 Kehmia Titanji
Kehmia Titanji- Division of Endocrinology, Metabolism and Lipids, Department of Medicine, Emory University School of Medicine, Atlanta, GA, United States
HIV infection leads to severe B cell dysfunction, which manifests as impaired humoral immune response to infection and vaccinations and is not completely reversed by otherwise effective antiretroviral therapy (ART). Despite its inability to correct HIV-induced B cell dysfunction, ART has led to significantly increased lifespans in people living with HIV/AIDS. This has in turn led to escalating prevalence of non-AIDS complications in aging HIV-infected individuals, including malignancies, cardiovascular disease, bone disease, and other end-organ damage. These complications, typically associated with aging, are a significant cause of morbidity and mortality and occur significantly earlier in HIV-infected individuals. Understanding the pathophysiology of these comorbidities and delineating clinical management strategies and potential cures is gaining in importance. Bone loss and osteoporosis, which lead to increase in fragility fracture prevalence, have in recent years emerged as important non-AIDS comorbidities in patients with chronic HIV infection. Interestingly, ART exacerbates bone loss, particularly within the first couple of years following initiation. The mechanisms underlying HIV-induced bone loss are multifactorial and complicated by the fact that HIV infection is linked to multiple risk factors for osteoporosis and fracture, but a very interesting role for B cells in HIV-induced bone loss has recently emerged. Although best known for their important antibody-producing capabilities, B cells also produce two cytokines critical for bone metabolism: the key osteoclastogenic cytokine receptor activator of NF-κB ligand (RANKL) and its physiological inhibitor osteoprotegerin (OPG). Dysregulated B cell production of OPG and RANKL was shown to be a major contributor to increased bone loss and fracture risk in animal models and HIV-infected humans. This review will summarize our current knowledge of the role of the OPG/RANK–RANKL pathway in B cells in health and disease, and the contribution of B cells to HIV-induced bone loss. Data from mouse studies indicate that RANKL and OPG may also play a role in B cell function and the implications of these findings for human B cell biology, as well as therapeutic strategies targeting the OPG/RANK–RANKL pathway, will be discussed.
Introduction
Rising incidences of bone loss in the form of low bone mineral density (BMD), osteopenia, and osteoporosis, and the resulting increased risk of fracture have over the past decade emerged as important non-AIDS comorbidities affecting HIV-infected individuals (1–6). Successful antiretroviral therapy (ART) over the past couple of decades has been instrumental in significantly extending the life expectancies of HIV-infected individuals to levels comparable to those of the general population (7). A significant proportion of people currently living with HIV in Europe and North America are over the age of 50 (8–10), and it is estimated that by 2030 as many as >70% of HIV-positive individuals will fall within this demographic. Similar to cardiovascular, liver and chronic kidney disease, and other comorbidities, bone loss occurs earlier and at a higher prevalence in HIV-positive individuals than in the HIV negative population (1, 8, 11). This raises concerns of a potential impending epidemic of fragility fractures and other age-associated comorbidities in this population (8, 12).
The underlying mechanisms of HIV-associated bone loss are multifactorial, given that most of the traditional risk factors for bone loss including low body mass index (BMI), older age, tobacco use, metabolic diseases, alcohol, and substance abuse are more prevalent in the HIV-infected population (10, 13). HIV infection is now however clearly established as one of the independent risk factors for bone loss (11, 14, 15), driven by the prevalence of HIV-associated risk factors including chronic inflammation, co-infection with hepatitis B or C, and paradoxically, ART (8, 10, 13). More recently, osteoimmunology has revealed the prominent role the immune system plays in bone metabolism (16) and consequently revealed that HIV-induced immune dysfunction is one of the most important contributors to bone loss.
Osteoimmunology, a term originally coined to describe studies involving the interface between the immune and skeletal systems (17), has been instrumental in our understanding of the numerous ways both organ systems are intertwined. It is now known that in various inflammatory pathological conditions characterized by bone loss, including periodontal disease (PD) and rheumatoid arthritis (RA), both cellular and soluble immune effectors can contribute to bone loss (18, 19). T cells are major contributors to bone loss in RA (20) and PD (21, 22) but their role in HIV-induced bone loss has not been elucidated. Emerging evidence now shows that B cells play an important role in bone biology in health and disease (23–25) and HIV-induced B cell dysfunction significantly contributes to HIV-induced bone loss (26).
Bone homeostasis, which is essential for maintaining skeletal integrity and strength, is regulated by a balance of bone formation by osteoblasts and resorption by osteoclasts and disruption of this balance results in bone disease (18, 27, 28). Osteoclasts are generated in a process known as osteoclastogenesis, which is driven by the key osteoclastogenic cytokine receptor activator of NF-κB ligand (RANKL). Osteoclasts originate from cells of the myeloid lineage, which in the presence of M-CSF and RANKL differentiate into receptor activator of NF-κB (RANK)-expressing pre-osteoclasts which proliferate and fuse to form giant multinucleated osteoclasts capable of resorbing bone (15, 29).
Excessive osteoclast activity, as occurs in osteoporosis, results in loss of bone mass and increased susceptibility to fracture (12, 28). The effects of B and T cells on bone are mediated by several key cytokine regulators of bone metabolism (11, 18), including the inflammatory cytokines tumor necrosis factor-α (TNF-α) and interferon-γ, which have been implicated in bone loss in RA, periodontitis, postmenopausal osteoporosis, and HIV (30). Most importantly, RANKL and OPG (18) play important roles in both organ systems and perfectly illustrate the intersection of bone biology and immunity. The OPG/RANK–RANKL pathway also mediates physiological processes in the vascular system, thus intersecting with the skeletal and immune system at this axis (Figure 1).
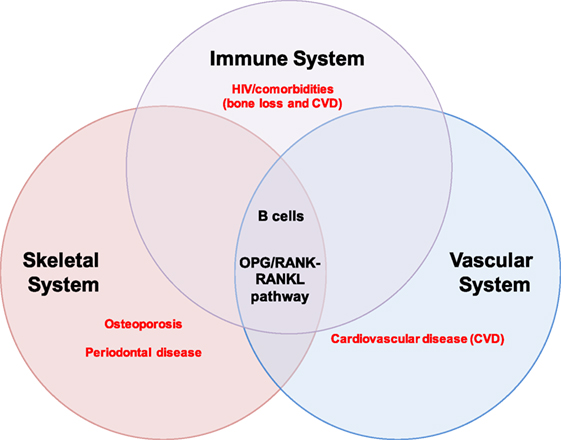
Figure 1. B cells and the OPG/RANK-RANKL pathway at the intersection of the immune, skeletal, and vascular organ systems. B cells mediate biological processes in health and disease via the OPG/RANK–RANKL pathway in three major organ systems in humans: the immune, skeletal, and vascular systems. The extensive intertwining of the immune and skeletal systems has given rise to a whole new field of study called osteoimmunology; some major pathologies implicating B cells and the OPG/RANK–RANKL pathway are highlighted in red and include osteoporosis and periodontal disease in the skeletal system, cardiovascular disease (CVD) in the vascular system, and HIV/comorbidities (bone loss and CVD) in the immune system.
This review will summarize our current knowledge of the role of the OPG/RANK–RANKL pathway in B cells in health and disease, and the contribution of B cells to HIV-induced bone loss. Data from mouse studies indicate that RANKL and OPG may also play a role in B cell function and the implications of these findings for human B cell biology as well as therapeutic strategies targeting the OPG/RANK–RANKL pathway will be discussed.
The OPG/RANK–RANKL Pathway and B Cells in Health
B cells are inextricably linked to bone, from their development in the bone marrow to the homing of terminally differentiated plasma cells back to the bone marrow (30, 31) and the bidirectional regulation of the skeletal system by B cells (23, 30, 32). Osteoblasts and bone marrow stromal cells regulate B lymphopoiesis through the production of IL-7, a critical cytokine for the differentiation of early-stage B cells in the bone marrow (33, 34). Another major interaction between the skeletal system and B cells revolves around the OPG/RANK–RANKL pathway.
B Cells and Osteoprotegerin (OPG)
The identification and characterization of OPG as a humoral regulator of bone resorption 20 years ago (35, 36) represents a major turning point in our understanding of the physiology of bone homeostasis (37, 38). OPG, named for its ability to protect bone by inhibiting osteoclast differentiation and activity, is a tumor necrosis factor receptor (TNFR) superfamily member which lacks transmembrane-spanning sequences and is secreted as a soluble protein (35, 36). OPG is the natural circulating inhibitor/decoy receptor of RANKL and can inhibit osteoclastogenesis by binding to RANKL, thus preventing bone resorption (35, 37). OPG mRNA is expressed by various tissues, including bone, brain, lung, heart, and kidney (35, 36). In the immune system, OPG is expressed in lymph nodes, B cells, and dendritic cells (DCs) and ligation of CD40 upregulates its expression (39).
Osteoblasts and their precursors were previously considered to be the primary source of OPG in the bone marrow (40, 41) but B lineage cells are now known to account for over 60% of total bone marrow OPG production (25). B cell knockout (KO) mice were osteoporotic and deficient in bone marrow OPG, confirming the critical role of B cells in the preservation of bone homeostasis and attainment of peak bone mass (25).
Unlike its role in bone homeostasis, the role of OPG in B cell function is less well documented. OPG KO mice develop severe osteoporosis due to unchecked osteoclastogenesis and bone resorption (42, 43). Interestingly, OPG-deficient mice also accumulated transitional/immature B cells in their spleens, and generated impaired antibody (Ab) responses to a T cell-dependent (DNP-KLH) antigen (Ag) challenge, suggesting that OPG may regulate B cell maturation and development of efficient Ab responses (44).
B Cells and RANKL
The ligand for OPG is identical to a TNFR family member called TNF-related activation-induced cytokine or RANKL (37, 45). Human RANKL exists in two forms: a cellular, membrane-bound form and a soluble form, and both forms were shown to be biologically capable of promoting osteoclast formation (46, 47). RANKL is also expressed in a variety of tissues, including bone marrow and lymphoid tissues (36, 47, 48). RANKL is best known for its indispensable role in the complete differentiation of mature osteoclasts (36, 37, 47). Unlike OPG, resting B cells have not been conclusively shown to produce significant amounts of RANKL, but activated B cells are an important source (23), particularly in inflammatory disease states.
B Cells and RANK
The receptor for RANKL, RANK, was initially identified on DCs (48) and later discovered to be expressed on preosteoclastic cells (37, 46, 49, 50) and B cells (39, 51). The binding of RANKL to RANK stimulates osteoclastogenesis, resulting in bone-resorbing osteoclasts (47).
Lack of functional RANK in both humans and mice results in osteopetrosis due to the absence of osteoclasts (19, 49, 52). Mice deficient in RANK had defects in B cell development which resulted in reduced numbers of mature B cells in the periphery (49). Humans with mutations in RANK also had B cell defects including hypogammaglobulinemia and impaired Ag-specific Ab responses (52).
The OPG/RANK–RANKL Pathway and B Cells in Non-HIV Disease
Osteoprotegerin, RANK, and RANKL are produced by a wide variety of cells and tissues in three major organ systems: the vascular, immune, and skeletal systems and are thus implicated in the pathogenesis of various diseases in these organs (15, 38) (Figure 1). Although best known for its involvement in the pathogenesis of osteoporosis and other bone diseases such as Paget’s disease of bone (53–55) and PD (38, 56), the OPG/RANK–RANKL pathway has also been implicated in other diseases including RA (14, 38, 57) and CVD (58–60).
Rheumatoid Arthritis
The bone and joint destruction that occurs in the autoimmune disorder RA results from increased RANKL-induced osteoclastic bone resorption in the synovial joints (57, 61, 62). Several immune cells have been identified as the sources of RANKL in the arthritic synovium, including Th17 cells (63), macrophages, DCs (57), and activated B cells (64). Targeted B cell depletion therapy for RA using the anti-CD20 Ab rituximab suggests that B cells play a critical role in RA-associated joint damage (64–66). B cells were shown to contribute to RA pathogenesis through their Ag-presenting function, autoantibody production, and cytokine secretion (66, 67). A link between B cells and joint destruction in RA has been confirmed by studies demonstrating that Rituximab significantly reduces RANKL levels in the synovium (68, 69). This link has recently been confirmed by studies identifying pro-inflammatory B cells as major sources of RANKL in RA (64, 66). These findings highlight the importance of Ab-independent (cytokine-producing) B cell functions in the pathogenesis of disease and make a case for the therapeutic potential of targeting the B cell OPG/RANK–RANKL pathway in RA and other diseases.
In contrast to RANKL, multiple studies have demonstrated that serum levels of OPG are elevated in RA, resulting in a decreased RANKL/OPG ratio (70, 71). Elevated OPG levels were independently associated with RA disease severity and CVD, and it has been suggested that OPG concentration could be used as a predictive marker for assessing RA-associated CVD risk (72, 73). Data on the role of B cell-produced OPG in the pathophysiology of RA are however lacking.
Cardiovascular Disease
A role for the OPG/RANK–RANKL pathway in the pathogenesis of vascular calcification and CVDs has been established for over a decade now. Both OPG and RANKL have been detected in atherosclerotic plaques (74) and an increased RANKL/OPG ratio is associated with atherosclerosis (59). Transgenic expression of OPG in OPG KO mice prevented the development of arterial calcification but exogenous OPG administration did not reverse existing calcification, suggesting that similar to bone, OPG is a protective factor in the cardiovascular system (75, 76). Results in human studies however seem to conflict with the animal studies, with higher OPG levels consistently associated with CVD incidence (76, 77). The contribution of B cells to OPG/RANK–RANKL-linked CVD has however not been clearly elucidated. Low-density lipoprotein (LDL) receptor KO mice (LDLR−/−) were B cell deficient and developed atherosclerosis, suggesting that B cells and/or antibodies are protective against atherosclerosis (78); it is conceivable that OPG produced by B cells mediates this protective effect.
Bone Diseases
Osteoporosis
Osteoporosis is characterized by loss of bone mass and mineral density resulting from an excess of bone resorption by osteoclasts relative to bone formation by osteoblasts (18, 27, 28). The role of the OPG/RANK–RANKL pathway in the pathogenesis of osteoporosis has been well documented and extensively reviewed (15, 37, 38, 62); the role of B cells is however still being elucidated.
Postmenopausal osteoporosis, the most common form of osteoporosis, arises from decreased estrogen levels (62) and was shown in both human patients and an animal model to be linked to increased RANKL expression by B cells (79). Mice subjected to ovariectomy, commonly used as an animal model of estrogen deficiency, have increased numbers of B cells, suggesting that B cells may play a role in estrogen-deficiency osteoporosis (79–81). Data on the contribution of B cells to ovariectomy-induced bone loss is however conflicting. Some studies have demonstrated that ovariectomy-induced bone loss occurs independently of mature B cells (82) and others show that ovariectomy-induced bone loss is linked to RANKL expression on immature B cells (79). Given the fact that B cells are able to express RANKL at various stages in their differentiation, this raises the possibility that the contribution of B lineage cells to estrogen-deficiency osteoporosis is dependent on the differentiation/maturation stage of the B cell. Beyond the differentiation stage however, the activation status of B cells seems to be a better indicator of their ability to produce bone-damaging RANKL (23). This is especially relevant in the context of inflammatory diseases like RA, PD, and HIV-induced bone loss.
Periodontal Disease
Periodontal diseases are inherited or acquired disorders affecting the supporting structures of the teeth and affect as many as 50–90% of the world’s population (83). The underlying microbial infections were traditionally the focus of majority of the research on the pathogenesis of PDs but in recent years the focus has shifted to the role of the host response/factors in pathogenesis (83, 84). Host immune/inflammatory responses are critical for pathogenesis and inflammation (84) and the term PD generally refers to inflammation-induced disorders, ranging from the mildest form (gingivitis) to the more invasive severe periodontitis (83). Unlike gingivitis which is completely reversible by effective regular oral hygiene, periodontitis extends deeper into the tissue and can result in the permanent loss of the supporting structures of the teeth and alveolar bone (83).
One of the microorganisms most commonly implicated in PD pathogenesis is Actinobacillus actinomycetemcomitans (Aa), which induces RANKL expression on a variety of cell types infiltrating in PD lesions (84). While the RANKL levels in PD lesions are consistently elevated in most clinical studies, some studies found lower (22) or unchanged (24) OPG levels in lesions, which both resulted in higher RANKL/OPG ratios in periodontitis compared to healthy controls (22, 24, 84). Activated B and T cells were shown to be the primary source of RANKL in gingival tissues from individuals with periodontitis (24, 85). B cell percentages in chronic PD lesions were associated with disease severity, suggesting that B cells promote PD (86) and interestingly, PD lesion-infiltrating B cells in humans were activated transitional CD5+ cells (86, 87). Using a rat model, it was also demonstrated that B cells contributed to osteoclast formation and periodontal bone loss by secreting RANKL following activation by Aa in a T cell-independent manner (85).
B Cells, The OPG/RANK–RANKL Pathways, and HIV-Induced Bone Loss
With the availability of ever-improving treatment regimens, ART is enabling HIV-infected individuals to live longer than ever before, but life expectancies of patients remain lower than those of the general population (7, 12, 15, 16, 59). Over 33% of people currently living with HIV in Europe are >50 years of age and this percentage is expected to increase to >70% by 2030 (88); in the US, the same demographic is estimated to constitute up to 50% of the HIV-positive population (89). This increased longevity is however accompanied by earlier occurrence and higher prevalence of several non-AIDS end-organ comorbidities including cardiovascular and bone diseases (90–92), which in turn imposes significant disease burdens on the patients, healthcare systems, and society.
As discussed above, under inflammatory conditions, B cells produce higher amounts of RANKL, leading to an increased RANKL/OPG ratio, which drives disease progression (26, 29) in inflammatory diseases such as RA. HIV infection is associated with persistent inflammation (93) and the success of B cell-targeted/depleting therapies in reducing inflammation in autoimmune disorders such as RA suggest that B cells may contribute to persistent inflammation (94, 95). Given the pivotal role this pathway plays in osteoclastogenesis and bone loss, its role in B cells and HIV is perfectly illustrated by its contribution to inflammation-driven HIV-induced bone loss.
A hallmark of chronic HIV infection is the altered distribution of subsets in the B cell compartment (93), notably the loss of resting memory B cells (26) and the expansion of exhausted/tissue-like memory B cells (26, 93, 96). Interestingly, OPG expression was lowest in the HIV-expanded tissue-like memory B cell subset, which conversely showed higher RANKL expression (26) (Figure 2). This tissue-like memory B cell subset was also previously shown to express the inhibitory receptor FcRL4 (96), which in RA defined a pro-inflammatory RANKL-producing B cells subset (66). Taken together, this suggests that inflammation does drive B cell subset RANKL expression in HIV infection.
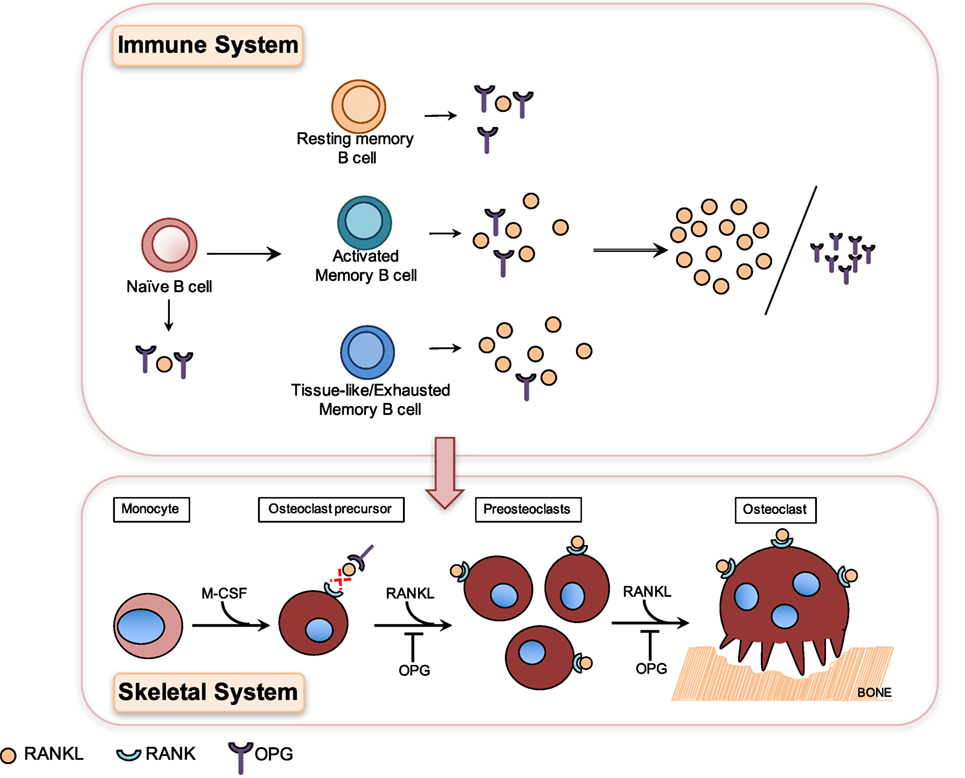
Figure 2. Differential production of osteoprotegerin (OPG) and receptor-activator of NF-κB ligand (RANKL) by B cell subsets results in higher RANKL/OPG ratio, which contributes to HIV-induced osteoclastogenesis and bone loss. Osteoclasts are generated in a process known as osteoclastogenesis, which is driven by the key osteoclastogenic cytokine RANKL. Osteoclasts originate from cells of the myeloid lineage, which in the presence of M-CSF and RANKL differentiate into receptor-activator of NF-κB (RANK)-expressing pre-osteoclasts, which proliferate and fuse to form giant multinucleated osteoclasts capable of resorbing bone. HIV infection leads to the depletion of resting memory B cells and expansion of activated B cell subsets including activated memory and tissue-like memory B cells. Resting memory B cells produce the highest amounts of OPG and tissue-like memory B cells conversely the lowest amounts of OPG and the highest amounts of RANKL (26). HIV-induced B cell subset changes therefore translate into higher RANKL/OPG ratios, which contribute to increased osteoclastogenesis and bone loss in HIV-infected patients.
Low BMD increases the risk of fragility fractures and is widely prevalent in HIV-infected individuals, with as many as 67% presenting with osteopenia and ~15% with osteoporosis (91). Increased osteopenia and osteoporosis rates translate into significantly elevated fracture risk, and studies show that HIV-infected individuals do indeed suffer more fragility fractures, at younger ages, than the general population (1). The ubiquitous presence of traditional risk factors for low BMD such as increased smoking and low BMI in most HIV-infected cohorts complicates efforts to understand and elucidate the mechanisms underlying HIV-induced bone loss (11, 26, 97). HIV infection in itself is now recognized as a risk factor for bone loss (97).
HIV transgenic rats almost perfectly mimic the clinical hallmarks of human HIV-induced bone disease, including profound skeletal damage. Bone loss in this model was driven by increased B cell RANKL expression concurrent with decreased OPG expression, which in turn resulted in increased RANKL/OPG ratio and thus osteoclastogenesis and bone loss (98). This mechanism of HIV-induced B cell dysfunction-driven bone loss was later confirmed in a clinical study of untreated HIV-infected individuals where it was demonstrated that increased B cell RANKL/OPG was indeed associated with increased bone resorption (26). This demonstrated for the first time that the OPG/RANK–RANKL pathway is indeed a key pathway utilized by B cells to effect skeletal damage in HIV infection. This demonstrates clearly how HIV-induced B cell changes in the immune system translate directly into dysfunction and bone loss in the skeletal system (Figure 2).
Regulatory Effects of the OPG/RANK–RANKL Pathway on B Cells and Humoral Immune Responses
Due to the expression of OPG, RANK, and RANKL on a wide variety of immune cell types, the pathway is thought to play an important role in immune cell biology. Despite the involvement of B cell-expressed OPG and RANKL in the normal function of the immune, skeletal, and vascular systems and in the pathogenesis of multiple diseases, the effect of these molecules on B cell physiology has not been extensively described.
Receptor-activator of NF-κB ligand plays an important role in the development of secondary lymphoid organs. RANK- and RANKL-deficient mice had poorly developed or completely lacked secondary lymphoid tissues including lymph nodes, Peyer’s patches, cryptopatches, and spleen (46, 49, 62).
The role of this pathway in B cell function has also been investigated in a few mouse studies. OPG-deficient mice accumulated transitional/immature B cells in their spleens and generated impaired Ab responses to a T cell-dependent (DNP-KLH) Ag challenge, suggesting that OPG may regulate B cell maturation and development of efficient Ab responses (44). Conversely, B cell development was impaired in RANKL-deficient mice, suggesting that OPG regulates B cell development.
In another study (99), OPG was used to treat mice induced to develop different types of cellular and humoral immune responses through: (1) infection with Mycobacterium bovis Bacillus Calmette and Guerin (BCG) followed by OPG-Fc treatment, (2) immunization with KLH in Freund’s adjuvant or by i.p. injection of a Pneumococcal Vaccine Polyvalent (Pneumovax®23, Merck) (3) immunization with Keyhole Limpet Hemocyanin (KLH) in vivo followed by OPG-Fc treatment, and (4) In a bid to induce contact hypersensitivity, mice were also sensitized with the hapten oxazolone, followed by treatment with OPG-Fc. T and B cells were also exposed to OPG in vitro. OPG treatment did not affect cell-mediated responses including contact hypersensitivity but increased humoral immune responses to KLH and the pneumococcal vaccine. In vitro, OPG modestly stimulated T cells but not the proliferation of B cells. These results demonstrated that OPG has modest regulatory effects on humoral immune responses to certain Ags. The potential impact of the OPG/RANK–RANKL on the generation of human humoral immune responses is not clear and definitely merits further study.
Therapeutic Strategies Targeting the OPG/RANK–RANKL Pathway
Although initially described in the context of bone disease, the OPG/RANK–RANKL pathway is now known to influence normal physiology and pathology in the immune, skeletal, and vascular systems. This opens up the potential for a lot of cross application of potential therapeutic strategies targeting this pathway.
One such strategy involves RANKL inhibition; E. coli-derived Fc-OPG showed great promise in phase I trials, causing rapid decline in bone turnover markers in postmenopausal women (100), also serving as a proof of concept that RANKL blockade could meaningfully impact bone turnover in humans (46). Perhaps the best known RANKL inhibitor in clinical use to date is denosumab, a fully human IgG2 Ab which binds RANKL with high affinity and unlike Fc-OPG does not bind to mouse and rat RANKL and TRAIL (46). In clinical use, denosumab effectively reduces fracture risk by reducing bone resorption and was shown to be superior to bisphosphonates in its ability to increase BMD in postmenopausal women (46). When used to treat cancer-induced bone disease, denosumab effectively reduced levels of bone turnover markers in patients with solid tumor (breast, prostate, and lung) metastases to bone and prolonged bone metastasis-free survival and delayed the onset of first metastasis in certain prostate cancers (101). Denosumab was also well-tolerated and no significant changes in B cell numbers were noted (102, 103). The effect of denosumab on B cell function is not fully elucidated; in one study investigating its utility as a postmenopausal osteoporosis treatment (104), 2/412 women developed transient non-neutralizing anti-denosumab antibodies, which did not adversely affect the skeleton but did appear to alter the effectiveness of the drug (104). Due to the wide pattern of expression of RANKL, including on lymphocytes, and in the vascular and skeletal systems, RANKL inhibition using denosumab could potentially increase susceptibility to infections and neoplasias (105), particularly in immunocompromised patients. No significant alterations in inflammation and immunity have however been observed in preclinical and clinical studies of denosumab, although rare cases of severe skin infections of the lower extremities were reported (106). To date, no data are available on the use of denosumab in HIV infection and the effect of RANKL blockade on humoral immune responses in HIV-infected individuals remains to be elucidated.
Conclusion
In summary, B cells are intricately intertwined with the OPG/RANK–RANKL pathway, plays important roles in the immune, skeletal, and vascular systems, and much remains to be discovered about the influence of this pathway on human humoral immune responses.
Author Contributions
KT designed, drafted, and revised the manuscript and is accountable for all aspects of the manuscript.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The author acknowledges grant support from the NHLBI (1K01HL131333-01).
References
1. Triant VA, Brown TT, Lee H, Grinspoon SK. Fracture prevalence among human immunodeficiency virus (HIV)-infected versus non-HIV-infected patients in a large U.S. healthcare system. J Clin Endocrinol Metab (2008) 93(9):3499–504. doi:10.1210/jc.2008-0828
2. Womack JA, Goulet JL, Gibert C, Brandt C, Chang CC, Gulanski B, et al. Increased risk of fragility fractures among HIV infected compared to uninfected male veterans. PLoS One (2011) 6(2):e17217. doi:10.1371/journal.pone.0017217
3. Young B, Dao CN, Buchacz K, Baker R, Brooks JT, HIV Outpatient Study (HOPS) Investigators. Increased rates of bone fracture among HIV-infected persons in the HIV Outpatient Study (HOPS) compared with the US general population, 2000-2006. Clin Infect Dis (2011) 52(8):1061–8. doi:10.1093/cid/ciq242
4. Brown TT, Ruppe MD, Kassner R, Kumar P, Kehoe T, Dobs AS, et al. Reduced bone mineral density in human immunodeficiency virus-infected patients and its association with increased central adiposity and postload hyperglycemia. J Clin Endocrinol Metab (2004) 89(3):1200–6. doi:10.1210/jc.2003-031506
5. Bruera D, Luna N, David DO, Bergoglio LM, Zamudio J. Decreased bone mineral density in HIV-infected patients is independent of antiretroviral therapy. AIDS (2003) 17(13):1917–23. doi:10.1097/00002030-200309050-00010
6. Prior J, Burdge D, Maan E, Milner R, Hankins C, Klein M, et al. Fragility fractures and bone mineral density in HIV positive women: a case-control population-based study. Osteoporos Int (2007) 18(10):1345–53. doi:10.1007/s00198-007-0428-7
7. van Sighem AI, Gras LA, Reiss P, Brinkman K, de Wolf F, ATHENA National Observational Cohort Study. Life expectancy of recently diagnosed asymptomatic HIV-infected patients approaches that of uninfected individuals. AIDS (2010) 24(10):1527–35. doi:10.1097/QAD.0b013e32833a3946
8. Cotter AG, Mallon PW. The effects of untreated and treated HIV infection on bone disease. Curr Opin HIV AIDS (2014) 9(1):17–26. doi:10.1097/COH.0000000000000028
9. Warriner AH, Mugavero M, Overton ET. Bone alterations associated with HIV. Curr HIV/AIDS Rep (2014) 11(3):233–40. doi:10.1007/s11904-014-0216-x
10. Compston J. HIV infection and bone disease. J Intern Med (2016) 280(4):350–8. doi:10.1111/joim.12520
11. Schafer JJ, Manlangit K, Squires KE. Bone health and human immunodeficiency virus infection. Pharmacotherapy (2013) 33(6):665–82. doi:10.1002/phar.1257
13. Guaraldi G, Zona S, Brothers TD, Carli F, Stentarelli C, Dolci G, et al. Aging with HIV vs. HIV seroconversion at older age: a diverse population with distinct comorbidity profiles. PLoS One (2015) 10(4):e0118531. doi:10.1371/journal.pone.0118531
14. Takayanagi H. Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol (2009) 5(12):667–76. doi:10.1038/nrrheum.2009.217
15. Weitzmann MN, Ofotokun I. Physiological and pathophysiological bone turnover – role of the immune system. Nat Rev Endocrinol (2016) 12(9):518–32. doi:10.1038/nrendo.2016.91
16. Ofotokun I, McIntosh E, Weitzmann MN. HIV: inflammation and bone. Curr HIV/AIDS Rep (2012) 9(1):16–25. doi:10.1007/s11904-011-0099-z
17. Arron JR, Choi Y. Bone versus immune system. Nature (2000) 408(6812):535–6. doi:10.1038/35046196
18. Rho J, Takami M, Choi Y. Osteoimmunology: interactions of the immune and skeletal systems. Mol Cells (2004) 17(1):1–9.
19. Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature (1999) 402(6759):304–9. doi:10.1038/35005552
20. O’Gradaigh D, Compston JE. T-cell involvement in osteoclast biology: implications for rheumatoid bone erosion. Rheumatology (2004) 43(2):122–30. doi:10.1093/rheumatology/keg447
21. Taubman MA, Kawai T. Involvement of T-lymphocytes in periodontal disease and in direct and indirect induction of bone resorption. Crit Rev Oral Biol Med (2001) 12(2):125–35. doi:10.1177/10454411010120020301
22. Crotti T, Smith MD, Hirsch R, Soukoulis S, Weedon H, Capone M, et al. Receptor activator NF kappaB ligand (RANKL) and osteoprotegerin (OPG) protein expression in periodontitis. J Periodontal Res (2003) 38(4):380–7. doi:10.1034/j.1600-0765.2003.00615.x
23. Horowitz MC, Fretz JA, Lorenzo JA. How B cells influence bone biology in health and disease. Bone (2010) 47(3):472–9. doi:10.1016/j.bone.2010.06.011
24. Kawai T, Matsuyama T, Hosokawa Y, Makihira S, Seki M, Karimbux NY, et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am J Pathol (2006) 169(3):987–98. doi:10.2353/ajpath.2006.060180
25. Li Y, Toraldo G, Li A, Yang X, Zhang H, Qian WP, et al. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood (2007) 109(9):3839–48. doi:10.1182/blood-2006-07-037994
26. Titanji K, Vunnava A, Sheth AN, Delille C, Lennox JL, Sanford SE, et al. Dysregulated B cell expression of RANKL and OPG correlates with loss of bone mineral density in HIV infection. PLoS Pathog (2014) 10(10):e1004497. doi:10.1371/journal.ppat.1004497
27. Tanaka Y, Nakayamada S, Okada Y. Osteoblasts and osteoclasts in bone remodeling and inflammation. Curr Drug Targets Inflamm Allergy (2005) 4(3):325–8. doi:10.2174/1568010054022015
28. Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet (2003) 4(8):638–49. doi:10.1038/nrg1122
29. Weitzmann MN. The role of inflammatory cytokines, the RANKL/OPG axis, and the immunoskeletal interface in physiological bone turnover and osteoporosis. Scientifica (Cairo) (2013) 2013:125705. doi:10.1155/2013/125705
30. Pietschmann P, Mechtcheriakova D, Meshcheryakova A, Foger-Samwald U, Ellinger I. Immunology of osteoporosis: a mini-review. Gerontology (2016) 62(2):128–37. doi:10.1159/000431091
31. Nagasawa T. Microenvironmental niches in the bone marrow required for B-cell development. Nat Rev Immunol (2006) 6(2):107–16. doi:10.1038/nri1780
32. Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol (2001) 19:595–621. doi:10.1146/annurev.immunol.19.1.595
33. Wu JY, Purton LE, Rodda SJ, Chen M, Weinstein LS, McMahon AP, et al. Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proc Natl Acad Sci U S A (2008) 105(44):16976–81. doi:10.1073/pnas.0802898105
34. Namen AE, Lupton S, Hjerrild K, Wignall J, Mochizuki DY, Schmierer A, et al. Stimulation of B-cell progenitors by cloned murine interleukin-7. Nature (1988) 333(6173):571–3. doi:10.1038/333571a0
35. Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell (1997) 89(2):309–19. doi:10.1016/S0092-8674(00)80209-3
36. Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology (1998) 139(3):1329–37. doi:10.1210/endo.139.3.5837
37. Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology (2001) 142(12):5050–5. doi:10.1210/endo.142.12.8536
38. Theoleyre S, Wittrant Y, Tat SK, Fortun Y, Redini F, Heymann D. The molecular triad OPG/RANK/RANKL: involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev (2004) 15(6):457–75. doi:10.1016/j.cytogfr.2004.06.004
39. Yun TJ, Chaudhary PM, Shu GL, Frazer JK, Ewings MK, Schwartz SM, et al. OPG/FDCR-1, a TNF receptor family member, is expressed in lymphoid cells and is up-regulated by ligating CD40. J Immunol (1998) 161(11):6113–21.
40. Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Boyle WJ, Riggs BL. The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res (2000) 15(1):2–12. doi:10.1359/jbmr.2000.15.1.2
41. Rifas L, Arackal S, Weitzmann MN. Inflammatory T cells rapidly induce differentiation of human bone marrow stromal cells into mature osteoblasts. J Cell Biochem (2003) 88(4):650–9. doi:10.1002/jcb.10436
42. Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev (1998) 12(9):1260–8. doi:10.1101/gad.12.9.1260
43. Mizuno A, Amizuka N, Irie K, Murakami A, Fujise N, Kanno T, et al. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun (1998) 247(3):610–5. doi:10.1006/bbrc.1998.8697
44. Yun TJ, Tallquist MD, Aicher A, Rafferty KL, Marshall AJ, Moon JJ, et al. Osteoprotegerin, a crucial regulator of bone metabolism, also regulates B cell development and function. J Immunol (2001) 166(3):1482–91. doi:10.4049/jimmunol.166.3.1482
45. Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem (1997) 272(40):25190–4. doi:10.1074/jbc.272.40.25190
46. Lacey DL, Boyle WJ, Simonet WS, Kostenuik PJ, Dougall WC, Sullivan JK, et al. Bench to bedside: elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat Rev Drug Discov (2012) 11(5):401–19. doi:10.1038/nrd3705
47. Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell (1998) 93(2):165–76. doi:10.1016/S0092-8674(00)81569-X
48. Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature (1997) 390(6656):175–9. doi:10.1038/36593
49. Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, et al. RANK is essential for osteoclast and lymph node development. Genes Dev (1999) 13(18):2412–24. doi:10.1101/gad.13.18.2412
50. Hsu H, Lacey DL, Dunstan CR, Solovyev I, Colombero A, Timms E, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci U S A (1999) 96(7):3540–5. doi:10.1073/pnas.96.7.3540
51. Perlot T, Penninger JM. Development and function of murine B cells lacking RANK. J Immunol (2012) 188(3):1201–5. doi:10.4049/jimmunol.1102063
52. Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A, et al. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet (2008) 83(1):64–76. doi:10.1016/j.ajhg.2008.06.015
53. Hofbauer LC, Schoppet M. Osteoprotegerin deficiency and juvenile Paget’s disease. N Engl J Med (2002) 347(20):1622–1623; author reply1622–1623. doi:10.1056/NEJM200211143472015
54. Menaa C, Reddy SV, Kurihara N, Maeda H, Anderson D, Cundy T, et al. Enhanced RANK ligand expression and responsivity of bone marrow cells in Paget’s disease of bone. J Clin Invest (2000) 105(12):1833–8. doi:10.1172/JCI9133
55. Whyte MP, Obrecht SE, Finnegan PM, Jones JL, Podgornik MN, McAlister WH, et al. Osteoprotegerin deficiency and juvenile Paget’s disease. N Engl J Med (2002) 347(3):175–84. doi:10.1056/NEJMoa013096
56. Taubman MA, Valverde P, Han X, Kawai T. Immune response: the key to bone resorption in periodontal disease. J Periodontol (2005) 76(11 Suppl):2033–41. doi:10.1902/jop.2005.76.11-S.2033
57. Geusens P. The role of RANK ligand/osteoprotegerin in rheumatoid arthritis. Ther Adv Musculoskelet Dis (2012) 4(4):225–33. doi:10.1177/1759720X12438080
58. Kaden JJ, Bickelhaupt S, Grobholz R, Haase KK, Sarikoc A, Kilic R, et al. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulate aortic valve calcification. J Mol Cell Cardiol (2004) 36(1):57–66. doi:10.1016/j.yjmcc.2003.09.015
59. Kelesidis T, Currier JS, Yang OO, Brown TT. Role of RANKL-RANK/osteoprotegerin pathway in cardiovascular and bone disease associated with HIV infection. AIDS Rev (2014) 16(3):123–33.
60. Kiechl S, Schett G, Wenning G, Redlich K, Oberhollenzer M, Mayr A, et al. Osteoprotegerin is a risk factor for progressive atherosclerosis and cardiovascular disease. Circulation (2004) 109(18):2175–80. doi:10.1161/01.CIR.0000127957.43874.BB
61. Takayanagi H. New developments in osteoimmunology. Nat Rev Rheumatol (2012) 8(11):684–9. doi:10.1038/nrrheum.2012.167
62. Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG system in immunity, bone, and beyond. Front Immunol (2014) 5:511. doi:10.3389/fimmu.2014.00511
63. Okamoto K, Takayanagi H. Regulation of bone by the adaptive immune system in arthritis. Arthritis Res Ther (2011) 13(3):219. doi:10.1186/ar3323
64. Yeo L, Toellner KM, Salmon M, Filer A, Buckley CD, Raza K, et al. Cytokine mRNA profiling identifies B cells as a major source of RANKL in rheumatoid arthritis. Ann Rheum Dis (2011) 70(11):2022–8. doi:10.1136/ard.2011.153312
65. Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med (2004) 350(25):2572–81. doi:10.1056/NEJMoa032534
66. Yeo L, Lom H, Juarez M, Snow M, Buckley CD, Filer A, et al. Expression of FcRL4 defines a pro-inflammatory, RANKL-producing B cell subset in rheumatoid arthritis. Ann Rheum Dis (2015) 74(5):928–35. doi:10.1136/annrheumdis-2013-204116
67. Anolik JH, Looney RJ, Lund FE, Randall TD, Sanz I. Insights into the heterogeneity of human B cells: diverse functions, roles in autoimmunity, and use as therapeutic targets. Immunol Res (2009) 45(2–3):144–58. doi:10.1007/s12026-009-8096-7
68. Boumans MJ, Thurlings RM, Yeo L, Scheel-Toellner D, Vos K, Gerlag DM, et al. Rituximab abrogates joint destruction in rheumatoid arthritis by inhibiting osteoclastogenesis. Ann Rheum Dis (2012) 71(1):108–13. doi:10.1136/annrheumdis-2011-200198
69. Tak PP, Rigby WF, Rubbert-Roth A, Peterfy CG, van Vollenhoven RF, Stohl W, et al. Inhibition of joint damage and improved clinical outcomes with rituximab plus methotrexate in early active rheumatoid arthritis: the IMAGE trial. Ann Rheum Dis (2011) 70(1):39–46. doi:10.1136/ard.2010.137703
70. Wang P, Li S, Liu LN, Lv TT, Li XM, Li XP, et al. Circulating osteoprotegerin levels are elevated in rheumatoid arthritis: a systematic review and meta-analysis. Clin Rheumatol (2017) 36(10):2193–200. doi:10.1007/s10067-017-3747-x
71. Remuzgo-Martinez S, Genre F, Lopez-Mejias R, Ubilla B, Mijares V, Pina T, et al. Expression of osteoprotegerin and its ligands, RANKL and TRAIL, in rheumatoid arthritis. Sci Rep (2016) 6:29713. doi:10.1038/srep29713
72. Dessein PH, Lopez-Mejias R, Gonzalez-Juanatey C, Genre F, Miranda-Filloy JA, Llorca J, et al. Independent relationship of osteoprotegerin concentrations with endothelial activation and carotid atherosclerosis in patients with severe rheumatoid arthritis. J Rheumatol (2014) 41(3):429–36. doi:10.3899/jrheum.131037
73. Lopez-Mejias R, Ubilla B, Genre F, Corrales A, Hernandez JL, Ferraz-Amaro I, et al. Osteoprotegerin concentrations relate independently to established cardiovascular disease in rheumatoid arthritis. J Rheumatol (2015) 42(1):39–45. doi:10.3899/jrheum.140690
74. Golledge J, McCann M, Mangan S, Lam A, Karan M. Osteoprotegerin and osteopontin are expressed at high concentrations within symptomatic carotid atherosclerosis. Stroke (2004) 35(7):1636–41. doi:10.1161/01.STR.0000129790.00318.a3
75. Min H, Morony S, Sarosi I, Dunstan CR, Capparelli C, Scully S, et al. Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis. J Exp Med (2000) 192(4):463–74. doi:10.1084/jem.192.4.463
76. Venuraju SM, Yerramasu A, Corder R, Lahiri A. Osteoprotegerin as a predictor of coronary artery disease and cardiovascular mortality and morbidity. J Am Coll Cardiol (2010) 55(19):2049–61. doi:10.1016/j.jacc.2010.03.013
77. Abedin M, Omland T, Ueland T, Khera A, Aukrust P, Murphy SA, et al. Relation of osteoprotegerin to coronary calcium and aortic plaque (from the Dallas Heart Study). Am J Cardiol (2007) 99(4):513–8. doi:10.1016/j.amjcard.2006.08.064
78. Major AS, Fazio S, Linton MF. B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol (2002) 22(11):1892–8. doi:10.1161/01.ATV.0000039169.47943.EE
79. Onal M, Xiong J, Chen X, Thostenson JD, Almeida M, Manolagas SC, et al. Receptor activator of nuclear factor kappaB ligand (RANKL) protein expression by B lymphocytes contributes to ovariectomy-induced bone loss. J Biol Chem (2012) 287(35):29851–60. doi:10.1074/jbc.M112.377945
80. Masuzawa T, Miyaura C, Onoe Y, Kusano K, Ohta H, Nozawa S, et al. Estrogen deficiency stimulates B lymphopoiesis in mouse bone marrow. J Clin Invest (1994) 94(3):1090–7. doi:10.1172/JCI117424
81. Miyaura C, Onoe Y, Inada M, Maki K, Ikuta K, Ito M, et al. Increased B-lymphopoiesis by interleukin 7 induces bone loss in mice with intact ovarian function: similarity to estrogen deficiency. Proc Natl Acad Sci U S A (1997) 94(17):9360–5. doi:10.1073/pnas.94.17.9360
82. Li Y, Li A, Yang X, Weitzmann MN. Ovariectomy-induced bone loss occurs independently of B cells. J Cell Biochem (2007) 100(6):1370–5. doi:10.1002/jcb.21121
83. Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet (2005) 366(9499):1809–20. doi:10.1016/S0140-6736(05)67728-8
84. Cochran DL. Inflammation and bone loss in periodontal disease. J Periodontol (2008) 79(8 Suppl):1569–76. doi:10.1902/jop.2008.080233
85. Han X, Kawai T, Eastcott JW, Taubman MA. Bacterial-responsive B lymphocytes induce periodontal bone resorption. J Immunol (2006) 176(1):625–31. doi:10.4049/jimmunol.176.1.625
86. Nikolajczyk BS. B cells as under-appreciated mediators of non-auto-immune inflammatory disease. Cytokine (2010) 50(3):234–42. doi:10.1016/j.cyto.2010.02.022
87. Berglundh T, Liljenberg B, Tarkowski A, Lindhe J. The presence of local and circulating autoreactive B cells in patients with advanced periodontitis. J Clin Periodontol (2002) 29(4):281–6. doi:10.1034/j.1600-051X.2002.290402.x
88. Smit M, Brinkman K, Geerlings S, Smit C, Thyagarajan K, Sighem A, et al. Future challenges for clinical care of an ageing population infected with HIV: a modelling study. Lancet Infect Dis (2015) 15(7):810–8. doi:10.1016/S1473-3099(15)00056-0
89. Effros RB, Fletcher CV, Gebo K, Halter JB, Hazzard WR, Horne FM, et al. Aging and infectious diseases: workshop on HIV infection and aging: what is known and future research directions. Clin Infect Dis (2008) 47(4):542–53. doi:10.1086/590150
91. Brown TT, Qaqish RB. Antiretroviral therapy and the prevalence of osteopenia and osteoporosis: a meta-analytic review. AIDS (2006) 20(17):2165–74. doi:10.1097/QAD.0b013e32801022eb
92. Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab (2007) 92(7):2506–12. doi:10.1210/jc.2006-2190
93. Moir S, Fauci AS. B cells in HIV infection and disease. Nat Rev Immunol (2009) 9(4):235–45. doi:10.1038/nri2524
94. Edwards JC, Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol (2006) 6(5):394–403. doi:10.1038/nri1838
95. Sanz I, Anolik JH, Looney RJ. B cell depletion therapy in autoimmune diseases. Front Biosci (2007) 12:2546–67. doi:10.2741/2254
96. Moir S, Ho J, Malaspina A, Wang W, DiPoto AC, O’Shea MA, et al. Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J Exp Med (2008) 205(8):1797–805. doi:10.1084/jem.20072683
97. McComsey GA, Tebas P, Shane E, Yin MT, Overton ET, Huang JS, et al. Bone disease in HIV infection: a practical review and recommendations for HIV care providers. Clin Infect Dis (2010) 51(8):937–46. doi:10.1086/656412
98. Vikulina T, Fan X, Yamaguchi M, Roser-Page S, Zayzafoon M, Guidot DM, et al. Alterations in the immuno-skeletal interface drive bone destruction in HIV-1 transgenic rats. Proc Natl Acad Sci U S A (2010) 107(31):13848–53. doi:10.1073/pnas.1003020107
99. Stolina M, Guo J, Faggioni R, Brown H, Senaldi G. Regulatory effects of osteoprotegerin on cellular and humoral immune responses. Clin Immunol (2003) 109(3):347–54. doi:10.1016/j.clim.2003.09.001
100. Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res (2001) 16(2):348–60. doi:10.1359/jbmr.2001.16.2.348
101. Smith MR, Saad F, Coleman R, Shore N, Fizazi K, Tombal B, et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial. Lancet (2012) 379(9810):39–46. doi:10.1016/S0140-6736(11)61226-9
102. Bekker PJ, Holloway DL, Rasmussen AS, Murphy R, Martin SW, Leese PT, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res (2004) 19(7):1059–66. doi:10.1359/JBMR.040305
103. McClung M. Role of RANKL inhibition in osteoporosis. Arthritis Res Ther (2007) 9(Suppl 1):S3. doi:10.1186/ar2167
104. McClung MR, Lewiecki EM, Cohen SB, Bolognese MA, Woodson GC, Moffett AH, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med (2006) 354(8):821–31. doi:10.1056/NEJMoa044459
105. George S, Brenner A, Sarantopoulos J, Bukowski RM. RANK ligand: effects of inhibition. Curr Oncol Rep (2010) 12(2):80–6. doi:10.1007/s11912-010-0088-1
Keywords: B cells, HIV, bone loss, comorbidities, cytokines, end-organ damage
Citation: Titanji K (2017) Beyond Antibodies: B Cells and the OPG/RANK-RANKL Pathway in Health, Non-HIV Disease and HIV-Induced Bone Loss. Front. Immunol. 8:1851. doi: 10.3389/fimmu.2017.01851
Received: 30 September 2017; Accepted: 06 December 2017;
Published: 22 December 2017
Edited by:
Gabriella Scarlatti, San Raffaele Hospital (IRCCS), ItalyReviewed by:
Martyn Andrew French, University of Western Australia, AustraliaAntonella Caputo, Università degli Studi di Padova, Italy
Copyright: © 2017 Titanji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kehmia Titanji, ktitanj@emory.edu