cost drugs or vaccines are available, they are unlikely to be used without significant government or philanthropic subsidization. Hence, a practical challenge is to supply relatively cheap drugs in resource poor settings, and this will require the participation of regulatory bodies, as well as public and private sector partnerships. A successful example of this was the implementation of the wider use of Ambisome for VL treatment. This is normally a high-cost drug, but has been substantially reduced in cost for the distribution through the public sector agencies in developing countries, by an agreement negotiated between WHO and the manufacturer (187). However, other issues can complicate such arrangements. For example, the requirement of a reliable cold chain for Ambisome implementation can result in a failure to provide the drug or the use of drug stored under conditions not consistent with storage advice. Therefore, the introduction of new treatment regimes for diseases such as VL will need to consider multiple aspects of drug development, formulation, storage, and delivery. One approach for reducing development costs is to repurpose drugs already licensed for other indications, as was the case with Ambisome, which was first licensed as an anti-fungal drug. Another way of reducing cost is to develop cheaper small molecules rather than more expensive biologics to target parasites or host responses. In addition, as well as usual safety considerations, the stability of drugs in areas of unreliable cold-chain must be considered, as well as ease of manufacturing. Therefore, when considering strategies to promote host directed therapy, regardless whether this be targeting specific immune check points or stimulating microbicidal mechanisms, small molecules are likely to be more cost-effective than antibodies.
Leishmaniasis has clear, unmet medical needs. These differ, depending on the disease in question. However, the host immune response to infection is a central component of each of these diseases, whether it is immune dysfunction in the case of VL and DCL or immune-mediated tissue pathology in the case of severe CL and MCL. Therefore, targeting these host responses, as is increasingly occurring in other chronic diseases, such as cancer and autoimmunity, offers promising new opportunities to either improve the efficacy of vaccine candidates or drug treatment protocols. Given the long time lines for vaccine development, the latter approach may have a greater impact in the short term. Furthermore, combining host-directed therapy with antiparasitic drug, offers the added advantage of further reducing parasite loads in treated individuals and improving long-term protective immunity. These outcomes will greatly benefit current disease elimination programs.
RK, SC, SN, SS, and CE wrote the paper. SN and SC prepared the figures and table.
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank staff in the Immunology and Infection laboratory at QIMR Berghofer and in Professor Shyam Sundar’s research group at Banaras Hindu University for helpful discussions.
This work was made possible through Queensland State Government funding. The research was supported by grants and fellowships from the National Health and Medical Research Council of Australia, a NIAID Tropical Medicine Research Centre grant (2U19AI074321), as well as an Australian Post-graduate Awards through Griffith University and an INSPIRE Fellowship to Rajiv Kumar provided by the Indian government Department of Science and Technology.
1. Pearson RD, Sousa AQ. Clinical spectrum of leishmaniasis. Clin Infect Dis (1996) 22:1–13. doi:10.1086/516284
2. Alvar J, Vélez ID, Bern C, Herrero M, Desjeux P, Cano J, et al. Leishmaniasis worldwide and global estimates of its incidence. PLoS One (2012) 7:e35671. doi:10.1371/journal.pone.0035671
3. Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, Rw P, et al. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat Rev Microbiol (2007) 5:873–82. doi:10.1038/nrmicro1748
4. Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis (2004) 27:305–18. doi:10.1016/j.cimid.2004.03.004
6. Al-Salem W, Herricks JR, Hotez PJ. A review of visceral leishmaniasis during the conflict in South Sudan and the consequences for East African countries. Parasit Vectors (2016) 9:460. doi:10.1186/s13071-016-1743-7
7. Ganguly S, Das NK, Barbhuiya JN, Chatterjee M. Post-kala-azar dermal leishmaniasis – an overview. Int J Dermatol (2010) 49:921–31. doi:10.1111/j.1365-4632.2010.04558.x
8. Zijlstra EE, Musa AM, Khalil EAG, El Hassan IM, El-Hassan AM. Post-kala-azar dermal leishmaniasis. Lancet Infect Dis (2003) 3:87–98. doi:10.1016/S1473-3099(03)00517-6
9. Ramesh V, Mukherjee A. Post-kala-azar dermal leishmaniasis. Int J Dermatol (1995) 34:85–91. doi:10.1111/j.1365-4362.1995.tb03584.x
10. Reithinger R, Dujardin JC, Louzir H, Pirmez C, Alexander B, Brooker S. Cutaneous leishmaniasis. Lancet Infect Dis (2007) 7:581–96. doi:10.1016/S1473-3099(07)70209-8
11. Rosas LE, Snider HM, Barbi J, Satoskar AA, Lugo-Villarino G, Keiser T, et al. Cutting edge: STAT1 and T-bet play distinct roles in determining outcome of visceral leishmaniasis caused by Leishmania donovani. J Immunol (2006) 177:22–5. doi:10.4049/jimmunol.177.1.22
12. Ansari NA, Saluja S, Salotra P. Elevated levels of interferon-γ, interleukin-10, and interleukin-6 during active disease in Indian kala azar. Clin Immunol (2006) 119:339–45. doi:10.1016/j.clim.2006.01.017
13. Caldas A, Favali C, Aquino D, Vinhas V, van Weyenbergh J, Brodskyn C, et al. Balance of IL-10 and interferon-γ plasma levels in human visceral leishmaniasis: implications in the pathogenesis. BMC Infect Dis (2005) 5:113. doi:10.1186/1471-2334-5-113
14. Karp CL, El-Safi SH, Wynn TA, Satti MMH, Kordofani AM, Hashim FA, et al. In vivo cytokine profiles in patients with kala-azar. Marked elevation of both interleukin-10 and interferon-gamma. J Clin Invest (1993) 91:1644–8. doi:10.1172/JCI116372
15. Nylen S, Maurya R, Eidsmo L, Manandhar KD, Sundar S, Sacks D. Splenic accumulation of IL-10 mRNA in T cells distinct from CD4+CD25+ (Foxp3) regulatory T cells in human visceral leishmaniasis. J Exp Med (2007) 204:805–17. doi:10.1084/jem.20061141
16. Kumar R, Singh N, Gautam S, Singh OP, Gidwani K, Rai M, et al. Leishmania specific CD4 T cells release IFNγ that limits parasite replication in patients with visceral leishmaniasis. PLoS Negl Trop Dis (2014) 8:e3198. doi:10.1371/journal.pntd.0003198
17. Ghalib HW, Piuvezam MR, Skeiky YA, Siddig M, Hashim FA, el-Hassan AM, et al. Interleukin 10 production correlates with pathology in human Leishmania donovani infections. J Clin Invest (1993) 92:324. doi:10.1172/JCI116570
18. Holaday BJ, Pompeu MM, Jeronimo S, Texeira MJ, de Sousa A, Vasconcelos AW, et al. Potential role for interleukin-10 in the immunosuppression associated with kala azar. J Clin Invest (1993) 92:2626. doi:10.1172/JCI116878
19. Stäger S, Maroof A, Zubairi S, Sanos SL, Kopf M, Kaye PM. Distinct roles for IL-6 and IL-12p40 in mediating protection against Leishmania donovani and the expansion of IL-10+ CD4+ T cells. Eur J Immunol (2006) 36:1764–71. doi:10.1002/eji.200635937
20. Anderson CF, Oukka M, Kuchroo VJ, Sacks D. CD4+ CD25− Foxp3− Th1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. J Exp Med (2007) 204:285–97. doi:10.1084/jem.20061886
21. Engwerda CR, Ng SS, Bunn PT. The regulation of CD4+ T cell responses during protozoan infections. Front Immunol (2014) 5:498. doi:10.3389/fimmu.2014.00498
22. Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol (2010) 10:170–81. doi:10.1038/nri2711
23. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 cells and the counter-regulation of immunity: natural mechanisms and therapeutic applications. Curr Top Microbiol Immunol (2014) 380:39–68. doi:10.1007/978-3-662-43492-5_3
24. Gautam S, Kumar R, Maurya R, Nylén S, Ansari N, Rai M, et al. IL-10 neutralization promotes parasite clearance in splenic aspirate cells from patients with visceral leishmaniasis. J Infect Dis (2011) 204:1134–7. doi:10.1093/infdis/jir461
25. Nylen S, Sacks D. Interleukin-10 and the pathogenesis of human visceral leishmaniasis. Trends Immunol (2007) 28:378–84. doi:10.1016/j.it.2007.07.004
26. Murphy ML, Wille U, Villegas EN, Hunter CA, Farrell JP. IL-10 mediates susceptibility to Leishmania donovani infection. Eur J Immunol (2001) 31:2848–56. doi:10.1002/1521-4141(2001010)31:10<2848:AID-IMMU2848>3.0.CO;2-T
27. Murray HW, Flanders KC, Donaldson DD, Sypek JP, Gotwals PJ, Liu J, et al. Antagonizing deactivating cytokines to enhance host defense and chemotherapy in experimental visceral leishmaniasis. Infect Immun (2005) 73:3903–11. doi:10.1128/IAI.73.7.3903-3911.2005
28. Ansari NA, Kumar R, Gautam S, Nylén S, Singh OP, Sundar S, et al. IL-27 and IL-21 are associated with T cell IL-10 responses in human visceral leishmaniasis. J Immunol (2011) 186:3977–85. doi:10.4049/jimmunol.1003588
29. Ato M, Stäger S, Engwerda CR, Kaye PM. Defective CCR7 expression on dendritic cells contributes to the development of visceral leishmaniasis. Nat Immunol (2002) 3:1185–91. doi:10.1038/ni861
30. Ghalib HW, Whittle JA, Kubin M, Hashim FA, el-Hassan AM, Grabstein KH, et al. IL-12 enhances Th1-type responses in human Leishmania donovani infections. J Immunol (1995) 154:4623–9.
31. Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MBA. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med (2006) 12:1301–9. doi:10.1038/nm1492
32. Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, Crotty S, et al. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med (2006) 203:2461–72. doi:10.1084/jem.20061462
33. Gomes NA, Gattass CR, Barreto-de-Souza V, Wilson ME, DosReis GA. TGF-β mediates CTLA-4 suppression of cellular immunity in murine kalaazar. J Immunol (2000) 164:2001–8. doi:10.4049/jimmunol.164.4.2001
34. Murphy ML, Engwerda CR, Gorak PM, Kaye PM. B7-2 blockade enhances T cell responses to Leishmania donovani. J Immunol (1997) 159:4460–6.
35. Murphy ML, Cotterell SEJ, Gorak PMA, Engwerda CR, Kaye PM. Blockade of CTLA-4 enhances host resistance to the intracellular pathogen, Leishmania donovani. J Immunol (1998) 161:4153–60.
36. Murray HW, Lu CM, Brooks EB, Fichtl RE, DeVecchio JL, Heinzel FP. Modulation of T-cell costimulation as immunotherapy or immunochemotherapy in experimental visceral leishmaniasis. Infect Immun (2003) 71:6453–62. doi:10.1128/IAI.71.11.6453-6462.2003
37. Zubairi S, Sanos SL, Hill S, Kaye PM. Immunotherapy with OX40L-Fc or anti-CTLA-4 enhances local tissue responses and killing of Leishmania donovani. Eur J Immunol (2004) 34:1433–40. doi:10.1002/eji.200324021
38. Esch KJ, Juelsgaard R, Martinez PA, Jones DE, Petersen CA. Programmed death 1-mediated T cell exhaustion during visceral leishmaniasis impairs phagocyte function. J Immunol (2013) 191:5542–50. doi:10.4049/jimmunol.1301810
39. Medina-Colorado AA, Osorio EY, Saldarriaga OA, Travi BL, Kong F, Spratt H, et al. Splenic CD4+ T cells in progressive visceral leishmaniasis show a mixed effector-regulatory phenotype and impair macrophage effector function through inhibitory receptor expression. PLoS One (2017) 12:e0169496. doi:10.1371/journal.pone.0169496
40. Chiku VM, Silva KLO, de Almeida BFM, Venturin GL, Leal AAC, de Martini CC, et al. PD-1 function in apoptosis of T lymphocytes in canine visceral leishmaniasis. Immunobiology (2016) 221:879–88. doi:10.1016/j.imbio.2016.03.007
41. Faleiro RJ, Kumar R, Bunn PT, Singh N, Chauhan SB, Sheel M, et al. Combined immune therapy for the treatment of visceral leishmaniasis. PLoS Negl Trop Dis (2016) 10:e0004415. doi:10.1371/journal.pntd.0004415
42. Belkaid Y, Hoffmann KF, Mendez S, Kamhawi S, Udey MC, Wynn TA, et al. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J Exp Med (2001) 194:1497–506. doi:10.1084/jem.194.10.1497
43. Kane MM, Mosser DM. The role of IL-10 in promoting disease progression in leishmaniasis. J Immunol (2001) 166:1141–7. doi:10.4049/jimmunol.166.2.1141
44. Castellano LR, Argiro L, Dessein H, Dessein A, da Silva MV, Correia D, et al. Potential use of interleukin-10 blockade as a therapeutic strategy in human cutaneous leishmaniasis. J Immunol Res (2015) 2015:152741. doi:10.1155/2015/152741
45. Salhi A, Rodrigues V Jr, Santoro F, Dessein H, Romano A, Castellano LR, et al. Immunological and genetic evidence for a crucial role of IL-10 in cutaneous lesions in humans infected with Leishmania braziliensis. J Immunol (2008) 180:6139–48. doi:10.4049/jimmunol.180.9.6139
46. Joshi T, Rodriguez S, Perovic V, Cockburn IA, Stäger S. B7-H1 blockade increases survival of dysfunctional CD8+ T cells and confers protection against Leishmania donovani infections. PLoS Pathog (2009) 5:e1000431. doi:10.1371/journal.ppat.1000431
47. Bankoti R, Stäger S. Differential regulation of the immune response in the spleen and liver of mice infected with Leishmania donovani. J Trop Med (2012) 2012:639304. doi:10.1155/2012/639304
48. Roy S, Gupta P, Palit S, Basu M, Ukil A, Das PK. The role of PD-1 in regulation of macrophage apoptosis and its subversion by Leishmania donovani. Clin Transl Immunology (2017) 6:e137. doi:10.1038/cti.2017.12
49. Liang SC, Greenwald RJ, Latchman YE, Rosas L, Satoskar A, Freeman GJ, et al. PD-L1 and PD-L2 have distinct roles in regulating host immunity to cutaneous leishmaniasis. Eur J Immunol (2006) 36:58–64. doi:10.1002/eji.200535458
50. Hernández-Ruiz J, Salaiza-Suazo N, Carrada G, Escoto S, Ruiz-Remigio A, Rosenstein Y, et al. CD8 cells of patients with diffuse cutaneous leishmaniasis display functional exhaustion: the latter is reversed, in vitro, by TLR2 agonists. PLoS Negl Trop Dis (2010) 4:e871. doi:10.1371/journal.pntd.0000871
51. Gautam S, Kumar R, Singh N, Singh AK, Rai M, Sacks D, et al. CD8 T cell exhaustion in human visceral leishmaniasis. J Infect Dis (2013) 209:290–9. doi:10.1093/infdis/jit401
52. Gomes NA, Barreto-de-Souza V, Wilson ME, DosReis GA. Unresponsive CD4+ T lymphocytes from Leishmania chagasi-infected mice increase cytokine production and mediate parasite killing after blockade of B7-1/CTLA-4 molecular pathway. J Infect Dis (1998) 178:1847–51. doi:10.1086/314520
53. Ishii N, Ndhlovu LC, Murata K, Sato T, Kamanaka M, Sugamura K. OX40 (CD134) and OX40 ligand interaction plays an adjuvant role during in vivo Th2 responses. Eur J Immunol (2003) 33:2372–81. doi:10.1002/eji.200324031
54. Akiba H, Miyahira Y, Atsuta M, Takeda K, Nohara C, Futagawa T, et al. Critical contribution of OX40 ligand to T helper cell type 2 differentiation in experimental leishmaniasis. J Exp Med (2000) 191:375–80. doi:10.1084/jem.191.2.375
55. Martin S, Agarwal R, Murugaiyan G, Saha B. CD40 expression levels modulate regulatory T cells in Leishmania donovani infection. J Immunol (2010) 185:551–9. doi:10.4049/jimmunol.0902206
56. Mathur RK, Awasthi A, Wadhone P, Ramanamurthy B, Saha B. Reciprocal CD40 signals through p38MAPK and ERK-1/2 induce counteracting immune responses. Nat Med (2004) 10:540–5. doi:10.1038/nm1045
57. Murray HW. Prevention of relapse after chemotherapy in a chronic intracellular infection: mechanisms in experimental visceral leishmaniasis. J Immunol (2005) 174:4916–23. doi:10.4049/jimmunol.174.8.4916
58. Brown JA, Titus RG, Nabavi N, Glimcher LH. Blockade of CD86 ameliorates Leishmania major infection by down-regulating the Th2 response. J Infect Dis (1996) 174:1303–8. doi:10.1093/infdis/174.6.1303
59. Elloso MM, Scott P. Expression and contribution of B7-1 (CD80) and B7-2 (CD86) in the early immune response to Leishmania major infection. J Immunol (1999) 162:6708–15.
60. Corry DB, Reiner SL, Linsley PS, Locksley RM. Differential effects of blockade of CD28-B7 on the development of Th1 or Th2 effector cells in experimental leishmaniasis. J Immunol (1994) 153:4142–8.
61. Brown JA, Greenwald RJ, Scott S, Schweitzer AN, Satoskar AR, Chung C, et al. T helper differentiation in resistant and susceptible B7-deficient mice infected with Leishmania major. Eur J Immunol (2002) 32:1764–72. doi:10.1002/1521-4141(200206)32:6<1764::AID-IMMU1764>3.0.CO;2-V
62. Greenwald RJ, McAdam AJ, Van der Woude D, Satoskar AR, Sharpe AH. Cutting edge: inducible costimulator protein regulates both Th1 and Th2 responses to cutaneous leishmaniasis. J Immunol (2002) 168:991–5. doi:10.4049/jimmunol.168.3.991
63. Stumhofer JS, Hunter CA. Advances in understanding the anti-inflammatory properties of IL-27. Immunol Lett (2008) 117:123–30. doi:10.1016/j.imlet.2008.01.011
64. Yoshida H, Yoshiyuki M, Hiroki Yoshida MY. Regulation of immune responses by interleukin-27. Immunol Rev (2008) 226:234–47. doi:10.1111/j.1600-065X.2008.00710.x
65. Spolski R, Kim H-P, Zhu W, Levy DE, Leonard WJ. IL-21 mediates suppressive effects via its induction of IL-10. J Immunol (2009) 182:2859–67. doi:10.4049/jimmunol.0802978
66. Pot C, Jin H, Awasthi A, Liu SM, Lai C-Y, Madan R, et al. Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J Immunol (2009) 183:797–801. doi:10.4049/jimmunol.0901233
67. Rosas LE, Satoskar AA, Roth KM, Keiser TL, Barbi J, Hunter C, et al. Interleukin-27R (WSX-1/T-cell cytokine receptor) gene-deficient mice display enhanced resistance to Leishmania donovani infection but develop severe liver immunopathology. Am J Pathol (2006) 168:158–69. doi:10.2353/ajpath.2006.050013
68. Murugaiyan G, Mittal A, Lopez-Diego R, Maier LM, Anderson DE, Weiner HL. IL-27 is a key regulator of IL-10 and IL-17 production by human CD4+ T cells. J Immunol (2009) 183:2435–43. doi:10.4049/jimmunol.0900568
69. Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, et al. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol (2007) 8:1363–71. doi:10.1038/ni1537
70. Pitta MGR, Romano A, Cabantous S, Henri S, Hammad A, Kouriba B, et al. IL-17 and IL-22 are associated with protection against human kala azar caused by Leishmania donovani. J Clin Invest (2009) 119:2379. doi:10.1172/JCI38813
71. Sheel M, Beattie L, Frame TCM, de Labastida Rivera F, Faleiro RJ, Bunn PT, et al. IL-17A-producing γδ T cells suppress early control of parasite growth by monocytes in the liver. J Immunol (2015) 195:5707–17. doi:10.4049/jimmunol.1501046
72. Nascimento MSL, Carregaro V, Lima-Júnior DS, Costa DL, Ryffel B, Duthie MS, et al. Interleukin 17A acts synergistically with interferon γ to promote protection against Leishmania infantum infection. J Infect Dis (2014) 211:1015–26. doi:10.1093/infdis/jiu531
73. Plitas G, Rudensky AY. Regulatory T cells: differentiation and function. Cancer Immunol Res (2016) 4:721–5. doi:10.1158/2326-6066.CIR-16-0193
74. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res (2017) 27:109–18. doi:10.1038/cr.2016.151
75. Coombes JL, Robinson NJ, Maloy KJ, Uhlig HH, Powrie F. Regulatory T cells and intestinal homeostasis. Immunol Rev (2005) 204:184–94. doi:10.1111/j.0105-2896.2005.00250.x
76. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155:1151–64.
77. Belkaid Y. Regulatory T cells and infection: a dangerous necessity. Nat Rev Immunol (2007) 7:875. doi:10.1038/nri2189
78. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+ CD25+ Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8:1353. doi:10.1038/ni1536
79. Maurya R, Kumar R, Prajapati VK, Manandhar KD, Sacks D, Sundar S, et al. Human visceral leishmaniasis is not associated with expansion or accumulation of Foxp3+ CD4 cells in blood or spleen. Parasite Immunol (2010) 32:479–83. doi:10.1111/j.1365-3024.2010.01219.x
80. Rai AK, Thakur CP, Singh A, Seth T, Srivastava SK, Singh P, et al. Regulatory T cells suppress T cell activation at the pathologic site of human visceral leishmaniasis. PLoS One (2012) 7:e31551. doi:10.1371/journal.pone.0031551
81. Bhattacharya P, Ghosh S, Ejazi SA, Rahaman M, Pandey K, Das VNR, et al. Induction of IL-10 and TGFβ from CD4+ CD25+ FoxP3+ T cells correlates with parasite load in Indian kala-azar patients infected with Leishmania donovani. PLoS Negl Trop Dis (2016) 10:e0004422. doi:10.1371/journal.pntd.0004422
82. Rodrigues OR, Marques C, Soares-Clemente M, Ferronha MH, Santos-Gomes GM. Identification of regulatory T cells during experimental Leishmania infantum infection. Immunobiology (2009) 214:101–11. doi:10.1016/j.imbio.2008.07.001
83. Somanna A, Mundodi V, Gedamu L. Functional analysis of cathepsin B-like cysteine proteases from Leishmania donovani complex. Evidence for the activation of latent transforming growth factor β. J Biol Chem (2002) 277:25305–12. doi:10.1074/jbc.M203034200
84. Boutard V, Havouis R, Fouqueray B, Philippe C, Moulinoux J-P, Baud L. Transforming growth factor-beta stimulates arginase activity in macrophages. Implications for the regulation of macrophage cytotoxicity. J Immunol (1995) 155:2077–84.
85. Iniesta V, Carlos Gómez-Nieto L, Molano I, Mohedano A, Carcelen J, Miron C, et al. Arginase I induction in macrophages, triggered by Th2-type cytokines, supports the growth of intracellular Leishmania parasites. Parasite Immunol (2002) 24:113–8. doi:10.1046/j.1365-3024.2002.00444.x
86. Boonstra A, Rajsbaum R, Holman M, Marques R, Asselin-Paturel C, Pereira JP, et al. Macrophages and myeloid dendritic cells, but not plasmacytoid dendritic cells, produce IL-10 in response to MyD88-and TRIF-dependent TLR signals, and TLR-independent signals. J Immunol (2006) 177:7551–8. doi:10.4049/jimmunol.177.11.7551
87. Owens BMJ, Beattie L, Moore JWJ, Brown N, Mann JL, Dalton JE, et al. IL-10-producing Th1 cells and disease progression are regulated by distinct CD11c+ cell populations during visceral leishmaniasis. PLoS Pathog (2012) 8:e1002827. doi:10.1371/journal.ppat.1002827
88. Svensson M, Maroof A, Ato M, Kaye PM. Stromal cells direct local differentiation of regulatory dendritic cells. Immunity (2004) 21:805–16. doi:10.1016/j.immuni.2004.10.012
89. Wakkach A, Fournier N, Brun V, Breittmayer J-P, Cottrez F, Groux H. Characterization of dendritic cells that induce tolerance and T regulatory 1 cell differentiation in vivo. Immunity (2003) 18:605–17. doi:10.1016/S1074-7613(03)00113-4
90. Colmenares M, Kima PE, Samoff E, Soong L, McMahon-Pratt D. Perforin and gamma interferon are critical CD8+ T-cell-mediated responses in vaccine-induced immunity against Leishmania amazonensis infection. Infect Immun (2003) 71:3172–82. doi:10.1128/IAI.71.6.3172-3182.2003
91. Stenger S, Hanson DA, Teitelbaum R, Dewan P, Niazi KR, Froelich CJ, et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science (1998) 282:121–5. doi:10.1126/science.282.5386.121
92. Ansari NA, Ramesh V, Salotra P. Interferon (IFN)-γ, tumor necrosis factor-α, interleukin-6, and IFN-γ receptor 1 are the major immunological determinants associated with post-kala azar dermal leishmaniasis. J Infect Dis (2006) 194:958–65. doi:10.1086/506624
93. Ansari NA, Katara GK, Ramesh V, Salotra P. Evidence for involvement of TNFR1 and TIMPs in pathogenesis of post-kala-azar dermal leishmaniasis. Clin Exp Immunol (2008) 154:391–8. doi:10.1111/j.1365-2249.2008.03761.x
94. Mohamed HS, Ibrahim ME, Miller EN, Peacock CS, Khalil EAG, Cordell HJ, et al. Genetic susceptibility to visceral leishmaniasis in The Sudan: linkage and association with IL4 and IFNGR1. Genes Immun (2003) 4:351. doi:10.1038/sj.gene.6363977
95. Salih MA, Ibrahim ME, Blackwell JM, Miller EN, Khalil EAG, ElHassan AM, et al. IFNG and IFNGR1 gene polymorphisms and susceptibility to post kala-azar dermal leishmaniasis in Sudan. Genes Immun (2007) 8:75. doi:10.1038/sj.gene.6364353
96. Katara GK, Ansari NA, Verma S, Ramesh V, Salotra P. Foxp3 and IL-10 expression correlates with parasite burden in lesional tissues of post kala azar dermal leishmaniasis (PKDL) patients. PLoS Negl Trop Dis (2011) 5:e1171. doi:10.1371/journal.pntd.0001171
97. Ismail A, El Hassan AM, Kemp K, Gasim S, Kadaru AEGMY, Møller T, et al. Immunopathology of post kala-azar dermal leishmaniasis (PKDL): T-cell phenotypes and cytokine profile. J Pathol (1999) 189:615–22. doi:10.1002/(SICI)1096-9896(199912)189:4<615::AID-PATH466>3.0.CO;2-Z
98. Gasim S, Elhassan AM, Khalil EAG, Ismail A, Kadaru AMY, Kharazmi A, et al. High levels of plasma IL-10 and expression of IL-10 by keratinocytes during visceral leishmaniasis predict subsequent development of post-kala-azar dermal leishmaniasis. Clin Exp Immunol (1998) 111:64–9. doi:10.1046/j.1365-2249.1998.00468.x
99. Ganguly S, Das NK, Panja M, Pal S, Modak D, Rahaman M, et al. Increased levels of interleukin-10 and IgG3 are hallmarks of Indian post-kala-azar dermal leishmaniasis. J Infect Dis (2008) 197:1762–71. doi:10.1086/588387
100. Ganguly S, Mukhopadhyay D, Das NK, Chaduvula M, Sadhu S, Chatterjee U, et al. Enhanced lesional Foxp3 expression and peripheral anergic lymphocytes indicate a role for regulatory T cells in Indian post-kala-azar dermal leishmaniasis. J Invest Dermatol (2010) 130:1013–22. doi:10.1038/jid.2009.393
101. Katara GK, Ansari NA, Singh A, Ramesh V, Salotra P. Evidence for involvement of Th17 type responses in post kala azar dermal leishmaniasis (PKDL). PLoS Negl Trop Dis (2012) 6:e1703. doi:10.1371/journal.pntd.0001703
102. Scott P, Novais FO. Cutaneous leishmaniasis: immune responses in protection and pathogenesis. Nat Rev Immunol (2016) 16:581–92. doi:10.1038/nri.2016.72
103. Mosmann TR, Coffman RL. Two types of mouse helper T-cell clone: implications for immune regulation. Immunol Today (1987) 8:223–7. doi:10.1016/0167-5699(87)90171-X
104. Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, et al. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science (2008) 321:970–4. doi:10.1126/science.1159194
105. Ribeiro-Gomes FL, Peters NC, Debrabant A, Sacks DL. Efficient capture of infected neutrophils by dendritic cells in the skin inhibits the early anti-Leishmania response. PLoS Pathog (2012) 8:e1002536. doi:10.1371/journal.ppat.1002536
106. Romano A, Carneiro MBH, Doria NA, Roma EH, Ribeiro-Gomes FL, Inbar E, et al. Divergent roles for Ly6C+ CCR2+ CX3CR1+ inflammatory monocytes during primary or secondary infection of the skin with the intra-phagosomal pathogen Leishmania major. PLoS Pathog (2017) 13:e1006479. doi:10.1371/journal.ppat.1006479
107. Charmoy M, Brunner-Agten S, Aebischer D, Auderset F, Launois P, Milon G, et al. Neutrophil-derived CCL3 is essential for the rapid recruitment of dendritic cells to the site of Leishmania major inoculation in resistant mice. PLoS Pathog (2010) 6:e1000755. doi:10.1371/journal.ppat.1000755
108. van Gisbergen KPJM, Sanchez-Hernandez M, Geijtenbeek TBH, van Kooyk Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J Exp Med (2005) 201:1281–92. doi:10.1084/jem.20041276
109. Goncalves R, Zhang X, Cohen H, Debrabant A, Mosser DM. Platelet activation attracts a subpopulation of effector monocytes to sites of Leishmania major infection. J Exp Med (2011) 208:1253–65. doi:10.1084/jem.20101751
110. León B, López-Bravo M, Ardavín C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity (2007) 26:519–31. doi:10.1016/j.immuni.2007.01.017
111. León B, Ardavín C. Monocyte-derived dendritic cells in innate and adaptive immunity. Immunol Cell Biol (2008) 86:320. doi:10.1038/icb.2008.14
112. De Trez C, Magez S, Akira S, Ryffel B, Carlier Y, Muraille E. iNOS-producing inflammatory dendritic cells constitute the major infected cell type during the chronic Leishmania major infection phase of C57BL/6 resistant mice. PLoS Pathog (2009) 5:e1000494. doi:10.1371/journal.ppat.1000494
113. Scharton TM, Scott P. Natural killer cells are a source of interferon gamma that drives differentiation of CD4+ T cell subsets and induces early resistance to Leishmania major in mice. J Exp Med (1993) 178:567–77. doi:10.1084/jem.178.2.567
114. Maroof A, Beattie L, Zubairi S, Svensson M, Stager S, Kaye PM. Posttranscriptional regulation of il10 gene expression allows natural killer cells to express immunoregulatory function. Immunity (2008) 29:295–305. doi:10.1016/j.immuni.2008.06.012
115. Uzonna JE, Joyce KL, Scott P. Low dose Leishmania major promotes a transient T helper cell type 2 response that is down-regulated by interferon γ-producing CD8+ T cells. J Exp Med (2004) 199:1559–66. doi:10.1084/jem.20040172
116. Belkaid Y, Von Stebut E, Mendez S, Lira R, Caler E, Bertholet S, et al. CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J Immunol (2002) 168:3992–4000. doi:10.4049/jimmunol.168.8.3992
117. Okwor IB, Jia P, Mou Z, Onyilagha C, Uzonna JE. CD8+ T cells are preferentially activated during primary low dose Leishmania major infection but are completely dispensable during secondary anti-Leishmania immunity. PLoS Negl Trop Dis (2014) 8:e3300. doi:10.1371/journal.pntd.0003300
118. Novais FO, Scott P. CD8+ T cells in cutaneous leishmaniasis: the good, the bad, and the ugly. Semin Immunopathol (2015) 37:251–9. doi:10.1007/s00281-015-0475-7
119. Reiner SL, Locksley RM. The regulation of immunity to Leishmania major. Annu Rev Immunol (1995) 13:151–77. doi:10.1146/annurev.iy.13.040195.001055
120. Castellano LR, Correia Filho D, Argiro L, Dessein H, Prata A, Dessein A, et al. Th1/Th2 immune responses are associated with active cutaneous leishmaniasis and clinical cure is associated with strong interferon-γ production. Hum Immunol (2009) 70:383–90. doi:10.1016/j.humimm.2009.01.007
121. Sacks D, Noben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol (2002) 2:845. doi:10.1038/nri933
122. Belkald Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+ CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature (2002) 420:502. doi:10.1038/nature01152
123. Peters NC, Pagán AJ, Lawyer PG, Hand TW, Roma EH, Stamper LW, et al. Chronic parasitic infection maintains high frequencies of short-lived Ly6C+ CD4+ effector T cells that are required for protection against re-infection. PLoS Pathog (2014) 10:e1004538. doi:10.1371/journal.ppat.1004538
124. Uzonna JE, Wei G, Yurkowski D, Bretscher P. Immune elimination of Leishmania major in mice: implications for immune memory, vaccination, and reactivation disease. J Immunol (2001) 167:6967–74. doi:10.4049/jimmunol.167.12.6967
125. Zaph C, Uzonna J, Beverley SM, Scott P. Central memory T cells mediate long-term immunity to Leishmania major in the absence of persistent parasites. Nat Med (2004) 10:1104. doi:10.1038/nm1108
126. Pakpour N, Zaph C, Scott P. The central memory CD4+ T cell population generated during Leishmania major infection requires IL-12 to produce IFN-γ. J Immunol (2008) 180:8299–305. doi:10.4049/jimmunol.180.12.8299
127. Colpitts SL, Scott P. The early generation of a heterogeneous CD4+ T cell response to Leishmania major. J Immunol (2010) 185:2416–23. doi:10.4049/jimmunol.1000483
128. Glennie ND, Volk SW, Scott P. Skin-resident CD4+ T cells protect against Leishmania major by recruiting and activating inflammatory monocytes. PLoS Pathog (2017) 13:e1006349. doi:10.1371/journal.ppat.1006349
129. Charmoy M, Hurrell BP, Romano A, Lee SH, Ribeiro-Gomes F, Riteau N, et al. The Nlrp3 inflammasome, IL-1β, and neutrophil recruitment are required for susceptibility to a nonhealing strain of Leishmania major in C57BL/6 mice. Eur J Immunol (2016) 46:897–911. doi:10.1002/eji.201546015
130. Gurung P, Karki R, Vogel P, Watanabe M, Bix M, Lamkanfi M, et al. An NLRP3 inflammasome-triggered Th2-biased adaptive immune response promotes leishmaniasis. J Clin Invest (2015) 125:1329. doi:10.1172/JCI79526
131. Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva ALN, Mineo TWP, et al. Inflammasome-derived IL-1 [beta] production induces nitric oxide-mediated resistance to Leishmania. Nat Med (2013) 19:909–15. doi:10.1038/nm.3221
132. Bacellar O, Lessa H, Schriefer A, Machado P, de Jesus AR, Dutra WO, et al. Up-regulation of Th1-type responses in mucosal leishmaniasis patients. Infect Immun (2002) 70:6734–40. doi:10.1128/IAI.70.12.6734-6740.2002
133. BARRAL A, Jesus AR, Almeida RP, Carvalho EM, Barral-Netto M, Costa JML, et al. Evaluation of T-cell subsets in the lesion infiltrates of human cutaneous and mucocutaneous leishmaniasis. Parasite Immunol (1987) 9:487–97. doi:10.1111/j.1365-3024.1987.tb00525.x
134. Faria DR, Gollob KJ, Barbosa J, Schriefer A, Machado PRL, Lessa H, et al. Decreased in situ expression of interleukin-10 receptor is correlated with the exacerbated inflammatory and cytotoxic responses observed in mucosal leishmaniasis. Infect Immun (2005) 73:7853–9. doi:10.1128/IAI.73.12.7853-7859.2005
135. Pirmez C, Yamamura M, Uyemura K, Paes-Oliveira M, Conceição-Silva F, Modlin RL. Cytokine patterns in the pathogenesis of human leishmaniasis. J Clin Invest (1993) 91:1390. doi:10.1172/JCI116341
136. Faria DR, Souza PEA, Duraes FV, Carvalho EM, Gollob KJ, Machado PR, et al. Recruitment of CD8+ T cells expressing granzyme A is associated with lesion progression in human cutaneous leishmaniasis. Parasite Immunol (2009) 31:432–9. doi:10.1111/j.1365-3024.2009.01125.x
137. Novais FO, Carvalho LP, Graff JW, Beiting DP, Ruthel G, Roos DS, et al. Cytotoxic T cells mediate pathology and metastasis in cutaneous leishmaniasis. PLoS Pathog (2013) 9:e1003504. doi:10.1371/journal.ppat.1003504
138. Novais FO, Carvalho AM, Clark ML, Carvalho LP, Beiting DP, Brodsky IE, et al. CD8+ T cell cytotoxicity mediates pathology in the skin by inflammasome activation and IL-1β production. PLoS Pathog (2017) 13:e1006196. doi:10.1371/journal.ppat.1006196
139. Boaventura VS, Santos CS, Cardoso CR, de Andrade J, Dos Santos WLC, Clarêncio J, et al. Human mucosal leishmaniasis: neutrophils infiltrate areas of tissue damage that express high levels of Th17-related cytokines. Eur J Immunol (2010) 40:2830–6. doi:10.1002/eji.200940115
140. Convit J, Kerdel-Vegas F, Gordon B. Disseminated anergic cutaneous leishmaniasis. Br J Dermatol (1962) 74:132–5. doi:10.1111/j.1365-2133.1962.tb13475.x
141. Hashiguchi Y, Gomez EL, Kato H, Martini LR, Velez LN, Uezato H. Diffuse and disseminated cutaneous leishmaniasis: clinical cases experienced in Ecuador and a brief review. Trop Med Health (2016) 44:2. doi:10.1186/s41182-016-0002-0
142. Petersen EA, Neva FA, Oster CN, Diaz HB. Specific inhibition of lymphocyte-proliferation responses by adherent suppressor cells in diffuse cutaneous leishmaniasis. N Engl J Med (1982) 306:387–92. doi:10.1056/NEJM198202183060702
143. Bomfim G, Nascimento C, Costa J, Carvalho EM, Barral-Netto M, Barral A. Variation of cytokine patterns related to therapeutic response in diffuse cutaneous leishmaniasis. Exp Parasitol (1996) 84:188–94. doi:10.1006/expr.1996.0104
144. Scorza BM, Carvalho EM, Wilson ME. Cutaneous manifestations of human and murine leishmaniasis. Int J Mol Sci (2017) 18:1296. doi:10.3390/ijms18061296
145. Franke ED, Lucas CM, Tovar AA, Kruger JH, De Rivera MVS, Wignall FS. Diffuse cutaneous leishmaniasis acquired in Peru. Am J Trop Med Hyg (1990) 43:260–2. doi:10.4269/ajtmh.1990.43.260
146. Convit J, Castellanos PL, Ulrich M, Castés M, Rondón A, Pinardi ME, et al. Immunotherapy of localized, intermediate, and diffuse forms of American cutaneous leishmaniasis. J Infect Dis (1989) 160:104–15. doi:10.1093/infdis/160.1.104
147. Badaró R, Johnson WD Jr. The role of interferon-γ in the treatment of visceral and diffuse cutaneous leishmaniasis. J Infect Dis (1993) 167:S13–7. doi:10.1093/infdis/167.Supplement_1.S13
148. Fernández-Figueroa EA, Rangel-Escareño C, Espinosa-Mateos V, Carrillo-Sánchez K, Salaiza-Suazo N, Carrada-Figueroa G, et al. Disease severity in patients infected with Leishmania mexicana relates to IL-1β. PLoS Negl Trop Dis (2012) 6:e1533. doi:10.1371/journal.pntd.0001533
149. Kellina OI. Changes in virulence of strains of Leishmania trópica major. Meditsinskaya Parazitol i Parazit Bolezn (1965) 34:701–8.
150. Nadim A, Javadian E, Tahvildar-Bidruni G, Ghorbani M. Effectiveness of leishmanization in the control of cutaneous leishmaniasis. Bull Soc Pathol Exot Filiales (1983) 76:377–83.
151. Romano A, Doria NA, Mendez J, Sacks DL, Peters NC. Cutaneous infection with Leishmania major mediates heterologous protection against visceral infection with Leishmania infantum. J Immunol (2015) 195:3816–27. doi:10.4049/jimmunol.1500752
152. Zijlstra EE, El-Hassan AM, Ismael A, Ghalib HW. Endemic kala-azar in eastern Sudan: a longitudinal study on the incidence of clinical and subclinical infection and post-kala-azar dermal leishmaniasis. Am J Trop Med Hyg (1994) 51:826–36. doi:10.4269/ajtmh.1994.51.826
153. Khalil EAG, Zijlstra EE, Kager PA, El Hassan AM. Epidemiology and clinical manifestations of Leishmania donovani infection in two villages in an endemic area in eastern Sudan. Trop Med Int Health (2002) 7:35–44. doi:10.1046/j.1365-3156.2002.00832.x
154. Ranasinghe S, Zhang W-W, Wickremasinghe R, Abeygunasekera P, Chandrasekharan V, Athauda S, et al. Leishmania donovani zymodeme MON-37 isolated from an autochthonous visceral leishmaniasis patient in Sri Lanka. Pathog Glob Health (2012) 106:421–4. doi:10.1179/2047773212Y.0000000054
155. de Oca MM, Kumar R, de Labastida Rivera F, Amante FH, Sheel M, Faleiro RJ, et al. Correction: blimp-1-dependent IL-10 production by Tr1 cells regulates TNF-mediated tissue pathology. PLoS Pathog (2016) 12:e1005460. doi:10.1371/journal.ppat.1005460
156. Rts S. Efficacy and safety of RTS, S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial. Lancet (2015) 386:31–45. doi:10.1016/S0140-6736(15)60721-8
157. Ockenhouse CF, Regules J, Tosh D, Cowden J, Kathcart A, Cummings J, et al. Ad35. CS. 01-RTS, S/AS01 heterologous prime boost vaccine efficacy against sporozoite challenge in healthy malaria-naive adults. PLoS One (2015) 10:e0131571. doi:10.1371/journal.pone.0131571
158. Fine PEM. Variation in protection by BCG: implications of and for heterologous immunity. Lancet (1995) 346:1339–45. doi:10.1016/S0140-6736(95)92348-9
159. Nguipdop-Djomo P, Heldal E, Rodrigues LC, Abubakar I, Mangtani P. Duration of BCG protection against tuberculosis and change in effectiveness with time since vaccination in Norway: a retrospective population-based cohort study. Lancet Infect Dis (2016) 16:219–26. doi:10.1016/S1473-3099(15)00400-4
160. Mendonça VRR, Queiroz ATL, Lopes FM, Andrade BB, Barral-Netto M. Networking the host immune response in Plasmodium vivax malaria. Malar J (2013) 12:69. doi:10.1186/1475-2875-12-69
161. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252. doi:10.1038/nrc3239
162. Haldar AK, Sen P, Roy S. Use of antimony in the treatment of leishmaniasis: current status and future directions. Mol Biol Int (2011) 2011:571242. doi:10.4061/2011/571242
163. Stauch A, Duerr H-P, Dujardin J-C, Vanaerschot M, Sundar S, Eichner M. Treatment of visceral leishmaniasis: model-based analyses on the spread of antimony-resistant L. donovani in Bihar, India. PLoS Negl Trop Dis (2012) 6:e1973. doi:10.1371/journal.pntd.0001973
164. Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clin Microbiol Rev (2006) 19:111–26. doi:10.1128/CMR.19.1.111-126.2006
165. Singh OP, Singh B, Chakravarty J, Sundar S. Current challenges in treatment options for visceral leishmaniasis in India: a public health perspective. Infect Dis Poverty (2016) 5:19. doi:10.1186/s40249-016-0112-2
166. Sundar S, Sinha PK, Rai M, Verma DK, Nawin K, Alam S, et al. Comparison of short-course multidrug treatment with standard therapy for visceral leishmaniasis in India: an open-label, non-inferiority, randomised controlled trial. Lancet (2011) 377:477–86. doi:10.1016/S0140-6736(10)62050-8
167. Sundar S, Chakravarty J. Leishmaniasis: an update of current pharmacotherapy. Expert Opin Pharmacother (2013) 14:53–63. doi:10.1517/14656566.2013.755515
168. García-Hernández R, Manzano JI, Castanys S, Gamarro F. Leishmania donovani develops resistance to drug combinations. PLoS Negl Trop Dis (2012) 6:e1974. doi:10.1371/journal.pntd.0001974
169. Gupta G, Oghumu S, Satoskar AR. Mechanisms of immune evasion in leishmaniasis. Adv Appl Microbiol (2013) 82:155. doi:10.1016/B978-0-12-407679-2.00005-3
170. Yazdanbakhsh M, Sacks DL. Why does immunity to parasites take so long to develop? Nat Rev Immunol (2010) 10:80. doi:10.1038/nri2673
171. Singh OP, Hasker E, Sacks D, Boelaert M, Sundar S. Asymptomatic Leishmania infection: a new challenge for Leishmania control. Clin Infect Dis (2014) 58:1424–9. doi:10.1093/cid/ciu102
172. Singh OP, Hasker E, Boelaert M, Sundar S. Elimination of visceral leishmaniasis on the Indian subcontinent. Lancet Infect Dis (2016) 16:e304–9. doi:10.1016/S1473-3099(16)30140-2
173. Singh OP, Sundar S. Immunotherapy and targeted therapies in treatment of visceral leishmaniasis: current status and future prospects. Front Immunol (2014) 5:296. doi:10.3389/fimmu.2014.00296
174. Murray HW, Montelibano C, Peterson R, Sypek JP. Interleukin-12 regulates the response to chemotherapy in experimental visceral leishmaniasis. J Infect Dis (2000) 182:1497–502. doi:10.1086/315890
175. Sundar S, Rosenkaimer F, Lesser ML, Murray HW. Immunochemotherapy for a systemic intracellular infection: accelerated response using interferon-γ in visceral leishmaniasis. J Infect Dis (1995) 171:992–6. doi:10.1093/infdis/171.4.992
176. Sundar S, Murray HW. Effect of treatment with interferon-γ alone in visceral leishmaniasis. J Infect Dis (1995) 172:1627–9. doi:10.1093/infdis/172.6.1627
177. Badaro R, Falcoff E, Badaro FS, Carvalho EM, Pedral-Sampaio D, Barral A, et al. Treatment of visceral leishmaniasis with pentavalent antimony and interferon gamma. N Engl J Med (1990) 322:16–21. doi:10.1056/NEJM199001043220104
178. Squires KE, Rosenkaimer F, Sherwood JA, Forni AL, Were JBO, Murray HW. Immunochemotherapy for visceral leishmaniasis: a controlled pilot trial of antimony versus antimony plus interferon-gamma. Am J Trop Med Hyg (1993) 48:666–9. doi:10.4269/ajtmh.1993.48.666
179. Mukhopadhyay D, Das NK, Roy S, Kundu S, Barbhuiya JN, Chatterjee M. Miltefosine effectively modulates the cytokine milieu in Indian post kala-azar dermal leishmaniasis. J Infect Dis (2011) 204:1427–36. doi:10.1093/infdis/jir551
180. Tokuda Y, Tsuji M, Yamazaki M, Kimura S, Abe S, Yamaguchi H. Augmentation of murine tumor necrosis factor production by amphotericin B in vitro and in vivo. Antimicrob Agents Chemother (1993) 37:2228–30. doi:10.1128/AAC.37.10.2228
181. Ghezzi MC. The release of TNF-α and IL-6 from human monocytes stimulated by filtrates of Candida albicans after treatment with amphotericin B. J Antimicrob Chemother (1994) 33:1039–43. doi:10.1093/jac/33.5.1039
182. Wadhone P, Maiti M, Agarwal R, Kamat V, Martin S, Saha B. Miltefosine promotes IFN-γ-dominated anti-leishmanial immune response. J Immunol (2009) 182:7146–54. doi:10.4049/jimmunol.0803859
183. Neves BM, Silvestre R, Resende M, Ouaissi A, Cunha J, Tavares J, et al. Activation of phosphatidylinositol 3-kinase/akt and impairment of nuclear factor-κB: molecular mechanisms behind the arrested maturation/activation state of Leishmania infantum-infected dendritic cells. Am J Pathol (2010) 177:2898–911. doi:10.2353/ajpath.2010.100367
184. Olivier M, Romero-Gallo B-J, Matte C, Blanchette J, Posner BI, Tremblay MJ, et al. Modulation of interferon-γ-induced macrophage activation by phosphotyrosine phosphatases inhibition. Effect on murine leishmaniasis progression. J Biol Chem (1998) 273:13944–9. doi:10.1074/jbc.273.22.13944
185. Matte C, Marquis J-F, Blanchette J, Gros P, Faure R, Posner BI, et al. Peroxovanadium-mediated protection against murine leishmaniasis: role of the modulation of nitric oxide. Eur J Immunol (2000) 30:2555–64. doi:10.1002/1521-4141(200009)30:9<2555::AID-IMMU2555>3.0.CO;2-X
186. Boelaert M, Meheus F, Sanchez A, Singh SP, Vanlerberghe V, Picado A, et al. The poorest of the poor: a poverty appraisal of households affected by visceral leishmaniasis in Bihar, India. Trop Med Int Health (2009) 14:639–44. doi:10.1111/j.1365-3156.2009.02279.x
Keywords: leishmaniasis, immunoregulation, immunotherapy, pathology, cell-mediated immunity
Citation: Kumar R, Chauhan SB, Ng SS, Sundar S and Engwerda CR (2017) Immune Checkpoint Targets for Host-Directed Therapy to Prevent and Treat Leishmaniasis. Front. Immunol. 8:1492. doi: 10.3389/fimmu.2017.01492
Received: 08 September 2017; Accepted: 23 October 2017;
Published: 08 November 2017
Edited by:
Heinrich Korner, University of Tasmania, AustraliaReviewed by:
Gerald Spaeth, Institut Pasteur, FranceCopyright: © 2017 Kumar, Chauhan, Ng, Sundar and Engwerda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajiv Kumar, rajiv082@yahoo.com;
Christian R. Engwerda, chrisE@qimr.edu.au
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.