 Niamh C. Williams
Niamh C. Williams Luke A. J. O’Neill*
Luke A. J. O’Neill*
- School of Biochemistry and Immunology, Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin, Ireland
Metabolism in immune cells is no longer thought of as merely a process for adenosine triphosphate (ATP) production, biosynthesis, and catabolism. The reprogramming of metabolic pathways upon activation is also for the production of metabolites that can act as immune signaling molecules. Activated dendritic cells (DCs) and macrophages have an altered Krebs cycle, one consequence of which is the accumulation of both citrate and succinate. Citrate is exported from the mitochondria via the mitochondrial citrate- carrier. Cytosolic metabolism of citrate to acetyl-coenzyme A (acetyl-CoA) is important for both fatty-acid synthesis and protein acetylation, both of which have been linked to macrophage and DC activation. Citrate-derived itaconate has a direct antibacterial effect and also has been shown to act as an anti-inflammatory agent, inhibiting succinate dehydrogenase. These findings identify citrate as an important metabolite for macrophage and DC effector function.
Metabolic Reprogramming in Macrophages and Dendritic Cells (DCs)
The innate immune system is the first line of defense against infection. Cells of the innate immune system have a range of germline encoded receptors, pathogen recognition receptors that allow for the recognition of pathogen-associated molecular patterns, and danger-associated molecular patterns from damaged cells or tissues (1, 2). Macrophages and DCs play an important role in the initiation and resolution of the immune response. Both can produce inflammatory mediators, phagocytose pathogens and release chemokines to recruit other immune cells to the site of infection (3). DCs are also important in the activation of naive T cells as they can present antigen to the T cell initiating an adaptive immune response (4).
The dual role played by macrophages in initiation and resolution of inflammation requires cells to adopt different processes. In macrophages, this can be broadly described in terms of M1, lipopolysaccharide (LPS)- or classically activated macrophages, and M2, IL-4-activated macrophages. M1 macrophages are more pro-inflammatory and will produce inflammatory mediators, such as nitric oxide (NO) and reactive oxygen species (ROS) (3, 5). M2 macrophages are important in helminth infection and the resolution of inflammation, secreting growth factors to aid in tissue repair and regeneration and cytokines such as IL-10 that can dampen the immune response (3, 6).
Both macrophages and DCs must be able to switch rapidly from a resting to an activated state. A hallmark of immune cell activation is a change in their metabolism. M1 macrophages upregulate glycolysis and the pentose phosphate pathway (PPP) while the Krebs cycle is broken at two points and the fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) are downregulated (5). Toll-like receptor (TLR)-activated DCs also have increased aerobic glycolysis and decreased OXPHOS and FAO (7). This inhibition of mitochondrial respiration in murine DCs is due to NO and long-term activation of glycolysis in activated DCs serves to produce adenosine triphosphate (ATP) to compensate for the collapse in mitochondrial function, maintain the mitochondrial membrane potential (ΔψM) and prevent cell death (8). The high rate of glycolysis is similar to that seen in tumor cells (3). Murine M2 macrophages also upregulate glycolysis, but the Krebs cycle is intact and OXPHOS is functioning (5). A general theme exists among immune cells where a reliance on aerobic glycolysis is important for cells, such as M1 macrophages and DCs, whereas immunomodulatory cells, such as M2 macrophages and regulatory T cells (Tregs), make use of OXPHOS (9).
Recent work has shed light on several of the key determinants of metabolic reprogramming in M1 macrophages and DCs. The upregulation of inducible nitric oxide synthase (iNOS) and resulting generation of NO causes inhibition of mitochondrial respiration in murine cells (10–12). Hypoxia-inducible factor-1α (HIF1α) can be induced under normoxic conditions in immune cells and this is crucial for upregulation of glycolysis (13). HIF-target genes included those encoding for glycolytic enzymes, the glucose transporter GLUT1, and lactate dehydrogenase (LDH), as well as inflammatory factors such as interleukin-1β (IL1β) (14–16). LPS-treatment of macrophages or DCs activates the mammalian target of rapamycin (mTOR), a central regulator of metabolism, which in turn boosts expression and activity of HIF1α (17). Finally AMP-activated protein kinase (AMPK) is inhibited by LPS treatment (18). AMPK senses the energy status of a cell, and when that is low this inhibits anabolic pathways and drives catabolic ones such as FAO, while also inhibiting mTOR and nuclear factor-κB (NF-κB) signaling. Its inhibition by LPS allows changes necessary for a pro-inflammatory response to occur. Several processes that are upregulated in M1 macrophages are downregulated in M2 macrophages. In mouse macrophages, iNOS expression is decreased and arginase-1 (ARG1) is highly upregulated, and so arginine is preferentially metabolized to proline and polyamines (19). mTOR is inhibited by activation of upstream repressors TSC1 and TSC2 while AMPK activity is high (18, 20).
It is possible that the upregulation and reliance on aerobic glycolysis is in part due to the rate of response required of these cells for an effective immune system. Glycolysis, though less efficient at producing ATP, is able to do so more rapidly than OXPHOS. However, there is evidence that upregulation of metabolic pathways is more nuanced than that. Metabolic changes are important not only in terms of generating biosynthetic precursors and for ATP production, but it also has emerged that metabolites themselves can act as signaling molecules and affect important inflammatory pathways (21, 22). Of particular interest are metabolites of the Krebs cycle. The oxidation of succinate by succinate dehydrogenase (SDH) has been shown to be of importance in the classical activation of macrophages (23). This leads to reverse electron transport (RET) in complex I of the electron transport chain (ETC) driving the production of ROS, which in turn leads to activation of HIF1α. Increased levels of cytosolic succinate can inhibit the prolyl hydroxylase domain enzymes via product inhibition, also potentiating HIF1α stabilization (24). This prevents the hydroxylation of proline residues on HIF1α, and so it is not ubquitinated and targeted for proteasomal degradation (25–28). Instead, it can heterodimerize with its binding partner the aryl hydrocarbon nuclear translocator (ARNT/HIF-1β). The HIF-1 complex can translocate to the nucleus and bind hypoxia response elements in the promoters of HIF target genes (29). HIF also represses mitochondrial function through upregulation of pyruvate dehydrogenase kinase 1 (PDK1) (30). PDK1 phosphorylates and inhibits pyruvate dehydrogenase (PDH) and so pyruvate cannot be converted into acetyl-CoA in order to enter the mitochondria and feed the Krebs cycle (31).
The fragmented Krebs cycle in macrophages is not only due to the break after succinate. A second breakpoint, at isocitrate dehydrogenase (IDH), allows for the withdrawal of citrate from the cycle. This proves not only to be important for lipid biosynthesis in macrophages and DCs, but also for the production of both pro- and anti-inflammatory mediators (32, 33). Glycolysis is rapidly upregulated in LPS-activated DCs for the production of citrate. This is necessary for the upregulation of fatty acid synthesis to allow for membrane expansion which is crucial for antigen presentation (34). Once exported to the cytosol citrate can be broken down to provide a source of acetyl-CoA for acetylation of both histone and non-histone proteins (35). Citrate metabolism provides a connection between carbohydrate metabolism, fatty acid metabolism, and epigenetic reprogramming and so changes in flux through this pathway may have wide ranging effects. This review will describe the role of citrate in innate immune cell function.
Citrate Provides a Bridge Between Carbohydrate and Fatty Acid Metabolism
Citrate is produced in the Krebs cycle (also known as the citric acid cycle or TCA cycle) from the aldol condensation of oxaloacetate, the end product of a previous turn of the cycle, and acetyl-CoA (Figure 1) (36). Acetyl-CoA may be derived from glucose via the glycolytic pathway, entering the mitochondria as pyruvate or from fatty acids that have undergone β-oxidation (36). In the Krebs cycle, citrate is converted into isocitrate via cis-aconitate by aconitase (36). IDH will then convert isocitrate to α-ketogluterate (αKG) in a decarboxylation reaction (36). The Krebs cycle continues and provides a major source of cellular ATP and also reducing equivalents that feed the electron transfer chain (36).
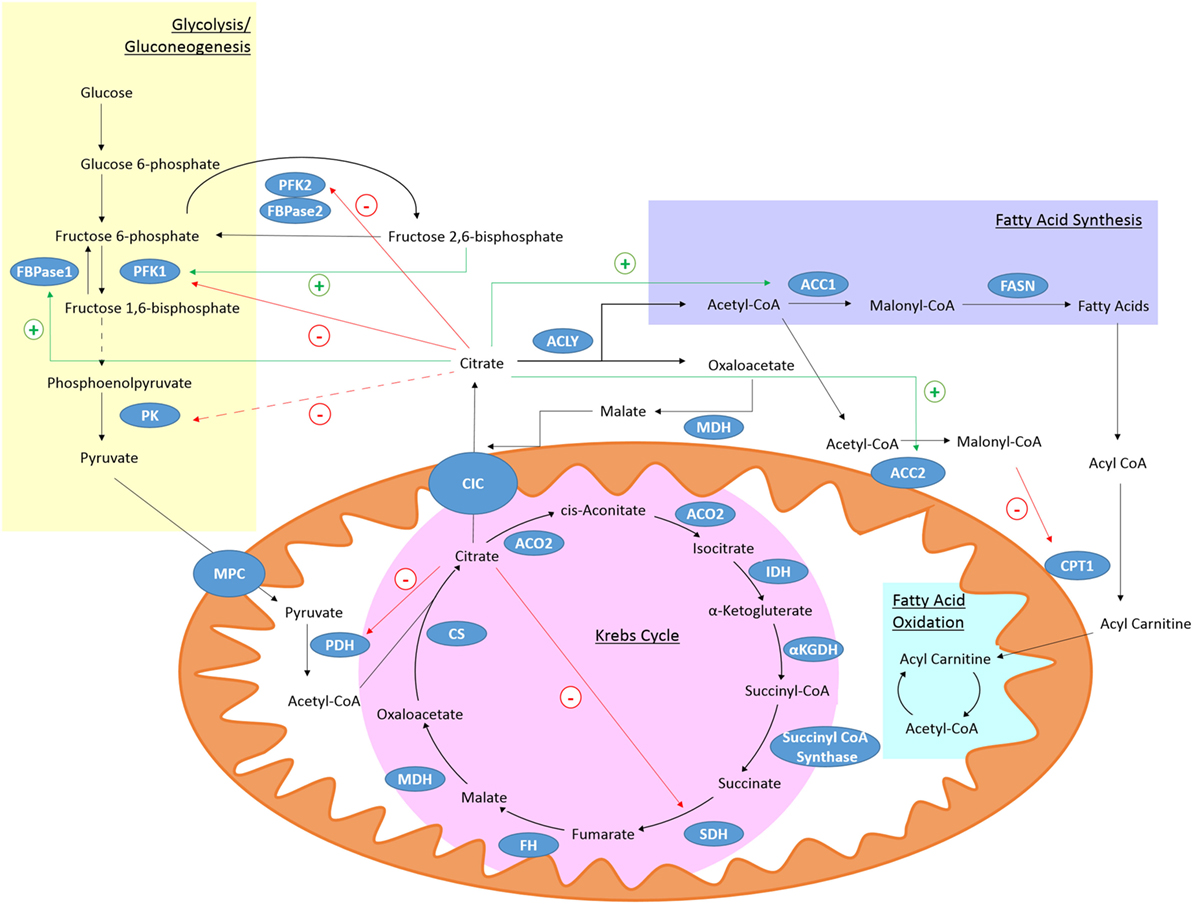
Figure 1. Citrate metabolism. Citrate is produced in the Krebs cycle from oxaloacetate and acetyl-CoA by citrate synthase (CS). It can be exported from the mitochondria through citrate carrier (CIC). Cytosolic citrate is broken down by ACLY to oxaloacetate and acetyl-CoA. Acetyl-CoA can be used as a substrate for fatty acid synthesis. High levels of cytosolic citrate can directly inhibit the glycolytic enzymes PFK1 and PFK2. PFK1 is also indirectly inhibited by decreased levels of fructose 2,6-bisphosphate and pyruvate kinase (PK) is indirectly inhibited (broken line) due to reduced levels of its activator, fructose-1,6-bisphosphate. Mitochondrial citrate can inhibit pyruvate dehydrogenase (PDH) and succinate dehydrogenase (SDH). Citrate-derived malonyl-CoA can block fatty acid oxidation (FAO) by inhibiting carnitine palmitoyltransferase 1 (CPT1). Citrate can activate the gluconeogenic enzyme fructose 1,6-bisphosphatase (FBPase1) and ACC, and so stimulate fatty acid synthesis.
The mitochondrial citrate carrier (CIC), also known as solute carrier family 25 member 1 (Slc25a1), can export citrate from the mitochondria in exchange for malate (37). Once in the cytosol citrate is broken down by ATP-Citrate lyase (ACLY) into acetyl-CoA and oxaloacetate (37). Oxaloacetate can be converted to malate by malate dehydrogenase (MDH) which can re-enter the mitochondria through CIC (37). Acetyl-CoA is further processed into malonyl-coenzyme A (malonyl-CoA) by acetyl-coA carboxylase (ACC) (38). Malonyl-CoA can be incorporated into cholesterol or fatty acids (38). The fatty acids are incorporated into phospholipids. Malonyl-CoA can also limit the β-oxidation of fatty acids as high levels can inhibit carnitine palmitoyltransferase 1 (CPT1) (39). Two isoforms of ACC exist, ACC1 and ACC2 (40). ACC2 is associated with the outer mitochondrial membrane and so can control the concentration of malonyl-CoA near CPT1 and regulate its activity (38). Acetyl-CoA can also be a substrate for protein and histone acetylation and so can have a wide ranging role in many cellular processes (41).
Citrate itself is known to inhibit several key glycolytic enzymes as part of a negative feedback loop. Phosphofructokinase (PFK) 1 and 2 are directly inhibited by citrate while pyruvate kinase (PK) is indirectly inhibited as citrate decreases levels of fructose-1,6-bisphosphate, which is a PK activator (42). PDH (43) and SDH (44), and therefore the Krebs cycle, can also be inhibited by high citrate levels. While citrate inhibits pathways producing ATP, it stimulates those that consume it. Citrate can allosterically activate ACC (45) and the gluconeogenic enzyme fructose-1,6-bisphosphatase (46). With citrate occupying a position that links many metabolic and cellular processes, it is not surprising that the metabolism of citrate may be of importance in the immune response.
Citrate as an Inflammatory Signal
While the role of other metabolites as inflammatory signals has been well discussed (21, 22) citrate also plays a role in key inflammatory pathways (Figure 2). In M1 macrophages, there is an increased isocitrate:αKG ratio and transcriptional downregulation of Idh1 (47). This break was also seen in DCs (34). With increased glycolytic flux in both activated DCs and macrophages and a break in the Krebs cycle, pyruvate derived from glucose feeds into Krebs cycle but cannot continue past citrate/isocitrate. An increase in the levels of citrate is detected in both mouse (LPS-stimulated) and human [tumour necrosis factor α (TNFα)- or interferon-γ (IFNγ)-stimulated] macrophages (24, 48). This coincides with upregulation of CIC and ACLY, both of which occur in an NF-κB-dependent manner where LPS or TNFα is used to activate the cells, or IFNγ can also induce CIC and ACLY via STAT1 (32, 49). The export and breakdown of mitochondrial citrate has been linked to the production of several important pro-inflammatory mediators in macrophages, namely NO, ROS, and prostaglandin E2 (PGE2) production in human macrophages (32, 48, 49). Inhibition of CIC activity or its genetic silencing with siRNA leads to a marked reduction in NO, ROS, and PGE2 production in LPS and cytokine-stimulated macrophages. Infantino et al. suggest that the decrease in PGE2 production is due to a decreased availability of precursors for PGE2 synthesis as adding exogenous acetate rescues the effect of CIC inhibition on PGE2 productions. Acetate can be converted to acetyl-CoA by acetyl-CoA synthase (ACSS) (50). Inhibition of fatty acid synthase (FASN) with C75 in DCs also reduced LPS-induced PGE2 (34). Endogenous PGE2 is essential for the production of LPS-induced pro-IL1β (51). This implies that citrate may be critical to IL1β production.
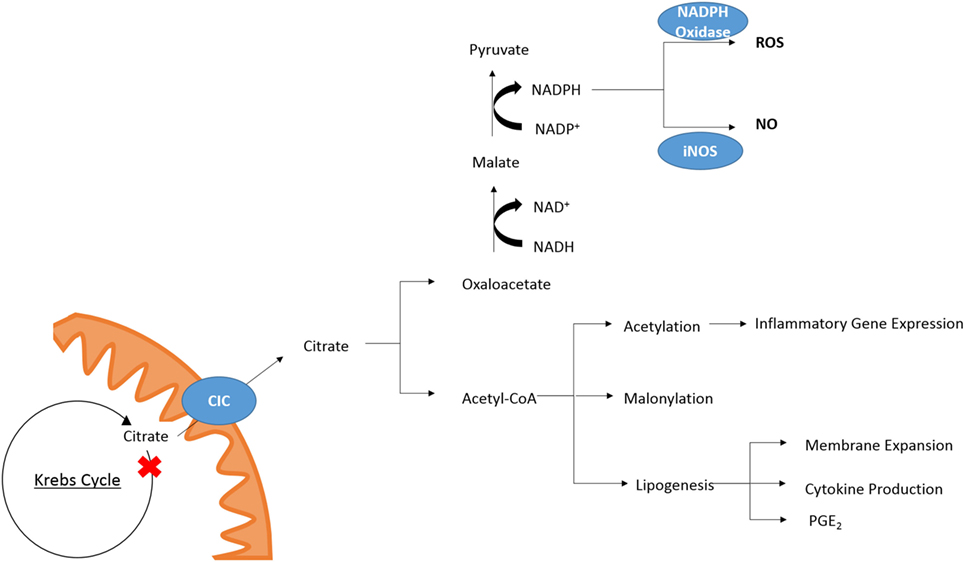
Figure 2. Citrate in inflammation. Cytosolic citrate is broken down into acetyl-CoA and oxaloacetate. Further processing of oxaloacetate can provide a source of NADPH which is required by NAPDH oxidase and iNOS for the production of ROS and NO, respectively. Acetyl-CoA is required for acetylation of proteins and can be converted to malonyl-CoA for lysine-malonylation. Citrate-derived lipid synthesis has been linked to membrane expansion necessary for DC activation and cytokine production. In macrophages, the production of PGE2 has been linked to citrate metabolism.
While Infantino et al. speculated that the effect of CIC inhibition on the production of NO and ROS was due to decreased production of NADPH by the breakdown of citrate-derived oxaloacetate to pyruvate by malic enzyme, a follow-up study reports that the NADP+/NAPDH ratio is unchanged in cytokine-activated macrophages treated with the CIC inhibitor 4-chloro-3-[(3-nitrophenyl)amino]sulfonyl benzoic acid (CNSB) versus untreated when they are in a glucose rich environment (52). When the same experiment was carried out in glucose-deprived conditions, there was a marked increase in the NADP+/NADPH ratio in activated, CNSB-treated macrophages compared to control cells. Citrate export from the mitochondria can provide a source of NADPH via two routes. The first is from oxaloacetate as mentioned, and the second is via the cytosolic, NADP+-dependent IDH (IDH1). IDH1 is known to be suppressed in cells activated by a pro-inflammatory stimuli (53), however, in glucose-deprived conditions the authors show IDH1 mRNA expression is increased in activated macrophages (52). While this could provide a mechanism for the decreased production of NO and ROS in glucose-deprived cells other processes such as the PPP may be compensating for decreased production of NADPH in macrophages treated with a CIC inhibitor but grown in a glucose-rich environment.
The processing of citrate by ACLY has also been suggested as an anti-cancer target as ACLY has been found to be overexpressed in several cancer types (54). Inhibition of ACLY with siRNA or pharmacologically induces cell death (55–58). One study has suggested that the anti-cancer effect of ACLY inhibition is directly due to its role in lipid synthesis (55). In several cancer cell lines, depletion of ACLY was observed to induce apoptosis, accompanied by increased levels of ROS. This increase in ROS was suggested to initiate cell death by activating AMPK (59). Another study has suggested that enzymatic activity alone is not enough to explain the initiation of growth arrest in tumor cells, and has shown that ACLY may be able to directly interact with AMPK (60). While ACLY has not been linked to the production of ROS in immune cells, an exact mechanism for the role of citrate metabolism in macrophages has not been defined and it would be an interesting avenue of investigation.
Similarly, citrate has been shown to be important for DC activation. Mitochondrial respiration in blood monocyte-derived DCs will begin to collapse in TLR-activated DCs 6 h after treatment, and by 24 h, there will be no detectable consumption of oxygen by the mitochondria (8). However, before 6 h the mitochondria are still functioning. Up until this time, the increase in glycolysis is important for DC activation but the increase does not fuel a greater rate of OXPHOS, inhibiting ATP synthase with oligomycin has no effect on early DC activation. Instead citrate is withdrawn from the Krebs cycle and used to support de novo fatty acid synthesis (34). Inhibition of ACC or FASN, with TOFA and C75, respectively, or blocking expression of Slc25a1 by retroviral introduction of shRNA diminished the early activation of DCs differentiated from bone marrow in the presence of the growth factor GM-CSF (GM-DCs). Production of fatty acids is required for membrane synthesis for the expansion of both the endoplasmic reticulum and Golgi. Their expansion allows for increased synthesis and secretion of various proteins important following TLR-activation. The PPP is also an important process in early DC activation, as it creates the reducing equivalent, NADPH, required not only for nucleotide synthesis and redox balance but also as a cofactor for lipogenesis. While the long-term commitment to glycolysis and collapse in mitochondrial respiration seen in activated DCs is dependent on a phosphatidylinositide-3-kinase (PI3K)/Akt pathway, the rapid upregulation of glycolysis was due to TANK-binding kinase 1 (TBK1)- and IκB Kinase ε (IKKε)-dependent activation of Akt. The PI3K/Akt pathway lead to production of NO, and inhibition of OXPHOS, forcing the cells to rely on increased glycolysis for activation and survival but the early TBK1-IKKε/Akt pathway increased glycolysis by promoting the association of hexokinase 2 (HK2) with voltage-dependant anion channels on the outer mitochondrial membrane.
While the same breakpoints in the Krebs cycle have not been described in activated natural killer cells (NK cells), the citrate–malate shuttle has recently been shown to be of importance in the metabolic reprogramming that occurs following NK cell activation (61). The citrate–malate shuttle refers to the export of citrate into the cytosol via CIC, and its breakdown by ACLY and malate dehydrogenase 1 (MDH1) yielding cytosolic malate which CIC exchanges for citrate. Assmann et al. show that increased expression of Slc25a1 and ACLY mRNA in activated NK cells is dependent on sterol regulatory element-binding protein (SREBP) activity, which is consistent with reports in other cell types including macrophages (62–66). Activated NK cells have increased glycolysis and OXPHOS which is crucial for their activation and growth. Pharmacological inhibition of SREBP activation or ACLY activity (therefore, inhibiting the citrate–malate shuttle) reduced cytokine-induced granzyme B expression and IFN-γ production. Similar results were seen with genetic inhibition of SREBP using SCAP−/− mice. This effect is independent of lipid and cholesterol synthesis downstream of ACLY as inhibition of ACC or FASN with TOFA or C75, respectively, does not affect NK cell activation. The authors speculate that the citrate–malate shuttle serves to convert cytosolic NADH to mitochondrial NADH, therefore, fueling OXPHOS and mitochondrial ATP synthesis while also replenishing the cytosolic pool of NAD+, which is an important cofactor for glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Inhibition of the malate–aspartate shuttle, which would be more commonly thought of as a means of replenishing mitochondrial NADH, did not affect OXPHOS in activated NK cells and neither did inhibition of the Krebs cycle enzyme SDH with dimethyl-malonate. This suggests that in NK cells elevated OXPHOS is maintained by the citrate–malate shuttle. While these studies show the cytosolic processing of citrate to be generally a pro-inflammatory event, citrate itself has been shown to inhibit the HIF asparaginyl hydroxylase [factor inhibiting HIF (FIH)] (67). FIH hydroxylates asparagine residues on HIF1α, preventing it from interacting with transcriptional coactivators such as CRE binding protein/p300 (67, 68). Increased flux through CIC and the cytosolic processing of citrate has, therefore, been shown to be of importance in the activation of macrophages, DCs, and NK cells.
Histone and Post-Translational Modifications by Citrate-Derived Acetylation
Acetyl-CoA is not only a substrate for de novo lipogenesis, it also is an important cofactor for the acetylation of histones and non-histone proteins (69). Acetylation can be co-translational, effecting the α-amino group of a protein’s N-terminal residue, or post-translational which concerns the ε-amino group of lysine residues (69, 70). Lysine acetylation is reversible and so provides a very useful mechanism for the regulation of gene expression and general protein function (69). Acetyl-CoA cannot travel across cell membranes, and so to exert its effects it must be generated in different cellular compartments (70). In the mitochondria, acetyl-CoA is present due to the β-oxidation of fatty acids or is generated from pyruvate by the PDH complex (41). In the cytosol, acetyl-CoA can be derived from citrate as previously discussed or from acetate by ACSS (71) or the degradation or N-acetylaspartate (NAA) by aspartoacylase (72). The generation of acetyl-CoA from NAA is more commonly associated with processes in the brain and it has also been shown to be a source of nuclear and cytosolic acetyl-CoA in brown adipose tissue (73).
ATP-citrate lyase links metabolism to histone acetylation as it converts glucose-derived citrate to acetyl-CoA and it has been found to localize to both nucleus and cytoplasm (35). Citrate is small enough to diffuse across nuclear pores allowing for acetyl-CoA to be produced in either cellular compartment, and siRNA-mediated knockdown of ACLY reduced global histone acetylation (35). In adipocytes mRNA expression of HK2, PFK1, and lactate dehydrogenase A (LDHA) were all reduced when ACLY was silenced (35). Since ACLY is upregulated in lipopolysaccharide (LPS)-stimulated macrophages (49), it would be interesting to see where ACLY localized to and if there was a direct effect on the expression of glycolytic genes due to changes in histone acetylation. ACLY has been shown to control glucose to acetate switch. ACLY-deficient cells upregulate ACSS2 allowing for the production of acetyl-CoA from acetate, ensuring cell viability and providing substrates for both fatty acid synthesis and histone acetylation (74).
As previously discussed, in glucose-deprived conditions, increased flux through CIC can sustain NADPH levels in glucose-deprived activated macrophages (52). Acetylation of CIC increases in glucose-deprived growth conditions compared to media containing glucose. By reconstituting liposomes with mitochondrial extracts, it was shown that the acetylation of CIC causes an increase in Vmax for citrate (52). In memory, CD8+ T cells GAPDH has been shown to be acetylated in an ACLY-dependent manner which increased its activity (75). Acetate is taken up and processed via the Krebs cycle to produce citrate. Citrate-derived acetyl-CoA was then used to acetylate GAPDH which may prevent it from binding IFNγ mRNA, and allowing its translation (76). Acetylated GAPDH had increased catalytic activity. While acetate can be used directly to generate acetyl-CoA, knockdown of the cytosolic ACSS (ACSS1) had no effect on IFNγ production, knockdown of ACLY, however, did decrease IFNγ production. It has also been shown that LDHA in T cells promotes the expression of IFNγ, independent of GAPDH regulation, as it ensures a high acetyl-CoA concentration produced via ACLY for histone acetylation (77). Though metabolic reprogramming differs in activated T cells compared to macrophages and DCs, this highlights the importance of the citrate pathway in control of both the metabolism of immune cells and their production of pro-inflammatory mediators, unlike in T cells. No work has yet been carried out to directly link citrate-derived acetylation in M1 macrophages or DCs, however, histone acetylation is important in macrophage activation and DC differentiation. IL-6 and IL-10 production are both regulated on histone and non-histone protein acetylation, respectively (78, 79). NF-κB activation is also dependent on acetylation of its RelA/p65 subunit (80) and a large number of enzymes involved in metabolic processes have been found to be acetylated in non-immune cells (78, 81). Therefore, it is likely that acetylation plays a role in the regulation of immune cell metabolism. Histone acetylation downstream of ACLY has been shown to be of importance in M2 macrophage activation (82). While STAT6 is the major regulator of IL4 induced genes a subset of genes important in the regulation of cellular proliferation and the production of chemokines are under additional control of an Akt–mTORC1 signaling pathway. Covarrubias et al. suggest a mechanism in which Akt regulates both protein levels and activity of ACLY to increase the acetyl-CoA pool for histone acetylation. They suggest that certain transcription factors and histone acetyltransferases, e.g., P300, are regulated by acetyl-CoA levels and that may link AKT/ACLY-dependent acetyl-CoA levels to specific gene induction.
Lysine-malonylation causes a net change in charge of the lysine residue from +1 to −1 and cause a change in mass of approximately 86 Da (83). Malonyl-CoA is the cofactor required (84). Malonyl-CoA can be produced in the cytosol from citrate-derived acetyl-CoA by ACC1/2 (38). The cytosolic pool of malonyl-CoA is regulated by both ACC and malonyl-CoA decarboxylase which catalyzes the reverse reaction from malonyl-CoA to acetyl-CoA (38). A malonyl-CoA pool also exists in the mitochondria, produced from acetyl-CoA by propionyl-CoA carboxylase or from malonate by acyl-CoA synthase family member 3 (ACSF3) (85). Lysine-malonylation has been shown to play a role in the regulation of mitochondrial function, FAO and glycolysis (86, 87). Notably histone malonylation does not occur at the N-terminal tail as happens with acetylation, suggesting that the regulatory role these two modification carry out may be very functionally different to acetylation (86). A large number of proteins involved in fatty acid metabolism are malonylated, including ACLY. However, no studies have yet been carried out regarding the functional consequence of lysine-malonylation in immune cells.
Itaconate
While the accumulation of citrate caused by the IDH1 breakpoint in the TCA cycle can be used to fuel fatty acid synthesis and histone acetylation, another fate of this citrate is the production of itaconate (Figure 3). First identified in 1836 as a product of the distillation of citric acid, itaconate has recently become a focus of the field of immunometabolism due to its potential role as an anti-inflammatory modulator. Itaconate is derived from citrate produced in the Krebs cycle and, in M1 macrophages, is one of the most highly induced metabolites following LPS treatment (33). Itaconate has also been shown to accumulate in LPS-treated DCs (34). Citrate is acted on by the mitochondrial aconitase 2 (ACO2) to produce cis-aconitate. Cis-aconitate is decarboxlylated by cis-aconitate decarboxylase, also known as immune-responsive gene 1 (IRG1), to produce itaconate. Itaconate has long been used in an industrial setting and is produced on an industrial scale as a fermentation product of Aspergillus terreus for use in the creation of polymer formation (88).
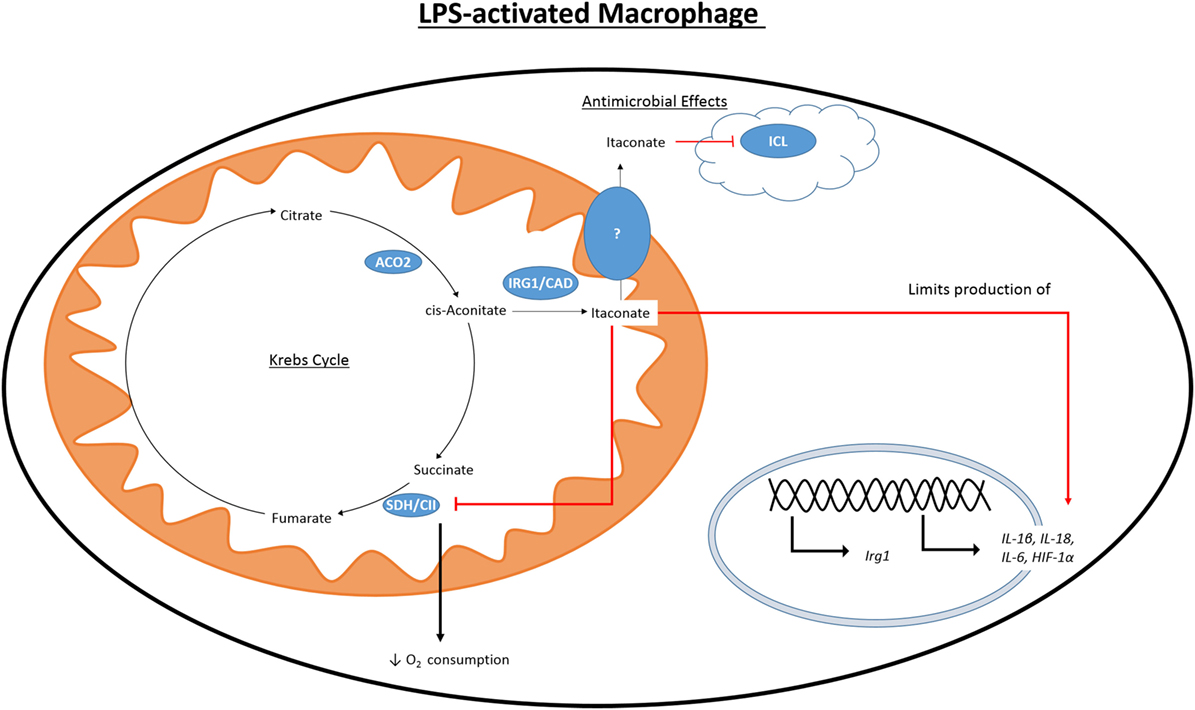
Figure 3. Citrate-derived itaconate. Citrate is converted to cis-aconitate by the Krebs cycle enzyme ACO2. Activation of macrophages with LPS causes induction of IRG1 which can produce itaconate by the decarboxylation of cis-aconitate. LPS also initiates the production of other pro-inflammatory mediators, such as HIF1 α, NO, and IL1β. Itaconate is toxic to microorganisms expressing ICL, a key component of the glycoxylate shunt in bacteria. Itaconate is also able to inhibit SDH and cause decreased production of NO, IL1β, IL18, and HIF1α.
In 1995, it was found that immunoresponsive gene 1 (Irg1) is highly upregulated in peritoneal macrophages following LPS stimulation (89), and has since been seen to be upregulated in the blood of human sepsis patients (90) and in the time during embryo implantation (91, 92). Despite lacking a sequence targeting it, there IRG1 has been found to associate with the mitochondria (93, 94). It was only in 2011 that itaconate was identified in multiple studies in an immune context and in 2013 that IRG1 and itaconate were connected (95). Itaconate was seen in the lungs of mice infected with Mycobacterium tuberculosis (MTB) and was not present in the lungs of control mice (96). In a separate study, itaconate was shown to be secreted by the macrophage cell line RAW264.7 following treatment with LPS (97). Michelucci et al. identified the role of IRG1 by siRNA-mediated silencing of Irg1 (officially renamed Acod1 after its function was discovered) and performing a metabolic screen to elucidate which metabolites were affected. They further showed by isotope-labeling that itaconate was derived from citrate. Genetic silencing of Irg1 causes macrophages to lose their bactericidal activity, which was due to decreased amounts of itaconate and the loss of its inhibitory effect on isocitrate lyase (ICL), a crucial enzyme of the glycoxylate shunt in bacteria (98). The glycoxylate shunt is a means for bacteria to survive in conditions of low glucose availability where acetate is the primary fuel source. Instead of the normal sequence of reaction in the Krebs cycle, the steps from αKG to fumarate are bypassed, and isocitrate is instead cleaved by ICL to glycoxylate and succinate in bacteria. Succinate enters the Krebs cycle and glycoxylate is then converted to malate by malate synthase. Malate can be processed to oxaloacetate by MDH as in the normal reactions of the Krebs cycle. Itaconate has been shown to inhibit growth of a number of ICL expressing microorganisms, including MTB, S. enterica and multidrug-resistant Staphylococcus aureus (MRSA) (95, 96, 99). Some bacteria are able to degrade itaconate, producing acetyl-CoA and pyruvate, due to the expression of genes that encode for itaconate-CoA transferase, itaconyl-CoA hydratase, and (S)-citramalyl-CoA ligase. Possession of these genes allows Pseudomonas aeruginosa and Yersinia pestis to survive in activated macrophages (100). There is a discussion as to the relevance of these studies due to differences in concentrations of itaconate used both in terms of the variety of concentrations used exogenously to inhibit bacterial growth and the range in reported intracellular concentrations (101, 102). It may be that intracellular itaconate is concentrated in vacuoles, and whole cell analysis will not adequately represent this, and that measuring the concentration of secreted itaconate in cell culture media does not determine what the local concentration would be.
While the effect of itaconate on bacterial survival has been well documented, more recent work has sought to elucidate the effect that a high intracellular concentration of itaconate has on the immune cells that produce it. Dimethyl itaconate (DMI) has been used in several studies as a cell permeable itaconate analog to boost the intracellular levels of itaconate. Pre-treatment of murine bone marrow-derived macrophages (BMDMs) with DMI prior to LPS stimulation reduced the reduced the expression of many pro-inflammatory genes including iNOS (33). DMI also impaired production of ROS, NO, IL1β, IL18, and IL6. While IL1β and IL18 mRNA expression was reduced by DMI it also caused a reduction in protein levels of both NLRP3 and ASC, indicating that inflammasome priming was also impaired. Irg1−/− BMDMs do not produce itaconate and have increased production of NO, IL1β, IL18, and IL6 when compared to WT BMDMs. HIF1α protein levels were also increased in LPS-activated Irg1−/− BMDMs while treatment with DMI inhibited its protein levels. Interestingly, Irg1−/− BMDMs show an altered profile of Krebs cycle metabolites than WT cells. WT BMDMs have increased levels of succinate, fumarate, and malate following LPS stimulation (47). LPS-stimulated Irg1−/− BMDMs have significantly higher levels of fumarate and malate than WT controls, while succinate levels are almost that of unstimulated cells. The authors suggest that this is due to the ability of itaconate to inhibit SDH, and they showed itaconate to inhibit a purified form of SDH. In contrast to the decrease in oxygen consumption rate (OCR) seen in WT BMDMs upon treatment with LPS, in Irg1−/− BMDMs OCR increases. As SDH also acts as complex II of the ETC, this highlights the ability of endogenous itaconate to regulate mitochondrial metabolism and is consistent with other reports of itaconate competitively inhibiting SDH, albeit weakly, the first of which was in 1949 (103–105). This led Lampropoulou et al. to suggest that LPS-induced Irg1 expression and corresponding increase in intracellular itaconate is responsible for the second break in the Krebs cycle at SDH. By inhibiting SDH itaconate would also prevent the generation of ROS through RET (106). When succinate accumulates and is oxidized by SDH, it will produce a large amount of coenzyme Q. Electrons are then forced back through complex I of the ETC-generating ROS. ROS is able to activate the inflammasome and, therefore, drive the production of IL1β and IL18 (107).
IRG1 has also been shown to play a role in the establishment of endotoxin tolerance in LPS-tolerized macrophages. siRNA-mediated knockdown of IRG1 in these macrophages was able to increase NF-κB and IRF3 activation, while the production of ROS and subsequent expression of the zinc-finger protein A20 were reduced (90). Heme oxygenase 1 and carbon monoxide were able to induce IRG1 and through IRG1 downregulated pro-inflammatory gene expression in LPS-treated RAW264.7 cells and in a LPS mouse sepsis model (108). Several other elements regulating IRG1 expression and, therefore, itaconate production have also recently been identified. Computational modeling coupled with siRNA knockdown identified interferon regulatory factor 1 as a regulator of IRG1 transcription in RAW264.7 macrophages and human PBMCs (94). Inhibition of branched-chain aminotransferase 1 in human monocyte-derived macrophages decreased levels of glycolysis and oxygen consumption while also reduced IRG1 mRNA and protein levels as well as itaconate production (109).
A major issue with the study of the functional effect of itaconate in macrophages to date has been the use of DMI. DMI was utilized as it is cell permeable, however, it has been shown that while DMI boosts the level of itaconate in the cell it is not itself metabolized to itaconate (110). This suggests that DMI somehow promotes LPS-driven synthesis of itaconate. El Azzouny et al. also confirm that LPS-stimulated macrophages have increased succinate levels and that this is not the result of itaconate being metabolized to succinate. The authors speculate that the effects of DMI on macrophage metabolism may be due to an ability to act as a cysteine alkylating agent or to alter redox homeostasis. They further suggest that, though one has not been identified, it is possible a cell surface receptor for itaconate exists that DMI would be able to bind. Other metabolites have been shown to signal through G-protein-coupled receptors, such as succinate through GPR91, which has been renamed SUCNR1 (111). While the effects of studies carried out utilizing DMI have been drawn into question, the body of work carried out using genetic inhibition or deletion of Irg1 and the striking amount by which Irg1 mRNA and itaconate synthesis are upregulated in activated immune cells still leaves it worthy of further investigation.
Conclusion
Our understanding of immune cell metabolism has come far since the early observations that activated macrophages were highly glycolytic (112, 113). It is now well accepted that these pathways play a part outside of their traditional energetic and biosynthetic roles. The discovery that the Krebs cycle is not complete in activated M1 macrophages and DCs highlights the importance of the withdrawal of citrate from the cycle for DC activation, the production of pro-inflammatory mediators and for the generation of itaconate. Citrate links many important cellular processes, bridging carbohydrate and fatty acid metabolism and protein modification. Its role in producing acetyl-CoA for the acetylation of histones may turn out to be its most striking role in regulating immune cell function. There is still much to be discovered regarding the regulation and consequences of metabolic reprogramming in immune cells, however, it is clear that a “citrate pathway” plays an important role in these processes and may be amenable to therapeutic targeting.
Author Contributions
NW wrote the manuscript. LO’N supervised and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was funded by Wellcome Trust UK and Science Foundation Ireland. Wellcome Trust (Award number(s): 205455/14107); Science Foundation Ireland (Award number(s): 12/IA/1531).
Abbreviations
ACC, acetyl-CoA carboxylase; ACLY, ATP-citrate lyase; ACO2, aconitase 2; ACSS, acetyl-CoA synthase; aKG, α-ketoglutarate; AMPK, AMP-activated protein kinase; ATP, adenosine triphosphate; BMDM, bone marrow-derived macrophage; CIC, citrate carrier; CPT1, carnitine palmitoyltransferase 1; CS, citrate synthase; DC, dendritic cell; DMI, dimethyl itaconate; ETC, electron transport chain; FAO, fatty acid oxidation; FASN, fatty acid synthase; FIH, factor inhibiting HIF; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HIF, hypoxia-inducible factor; HK2, hexokinase 2; ICL, isocitrate lyase; IDH, isocitrate dehydrogenase; IFN-γ, interferon-γ; IKKε, IκB kinase ε; iNOS, inducible nitric oxide synthase; IRF, interferon regulatory factor; IRG1, immune-responsive gene 1; LDHA, lactate dehydrogenase A; LPS, lipopolysaccharide; MDH, malate dehydrogenase; MPC, mitochondrial pyruvate carrier; MTB, Mycobacterium tuberculosis; mTOR, mammalian target of rapamycin; NAA, N-acetylaspartic acid; NF-κB, nuclear factor-κB; NK cell, natural killer cell; NO, nitric oxide; OCR, oxygen consumption rate; OXPHOS, oxidative phosphorylation; PDH, pyruvate dehydrogenase; PDK1, pyruvate dehydrogenase kinase 1; PFK, phosphofructokinase; PGE2, prostaglandin E2; PI3K, phosphatidylinositide-3-kinase; PK, pyruvate kinase; PPP, pentose phosphate pathway; RET, reverse electron transport; ROS, reactive oxygen species; SDH, succinate dehydrogenase; SREBP, sterol regulatory element-binding protein; TBK1, TANK-binding kinase 1; TLR, toll-like receptor; TNFα, tumour necrosis factor α.
References
1. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140(6):805–20. doi:10.1016/j.cell.2010.01.022
2. Barton GM. A calculated response: control of inflammation by the innate immune system. J Clin Invest (2008) 118(2):413–20. doi:10.1172/JCI34431
3. Kelly B, O’Neill LAJ. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res (2015) 25(7):771–84. doi:10.1038/cr.2015.68
4. Pearce EJ, Everts B. Dendritic cell metabolism. Nat Rev Immunol (2015) 15(1):18–29. doi:10.1038/nri3771
5. Galván-Peña S, O’Neill LAJ. Metabolic reprograming in macrophage polarization. Front Immunol (2014) 5:420. doi:10.3389/fimmu.2014.00420
6. Reece JJ, Siracusa MC, Scott AL. Innate immune responses to lung-stage helminth infection induce alternatively activated alveolar macrophages. Infect Immun (2006) 74(9):4970–81. doi:10.1128/IAI.00687-06
7. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood (2010) 115(23):4742–9. doi:10.1182/blood-2009-10-249540
8. Everts B, Amiel E, van der Windt GJW, Freitas TC, Chott R, Yarasheski KE, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood (2012) 120(7):1422–31. doi:10.1182/blood-2012-03-419747
9. O’Neill LAJ. Glycolytic reprogramming by TLRs in dendritic cells. Nat Immunol (2014) 15(4):314–5. doi:10.1038/ni.2852
10. Lorsbach RB, Murphy WJ, Lowenstein CJ, Snyder SH, Russell SW. Expression of the nitric oxide synthase gene in mouse macrophages activated for tumor cell killing. Molecular basis for the synergy between interferon-gamma and lipopolysaccharide. J Biol Chem (1993) 268(3):1908–13.
11. Lu L, Bonham CA, Chambers FG, Watkins SC, Hoffman RA, Simmons RL, et al. Induction of nitric oxide synthase in mouse dendritic cells by IFN-gamma, endotoxin, and interaction with allogeneic T cells: nitric oxide production is associated with dendritic cell apoptosis. J Immunol (1996) 157(8):3577–86.
12. Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci U S A (1998) 95(13):7631–6. doi:10.1073/pnas.95.13.7631
13. Blouin CC, Pagé EL, Soucy GM, Richard DE. Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1α. Blood (2004) 103(3):1124–30. doi:10.1182/blood-2003-07-2427
14. Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem (2001) 276(12):9519–25. doi:10.1074/jbc.M010144200
15. Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem (1996) 271(51):32529–37. doi:10.1074/jbc.271.51.32529
16. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction, and is a critical determinant of the Warburg effect in LPS-activated macrophages. Cell Metab (2015) 21(1):65–80. doi:10.1016/j.cmet.2015.01.017
17. Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD, et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun (2013) 4:2834. doi:10.1038/ncomms3834
18. Sag D, Carling D, Stout RD, Suttles J. Adenosine 5’-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol (2008) 181(12):8633–41. doi:10.4049/jimmunol.181.12.8633
19. Hesse M, Modolell M, La Flamme AC, Schito M, Fuentes JM, Cheever AW, et al. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-arginine metabolism. J Immunol (2001) 167(11):6533–44. doi:10.4049/jimmunol.167.11.6533
20. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell (2003) 115(5):577–90. doi:10.1016/S0092-8674(03)00929-2
21. McGettrick AF, O’Neill LAJ. How metabolism generates signals during innate immunity and inflammation. J Biol Chem (2013) 288(32):22893–8. doi:10.1074/jbc.R113.486464
22. Mills E, O’Neill LAJ. Succinate: a metabolic signal in inflammation. Trends Cell Biol (2014) 24(5):313–20. doi:10.1016/j.tcb.2013.11.008
23. Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell (2016) 167(2):457–70.e13. doi:10.1016/j.cell.2016.08.064
24. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1[bgr] through HIF-1[agr]. Nature (2013) 496(7444):238–42. doi:10.1038/nature11986
25. Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its α subunit. J Biol Chem (1996) 271(50):32253–9. doi:10.1074/jbc.271.50.32253
26. Epstein ACR, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by Prolyl hydroxylation. Cell (2001) 107(1):43–54. doi:10.1016/S0092-8674(01)00507-4
27. Bruick RK, McKnight SL. A conserved family of Prolyl-4-hydroxylases that modify HIF. Science (2001) 294(5545):1337–40. doi:10.1126/science.1066373
28. Maxwell PH, Wiesener MS, Chang G-W, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature (1999) 399(6733):271–5. doi:10.1038/20459
29. Corcoran SE, O’Neill LAJ. HIF1α and metabolic reprogramming in inflammation. J Clin Invest (2016) 126(10):3699–707. doi:10.1172/JCI84431
30. Kim J-W, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3(3):177–85. doi:10.1016/j.cmet.2006.02.002
31. Patel MS, Korotchkina LG. Regulation of mammalian pyruvate dehydrogenase complex by phosphorylation: complexity of multiple phosphorylation sites and kinases. Exp Mol Med (2001) 33:191–7. doi:10.1038/emm.2001.32
32. Infantino V, Convertini P, Cucci L, Panaro Maria A, Di Noia Maria A, Calvello R, et al. The mitochondrial citrate carrier: a new player in inflammation. Biochem J (2011) 438(3):433–6. doi:10.1042/BJ20111275
33. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab (2016) 24(1):158–66. doi:10.1016/j.cmet.2016.06.004
34. Everts B, Amiel E, Huang SC-C, Smith AM, Chang C-H, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKK[epsiv] supports the anabolic demands of dendritic cell activation. Nat Immunol (2014) 15(4):323–32. doi:10.1038/ni.2833
35. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science (2009) 324(5930):1076–80. doi:10.1126/science.1164097
36. Akram M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochem Biophys (2014) 68(3):475–8. doi:10.1007/s12013-013-9750-1
37. Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch (2004) 447(5):689–709. doi:10.1007/s00424-003-1099-7
38. Saggerson D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annu Rev Nutr (2008) 28(1):253–72. doi:10.1146/annurev.nutr.28.061807.155434
39. Paumen MB, Ishida Y, Muramatsu M, Yamamoto M, Honjo T. Inhibition of carnitine palmitoyltransferase I augments sphingolipid synthesis and palmitate-induced apoptosis. J Biol Chem (1997) 272(6):3324–9. doi:10.1074/jbc.272.6.3324
40. Brownsey RW, Zhande R, Boone AN. Isoforms of acetyl-CoA carboxylase: structures, regulatory properties and metabolic functions. Biochem Soc Trans (1997) 25(4):1232–8. doi:10.1042/bst0251232
41. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab (2015) 21(6):805–21. doi:10.1016/j.cmet.2015.05.014
42. Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp Mol Pathol (2009) 86(3):174–9. doi:10.1016/j.yexmp.2009.01.003
43. Taylor WM, Halperin ML. Regulation of pyruvate dehydrogenase in muscle. Inhibition by citrate. J Biol Chem (1973) 248(17):6080–3.
44. Hillar M, Lott V, Lennox B. Correlation of the effects of citric acid cycle metabolites on succinate oxidation by rat liver mitochondria and submitochondrial particles. J Bioenerg (1975) 7(1):1–16. doi:10.1007/BF01558459
45. Martin DB, Vagelos PR. The mechanism of tricarboxylic acid cycle regulation of fatty acid synthesis. J Biol Chem (1962) 237(6):1787–92.
46. Iacobazzi V, Infantino V. Citrate – new functions for an old metabolite. Biol Chem (2014) 395(4):387–99. doi:10.1515/hsz-2013-0271
47. Jha Abhishek K, Huang Stanley C-C, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity (2015) 42(3):419–30. doi:10.1016/j.immuni.2015.02.005
48. Infantino V, Iacobazzi V, Menga A, Avantaggiati ML, Palmieri F. A key role of the mitochondrial citrate carrier (SLC25A1) in TNFα- and IFNγ-triggered inflammation. Biochim Biophys Acta (2014) 1839(11):1217–25. doi:10.1016/j.bbagrm.2014.07.013
49. Infantino V, Iacobazzi V, Palmieri F, Menga A. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem Biophys Res Commun (2013) 440(1):105–11. doi:10.1016/j.bbrc.2013.09.037
50. Starai VJ, Escalante-Semerena JC. Acetyl-coenzyme A synthetase (AMP forming). Cell Mol Life Sci (2004) 61(16):2020–30. doi:10.1007/s00018-004-3448-x
51. Zasłona Z, Pålsson-McDermott EM, Menon D, Haneklaus M, Flis E, Prendeville H, et al. The induction of pro-IL-1β by lipopolysaccharide requires endogenous prostaglandin E2 production. J Immunol (2017) 198(9):3558–64. doi:10.4049/jimmunol.1602072
52. Palmieri EM, Spera I, Menga A, Infantino V, Porcelli V, Iacobazzi V, et al. Acetylation of human mitochondrial citrate carrier modulates mitochondrial citrate/malate exchange activity to sustain NADPH production during macrophage activation. Biochim Biophys Acta (2015) 1847(8):729–38. doi:10.1016/j.bbabio.2015.04.009
53. Haseeb A, Makki MS, Haqqi TM. Modulation of ten-eleven translocation 1 (TET1), isocitrate dehydrogenase (IDH) expression, α-ketoglutarate (α-KG), and DNA hydroxymethylation levels by interleukin-1β in primary human chondrocytes. J Biol Chem (2014) 289(10):6877–85. doi:10.1074/jbc.M113.512269
54. Wang Y, Wang Y, Shen L, Pang Y, Qiao Z, Liu P. Prognostic and therapeutic implications of increased ATP citrate lyase expression in human epithelial ovarian cancer. Oncol Rep (2012) 27(4):1156–62. doi:10.3892/or.2012.1638
55. Zaidi N, Royaux I, Swinnen JV, Smans K. ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell- and environment-dependent mechanisms. Mol Cancer Ther (2012) 11(9):1925. doi:10.1158/1535-7163.MCT-12-0095
56. Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, et al. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res (2008) 68(20):8547–54. doi:10.1158/0008-5472.CAN-08-1235
57. Wang D, Yin L, Wei J, Yang Z, Jiang G. ATP citrate lyase is increased in human breast cancer, depletion of which promotes apoptosis. Tumor Biol (2017) 39(4):1010428317698338. doi:10.1177/1010428317698338
58. Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Res (2012) 72(15):3709–14. doi:10.1158/0008-5472.CAN-11-4112
59. Migita T, Okabe S, Ikeda K, Igarashi S, Sugawara S, Tomida A, et al. Inhibition of ATP citrate lyase induces an anticancer effect via reactive oxygen species: AMPK as a predictive biomarker for therapeutic impact. Am J Pathol (2013) 182(5):1800–10. doi:10.1016/j.ajpath.2013.01.048
60. Lee J-H, Jang H, Lee S-M, Lee J-E, Choi J, Kim TW, et al. ATP-citrate lyase regulates cellular senescence via an AMPK- and p53-dependent pathway. FEBS J (2015) 282(2):361–71. doi:10.1111/febs.13139
61. Assmann N, O’Brien KL, Donnelly RP, Dyck L, Zaiatz-Bittencourt V, Loftus RM, et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat Immunol (2017) 18(11):1197–206. doi:10.1038/ni.3838
62. Infantino V, Iacobazzi V, Santis FD, Mastrapasqua M, Palmieri F. Transcription of the mitochondrial citrate carrier gene: role of SREBP-1, upregulation by insulin and downregulation by PUFA. Biochem Biophys Res Commun (2007) 356(1):249–54. doi:10.1016/j.bbrc.2007.02.114
63. Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga J-I, Tamura Y, et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J Biol Chem (1999) 274(50):35832–9. doi:10.1074/jbc.274.50.35832
64. Gnoni GV, Priore P, Geelen MJH, Siculella L. The mitochondrial citrate carrier: metabolic role and regulation of its activity and expression. IUBMB Life (2009) 61(10):987–94. doi:10.1002/iub.249
65. Shimomura I, Shimano H, Korn BS, Bashmakov Y, Horton JD. Nuclear sterol regulatory element-binding proteins activate genes responsible for the entire program of unsaturated fatty acid biosynthesis in transgenic mouse liver. J Biol Chem (1998) 273(52):35299–306. doi:10.1074/jbc.273.52.35299
66. Moon Y-A, Lee J-J, Park S-W, Ahn Y-H, Kim K-S. The roles of sterol regulatory element-binding proteins in the transactivation of the rat ATP citrate-lyase promoter. J Biol Chem (2000) 275(39):30280–6. doi:10.1074/jbc.M001066200
67. Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem (2007) 282(7):4524–32. doi:10.1074/jbc.M610415200
68. Zhang N, Fu Z, Linke S, Chicher J, Gorman JJ, Visk D, et al. The asparaginyl hydroxylase factor inhibiting HIF-1alpha is an essential regulator of metabolism. Cell Metab (2010) 11(5):364–78. doi:10.1016/j.cmet.2010.03.001
69. Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol (2014) 15(8):536–50. doi:10.1038/nrm3841
70. Shi L, Tu BP. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol (2015) 33:125–31. doi:10.1016/j.ceb.2015.02.003
71. Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell (2015) 27(1):57–71. doi:10.1016/j.ccell.2014.12.002
72. Mehta V, Namboodiri MA. N-acetylaspartate as an acetyl source in the nervous system. Brain Res Mol Brain Res (1995) 31(1–2):151–7. doi:10.1016/0169-328X(95)00044-S
73. Prokesch A, Pelzmann HJ, Pessentheiner AR, Huber K, Madreiter-Sokolowski CT, Drougard A, et al. N-acetylaspartate catabolism determines cytosolic acetyl-CoA levels and histone acetylation in brown adipocytes. Sci Rep (2016) 6:23723. doi:10.1038/srep23723
74. Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, et al. ATP-citrate lyase controls a glucose-to-acetate metabolic switch. Cell Rep (2016) 17(4):1037–52. doi:10.1016/j.celrep.2016.09.069
75. Balmer Maria L, Ma Eric H, Bantug Glenn R, Grählert J, Pfister S, Glatter T, et al. Memory CD8(+) T cells require increased concentrations of acetate induced by stress for optimal function. Immunity (2016) 44(6):1312–24. doi:10.1016/j.immuni.2016.03.016
76. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153(6):1239–51. doi:10.1016/j.cell.2013.05.016
77. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science (2016) 354(6311):481–4. doi:10.1126/science.aaf6284
78. Wang B, Rao YH, Inoue M, Hao R, Lai CH, Chen D, et al. Microtubule acetylation amplifies p38 kinase signalling and anti-inflammatory IL-10 production. Nat Commun (2014) 5:3479. doi:10.1038/ncomms4479
79. Hu L, Yu Y, Huang H, Fan H, Hu L, Yin C, et al. Epigenetic regulation of interleukin 6 by histone acetylation in macrophages and its role in paraquat-induced pulmonary fibrosis. Front Immunol (2017) 7:696. doi:10.3389/fimmu.2016.00696
80. Greene WC, Chen LF. Regulation of NF-kappaB action by reversible acetylation. Novartis Found Symp (2004) 259:208–17; discussion 18–25. doi:10.1002/0470862637.ch15
81. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, et al. Regulation of cellular metabolism by protein lysine acetylation. Science (2010) 327(5968):1000. doi:10.1126/science.1179689
82. Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife (2016) 5:e11612. doi:10.7554/eLife.11612
83. Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics (2011) 10(12):M111.012658. doi:10.1074/mcp.M111.012658
84. Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science (2011) 334(6057):806–9. doi:10.1126/science.1207861
85. Witkowski A, Thweatt J, Smith S. Mammalian ACSF3 protein is a malonyl-CoA synthetase that supplies the chain extender units for mitochondrial fatty acid synthesis. J Biol Chem (2011) 286(39):33729–36. doi:10.1074/jbc.M111.291591
86. Colak G, Pougovkina O, Dai L, Tan M, Te Brinke H, Huang H, et al. Proteomic and biochemical studies of lysine malonylation suggest its malonic aciduria-associated regulatory role in mitochondrial function and fatty acid oxidation. Mol Cell Proteomics (2015) 14(11):3056–71. doi:10.1074/mcp.M115.048850
87. Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, et al. SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol Cell (2015) 59(2):321–32. doi:10.1016/j.molcel.2015.05.022
88. Willke T, Vorlop KD. Biotechnological production of itaconic acid. Appl Microbiol Biotechnol (2001) 56(3–4):289–95. doi:10.1007/s002530100685
89. Lee CG, Jenkins NA, Gilbert DJ, Copeland NG, O’Brien WE. Cloning and analysis of gene regulation of a novel LPS-inducible cDNA. Immunogenetics (1995) 41(5):263–70. doi:10.1007/BF00172150
90. Li Y, Zhang P, Wang C, Han C, Meng J, Liu X, et al. Immune responsive gene 1 (IRG1) promotes endotoxin tolerance by increasing A20 expression in macrophages through reactive oxygen species. J Biol Chem (2013) 288(23):16225–34. doi:10.1074/jbc.M113.454538
91. Cheon Y-P, Xu X, Bagchi MK, Bagchi IC. Immune-responsive gene 1 is a novel target of progesterone receptor and plays a critical role during implantation in the mouse. Endocrinology (2003) 144(12):5623–30. doi:10.1210/en.2003-0585
92. Chen B, Zhang D, Pollard JW. Progesterone regulation of the mammalian ortholog of methylcitrate dehydratase (immune response gene 1) in the uterine epithelium during implantation through the protein kinase C pathway. Mol Endocrinol (2003) 17(11):2340–54. doi:10.1210/me.2003-0207
93. Degrandi D, Hoffmann R, Beuter-Gunia C, Pfeffer K. The proinflammatory cytokine-induced IRG1 protein associates with mitochondria. J Interferon Cytokine Res (2009) 29(1):55–67. doi:10.1089/jir.2008.0013
94. Tallam A, Perumal TM, Antony PM, Jager C, Fritz JV, Vallar L, et al. Gene regulatory network inference of immunoresponsive gene 1 (IRG1) identifies interferon regulatory factor 1 (IRF1) as its transcriptional regulator in mammalian macrophages. PLoS One (2016) 11(2):e0149050. doi:10.1371/journal.pone.0149050
95. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A (2013) 110(19):7820–5. doi:10.1073/pnas.1218599110
96. Shin J-H, Yang J-Y, Jeon B-Y, Yoon YJ, Cho S-N, Kang Y-H, et al. 1H NMR-based metabolomic profiling in mice infected with Mycobacterium tuberculosis. J Proteome Res (2011) 10(5):2238–47. doi:10.1021/pr101054m
97. Sugimoto M, Sakagami H, Yokote Y, Onuma H, Kaneko M, Mori M, et al. Non-targeted metabolite profiling in activated macrophage secretion. Metabolomics (2012) 8(4):624–33. doi:10.1007/s11306-011-0353-9
98. Berg IA, Filatova LV, Ivanovsky RN. Inhibition of acetate and propionate assimilation by itaconate via propionyl-CoA carboxylase in isocitrate lyase-negative purple bacterium Rhodospirillum rubrum. FEMS Microbiol Lett (2002) 216(1):49–54. doi:10.1111/j.1574-6968.2002.tb11413.x
99. Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, et al. IFNs modify the proteome of Legionella-containing vacuoles and restrict infection via IRG1-derived itaconic acid. PLoS Pathog (2016) 12(2):e1005408. doi:10.1371/journal.ppat.1005408
100. Sasikaran J, Ziemski M, Zadora PK, Fleig A, Berg IA. Bacterial itaconate degradation promotes pathogenicity. Nat Chem Biol (2014) 10(5):371–7. doi:10.1038/nchembio.1482
101. Luan Harding H, Medzhitov R. Food fight: role of itaconate and other metabolites in antimicrobial defense. Cell Metab (2016) 24(3):379–87. doi:10.1016/j.cmet.2016.08.013
102. Ryan DG, O’Neill LAJ. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett (2017) 591(19):2992–3006. doi:10.1002/1873-3468.12744
103. Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, Sousa C, et al. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J Biol Chem (2016) 291(27):14274–84. doi:10.1074/jbc.M115.685792
104. Ackermann WW, Potter VR. Enzyme inhibition in relation to chemotherapy. Proc Soc Exp Biol Med (1949) 72(1):1–9. doi:10.3181/00379727-72-17313
105. Nemeth B, Doczi J, Csete D, Kacso G, Ravasz D, Adams D, et al. Abolition of mitochondrial substrate-level phosphorylation by itaconic acid produced by LPS-induced Irg1 expression in cells of murine macrophage lineage. FASEB J (2016) 30(1):286–300. doi:10.1096/fj.15-279398
106. Murphy Michael P. How mitochondria produce reactive oxygen species. Biochem J (2009) 417(Pt 1):1–13. doi:10.1042/BJ20081386
107. Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol (2011) 187(2):613–7. doi:10.4049/jimmunol.1100613
108. Jamal Uddin M, Joe Y, Kim S-K, Oh Jeong S, Ryter SW, Pae H-O, et al. IRG1 induced by heme oxygenase-1/carbon monoxide inhibits LPS-mediated sepsis and pro-inflammatory cytokine production. Cell Mol Immunol (2016) 13(2):170–9. doi:10.1038/cmi.2015.02
109. Papathanassiu AE, Ko J-H, Imprialou M, Bagnati M, Srivastava PK, Vu HA, et al. BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Nat Commun (2017) 8:16040. doi:10.1038/ncomms16040
110. ElAzzouny M, Tom CTMB, Evans CR, Olson LL, Tanga MJ, Gallagher KA, et al. Dimethyl itaconate is not metabolized into itaconate intracellularly. J Biol Chem (2017) 292(12):4766–9. doi:10.1074/jbc.C117.775270
111. Sundstrom L, Greasley PJ, Engberg S, Wallander M, Ryberg E. Succinate receptor GPR91, a Galpha(i) coupled receptor that increases intracellular calcium concentrations through PLCbeta. FEBS Lett (2013) 587(15):2399–404. doi:10.1016/j.febslet.2013.05.067
112. Newsholme P, Curi R, Gordon S, Newsholme EA. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J (1986) 239(1):121–5. doi:10.1042/bj2390121
Keywords: immunometabolism, citrate, ATP-citrate lyase, itaconate, acetylation, metabolism, macrophages, Krebs cycle
Citation: Williams NC and O’Neill LAJ (2018) A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 9:141. doi: 10.3389/fimmu.2018.00141
Received: 06 October 2017; Accepted: 16 January 2018;
Published: 05 February 2018
Edited by:
Charles E. McCall, Wake Forest Baptist Medical Center, United StatesReviewed by:
Markus Munder, Johannes Gutenberg-Universität Mainz, GermanyXiaoyu Hu, Tsinghua University, China
Geanncarlo Lugo-Villarino, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), France
Copyright: © 2018 Williams and O’Neill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luke A. J. O’Neill, laoneill@tcd.ie