 Niloofar Karaji
Niloofar Karaji Quentin J. Sattentau*
Quentin J. Sattentau*
- The Sir William Dunn School of Pathology, The University of Oxford, Oxford, United Kingdom
The prompt and efficient clearance of unwanted and abnormal cells by phagocytes is termed efferocytosis and is crucial for organism development, maintenance of tissue homeostasis, and regulation of the immune system. Dying cells are recognized by phagocytes through pathways initiated via “find me” signals, recognition via “eat me” signals and down-modulation of regulatory “don’t eat me” signals. Pathogen infection may trigger cell death that drives phagocytic clearance in an immunologically silent, or pro-inflammatory manner, depending on the mode of cell death. In many cases, efferocytosis is a mechanism for eliminating pathogens and pathogen-infected cells; however, some pathogens have subverted this process and use efferocytic mechanisms to avoid innate immune detection and assist phagocyte infection. In parallel, phagocytes can integrate signals received from infected dying cells to elicit the most appropriate effector response against the infecting pathogen. This review focuses on pathogen-induced cell death signals that drive infected cell recognition and uptake by phagocytes, and the outcomes for the infected target cell, the phagocyte, the pathogen and the host.
Introduction
To maintain and protect themselves, multicellular organisms remove dead and dying cells arising during normal tissue development and function (1) or triggered by infection or sterile inflammation (2). At steady state, even within tissues with high constitutive rates of apoptosis, the number of detectable apoptotic cells is relatively low, indicating a high rate of removal (3–5). Efficient clearance is vital for the constant removal of approximately 106 cells/s undergoing apoptosis in various tissues in adult humans (6). Phagocytosis is defined as engulfment of particulate matter of >0.5–1 µm (7, 8) and is mediated by both professional and non-professional phagocytic cell types. Professional phagocytes are primarily macrophages and immature dendritic cells (DCs) resident in multiple tissues and tissue-infiltrating monocytes, neutrophils, and eosinophils. Non-professional phagocytes such as epithelial cells of mammary epithelium (9) and astrocytes in the brain (10) can also capture and engulf material including dying cells that are present in close proximity within tissue. “Efferocytosis” is a term describing the engulfment by phagocytes of dying and dead cells and their debris (11, 12) and demonstrates features of both conventional phagocytosis and the fluid-phase uptake mechanism macropinocytosis (13–15), resulting in uptake into “spaceous phagosomes” (15). However, although the term efferocytosis distinguishes recognition and engulfment of dead and dying cells from phagocytosis of other objects (12, 16), we are unaware of specific mechanistic differences that discriminate between the two processes. Efferocytosis is mediated by a variety of interactions between the phagocyte and its dying target cell that show substantial redundancy and many soluble and cell surface receptor–ligand interactions defined for phagocytosis are used in efferocytosis (described in more detail below). The initial definition of efferocytosis related to clearance of apoptotic cells (15), but this has more recently been widened to include other modes of cell death (17, 18).
Cell death has been broadly categorized into accidental (necrosis) and regulated (including apoptosis, pyroptosis, and necroptosis) (17). Accidental cell death occurs during severe physical or chemical insult, such as membrane shearing and rupture via extremes of pressure, temperature, osmolarity, pH, or exposure to agents such as detergents and bacterial toxins, and is insensitive to pharmacologic or genetic manipulation. Because accidental cell death results in uncontrolled release of cell contents including damage-associated molecular patterns (DAMPs), it is pro-inflammatory (19, 20). Regulated modes of cell death are implicated in post-embryonic ontogeny, tissue homeostasis, and during infection and immune responses, have a genetically programmed component, and can be modulated by altering pro- and anti-death signals (17). Pathogen infection is variously associated with all forms of regulated cell death (Figure 1A) and necrosis, and the type of cell death induced is directly linked to the type of infecting pathogen, its life cycle and its pathogen-associated molecular patterns (PAMPs) recognized by pattern recognition receptors (PRRs).
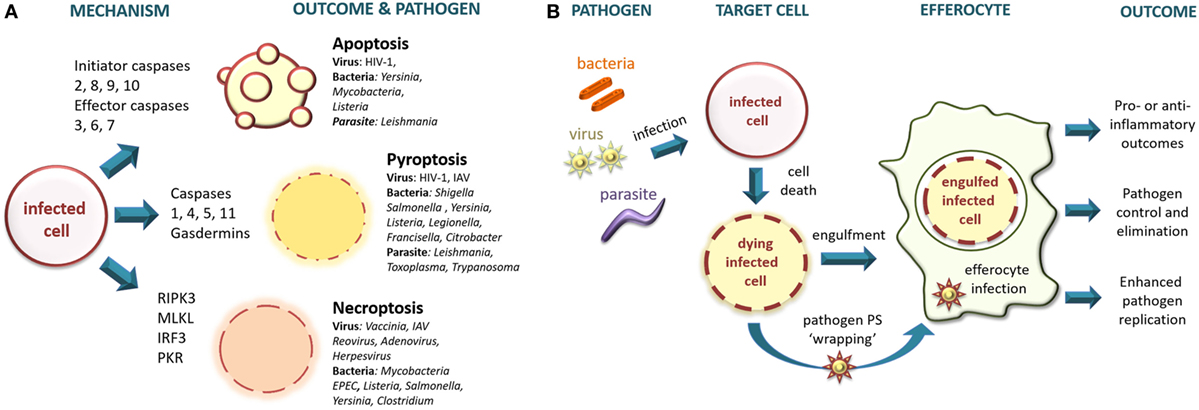
Figure 1. (A) Intracellular pathogens trigger regulated modes of cell death. Intracellular bacteria, viruses, and parasites infect target cells triggering different cell death pathways. Apoptosis is a non-inflammatory type of cell death in which the cellular contents remain membrane enclosed, whereas pyroptosis and necroptosis result in compromised membrane integrity leading to the release of intracellular contents that are pro-inflammatory. RIPK3, receptor-interacting protein kinase 3; MLKL, mixed lineage kinase domain-like; IRF3, interferon regulatory factor-3; PKR, protein kinase R; IAV, influenza A virus; EPEC, enteropathogenic Escherichia coli. (B) Pathogen infection modulates efferocytic outcomes. Intracellular bacteria, viruses, and parasites infect target cells inducing apoptosis. Dying target cells expose eat me signals and may down-modulate don’t eat me signals, leading to uptake and engulfment by efferocytes. Alternatively, the pathogen may escape from the infected cell wrapped in phosphatidylserine (PS)-containing membrane to deploy “apoptotic mimicry” for entry into the efferocyte. Efferocytosis may eliminate the pathogen, or may allow the pathogen to infect the efferocyte in a Trojan-horse type maneuver. The efferocyte will initiate anti-inflammatory or pro-inflammatory signaling depending upon the combined presence of immune-silencing signals (e.g., PS) and pro-inflammatory pathogen-associated molecular patterns and damage-associated molecular patterns.
Pathogen-Triggered Cell Death
Apoptosis is a caspase-dependent programmed form of regulated cell death resulting in a series of well-characterized morphologic and molecular changes culminating in membrane blebbing, DNA fragmentation and expression of signals designed to attract phagocytes and trigger engulfment and disposal of the apoptotic cargo (17). Apoptotic cells that are not rapidly efferocytosed become late apoptotic cells with a phenotype related to that of necrosis and are pro-inflammatory (21, 22). Apoptosis may be triggered in various cell types by a variety of pathogens, including intracellular bacteria (23), parasites (24), and viruses (25). Despite having evolved in part as a mechanism to limit pathogen replication and spread, apoptosis may have been subverted to contribute to pathogen survival and disease pathogenesis as discussed below. Pyroptosis is a regulated mode of cell death triggered principally by infection with intracellular pathogens (26, 27) and is linked to inflammasome activation driving caspase-1 or non-canonical caspase-11-triggering of the pore-forming effector gasdermin family (28, 29). Since pyroptosis results in plasma membrane permeabilization and eventual rupture with release of cytoplasmic contents, it has pro-inflammatory outcomes similar to accidental cell death and necroptosis. Necroptosis is triggered by infection with a variety of intracellular pathogens including viruses and bacteria (30, 31). Similar to pyroptosis, necroptosis is also a pro-inflammatory mode of regulated cell death initiated by cell surface receptors but is triggered in a caspase-independent RIPK3-dependent manner and may be modulated by caspase-8 activation toward apoptosis (17). Non-canonical forms of necroptosis activated by IRF3 and PKR-dependent pathways have also been described (31). The different modes of pathogen-initiated cell death and their mechanisms have been recently reviewed [e.g., Ref. (27, 30–33)] and so will not be further discussed here but are summarized in Figure 1A with some examples of pathogens implicated.
Efferocytic Signals
Phagocytes engage apoptotic cells via a defined set of markers termed “apoptotic cell-associated molecular patterns” or ACAMPs (16). ACAMPs include externalized phosphatidylserine, calreticulin, and modified carbohydrates that are recognized by a set of specific receptors and bridging molecules and will be described briefly below. Efferocytosis of dead and dying cells can be divided into four distinct stages (34, 35): (i) Detection of the target cell by release of chemotactic “find me” signals (36, 37) that include lysophosphatidylcholine, CX3CL1 (fractalkine), nucleotides adenosine triphosphate and uridine 5′ triphosphate, and sphingosine 1-phosphate (38–41). (ii) Exposure of “eat me” signals (42, 43), of which phosphatidylserine (PS) exposure on the outer leaflet of the plasma membrane is the best characterized, and although initially described in the context of apoptosis, appears to be shared between all modes of cell death (43–47). However, whereas PS externalization during apoptosis is mediated enzymatically, it becomes accessible during pyroptosis and necroptosis via membrane permeabilization. Eat me signaling is counter-balanced by expression levels of “don’t eat me” cell surface molecules such as CD47 (48, 49). Many eat me signals are recognized directly by phagocyte receptors such as members of the T-cell immunoglobulin domain and mucin domain (TIM) family, complement receptors CR3 and CR4, scavenger receptors SRA and CD36, mannose receptor (MR) and integrins α5β3 and α5β5, whereas others require bridging molecules such as collectins, complement C1q, mannose binding lectin, pentraxin3, ficolins, thrombospondin, and milk fat globule protein (MFG-8) for their recognition (18, 42, 50, 51). (iii) Following recognition of eat me signals, receptors such as antibody Fc (52) and complement (53) receptors signal to the cytoskeleton and are directly phagocytic, whereas others such as TIM-4 (54) are implicated only in tethering the target cell. However, a phagocyte will integrate signals from multiple receptors to inform the outcome of whether or not to engulf the target cell (12, 34, 55). (iv) Cellular material is fully internalized via cytoskeletal rearrangement of the plasma membrane (5, 12, 35) with processing of the engulfed cell usually (5, 55), but not exclusively (56), leading to its elimination within a phagolysosome-type compartment (57, 58). During the target cell recognition process, phagocytes may further evaluate the target’s chemical composition to estimate the threat posed by its contents and form such as size (59), geometry (60), and topography (61). This assessment determines (i) whether engulfment occurs or is replaced with, for example, neutrophil NETosis, an anti-microbial cell death mechanism whereby neutrophils eject chromatin extracellular traps (62); (ii) the fate of the target cell within the phagocyte; and (iii) whether the clearance process is immunologically silent, such as the efferocytosis of apoptotic cells (11, 63), or stimulates an inflammatory response such as the engulfment of most pathogens, pathogen-infected cells, and necrotic cells (8, 19, 20).
This review will not further consider find me and eat me signals involved in recognition and uptake of dead and dying cells, or the downstream signaling and cytoskeletal changes leading to phagocytic uptake, topics that have been very comprehensively reviewed recently (4, 6, 12, 16, 63–67). Instead, we will highlight recent discoveries regarding phagocyte recognition of cell death triggered by microbial infection and outcomes for the pathogen and infected target cell, the phagocyte, and the host.
Pathogen Infection Driving Cell Death and Efferocytosis
Infection by intracellular pathogens may lead to cell death by any of the regulated pathways described earlier (see Figure 1A), or by necrotic cell death in the case of some lytic infections. Microbial induction of regulated death is generally considered to be a mechanism evolved to reduce or prevent pathogen replication and spread (33). The beneficial outcomes for the host of pathogen-triggered cell death may comprise: (i) removal of the intracellular environment required for survival and replication; (ii) direct antipathogen effects of released intracellular components; (iii) initiation of an anti-microbial inflammatory response by release of DAMPs and PAMPs; and (iv) uptake and presentation of pathogen antigens by antigen-presenting cells. Induction of cell death may itself be sufficient to reduce and control pathogen replication or may be assisted or mediated by efferocytic mechanisms as has been described for several bacterial and viral pathogens (14). Conversely, some pathogens may use efferocytic mechanisms to invade the phagocyte in a “Trojan-horse” type of manoeuver and thereby perpetuate or enhance replication and dissemination (14, 68). These outcomes are discussed in more detail below and summarized in Figure 1B.
Efferocytosis in Pathogen Control and Its Subversion by Pathogens
Bacterial Infection
Gram-negative intracellular pathogenic bacteria including the Enterobacteriaceae Shigella (69, 70) and Salmonella (71) were initially proposed to induce apoptosis in infected macrophages. However, more recently, this view has been modified to take into account features of pyroptotic cell death including caspase-1 and inflammasome activation (26, 72, 73). Virulent strains of many gram-negative intracellular bacteria have evolved to evade pyroptotic cell death, for example, Shigella inhibition of caspase-4 activation (74), testifying to its importance as an innate immune antibacterial mechanism (26, 75, 76). Using an attenuated strain of Salmonella typhimurium that constitutively expresses flagellin and, therefore, activates NLRC4, Jorgensen et al. demonstrated that infected pyroptotic macrophages form “pore-induced intracellular traps” that capture bacteria within cellular debris without killing them (77). The bacterium-containing cell debris is then cleared by efferocytic neutrophils that are attracted by find me and eat me signals and kill the bacteria in a ROS-dependent manner (72, 77). Although apoptotic cell death of human monocytes or macrophages infected with wild-type (78–80) or attenuated (81) Mycobacterium tuberculosis is associated with reduced bacterial survival, the mechanism was until recently unclear. Apoptosis may impart some cell-intrinsic anti-M. tuberculosis activity to macrophages by enclosing the bacilli within apoptotic membrane vesicles, but efferocytosis appears to be an important adjunct mechanism to clear viable bacteria associated with apoptotic macrophages (82, 83). Efferocytosis of apoptotic M. tuberculosis-infected macrophages by uninfected macrophages results in their trafficking to a degradative phagolysosomal compartment (83). Similarly, in the zebrafish model, apoptotic Mycobacterium marinum-infected granulomatous macrophages were engulfed by neutrophils resulting in their death by oxidative-burst exposure (84). Not only does efferocytosis reduce mycobacterial viability in human cells but also allows cross-presentation by DC of mycobacterial antigens for MHC class-I and CD1 presentation to CD8+ T cells in mice, reinforcing adaptive anti-bacterial immunity (85). The importance of macrophage apoptosis as an anti-mycobacterial mechanism contrasts with the finding that some virulent forms of M. tuberculosis have evolved to divert apoptotic cell death toward a programmed necrotic pathway, which fails to inhibit bacterial growth and allows bacterial release from the disrupted cell, promoting pathogen dissemination (79, 81, 86, 87). Thus, the type of cell death induced directly influences the outcome for the pathogen.
Bacteria may use efferocytic pathways to escape from elimination or to assist their dissemination using Trojan-horse-type mechanisms in which dying cells carrying viable bacteria are engulfed leading to infection of the efferocyte. After the uptake of M. marinum by zebrafish macrophages, the infected macrophage undergoes apoptotic cell death. These apoptotic, infected macrophages are engulfed by other healthy macrophages, driving dissemination of infection via efferocytosis to increase granuloma burden and seed secondary granulomas in a manner dependent on the RD1 virulence factor (88). Eat me signals potentially involved in efferocytic mycobacterial infected cell uptake have been partially defined. Blocking of human macrophage cell surface MR using anti-MR antibody or pre-incubation with competitive soluble sugars (mannan and GlcNAc) (89), or blocking TIM-4 (83) significantly reduced the uptake of apoptotic M. tuberculosis-infected macrophages by uninfected macrophages. TIM-4 is also implicated in the subversion of efferocytic mechanisms by the gram-positive bacterial pathogen Listeria monocytogenes. The bacterium is phagocytosed by macrophages but avoids elimination by escaping the phagosome using the pore-forming toxin listeriolysin O (LLO) and recruits actin to drive cell-to-cell spread (90). The bacterium wraps itself in vesicles derived from the LLO-damaged host cell plasma membrane that exposes PS, which are in turn recognized by TIM-4 on healthy macrophages, leading to bacterial uptake and infection of a new host cell (91). Infection of a mouse strain lacking TIM-4 expression resulted in impaired bacterial growth, thus emphasizing its role in vivo. An efferocytic Trojan-horse mechanism of dissemination using neutrophils as a cellular vector is proposed for Chlamydia pneumoniae (92) and Yersinia pestis (93). In mice, both bacteria are initially phagocytosed by neutrophils at the site of inoculation, but the bacteria survive and, in the case of Y. pestis, replicate within the neutrophils. Subsequent infected neutrophil apoptosis and PS exposure triggered efferocytosis by macrophages, which were permissive for replication of both bacteria but elicited an anti-inflammatory cytokine response, potentially limiting anti-bacterial activity (92, 93). Macrophage recognition of C. pneumoniae in the context of apoptotic cells was shown to be important for bacterial replication, since inhibition of efferocytosis using annexin-V reduced macrophage infection (92).
Viral Infection
There is limited work on the role of efferocytosis in controlling viral replication. Influenza A virus infection of HeLa cells resulted in their apoptosis and rapid efferocytic engulfment and transit into phagosome-like structures within murine alveolar macrophages (94). The outcome of this was to limit viral release in the culture, suggesting that this may be a mechanism for suppressing replication in vivo (95). Of interest, the authors demonstrated that the eat me signals implicated in this efferocytic uptake were a combination of plasma membrane PS exposure and desialylation of surface glycans on the infected cells (96), consistent with other studies suggesting that loss of cell surface sialic acid during apoptosis is a novel eat me signal (97).
Recently, Baxter et al. observed that recognition and uptake of HIV-1-infected CD4+ T cells by human monocyte-derived macrophages led to enhanced macrophage infection when compared to incubation of these cells with cell-free virus (98). Macrophage infection by this cell-to-cell route was high multiplicity allowing robust infection even by weakly-macrophage–tropic viral strains (98). Although infected apparently healthy cells were weakly selectively captured, the strongest eat me signal came from dying HIV-1-infected cells, implying efferocytic signals. However inhibitors of PS–receptor interactions and other apoptotic cell death recognition receptor–ligand interactions failed to significantly reduce infected T-cell uptake, suggesting alternative signals that have yet to be defined (98). Macrophage phagocytosis of simian immunodeficiency virus-infected CD4+ T cells occurs in the macaque model, suggesting that this mode of viral spread may have in vivo relevance for immunodeficiency viruses, although it is unclear if the macrophages were productively infected or simply harbored infected efferocytosed cells (99, 100). This raises the general caveat that phagocytes may take up pathogen-infected cells giving the appearance of infection but without necessarily undergoing productive infection themselves (68). Thus, astrocytes, long proposed to undergo an atypical HIV-1 infection in the brain but which lack the primary HIV-1 receptor CD4, are phagocytic and engulf dying HIV-1-infected cells leading to markers of viral infection but are resistant to viral entry and infection (101). Finally, in an interesting twist to this paradigm, human papilloma virus (HPV) appears to have subverted efferocytosis to facilitate viral persistence in vivo. Efferocytosis of HPV-infected cervical cancer cells by primary human fibroblasts (102) led to expression of the HPV E6 gene within the fibroblasts and elicitation of a tumorigenic phenotype with implications for viral persistence (103).
While not formally efferocytosis, apoptotic cell mimicry achieved by the incorporation of PS into viral envelopes is a related phenomenon that has been described as enhancing infectivity for several enveloped virus families (104, 105) including HIV-1 (106), vaccinia virus (107), and lentiviral vectors pseudotyped with multiple viral envelope glycoproteins (105, 108). Non-enveloped picornaviruses also use this strategy, effected by wrapping themselves in PS-containing vesicles during cellular exit (109, 110). PS incorporated into the viral envelope during budding is recognized by a multitude of PS receptors on the target cell including TIM-1 and TIM-4 and TAM tyrosine kinase receptors TYRO3, AXL, and MER (11, 105, 108, 111) and may be further enhanced by bridging molecules such as MFG-E8 (105). Apoptotic mimicry has the major advantage of compromising pro-inflammatory programs by activating anti-inflammatory signaling cascades, which would otherwise trigger innate and adaptive immune responses against the invading virus, reducing viral replication in vivo (112). This immune evasion strategy is also used to good effect by parasites (see below).
Parasite Infection
Leishmania infection is transmitted to man by the sandfly, recruits a rapid neutrophil influx to the site of parasite entry, and replicates primarily within macrophages. The Leishmania major inoculum consists of a mixture of apoptotic and viable parasites, and depletion of apoptotic parasites reduces in vivo infectivity in a mouse model, proposed to be a consequence of loss of the anti-inflammatory TGF-β signal imparted on macrophages by the PS-expressing parasites (113). Using intravital microscopy of infected sandfly bites in mouse ear, it was observed that neutrophils are rapidly recruited to the site of the bite and engulf the parasites, many of which remain viable and infectious (114). Leishmania uptake by neutrophils can delay or accelerate neutrophil death in a manner dependent upon the experimental system. In vitro studies demonstrate that apoptosis of neutrophils is delayed for up to 2 days by L. major infection, thereby potentially serving as intracellular survival vectors for the parasites, during which time the neutrophils release MIP-1β, which attracts monocytes and macrophages (115). However, by contrast with in vitro studies, Leishmania-infected neutrophils analyzed ex vivo showed enhanced expression of PS, indicating accelerated apoptosis and potentially serving as an eat me signal for macrophage and DC uptake (116). Regardless of the underlying mechanism, macrophages may then efferocytose-infected apoptotic neutrophils becoming infected themselves in the process, the Trojan-horse mechanism (113). An alternative scenario, imaged by intravital microscopic analysis, is that rather than carrying parasites into the macrophage by efferocytosis, neutrophils may release viable parasites into regions densely populated by macrophages for subsequent macrophage engulfment and infection (114). Interestingly, in this study, depletion of neutrophils reduced L. major infection in mice (114), consistent with the idea that efferocytosis of apoptotic neutrophils imprinted an anti-inflammatory TGF-β signal on the neutrophils, preventing effective parasite elimination (115). A similar finding was also obtained when apoptotic neutrophils were engulfed by L. major-infected macrophages that produced the anti-inflammatory mediators TGF-β and prostaglandin PGE2 (117). However, these results must be evaluated in the context of the strain of mouse, the species of parasite, and the timing of macrophage encounter with apoptotic neutrophils in comparison with their encounter with the parasite. By contrast with the BALB/c mice used in the study above, engulfment of neutrophils by L. major-infected macrophages from parasite-resistant C57BL/6 mice reduced parasite load, concomitant with the secretion of TNF that most likely antagonized the anti-inflammatory signals released by uptake of apoptotic neutrophils (117, 118). Moreover, parasite killing may be parasite species dependent, since neutrophils from parasite-susceptible BALB/c mice triggered macrophage killing of Leishmania amazonensis and Leishmania braziliensis (119, 120). Finally, efferocytosis of apoptotic neutrophils by macrophages from resistant C57BL/6 mice 3 days prior to encounter with L. major led to enhanced permissivity to the parasite (121), a result that contrasts with studies in which infection took place prior to neutrophil exposure. Thus, in summary, whether efferocytosis of dying neutrophils results in advantageous or deleterious consequences for the parasite is complex and context dependent.
Trypanosma cruzi infection induces lymphocyte apoptosis in both experimentally infected mice (122, 123) and infected humans (124), and disease severity correlated with the degree of ex vivo apoptosis observed (124, 125). Mouse experiments support the concept that phagocyte uptake of apoptotic T lymphocytes results in the establishment of an anti-inflammatory response dictated by TGF-β and prostaglandin PGE2 that fuels parasite persistence and disease (126). Treatment of T. cruzi infected mice with inhibitors of apoptosis reversed the anti-inflammatory phenotype and reduced ex vivo parasite replication (123, 127), consistent with efferocytosis of apoptotic cells reducing macrophage anti-parasite activity and enhancing parasite persistence and disease.
Immune Consequences of Infected Cell Efferocytosis
The outcome for the phagocyte of engulfment of an infected dying cell is influenced both by the infecting pathogen and by the mode of death elicited in the target cell. PS exposed on apoptotic cells delivers an anti-inflammatory signal that is associated with defined receptor and signaling pathways and the production of regulatory cytokines such as TGF-β and IL-10 (11, 66) and is essential for rapid removal of apoptotic cells to avoid inflammatory and potential autoimmune consequences (19, 63). However, this is in the context of uninfected cells undergoing homeostatic apoptosis and immune-silent clearance. As described earlier, pathogen infection may induce more pro-inflammatory types of cell death via release of DAMPs and components of the pathogen present within the infected dying cell act as PAMPs to signal a pro-inflammatory response through PRRs (128). Thus, the phagocyte must integrate the pro- and anti-inflammatory signals to initiate the appropriate outcome, resulting in pathogen containment or clearance. Due to this complexity, experiments to probe the effects of specific pathogen infections of target cells on phagocyte pro- and anti-inflammatory programs are challenging to design and interpret. However, some information is available, particularly with regard to outcomes of efferocytosis of bacterial infection in the context of apoptotic cells. Torchinsky et al. (129) demonstrated that the combination of apoptotic and TLR-4-based signals presented to DCs by apoptotic neutrophils or B cells associated or not with E. coli or LPS triggered the release of TGF-β and IL-23 in the context of IL-6, a cytokine pattern, which favors the induction of Th17 effector Th cells. Th17 cells secrete the cytokine IL-17, which is important for the recruitment of neutrophils to resolve bacterial infections, and the combination of apoptotic cells and bacterial PAMPs was optimal for Th17 induction in the context of the model Citrobacter rodentium infection of mouse gut (129). LPS alone failed to induce biologically active TGF-β and also induced high levels of IL-12, favoring a Th1-type response rather than Th17-type response, an outcome that would be suboptimal for extracellular bacterial clearance. Similarly, comparison of DC-mediated efferocytosis of an uninfected or E. coli-infected macrophage line induced to undergo apoptosis resulted in distinct outcomes (130). DC efferocytosis of infected apoptotic cells showed increased CD86 and CCR7 expression associated with an enhanced migratory capacity compared to uninfected apoptotic cells and enhanced production of IL-6, TGF-β, and IL-23, indicative of Th17 differentiation capacity (130). This again suggests that combination of pathogen and dying cells integrates signals to elicit the most appropriate immune outcome to control the specific pathogen. However, infection associated with apoptosis may also lead to misdirected adaptive immunity resulting in autoimmune outcomes. Thus, using apoptotic B cells infected with L. monocytogenes, Campisi et al. showed that the combination of stimuli present within the phagocyte resulted in presentation of self-antigens in the context of a pro-inflammatory environment (131). Using an in vivo murine model, this translated into Th17-induced colitis in the context of a C. rodentium bacterial infection, confirming the potentially deleterious effects of co-presentation of apoptotic and inflammatory infection signals by DC.
The most obvious implication of efferocytic uptake of virally infected dying cells is in cross-presentation, since CD8+ T-cell priming against viral infections requires access of viral antigens to the MHC class-I processing and presentation apparatus. While viruses that infect antigen-presenting cells do this directly by cytoplasmic expression of their antigens, induction of immune responses to viruses that do not productively infect DCs rely upon efferocytosis of infected apoptotic cells followed by cross-presentation. The first demonstration of this was in the context of influenza virus infection of monocytes that led to their apoptosis and uptake by DCs, driving efficient CD8 T-cell priming against influenza antigens (132). Since then, multiple studies have reported on cross-presentation of viral antigens by efferocytic uptake of dying cells infected with vaccinia virus, HTLV-1, measles virus, CMV, and EBV (133, 134). Similar observations have been made for a series of intracellular bacterial pathogens including L. monocytogenes and M. tuberculosis (133). Although much of the cell biology of cross-presentation has been defined, what remains to be addressed is how the combination of cell death and pathogen-triggered signals influence the outcome of cross-presentation, as for example has been dissected for T-helper cell responses (128). The restriction factor SAMHD1 renders DC relatively resistant to HIV-1 infection and limits DC activation and antigen presentation (135). However, as recently demonstrated by Silvin et al. (136), DCs are heterogeneous with respect to viral infection and, while CD1c+ DCs are sensitive to HIV-1 and influenza virus infection resulting in DC death, CD141+ DCs are resistant. The authors provide evidence that in the absence of direct infection, CD141+ DC acquires viral antigen by efferocytosis of dying virus-infected cells including CD1c+ DC, allowing efficient cross-presentation. Also relevant to cross-presentation of infected dying cells, cells dying by necroptosis, rather than by necrosis or apoptosis, trigger a RIPK1-dependent NFκB transcriptional program-directing release of inflammatory cytokines that enhance cross-priming of CD8+ T cells by DC (137). Although the cells in this instance were not infected, the relationship between this mechanism and infections driving necroptosis is obvious and raises questions regarding the ability of pathogens to modulate or suppress NFκB activation and other pro-inflammatory programs in dying cells. HIV-1 infection is a weak trigger of type-I interferon responses in macrophages, considered in part to result from “shielding” of viral nucleic acid PAMPs from intracellular sensors (138, 139), although early entry events can elicit low interferon levels (140). HIV-1 infection of CD4+ T cells leads to their death by apoptosis (141) during productive infection or pyroptosis during abortive infection (142), and in vivo, there is likely to be a combination of these types of death associated with infection. Using model in vitro systems, Lepelley et al. showed that HIV-1-infected CD4+ T cells elicit robust type-I interferon release from plasmacytoid DCs that is partially elicited by TLR-7 sensing of viral RNA (143) but potentially also by DAMPs released from infected dying T cells. Thus, cell-associated viral PAMPs appear to elicit a stronger innate immune anti-viral response than the virus alone.
Concluding Remarks
It is evident that efferocytosis is both an essential element of tissue homeostasis and a mechanism for elimination of intracellular pathogens. However, as described earlier, subversion of efferocytic mechanisms via (i) the Trojan-horse type of strategy, (ii) cell-free microorganisms expressing or hijacking PS-containing membrane, and (iii) triggering of anti-inflammatory programs in macrophages by efferocytosis of apoptotic cells contributes to immune evasion and pathogen persistence. Therapeutic intervention in efferocytic pathways may well be a rational approach to reducing infection by certain pathogens, but care must of course be exercised to avoid perturbing the fine balance between homeostasis and deleterious inflammation. In vitro studies demonstrated a reduction in HIV-1 infectivity of macrophages in the presence of soluble recombinant annexin-V, suggesting a mechanism to target this viral reservoir (144). The anti-PS monoclonal antibody Bavituximab has been used in a number of clinical trials as an anti-cancer agent, and its use in targeting viral infections such as HIV-1 (145), Pichinde virus (as a model for Lassa fever virus), and CMV (146) has been investigated. Aside from directly targeting pathogen replication, chronic infections such as HIV-1 (147) and HCV (148) are associated with long-term inflammatory outcomes that predispose to disease even in the context of suppressive anti-viral regimens. Chronic inflammation in HIV-1 infection is linked to acute inflammatory events in the gut-associated lymphoid tissue (GALT) initiated by massive HIV-1 infection and death of CD4+ T cells (149) that predispose this tissue to translocation of microbial products from the lumen (150). Excessive CD4+ T-cell apoptotic death may saturate GALT efferocytic capacity driving neglect of apoptotic cells by phagocytes leading to secondary necrosis, a type of cell death associated with tissue infiltration of monocytes and neutrophils that may mediate further tissue damage and is linked to chronic inflammatory autoimmune conditions (22). Moreover, abortive HIV-1 infection of tissue CD4+ T cells has been implicated in pyroptotic cell death (142), which might directly promote pro-inflammatory programs in phagocytes. Whether modulating efferocytosis in conditions such as this may influence the inflammatory outcome is a question that requires attention. Very recently, it has been demonstrated that efferocytosis during drosophila development can reprogram macrophages via JNK signaling to increased expression of the damage receptor Draper for robust responses to subsequent tissue injury or infection (151). This type of priming leading to innate immune “memory” deserves further investigation and may potentially be targeted for either anti-pathogen or anti-inflammatory outcomes in the clinic.
Author Contributions
NK and QS conceptualized the article. NK wrote and revised the first draft. NK and QS revised and edited subsequent drafts.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The authors acknowledge funding by the Medical Research Council UK and the Edward Penley Abraham Trust, The Sir William Dunn School of Pathology.
References
1. Baehrecke EH. How death shapes life during development. Nat Rev Mol Cell Biol (2002) 3(10):779–87. doi:10.1038/nrm931
2. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol (2011) 11(11):762–74. doi:10.1038/nri3070
3. Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature (2001) 411(6834):207–11. doi:10.1038/35075603
4. Elliott MR, Ravichandran KS. The dynamics of apoptotic cell clearance. Dev Cell (2016) 38(2):147–60. doi:10.1016/j.devcel.2016.06.029
5. Zent CS, Elliott MR. Maxed out macs: physiologic cell clearance as a function of macrophage phagocytic capacity. FEBS J (2017) 284(7):1021–39. doi:10.1111/febs.13961
6. Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med (2010) 207(9):1807–17. doi:10.1084/jem.20101157
7. Gordon S. Phagocytosis: an immunobiologic process. Immunity (2016) 44(3):463–75. doi:10.1016/j.immuni.2016.02.026
8. Underhill DM, Goodridge HS. Information processing during phagocytosis. Nat Rev Immunol (2012) 12(7):492–502. doi:10.1038/nri3244
9. Monks J, Smith-Steinhart C, Kruk ER, Fadok VA, Henson PM. Epithelial cells remove apoptotic epithelial cells during post-lactation involution of the mouse mammary gland. Biol Reprod (2008) 78(4):586–94. doi:10.1095/biolreprod.107.065045
10. Loov C, Mitchell CH, Simonsson M, Erlandsson A. Slow degradation in phagocytic astrocytes can be enhanced by lysosomal acidification. Glia (2015) 63:1997–2009. doi:10.1002/glia.22873
11. Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ (2016) 23(6):962–78. doi:10.1038/cdd.2016.11
12. Henson PM. Cell removal: efferocytosis. Annu Rev Cell Dev Biol (2017) 33:127–44. doi:10.1146/annurev-cellbio-111315-125315
13. Hoffmann PR, deCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, et al. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J Cell Biol (2001) 155(4):649–59. doi:10.1083/jcb.200108080
14. Martin CJ, Peters KN, Behar SM. Macrophages clean up: efferocytosis and microbial control. Curr Opin Microbiol (2014) 17:17–23. doi:10.1016/j.mib.2013.10.007
15. deCathelineau AM, Henson PM. The final step in programmed cell death: phagocytes carry apoptotic cells to the grave. Essays Biochem (2003) 39:105–17. doi:10.1042/bse0390105
17. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ (2015) 22(1):58–73. doi:10.1038/cdd.2014.137
18. Poon IKH, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol (2014) 14(3):166–80. doi:10.1038/nri3607
19. Blander JM. The many ways tissue phagocytes respond to dying cells. Immunol Rev (2017) 277(1):158–73. doi:10.1111/imr.12537
20. Yatim N, Cullen S, Albert ML. Dying cells actively regulate adaptive immune responses. Nat Rev Immunol (2017) 17(4):262–75. doi:10.1038/nri.2017.9
21. Poon IKH, Hulett MD, Parish CR. Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ (2010) 17(3):381–97. doi:10.1038/cdd.2009.195
22. Sachet M, Liang YY, Oehler R. The immune response to secondary necrotic cells. Apoptosis (2017) 22:1189–204. doi:10.1007/s10495-017-1413-z
23. Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, et al. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol (2009) 10(8):899–906. doi:10.1038/ni.1758
24. Decote-Ricardo D, Nunes MP, Morrot A, Freire-de-Lima CG. Implication of apoptosis for the pathogenesis of Trypanosoma cruzi infection. Front Immunol (2017) 8:518. doi:10.3389/fimmu.2017.00518
25. Yu X, He S. The interplay between human herpes simplex virus infection and the apoptosis and necroptosis cell death pathways. Virol J (2016) 13:77. doi:10.1186/s12985-016-0528-0
26. Jorgensen I, Miao EA. Pyroptotic cell death defends against intracellular pathogens. Immunol Rev (2015) 265(1):130–42. doi:10.1111/imr.12287
27. Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev (2017) 277(1):61–75. doi:10.1111/imr.12534
28. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi:10.1038/nature15514
29. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526(7575):666–71. doi:10.1038/nature15541
30. Brault M, Oberst A. Controlled detonation: evolution of necroptosis in pathogen defense. Immunol Cell Biol (2017) 95(2):131–6. doi:10.1038/icb.2016.117
31. Sridharan H, Upton JW. Programmed necrosis in microbial pathogenesis. Trends Microbiol (2014) 22(4):199–207. doi:10.1016/j.tim.2014.01.005
32. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol (2017) 17(3):151–64. doi:10.1038/nri.2016.147
33. Stephenson HN, Herzig A, Zychlinsky A. Beyond the grave: when is cell death critical for immunity to infection? Curr Opin Immunol (2016) 38:59–66. doi:10.1016/j.coi.2015.11.004
34. Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol (2013) 5(1):a008748–008748. doi:10.1101/cshperspect.a008748
35. Fond AM, Ravichandran KS. Clearance of dying cells by phagocytes: mechanisms and implications for disease pathogenesis. Adv Exp Med Biol (2016) 930:133–49. doi:10.1007/978-3-319-39406-0
36. Peter C, Wesselborg S, Lauber K. Molecular suicide notes: last call from apoptosing cells. J Mol Cell Biol (2010) 2(2):78–80. doi:10.1093/jmcb/mjp045
37. Kroemer G. Dying cell recognition shapes the pathophysiology of cell death. Cell Death Differ (2016) 23(6):913–4. doi:10.1038/cdd.2016.30
38. Lauber K, Bohn E, Kröber SM, Xiao Y-J, Blumenthal SG, Lindemann RK, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell (2003) 113(6):717–30. doi:10.1016/S0092-8674(03)00422-7
39. Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood (2008) 112(13):5026–36. doi:10.1182/blood-2008-06-162404
40. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature (2009) 461(7261):282–6. doi:10.1038/nature08296
41. Gude DR, Alvarez SE, Paugh SW, Mitra P, Yu J, Griffiths R, et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J (2008) 22(8):2629–38. doi:10.1096/fj.08-107169
42. Li W. Eat-me signals: keys to molecular phagocyte biology and “appetite” control. J Cell Physiol (2012) 227(4):1291–7. doi:10.1002/jcp.22815
43. Segawa K, Nagata S. An apoptotic ‘Eat Me’ signal: phosphatidylserine exposure. Trends Cell Biol (2015) 25(11):639–50. doi:10.1016/j.tcb.2015.08.003
44. Wang Q, Imamura R, Motani K, Kushiyama H, Nagata S, Suda T. Pyroptotic cells externalize eat-me and release find-me signals and are efficiently engulfed by macrophages. Int Immunol (2013) 25(2):363–72. doi:10.1093/intimm/dxs161
45. Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir-Birin Y, Croker BA, et al. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol (2017) 15(6):e2002711. doi:10.1371/journal.pbio.2002711
46. Hirt UA, Leist M. Rapid, noninflammatory and PS-dependent phagocytic clearance of necrotic cells. Cell Death Differ (2003) 10(10):1156–64. doi:10.1038/sj.cdd.4401286
47. Brouckaert G, Kalai M, Krysko DV, Saelens X, Vercammen D, Ndlovu MN, et al. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol Biol Cell (2004) 15(3):1089–100. doi:10.1091/mbc.E03-09-0668
48. Brown GC, Neher JJ. Eaten alive! Cell death by primary phagocytosis: ‘phagoptosis’. Trends Biochem Sci (2012) 37(8):325–32. doi:10.1016/j.tibs.2012.05.002
49. Gardai SJ, Bratton DL, Ogden CA, Henson PM. Recognition ligands on apoptotic cells: a perspective. J Leukoc Biol (2006) 79(5):896–903. doi:10.1189/jlb.1005550
50. Park SY, Kim IS. Engulfment signals and the phagocytic machinery for apoptotic cell clearance. Exp Mol Med (2017) 49(5):e331. doi:10.1038/emm.2017.52
51. Biermann M, Maueroder C, Brauner JM, Chaurio R, Janko C, Herrmann M, et al. Surface code – biophysical signals for apoptotic cell clearance. Phys Biol (2013) 10(6):065007. doi:10.1088/1478-3975/10/6/065007
52. Anderson CL, Shen L, Eicher DM, Wewers MD, Gill JK. Phagocytosis mediated by three distinct Fc gamma receptor classes on human leukocytes. J Exp Med (1990) 171(4):1333–45. doi:10.1084/jem.171.4.1333
53. Ross GD, Reed W, Dalzell JG, Becker SE, Hogg N. Macrophage cytoskeleton association with CR3 and CR4 regulates receptor mobility and phagocytosis of iC3b-opsonized erythrocytes. J Leukoc Biol (1992) 51(2):109–17.
54. Park S-Y, Jung M-Y, Lee S-J, Kang K-B, Gratchev A, Riabov V, et al. Stabilin-1 mediates phosphatidylserine-dependent clearance of cell corpses in alternatively activated macrophages. J Cell Sci (2009) 122(18):3365–73. doi:10.1242/jcs.049569
55. Green DR, Oguin TH, Martinez J. The clearance of dying cells: table for two. Cell Death Differ (2016) 23(6):915–26. doi:10.1038/cdd.2015.172
56. Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell (2007) 131(5):966–79. doi:10.1016/j.cell.2007.10.040
57. Flannagan RS, Jaumouille V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol (2012) 7:61–98. doi:10.1146/annurev-pathol-011811-132445
58. Pauwels AM, Trost M, Beyaert R, Hoffmann E. Patterns, receptors, and signals: regulation of phagosome maturation. Trends Immunol (2017) 38(6):407–22. doi:10.1016/j.it.2017.03.006
59. Brewer JM, Pollock KGJ, Tetley L, Russell DG. Vesicle size influences the trafficking, processing, and presentation of antigens in lipid vesicles. J Immunol (2004) 173(10):6143–50. doi:10.4049/jimmunol.173.10.6143
60. Doshi N, Mitragotri S. Macrophages recognize size and shape of their targets. PLoS One (2010) 5(4):e10051. doi:10.1371/journal.pone.0010051
61. Champion JA, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci U S A (2006) 103(13):4930–4. doi:10.1073/pnas.0600997103
62. Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol (2014) 15(11):1017–25. doi:10.1038/ni.2987
63. Greenlee-Wacker MC. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev (2016) 273(1):357–70. doi:10.1111/imr.12453
64. Medina CB, Ravichandran KS. Do not let death do us part: ‘find-me’ signals in communication between dying cells and the phagocytes. Cell Death Differ (2016) 23(6):979–89. doi:10.1038/cdd.2016.13
65. Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity (2011) 35(4):445–55. doi:10.1016/j.immuni.2011.09.004
66. Penberthy KK, Ravichandran KS. Apoptotic cell recognition receptors and scavenger receptors. Immunol Rev (2016) 269(1):44–59. doi:10.1111/imr.12376
67. Kinchen JM, Ravichandran KS. Journey to the grave: signaling events regulating removal of apoptotic cells. J Cell Sci (2007) 120(Pt 13):2143–9. doi:10.1242/jcs.03463
68. Sattentau QJ, Stevenson M. Macrophages and HIV-1: an unhealthy constellation. Cell Host Microbe (2016) 19:304–10. doi:10.1016/j.chom.2016.02.013
69. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature (1992) 358(6382):167–9. doi:10.1038/358167a0
70. Hilbi H, Moss JE, Hersh D, Chen Y, Arondel J, Banerjee S, et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J Biol Chem (1998) 273(49):32895–900. doi:10.1074/jbc.273.49.32895
71. Monack DM, Raupach B, Hromockyj AE, Falkow S. Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc Natl Acad Sci U S A (1996) 93(18):9833–8. doi:10.1073/pnas.93.18.9833
72. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol (2010) 11(12):1136–42. doi:10.1038/ni.1960
73. Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog (2007) 3(8):e111. doi:10.1371/journal.ppat.0030111
74. Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, Ashida H, et al. The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe (2013) 13(5):570–83. doi:10.1016/j.chom.2013.04.012
75. Cunha LD, Zamboni DS. Subversion of inflammasome activation and pyroptosis by pathogenic bacteria. Front Cell Infect Microbiol (2013) 3:76. doi:10.3389/fcimb.2013.00076
76. Bleriot C, Lecuit M. The interplay between regulated necrosis and bacterial infection. Cell Mol Life Sci (2016) 73(11–12):2369–78. doi:10.1007/s00018-016-2206-1
77. Jorgensen I, Zhang Y, Krantz BA, Miao EA. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J Exp Med (2016) 213(10):2113–28. doi:10.1084/jem.20151613
78. Oddo M, Renno T, Attinger A, Bakker T, MacDonald HR, Meylan PR. Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J Immunol (1998) 160(11):5448–54.
79. Behar SM, Martin CJ, Booty MG, Nishimura T, Zhao X, Gan HX, et al. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol (2011) 4(3):279–87. doi:10.1038/mi.2011.3
80. Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, et al. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun (1997) 65(1):298–304.
81. Molloy A, Laochumroonvorapong P, Kaplan G. Apoptosis, but not necrosis, of infected monocytes is coupled with killing of intracellular bacillus Calmette-Guerin. J Exp Med (1994) 180(4):1499–509. doi:10.1084/jem.180.4.1499
82. Fratazzi C, Arbeit RD, Carini C, Remold HG. Programmed cell death of Mycobacterium avium serovar 4-infected human macrophages prevents the mycobacteria from spreading and induces mycobacterial growth inhibition by freshly added, uninfected macrophages. J Immunol (1997) 158(9):4320–7.
83. Martin CJ, Booty MG, Rosebrock TR, Nunes-Alves C, Desjardins DM, Keren I, et al. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe (2012) 12(3):289–300. doi:10.1016/j.chom.2012.06.010
84. Yang C-T, Cambier CJ, Davis JM, Hall CJ, Crosier PS, Ramakrishnan L. Neutrophils exert protection in the early tuberculous granuloma by oxidative killing of mycobacteria phagocytosed from infected macrophages. Cell Host Microbe (2012) 12(3):301–12. doi:10.1016/j.chom.2012.07.009
85. Schaible UE, Winau F, Sieling PA, Fischer K, Collins HL, Hagens K, et al. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat Med (2003) 9(8):1039–46. doi:10.1038/nm906
86. Duan L, Gan H, Golan DE, Remold HG. Critical role of mitochondrial damage in determining outcome of macrophage infection with Mycobacterium tuberculosis. J Immunol (2002) 169(9):5181–7. doi:10.4049/jimmunol.169.9.5181
87. Amaral EP, Ribeiro SC, Lanes VR, Almeida FM, de Andrade MR, Bomfim CC, et al. Pulmonary infection with hypervirulent mycobacteria reveals a crucial role for the P2X7 receptor in aggressive forms of tuberculosis. PLoS Pathog (2014) 10(7):e1004188. doi:10.1371/journal.ppat.1004188
88. Davis JM, Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell (2009) 136(1):37–49. doi:10.1016/j.cell.2008.11.014
89. Garcia-Aguilar T, Espinosa-Cueto P, Magallanes-Puebla A, Mancilla R. The mannose receptor is involved in the phagocytosis of mycobacteria-induced apoptotic cells. J Immunol Res (2016) 2016:1–14. doi:10.1155/2016/3845247
90. Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, Cossart P. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell (1992) 68(3):521–31. doi:10.1016/0092-8674(92)90188-I
91. Czuczman MA, Fattouh R, van Rijn JM, Canadien V, Osborne S, Muise AM, et al. Listeria monocytogenes exploits efferocytosis to promote cell-to-cell spread. Nature (2014) 509(7499):230–4. doi:10.1038/nature13168
92. Rupp J, Pfleiderer L, Jugert C, Moeller S, Klinger M, Dalhoff K, et al. Chlamydia pneumoniae hides inside apoptotic neutrophils to silently infect and propagate in macrophages. PLoS One (2009) 4(6):e6020. doi:10.1371/journal.pone.0006020
93. Spinner JL, Winfree S, Starr T, Shannon JG, Nair V, Steele-Mortimer O, et al. Yersinia pestis survival and replication within human neutrophil phagosomes and uptake of infected neutrophils by macrophages. J Leukoc Biol (2014) 95(3):389–98. doi:10.1189/jlb.1112551
94. Shiratsuchi A, Kaido M, Takizawa T, Nakanishi Y. Phosphatidylserine-mediated phagocytosis of influenza A virus-infected cells by mouse peritoneal macrophages. J Virol (2000) 74(19):9240–4. doi:10.1128/JVI.74.19.9240-9244.2000
95. Fujimoto I, Pan J, Takizawa T, Nakanishi Y. Virus clearance through apoptosis-dependent phagocytosis of influenza A virus-infected cells by macrophages. J Virol (2000) 74(7):3399–403. doi:10.1128/JVI.74.7.3399-3403.2000
96. Watanabe Y, Shiratsuchi A, Shimizu K, Takizawa T, Nakanishi Y. Role of phosphatidylserine exposure and sugar chain desialylation at the surface of influenza virus-infected cells in efficient phagocytosis by macrophages. J Biol Chem (2002) 277(20):18222–8. doi:10.1074/jbc.M201074200
97. Meesmann HM, Fehr E-M, Kierschke S, Herrmann M, Bilyy R, Heyder P, et al. Decrease of sialic acid residues as an eat-me signal on the surface of apoptotic lymphocytes. J Cell Sci (2010) 123(Pt 19):3347–56. doi:10.1242/jcs.066696
98. Baxter AE, Russell RA, Duncan CJ, Moore MD, Willberg CB, Pablos JL, et al. Macrophage infection via selective capture of HIV-1-infected CD4+ T cells. Cell Host Microbe (2014) 16(6):711–21. doi:10.1016/j.chom.2014.10.010
99. Calantone N, Wu F, Klase Z, Deleage C, Perkins M, Matsuda K, et al. Tissue myeloid cells in SIV-infected primates acquire viral DNA through phagocytosis of infected T cells. Immunity (2014) 41(3):493–502. doi:10.1016/j.immuni.2014.08.014
100. Deleage C, Wietgrefe SW, Del Prete G, Morcock DR, Hao XP, Piatak M Jr, et al. Defining HIV and SIV reservoirs in lymphoid tissues. Pathog Immun (2016) 1(1):68–106. doi:10.20411/pai.v1i1.100
101. Russell RA, Chojnacki J, Jones DM, Johnson E, Do T, Eggeling C, et al. Astrocytes resist HIV-1 fusion but engulf infected macrophage material. Cell Rep (2017) 18(6):1473–83. doi:10.1016/j.celrep.2017.01.027
102. Hermetet F, Jacquin E, Launay S, Gaiffe E, Couturier M, Hirchaud F, et al. Efferocytosis of apoptotic human papillomavirus-positive cervical cancer cells by human primary fibroblasts. Biol Cell (2016) 108(7):189–204. doi:10.1111/boc.201500090
103. Gaiffe E, Pretet JL, Launay S, Jacquin E, Saunier M, Hetzel G, et al. Apoptotic HPV positive cancer cells exhibit transforming properties. PLoS One (2012) 7(5):e36766. doi:10.1371/journal.pone.0036766
104. Amara A, Mercer J. Viral apoptotic mimicry. Nat Rev Microbiol (2015) 13(8):461–9. doi:10.1038/nrmicro3469
105. Morizono K, Chen IS. Role of phosphatidylserine receptors in enveloped virus infection. J Virol (2014) 88(8):4275–90. doi:10.1128/JVI.03287-13
106. Callahan MK, Popernack PM, Tsutsui S, Truong L, Schlegel RA, Henderson AJ. Phosphatidylserine on HIV envelope is a cofactor for infection of monocytic cells. J Immunol (2003) 170(9):4840–5. doi:10.4049/jimmunol.170.9.4840
107. Mercer J, Helenius A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science (2008) 320(5875):531–5. doi:10.1126/science.1155164
108. Jemielity S, Wang JJ, Chan YK, Ahmed AA, Li W, Monahan S, et al. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog (2013) 9(3):e1003232. doi:10.1371/journal.ppat.1003232
109. Chen YH, Du W, Hagemeijer MC, Takvorian PM, Pau C, Cali A, et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell (2015) 160(4):619–30. doi:10.1016/j.cell.2015.01.032
110. Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L, et al. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature (2013) 496(7445):367–71. doi:10.1038/nature12029
111. Gu L, Sims B, Krendelchtchikov A, Tabengwa E, Matthews QL. Differential binding of the HIV-1 envelope to phosphatidylserine receptors. Biochim Biophys Acta (2017) 1859(10):1962–6. doi:10.1016/j.bbamem.2017.06.007
112. Bhattacharyya S, Zagorska A, Lew ED, Shrestha B, Rothlin CV, Naughton J, et al. Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell Host Microbe (2013) 14(2):136–47. doi:10.1016/j.chom.2013.07.005
113. van Zandbergen G, Bollinger A, Wenzel A, Kamhawi S, Voll R, Klinger M, et al. Leishmania disease development depends on the presence of apoptotic promastigotes in the virulent inoculum. Proc Natl Acad Sci U S A (2006) 103(37):13837–42. doi:10.1073/pnas.0600843103
114. Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, et al. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science (2008) 321(5891):970–4. doi:10.1126/science.1159194
115. van Zandbergen G, Klinger M, Mueller A, Dannenberg S, Gebert A, Solbach W, et al. Cutting edge: neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J Immunol (2004) 173(11):6521–5. doi:10.4049/jimmunol.173.11.6521
116. Ribeiro-Gomes FL, Peters NC, Debrabant A, Sacks DL. Efficient capture of infected neutrophils by dendritic cells in the skin inhibits the early anti-Leishmania response. PLoS Pathog (2012) 8(2):e1002536. doi:10.1371/journal.ppat.1002536
117. Ribeiro-Gomes FL, Otero AC, Gomes NA, Moniz-De-Souza MC, Cysne-Finkelstein L, Arnholdt AC, et al. Macrophage interactions with neutrophils regulate Leishmania major infection. J Immunol (2004) 172(7):4454–62. doi:10.4049/jimmunol.172.7.4454
118. Ribeiro-Gomes FL, Moniz-de-Souza MC, Alexandre-Moreira MS, Dias WB, Lopes MF, Nunes MP, et al. Neutrophils activate macrophages for intracellular killing of Leishmania major through recruitment of TLR4 by neutrophil elastase. J Immunol (2007) 179(6):3988–94. doi:10.4049/jimmunol.179.6.3988
119. de Souza Carmo EV, Katz S, Barbieri CL. Neutrophils reduce the parasite burden in Leishmania (Leishmania) amazonensis-infected macrophages. PLoS One (2010) 5(11):e13815. doi:10.1371/journal.pone.0013815
120. Novais FO, Santiago RC, Bafica A, Khouri R, Afonso L, Borges VM, et al. Neutrophils and macrophages cooperate in host resistance against Leishmania braziliensis infection. J Immunol (2009) 183(12):8088–98. doi:10.4049/jimmunol.0803720
121. Filardy AA, Pires DR, Nunes MP, Takiya CM, Freire-de-Lima CG, Ribeiro-Gomes FL, et al. Proinflammatory clearance of apoptotic neutrophils induces an IL-12(low)IL-10(high) regulatory phenotype in macrophages. J Immunol (2010) 185(4):2044–50. doi:10.4049/jimmunol.1000017
122. Lopes MF, da Veiga VF, Santos AR, Fonseca ME, DosReis GA. Activation-induced CD4+ T cell death by apoptosis in experimental Chagas’ disease. J Immunol (1995) 154(2):744–52.
123. Cabral-Piccin MP, Guillermo LV, Vellozo NS, Filardy AA, Pereira-Marques ST, Rigoni TS, et al. Apoptotic CD8 T-lymphocytes disable macrophage-mediated immunity to Trypanosoma cruzi infection. Cell Death Dis (2016) 7:e2232. doi:10.1038/cddis.2016.135
124. Rodrigues V Jr, Agrelli GS, Leon SC, Silva Teixeira DN, Tostes S Jr, Rocha-Rodrigues DB. Fas/Fas-L expression, apoptosis and low proliferative response are associated with heart failure in patients with chronic Chagas’ disease. Microbes Infect (2008) 10(1):29–37. doi:10.1016/j.micinf.2007.09.015
125. Chaves AT, de Assis Silva Gomes Estanislau J, Fiuza JA, Carvalho AT, Ferreira KS, Fares RC, et al. Immunoregulatory mechanisms in Chagas disease: modulation of apoptosis in T-cell mediated immune responses. BMC Infect Dis (2016) 16:191. doi:10.1186/s12879-016-1523-1
126. Freire-de-Lima CG, Nascimento DO, Soares MB, Bozza PT, Castro-Faria-Neto HC, de Mello FG, et al. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature (2000) 403(6766):199–203. doi:10.1038/35003208
127. Silva EM, Guillermo LV, Ribeiro-Gomes FL, De Meis J, Nunes MP, Senra JF, et al. Caspase inhibition reduces lymphocyte apoptosis and improves host immune responses to Trypanosoma cruzi infection. Eur J Immunol (2007) 37(3):738–46. doi:10.1002/eji.200636790
128. Torchinsky MB, Garaude J, Blander JM. Infection and apoptosis as a combined inflammatory trigger. Curr Opin Immunol (2010) 22(1):55–62. doi:10.1016/j.coi.2010.01.003
129. Torchinsky MB, Garaude J, Martin AP, Blander JM. Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature (2009) 458(7234):78–82. doi:10.1038/nature07781
130. Penteado LA, Dejani NN, Verdan FF, Orlando AB, Nino VE, Dias FN, et al. Distinctive role of efferocytosis in dendritic cell maturation and migration in sterile or infectious conditions. Immunology (2017) 151(3):304–13. doi:10.1111/imm.12731
131. Campisi L, Barbet G, Ding Y, Esplugues E, Flavell RA, Blander JM. Apoptosis in response to microbial infection induces autoreactive TH17 cells. Nat Immunol (2016) 17(9):1084–92. doi:10.1038/ni.3512
132. Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature (1998) 392(6671):86–9. doi:10.1038/32183
133. Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol (2010) 10(6):403–14. doi:10.1038/nri2780
134. van Montfoort N, van der Aa E, Woltman AM. Understanding MHC class I presentation of viral antigens by human dendritic cells as a basis for rational design of therapeutic vaccines. Front Immunol (2014) 5:182. doi:10.3389/fimmu.2014.00182
135. Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature (2011) 474(7353):654–7. doi:10.1038/nature10117
136. Silvin A, Yu CI, Lahaye X, Imperatore F, Brault JB, Cardinaud S, et al. Constitutive resistance to viral infection in human CD141+ dendritic cells. Sci Immunol (2017) 2(13):eaai8071. doi:10.1126/sciimmunol.aai8071
137. Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science (2015) 350(6258):328–34. doi:10.1126/science.aad0395
138. Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature (2013) 503(7476):402–5. doi:10.1038/nature12769
139. Lahaye X, Satoh T, Gentili M, Cerboni S, Conrad C, Hurbain I, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity (2013) 39(6):1132–42. doi:10.1016/j.immuni.2013.11.002
140. Decalf J, Desdouits M, Rodrigues V, Gobert FX, Gentili M, Marques-Ladeira S, et al. Sensing of HIV-1 entry triggers a type I interferon response in human primary macrophages. J Virol (2017) 91(15):e00147-17. doi:10.1128/JVI.00147-17
141. Cooper A, García M, Petrovas C, Yamamoto T, Koup RA, Nabel GJ. HIV-1 causes CD4 cell death through DNA-dependent protein kinase during viral integration. Nature (2013) 498(7454):376–9. doi:10.1038/nature12274
142. Doitsh G, Galloway NLK, Geng X, Yang Z, Monroe KM, Zepeda O, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature (2014) 505:509–14. doi:10.1038/nature12940
143. Lepelley A, Louis S, Sourisseau M, Law HK, Pothlichet J, Schilte C, et al. Innate sensing of HIV-infected cells. PLoS Pathog (2011) 7(2):e1001284. doi:10.1371/journal.ppat.1001284
144. Munoz LE, Franz S, Pausch F, Furnrohr B, Sheriff A, Vogt B, et al. The influence on the immunomodulatory effects of dying and dead cells of Annexin V. J Leukoc Biol (2007) 81(1):6–14. doi:10.1189/jlb.0306166
145. Moody MA, Liao HX, Alam SM, Scearce RM, Plonk MK, Kozink DM, et al. Anti-phospholipid human monoclonal antibodies inhibit CCR5-tropic HIV-1 and induce beta-chemokines. J Exp Med (2010) 207(4):763–76. doi:10.1084/jem.20091281
146. Soares MM, King SW, Thorpe PE. Targeting inside-out phosphatidylserine as a therapeutic strategy for viral diseases. Nat Med (2008) 14(12):1357–62. doi:10.1038/nm.1885
147. Taiwo B, Barcena L, Tressler R. Understanding and controlling chronic immune activation in the HIV-infected patients suppressed on combination antiretroviral therapy. Curr HIV/AIDS Rep (2013) 10(1):21–32. doi:10.1007/s11904-012-0147-3
148. Naggie S. Hepatitis C virus, inflammation, and cellular aging: turning back time. Top Antivir Med (2017) 25(1):3–6.
149. Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med (2004) 200(6):749–59. doi:10.1084/jem.20040874
150. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med (2006) 12(12):1365–71. doi:10.1038/nm1511
Keywords: phagocytosis, efferocytosis, cell death, pathogen, bacteria, virus, parasite, inflammation
Citation: Karaji N and Sattentau QJ (2017) Efferocytosis of Pathogen-Infected Cells. Front. Immunol. 8:1863. doi: 10.3389/fimmu.2017.01863
Received: 02 October 2017; Accepted: 07 December 2017;
Published: 22 December 2017
Edited by:
Christel Vérollet, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceReviewed by:
Mark Marsh, University College London, United KingdomPhilippe Benaroch, Centre national de la recherche scientifique (CNRS), France
Serge Benichou, Centre national de la recherche scientifique (CNRS), France
Copyright: © 2017 Karaji and Sattentau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quentin J. Sattentau, quentin.sattentau@path.ox.ac.uk