 Susanta Pahari
Susanta Pahari Gurpreet Kaur
Gurpreet Kaur Shikha Negi
Shikha Negi Mohammad Aqdas
Mohammad Aqdas Deepjyoti K. Das
Deepjyoti K. Das Hilal Bashir
Hilal Bashir Sanpreet Singh
Sanpreet Singh Mukta Nagare
Mukta Nagare Junaid Khan
Junaid Khan Javed N. Agrewala*
Javed N. Agrewala*
- Immunology Laboratory, CSIR-Institute of Microbial Technology, Chandigarh, India
The mononuclear phagocyte system (MPS) constitutes dendritic cells, monocytes, and macrophages. This system contributes to various functions that are essential for maintaining homeostasis, activation of innate immunity, and bridging it with the adaptive immunity. Consequently, MPS is highly important in bolstering immunity against the pathogens. However, MPS is the frontline cells in destroying Mycobacterium tuberculosis (Mtb), yet the bacterium prefers to reside in the hostile environment of macrophages. Therefore, it may be very interesting to study the struggle between Mtb and MPS to understand the outcome of the disease. In an event when MPS predominates Mtb, the host remains protected. By contrast, the situation becomes devastating when the pathogen tames and tunes the host MPS, which ultimately culminates into tuberculosis (TB). Hence, it becomes extremely crucial to reinvigorate MPS functionality to overwhelm Mtb and eliminate it. In this article, we discuss the strategies to bolster the function of MPS by exploiting the molecules associated with the innate immunity and highlight the mechanisms involved to overcome the Mtb-induced suppression of host immunity. In future, such approaches may provide an insight to develop immunotherapeutics to treat TB.
Introduction
Despite of the fact that efficient anti-tuberculosis (TB) drugs are available, TB remains to ruin public health globally. Reports suggest that one-third of the populace is infected with Mycobacterium tuberculosis (Mtb), almost 10.4 million active cases and around 1.8 million deaths in 2016 (1). The occurrence of threat is further complicated due to acquired immunodeficiency syndrome pandemic, the appearance of multidrug-resistant (MDR), extensively drug-resistant, as well as totally drug-resistant Mtb strains (2). Vaccines are the most effective strategy to control and eliminate any disease (3, 4). Ironically, bacillus Calmette–Guérin (BCG) is the most controversial vaccine because of its variable efficacy worldwide (5). Moreover, it protects only children but not adults (6). Therefore, an urgent necessity and the challenge for the scientific society are to improve the current drug regimen or develop alternative stratagems against TB.
Our immune system is quite complex and complicated, comprising of innate as well as adaptive branch of immunity. Innate immunity is the primary and foremost line of defense against intruding pathogens (7). Innate immunity was initially believed to be non-specific and considered to be of lesser importance for the immune function. On the other hand, adaptive immunity is allied with the exclusion of intracellular pathogens in the subsequent stages of infection. It was considered as sentinel of the immune system owing to its specificity as well as immunological memory generation. Since the last few decades, innate immunity has gained enormous consideration due to the discovery of “germ line-encoded” pattern recognition receptors (PRRs), which makes the innate immunity capable of discriminating between self and an array of pathogens (8). PRRs are predominantly expressed by various antigen-presenting cells (APCs) such as monocytes, macrophages, and dendritic cells (DCs). These cells constitute the mononuclear phagocyte system (MPS). Mononuclear phagocyte cells (MPCs) are progenitors derived from bone marrow hematopoietic cell lineage (9). “Committed myeloid progenitor cells” can differentiate into blood monocytes, which then migrate to the bloodstream and subsequently enter in different tissues to develop into the resident tissue macrophages and DCs (10, 11). In the conventional sight of the MPS, cell division happens primarily in monoblasts and promonocytes. The expansion of mature macrophages provides the maintenance and number of resident tissue macrophages (10). MPCs mainly contribute in the recognition and eradication of pathogens and their related products. Furthermore, they contribute substantially in promoting innate immunity and subsequently stimulating, shaping, and expanding the adaptive immunity (12). Initiation of adaptive immunity not only depends on the direct detection of antigen by the receptors of MPCs but also relies on crucial signals delivered through costimulatory molecules, cytokines, and PRRs (13). Importantly, DCs contribute considerably in bridging innate and adaptive immunity (8, 14). DCs express a plentiful amount of costimulatory molecules and PRRs, which regulate several immune functions and signaling cascades that are crucial for the instigation of adaptive immune response (15). In addition, they successively alert other immune cells to accumulate at the infection site. Furthermore, they combat and resist Mtb in establishing infection and restrain them from becoming an active disease.
Based on the aforementioned investigations, MPS are considered as an important first line of defense against pathogen. Exploiting MPCs or their components, namely, PRRs, costimulatory molecules, cytokines, and chemokines as therapeutic agents may be an exciting line of study to control TB. Previously, our group has highlighted the importance of signaling through innate molecules in context with nasal and mucosal immunity to restrict Mtb entry and consequently prohibiting its infection. We discussed the role of several immunomodulators in vitro, in vivo, or in clinical studies to enhance the efficacy of anti-TB drugs in treating TB patients (16). Current review highlights the interaction between Mtb and MPS influencing the outcome of disease. Hence, as evidenced by published literature, we hypothesize a crucial strategy to reinvigorate MPS functionality to overwhelm Mtb and eliminate it. Furthermore, we discuss the strategies to bolster the function of MPS by exploiting the molecules associated with the innate immunity and highlight the mechanisms involved therein. It may be hypothesized that involving MPS in conjunction with drugs, as an adjunct therapy may lessen the dose as well as duration of ongoing drug regimen; and therefore, may reduce the chances of developing drug resistance by the pathogen.
Various Mononuclear Phagocytic Cells and Their Function in Innate and Adaptive Immunity
Mononuclear phagocyte cells located in various tissues differ in terms of their nomenclature and morphological appearance (17). For example, macrophages are called as histiocytes in subcutaneous tissues, Kupffer cells resides in liver, microglia present in nervous tissue, alveolar macrophages in lungs, osteoclasts in bones, etc. Besides phagocytosing pathogens and eliminating them from the blood, lymph, and tissues, MPS also clears the senescent cells and mounts immunity against the pathogens (18). MPS recognizes, captures, and internalizes the pathogenic determinants identified as pathogen-associated molecular patterns (PAMPs) through PRRs localized on their surface. This leads to the secretion of biologically active molecules such as free radicals, cytokines, and chemokines. The chemokines attract chiefly neutrophils from the bloodstream and initiate a pro-inflammatory response leading to the engulfment and destruction of Mtb.
Lung alveolar macrophages and myeloid DCs are some of the foremost cell types that get infected after aerosol challenge with Mtb. Subsequently, interstitial macrophages, monocytes, and neutrophils are recruited to infection site (19). MPCs capture Mtb and migrate to the local draining lymph nodes, then process and present antigens efficiently in context with MHCs to activate T cells (20). The intensity of MPCs and Mtb counterattack widely depends upon the host genetics as well as bacterial virulence factors. Accordingly, Mtb replicates within the host MPCs (21, 22) and manipulates function by impairing their ability to control infection (23, 24). Mtb can obstruct the antigen processing and presentation by MPCs to T cells (25–27). Macrophages and DCs that are not optimally activated cannot kill the intracellular Mtb and serve as a reservoir for the dissemination of the pathogen. In addition, due to their striking migratory potential, they play a key role in transmitting Mtb from the site of infection to other tissues (28).
Evasion Strategies Adopted by Mtb to Counteract Host Immunity
One of the major mechanisms through which Mtb obstructs MPS function is by inhibiting the fusion of phagosome with lysosome. Various mycobacterial lipids and glycolipids, proteins, and enzymes, namely, lipoarabinomannan (LAM) and trehalose-6,6’-dimycolate (TDM), protein tyrosine phosphatase A (PtpA), secretory acid phosphatase M, zinc-dependent metalloprotease 1, lipoamide dehydrogenase C, serine/threonine protein kinase G, and PEPGRS62 protein have been proved to play an important role in the capacity of Mtb to escape phagolysosome fusion (29–32).
Two signals are important for the optimal activation of T cells. Initial one is the engagement of TCR with MHC–peptide complex and subsequent upregulation of costimulatory molecules. Instead of getting activated, T cells get anergized (“a state of unresponsiveness”) in the absence of costimulatory molecules (33). Interestingly, Mtb has the ability to successfully down modulate the expression of costimulatory molecules. Furthermore, after infecting MPCs, Mtb upregulates the expression of immunosuppressive markers programmed cell death-1, Lymphocyte activation gene-3, and T-cell immunoglobulin mucin-domain containing-3, thus retaliating against the potential threat caused by T cells (34, 35).
Another mechanism is the deprivation of MPS nutrients by Mtb. The most common battle between host cells and the pathogen is for iron utilization. Mtb efficiently utilizes its siderophores for iron uptake and thus deprives host of its availability (36). Furthermore, carbon from various sugars and fatty acids are extracted by Mtb in host cells via its major enzymes, such as the polyphosphate glucokinase, isocitrate lyases (ICL1 and ICL2), and the phosphoenolpyruvate carboxykinase (37–39). Mtb favors the differentiation of macrophages toward M2 subtype (40). By contrast, it impairs the formation of M1 macrophages. M2 macrophages are responsible for suppression of inflammatory function. M1 subtype arises from type-1 inflammatory conditions and secretes pro-inflammatory cytokines and is endowed with microbicidal activity (41). Virulence factors of Mtb are known to preferably skew the generation of M2 macrophages (42). Therefore, Mtb is successful in creating an environment for its intracellular survival inside macrophages (43, 44). Furthermore, Mtb mainly skews the differentiation of CD4 T cells toward Th2 cells phenotype (45). Similarly, the generation of regulatory T cells that secrete TGF-β is promoted. Both Th2 cells and Tregs help in the TB progression. By contrast, the formation of Th1 cells and Th17 cells is suppressed by hijacked MPS, since they have potential to successfully control the Mtb infection (46).
MPS Helps in the Restoration of Host Immunity Impaired by Mtb
Mononuclear phagocyte system contributes significantly to the health and disease (47). One of the most imperative mechanism and early response of innate immunity is the generation of reactive oxygen species (ROS) by MPS, which not only destroys the pathogen but also plays a physiological role in maintaining and controlling the cellular functions. Clearance of colonized microorganisms and initiation of signaling pathways related to inflammation, cell proliferation, and induction of immunity is highly dependent on ROS (48). Two sources of ROS generation in the host upon microbial infection is membrane-bound NADPH oxidase complex as well as mitochondrial electron transport chain (49). Important PRRs associated with an intracellular pathogen is NOD-like receptors (NLRs), which makes cell attentive on pathogen interaction/invasion. Among many NLRs, NLRX1 moves to mitochondria and initiates the ROS production (50).
Role of MPS to Overcome the Modulation of Cellular Metabolism and Nutrient Acquisition by Mtb
Mycobacterium tuberculosis utilizes cholesterol for its survival and establishes infection in the host cells (43). This cholesterol is further converted into sterol, which is crucial for Mtb persistence in the host cells (51). Adenosine-5′-triphosphate (ATP) plays a decisive role in the host by acting directly on cell metabolism and signaling cascade. The ATP that comes out of the cell into the extracellular environment is known as extracellular ATP (eATP). It has been seen that eATP can activate the immune system by acting as a “danger signal” (52). Moreover, it is well known that eATP has a potential role in stimulating the release of pro-inflammatory cytokines. eATP induces IL-6 secretion from macrophages (53) and IL-1β production from LPS primed monocytes (54). Furthermore, it is noted that eATP signaling is not only implicated in the generation of ROS and pro-inflammatory cytokines but also plays a significant role in antigen presentation. Previous report demonstrated that eATP along with its putative receptor P2X7 on inflammasome activation induces the shedding of exosomes containing the MHC class II from macrophages (55). It is well-known fact that ROS signaling is involved in the inflammasome formation (56). Thus, it facilitates innate as well as adaptive immune response. Mtb-infected phagocytes release exosomes containing the MHC class II and Mtb Ag85B, which activates the T cells (57). The eATP–P2X7 receptor signaling plays an important role in clearing Mtb infection through multiple ways such as phospholipase-D (58), apoptosis (57), phagosome–lysosome fusion (59), and autophagy (60). Thus, MPS is recognized to play an appreciable role in neutralizing and eradication of Mtb from the host.
Involvement of Phagocytic Cells to Boost Immunity Against Mtb
Both DCs and macrophages play crucial roles in protection against mycobacterium. The presence of Mtb at infection site is sensed by macrophages through chemokine-mediated migration, as these macrophages express surface receptor for these chemokines known as G-protein-coupled receptors (61). Mtb is efficiently phagocytosed by these professional phagocytic cells. Studies on human macrophages have shown that phagocytosis is significantly improved in the presence of anti-Mtb antibodies and complement factors (62). Once Mtb is phagocytosed by the macrophage or DCs, it encounters a number of defense mechanisms operated through the innate immunity of the host. These include the formation of free radicals, namely, ROS, reactive nitrogen intermediates (RNI), cytokines, and chemokines. Moreover, MPS helps in the differentiation of T cells; DCs secrete IL-12, which results in the generation of Th1 cells. Moreover, Th1 cells mainly secrete IFN-γ that activates macrophages to release TNF-α (63). Similarly, IL-6 and TGF-β secreted by MPS helps to differentiate naïve T cells into Th17 phenotype (64). Th1 cells and Th17 cells can reciprocally regulate the function of Th2 cells and Tregs, respectively. Both Th2 cells and Tregs promote the progression of TB.
Mononuclear phagocyte system employs factors that are involved in basic metabolism of the body to fight against intracellular pathogens. One such important molecule is vitamin D3, which enhances the phagocytosis of MPS by upregulating the expression of CD14 and CD206 receptors (65). Toll-like receptor (TLR)-2 signaling in macrophage upregulates the expression of vitamin-D–1-hydroxylase and surface vitamin-D receptor, which stimulates the generation of antimicrobial peptide cathelicidin and contributes to resistance to Mtb (66).
PRRs-Mediated Bolstering of MPS Activity Against Mtb
Mononuclear phagocyte cells are the key sensory cells that reinforce the innate immunity. They express the plethora of innate receptors such as TLRs, NLRs, and C-type lectin receptors (CLRs), which are collectively called as PRRs that are present either on the cell surface or endocytic vesicles. The PRRs including TLRs (TLR-2, -3, -4, and -9) and non-TLRs [CLRs, NLRs such as nucleotide-binding oligomerization domain (NOD)-2, mannose receptors (MRs), Dectin-1, and DC-SIGN] recognize conserved PAMPs that are present on Mtb. PRRs have the capacity to recognize a broad range of structural components of pathogens grouped as PAMPs and DAMPs, which includes lipopeptides, lipoproteins, lipoteichoic acid, peptidoglycans, ssRNA, dsRNA, siRNA, mRNA, DNA, LPS, heat shock proteins, and flagellin. The role of several PPRs in protecting against Mtb has been widely studied (16, 67–70). The interaction of PRRs with PAMPs triggers a series of signaling pathways inside the MPCs (Table 1). PRRs activated MPCs acquire augmented expression of MHC I, MHC II, and costimulatory molecules on their surface (63), which leads to the better presentation of Mtb antigens to naïve T cells followed by generation of efficient T cell response against this pathogen. Mtb loaded macrophages, after activation, can mount bactericidal activities such as nitric oxide (NO) production, maturation of phagosomes toward phagolysosomes and autophagolysosomes (71). Recruitment and activation of many signaling molecules in cascade lead to nuclear translocation of NF-κB, which eventually causes the activation of MPCs. In a different setup, the activated macrophages have the capability to carry out macro-autophagy to take care of intracellular Mtb (72, 73). In a similar phenomenon known as “programmed necroptosis,” MPCs controls the intracellular replication of Mtb. This process speeds up the recruitment of neutrophils and thereby enhances the killing of mycobacterium (74). MPS activation is evident by the release of pro-inflammatory cytokines such as IL-1β, IL-6, IL-12, and TNF-α; which help in phagocytosis of the bacterium followed by activation of Mtb reactive T cells. These T cells play a cardinal role in controlling the Mtb growth.
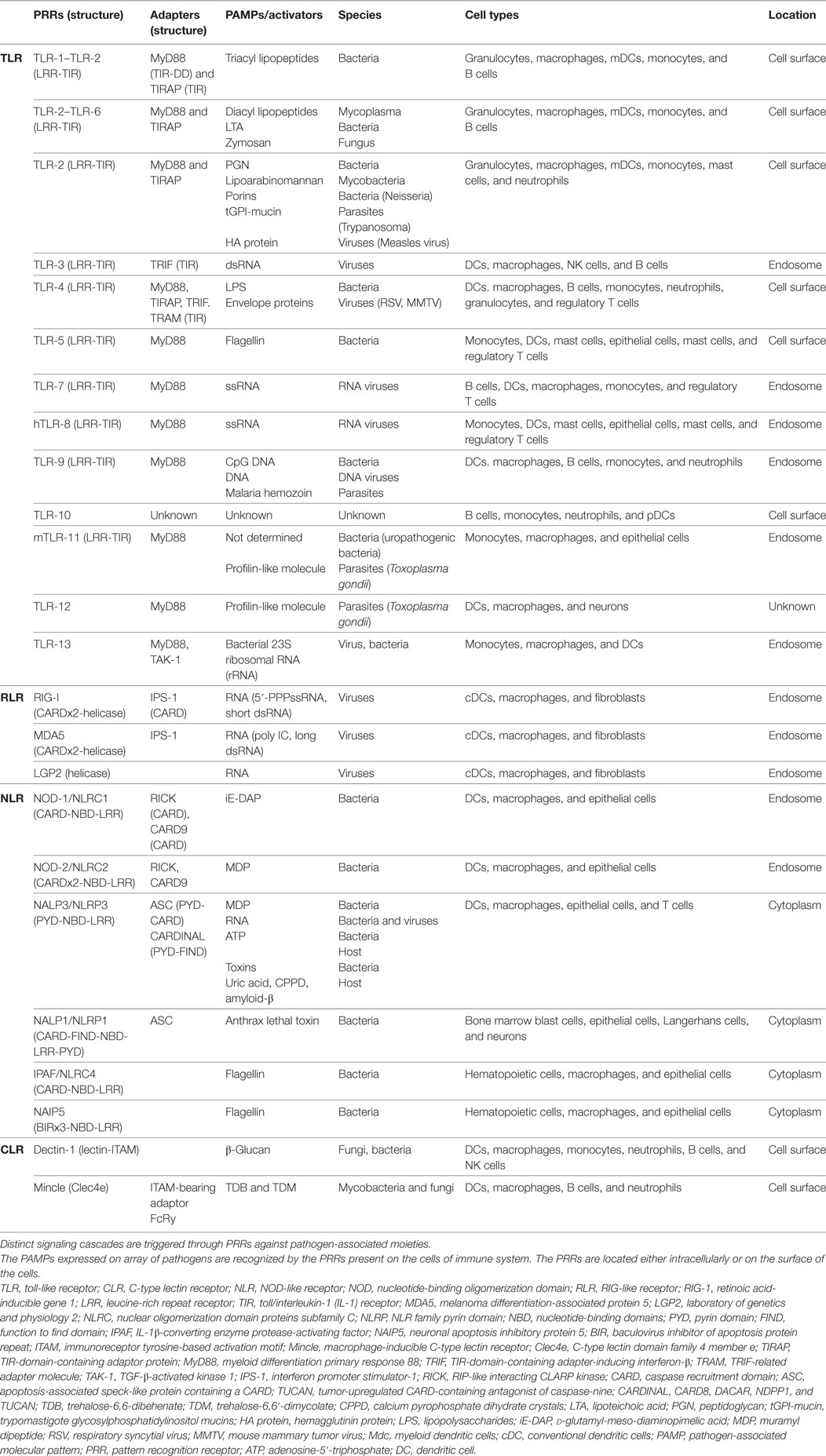
Table 1. Activation of PRRs through PAMPs.
Importance of PRRs signaling in the activation of an immune response against Mtb can be accounted by the evidence that MyD88−/− mice were more prone to Mtb infection. TLR-2-knockout mice showed low IL-12 and TNF-α yield on Mtb infection and more granulomas formation in the lungs (75). The 19 kDa lipoprotein of Mtb activates MPCs through TLR-2 triggering and induces IL-12 and NO production, and subsequently killing of Mtb (76). In humans, the interaction of 19 kDa lipoprotein with TLR-2 induces apoptosis of Mtb-infected macrophages (77). TLR-4 senses HSP60/65 and 38-kDa Mtb antigen inducing protective TNF-α production (78). In addition, another Mtb small heat shock protein X also recognized as α-crystallin-1 can specifically modulate the function of DCs at different maturation stages (79, 80). TLR-4 activation is known to induce macro-autophagy by recruitment of Beclin-1. Recently, we showed that cumulative signaling of DCs through TLR-4 and NOD-2 successfully inhibits the intracellular survival of Mtb through autophagy (70). Mycobacterial DNA interacts with TLR-9 and elicits macrophages to produce pro-inflammatory cytokines. Furthermore, TLR-9-knockout mice showed less release of IFN-γ and IL-12. Mtb-infected macrophages and DCs deficient in TLR-9 are less responsive to IL-12 (81). NLRs and CLRs can also influence the function of MPCs. NOD-2-deficient mice exhibit impairment in cytokine and NO release upon Mtb infection (82). Activation of Mtb-infected human macrophages through NOD-2 induces autophagy and restricts Mtb growth (83). Likewise, signaling of Mtb-infected DCs through Dectin-1 and macrophage-inducible C-type lectin (Mincle) influences the intracellular survival of the pathogen (84, 85). By contrast, DC-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) interaction with LAM of Mtb initiates the anti-inflammatory response by inducing the secretion of IL-10 (86). Overall, it signifies that the engagement of various PRRs on MPCs can differentially regulate their function toward Mtb.
Contribution of Inflammatory Response in Controlling Mtb Infection
Inflammatory response generated by cytokines helps to control Mtb infection directing the pathogenesis of disease. Diverse cytokines produced on Mtb infection determines the fate of host response. Cells of MPS recruited at the infection site trigger cascade of events necessary for the release of various pro-inflammatory cytokines such as IL-1β, IL-6, IL-12, TNF-α, and IL-18. Furthermore, the protective function of IL-1 during Mtb infection was first demonstrated in mice dually deficient in IL-1α/β or IL-1R1 signaling (87). Several findings have reported enhanced Mtb load and less survival of mice with defect in IL-1R1 signaling. By contrast, IL-1β and IL-18 are synthesized after processing by caspases-1 of pro-IL-1β and pro-IL-18, respectively. Besides caspase-1, four more caspases, caspases-11 and -12 of mouse and caspases-4 and -5 of human regulate the inflammatory processing. Inflammasome plays a decisive role in host defense, as mice lacking IL-1β, IL-1 receptor, or IL-18 was more prone to Mtb infection. Furthermore, ASC protein deficiency led to the severe form of disease in the murine model of TB. IL-1β production is known to rely on the early secreted antigenic target of 6 kDa (ESAT-6) secretion system 1 (ESX-1) of Mtb, which contributes to the expression of virulence genes encoded by region of difference (RD-1). Inflammasome formation mediated by ESX-1 relies on the host NLR family pyrin domain containing-3 (NLRP3) along with ASC protein (88). Based on the above observations, it can be speculated that failure of BCG to induce optimum protection in TB is attributed to the lack of IL-1β and IL-18 mediated by RD-1 (89).
We, therefore, postulate that adjunct therapy of BCG with innate ligands that can regulate inflammasome formation and can enhance its efficacy as a potential vaccine against Mtb. In addition, inflammasome regulates Mtb infection during early phase by activating innate immunity and also plays a decisive role in augmenting the adaptive immunity against the bacterium.
Activation of CD4 T Cells and CD8 T Cells by MPS in Restricting Mtb Growth
The onset of the adaptive immune response to Mtb generally takes 8–11 days subsequent to primary exposure of Mtb. This involves the transportation of the bacterium to the draining lymph nodes (90). Infected MPCs are involved in the priming and proliferation of Mtb-specific effector T cells and their subsequent migration to the lungs.
T cells play a fundamental role in conferring defense against Mtb. IL-12 secreted by MPS is a crucial regulator of the differentiation of naive CD4 T cells to Th1 cells. IL-6, TGF-β, and IL-1β released by MPCs help in the differentiation to Th17 cells (91). Inflammasome generated by MPS in synergy with IL-6 facilitates Th17 cells development via upregulation of IRF4 and RORγt. In absence of TGF-β signaling, IL-1β coordinates with IL-6 and IL-23 to generate pathogenic Th17 cells (92). A concerted action of both Th1 cells and Th17 cells is essential to control Mtb infection. IFN-γ released by Th1 cells plays a fundamental role in the activation of MPCs and the release of TNF-α, a cytokine responsible for inhibiting the growth of Mtb (93). Recent evidences highlight the role of Th17 cells producing IL-17 and IL-22 in restricting Mtb (93). Th17 cells mainly recruit monocytes and Th1 cells to the lungs that help to clear infection rapidly (93). Recent studies showed that Tregs can effectively diminish Th1 immunity (94) or hinder the effector T cells influx to the lungs during initial phase of Mtb infection (95). Similarly, Th2 cells that secrete mainly IL-4 and IL-13 significantly contribute to the progression of TB (96).
Although, CD8 T cells sufficiently provide immunity against Mtb, but their role is not adequately studied as has been done in the case of CD4 T cells. However, the importance of CD8 T cells has been established by the fact that their depletion leads to higher susceptibility toward Mtb in the experimental model of TB. Furthermore, β2m-knockout mice died rapidly on exposure to Mtb (97). CD8 T cells released IFN-γ and granulysin lyse the Mtb-infected macrophages, as well as can induce perforin (Pfn)-mediated cytotoxicity to kill the Mtb (98, 99). Furthermore, CD8 T cells in lung can directly lyse Mtb-infected macrophages in a Pfn-dependent manner (100). In addition, the presence of CD8 T cells expressing granzyme B has been observed in the TB patients and latent infection (101). However, CD8 T cells secrete IL-17, TNF-α, IL-10, and IL-2 but IFN-γ is considered to be a key mediator in defense against Mtb (101). Furthermore, IFN-γ augments the production of various chemokines CXCL9, CXCL10, and CXCL11 and therefore helps in recruiting the cells toward granulomas. Recently, it has been shown that the Mtb-infected macrophages undergoing apoptosis releases several antigens in apoptotic vesicles, thus permitting the access of these apoptotic bodies to bystander cells to present antigen in context with MHC class I molecules. This can be confirmed by inhibiting the formation of blebbing in a plasma membrane by caspase inhibitors. Consequently, it may hamper the CD8 T cell activation. Furthermore, unconventional CD1-restricted γδ-TCR T cells can specifically respond to Mtb glycolipids (102). The γδ T cells are the less abundant type of T cells population, which differ from common αβ T cells in having gamma delta (γδ) glycoprotein chains bearing TCR on T cells (102). Mycobacterial phospho-antigens are known as the potent activators of Vγ9Vδ2 T cell functions (103). These cells recognize Mtb-infected monocytes and alveolar macrophages in a non-MHC restricted manner (104). γδ T cells are responsible for initiating defense mechanism upon Mtb infection by generating cytotoxic function, cytokine secretion, and contact-dependent lysis of infected cells (105).
Involvement of Reactive Oxygen and Nitrogen Species in Controlling Mtb Infection
Antimicrobial ROS and RNI are critical in controlling Mtb infection. RNI and NO produced by MPCs are considered potent antimicrobial agents. Human alveolar macrophages can kill Mtb in an iNOs dependent manner. Whereas, macrophages obtained from healthy individuals that are latently infected with Mtb prevent the growth of bacterium by secreting NO (106). These results were validated in the murine model of TB, where the abrogation of inducible NO synthase activity resulted in a dramatic increase in the microbial burden (107). Moreover, disruption of Inos gene increases Mtb dissemination and mortality of the mice. Indeed, NO released by macrophages is critically dependent on IFN-γ (108). Although effector T cells are the key producer of IFN-γ but it takes few weeks for these cells to release IFN-γ. Nevertheless, the NO production by macrophages is noticed within 3 h of Mtb infection (109). NK and γδ T cells are the first to reach the infection site. They secrete IFN-γ that stimulates macrophages to produce NO. The antimycobacterial function of NO secreted by MPS is well documented in the case of mice. However, there are evidences that depict that NO secretion by human alveolar epithelial cells and macrophages inhibits the growth of Mtb, but the role of NO still needs to be fully authenticated in humans (110). Furthermore, NOS and NO are highly evident in the macrophages obtained from bronchoalveolar lavages of TB patients. Apparently, MDR patients secrete lesser NO, as compared with TB patients (111). In addition, it has been reported that NOS2-deficient mice are immunocompetent to cure Mtb infection (112). It has been demonstrated that ciprofloxacin elicits the release of NO to eliminate Mtb (113). Furthermore, NO regulates the secretion of many pro-inflammatory cytokines such as IL-1β, IL-8, and TNF-α (114).
However, Mtb has successfully developed immune evasion strategies to resist the intracellular killing by ROS and RNI produced by MPS. Mtb has phenolic glycolipid I, cyclopropanated mycolic acids, and LAM rich thick cell wall that is the effective scavenger of oxygen radicals providing resistance to ROS (115). Besides, Mtb produces various ROS scavenging enzymes such as KatG, superoxide dismutases (Sod A and C), peroxidase along with peroxynitrite reductase complex consisting of AhpC, AhpD, SucB (DlaT), and Lpd (116, 117). Interestingly, Lsr2 bound to Mtb DNA protects the pathogen from ROS mediated destruction (118). Truncated hemoglobin in M. smegmatis protects the bacterium from aerobic respiration by inhibiting the NO production (119). Surprisingly, despite the strong killing potential of RNI, ROS, and cytokines, Mtb has successfully learned to prevail in the host environment.
MPS Resist Mtb Inflicted Death
The apoptosis (programmed cell death) is a well-known event, where a cell undergoing death still retains its cytoplasmic material within membranous vesicles called as apoptotic bodies. MPCs eliminate apoptotic bodies through the mechanism known as “efferocytosis,” which critically contributes in boosting host immune response. The caspase family of serine proteases is the essential molecules that generate apoptosis in MPS. Apoptosis operates through several classical pathways. One of the essential pathways is the ligation of TNF receptor family-2, which activates caspases and subsequently induces the formation of apoptotic bodies. Primarily, apoptosis occurs due to the nutrients deprivation, oxidative stress, or intracellular stresses that ultimately alter the mitochondrial membrane permeability. Consequently, cytochrome c is translocated to cytosol, which leads to the caspases activation. Another pathway is interceded by the release of granzyme B from cytotoxic T cells as well as NK cells. Subsequent to Mtb infection, MPCs augment the production of TNF-α, which elicits apoptosis. This process confines the growth of Mtb by activating local MPS and insulating it into the apoptotic vesicles. Intriguingly, considerable inhibition in Mtb growth was demonstrated when cells undergoing apoptosis were cocultured with naïve macrophages. In addition, it has been demonstrated that antimicrobial effect performed by macrophages is through the involvement of IL-1 signaling and NO-dependent anti-mycobacterial activity (120). The level of apoptosis induced by virulent and avirulent Mtb is quite distinct (121). Few reports signify that virulent strain of Mtb triggers necrosis by avoiding host defensive strategy, while avirulent strain induces apoptosis (121, 122). Furthermore, the frequency of apoptosis is quite high in the macrophages infected with an attenuated strain of Mtb (123). Even though the production of TNF-α is commensurate, MPCs infected with non-virulent Mtb are found to be more susceptible to undergo apoptosis. The possible reasons suggested for the differences between virulent and avirulent Mtb may be the virulence factors, bacterial load and duration of exposure. H37Rv infected MPCs secretes IL-10, leading to the induction of TNFR. The soluble form of TNF-α and TNFR complex inhibits the apoptosis (124). Furthermore, ESAT-6 of Mtb, leads to apoptosis in THP-1 macrophages by diminishing the expression of antiapoptotic molecules (125).
Neutrophil plays an imperative role in preventing Mtb infection (126). These cells reach first at the infection site. Furthermore, neutrophils phagocytose Mtb and generate ROS and restrict Mtb growth. In addition, they help macrophages to eliminate the infection. Mtb-infected neutrophils can also undergo apoptosis. At the time of apoptosis, these cells display “eat-me or find-me” signal on their plasma membrane, which helps in recognizing the unwanted constituents of MPCs (127). Macrophages recognize “find-me” signals and then phagocytose these cells. Macrophages engulf neutrophils undergoing apoptosis and secrete TNF-α to control Mtb infection by granulomas formation (128). It will be an exciting line of investigation employing the concept of “eat-me” signal to develop a novel strategy for targeted delivery of immunomodulators along with anti-TB drugs for the clearance of Mtb hiding in a quiescent state inside the endosome (Figure 1).
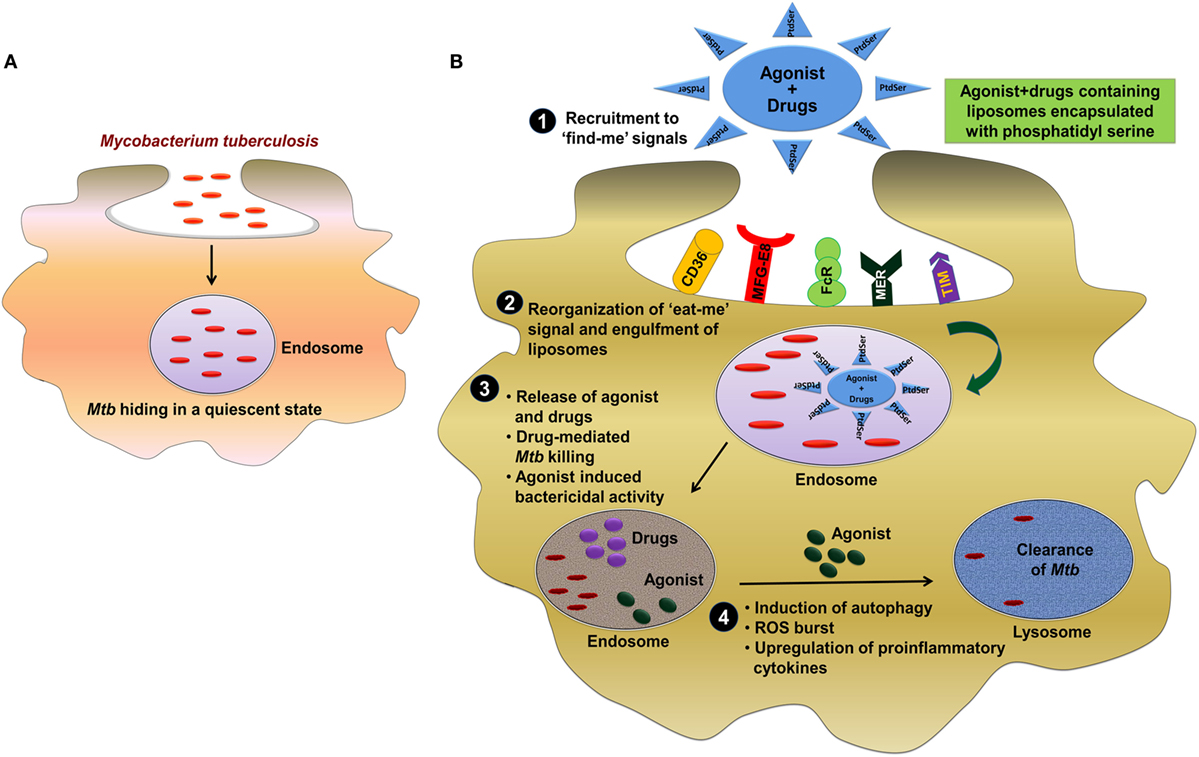
Figure 1. Involvement of “eat-me” signal in targeted delivery of immunomodulators along with anti-TB drug for the clearance of Mtb. (A) Mtb employs elegant strategy to impair the function of host cells by residing inside the endosome of macrophages in a quiescent state. (B) The most effective strategy to control and eliminate Mtb can be through targeting of pathogen-bearing MPCs by exploiting “eat-me” signal. One of the possible approach could be the engagement of phosphatidylserine (PtdSer) as an “eat-me” signal to deliver PRRs agonist/drug in liposome to target Mtb in the endosomes. (1, 2) Primarily, mononuclear phagocytic cells recognize “find-me” signal by several receptors such as CD36, MFG-E8, FcR, MER, TIM, and then phagocytose the liposomes through receptor-mediated endocytosis. (3) The direct clearance of Mtb in the endosome can be achieved by delivering the drug (rifampicin/isoniazid) to the site of infection. (4) However, the majority of Mtb would be eliminated but the eradication of residual bacterial population can be achieved by the agonist of TLRs (TLR-2, TLR-4, and TLR-9), NLRs (NOD-1 and NOD-2), and CLRs (Mincle, Dectin-1, and Dectin-2) mediated bactericidal mechanism and subsequently clearance of Mtb from lysosomes. Abbreviations: CD36, cluster of differentiation 36; MFG-E8, milk fat globule-EGF factor 8; FcR, Fc receptor; MER, membrane-bound receptor tyrosine kinase; TIM, T cell immunoglobulin and mucin domain; MPC, mononuclear phagocyte cell; NLR, NOD-like receptor; PtdSer, phosphatidylserine TLR, toll-like receptor; CLR, C-type lectin receptor; Mtb, Mycobacterium tuberculosis; TB, tuberculosis; NOD, nucleotide-binding oligomerization domain; Mincle, macrophage-inducible C-type lectin.
Role of ER Stress (ERS) in the Regulation of Innate Immunity During Mtb Infection
In humans, endoplasmic reticulum (ER) performs various functions such as metabolism of lipids, protein folding, and maintaining cellular homeostasis. Different factors such as accumulation of unfolded proteins, loss of oxygen or hypoxia, and bacterial infections are responsible for the unfolded protein response (UPR), which causes ERS. Uncontrolled ERS leads to apoptosis. Furthermore, UPR activates various innate signaling pathways, which result in the survival of intracellular pathogens such as Mtb (129, 130). Apoptosis of macrophages helps to prevent the spread of mycobacterial infection by activating innate immunity (131). However, growing number of findings suggest that Mtb has evolved various strategies to control the ERS for its survival in the host (132). Mtb can efficiently alter the structure of macrophage ER. It was shown that macrophages infected by virulent (H37Rv) along with avirulent (H37Ra) Mtb strains possess distinct ER phenotypes (133). Difference in the morphology of ER in macrophages targeted during Mtb infection is a crucial factor for initiation of apoptosis. Ca2+ is very important in different apoptotic pathways and is responsible for the phagosome–lysosome fusion. A smooth ER phenotype linked with avirulent Mtb-infected macrophages increases cytosolic Ca2+ levels and simultaneously increases the synthesis of phosphatidyl choline/phosphatidyl ethanolamine (PC/PE), which leads to apoptosis. However, H37Rv but not H37Ra manipulates rough ER of macrophages and disturbs the cholesterol homeostasis to inhibit the apoptosis and establishes its intracellular persistence.
Endoplasmic reticulum stress is already known to influence macrophages. It stimulates conversion of macrophages toward M1 phenotype and induces apoptosis, thereby aiding in Mtb clearance. On the other hand, polarization of M2 phenotype by Mtb infection aids its escape by suppressing apoptosis (134). At the site of granuloma formation during Mtb infection, ERS markers such as activating transcription factor-3, pIre1α, and eukaryotic initiation factor 2α levels are increased (135). Depending on ERS, macrophage apoptosis is influenced by various Mtb proteins such as ESAT-6, 38-kDa antigen and PE-PGRS33. Henceforth, ERS is crucial in imparting protection against Mtb and restricting it to advanced granulomas (63, 136).
MPCs Restrict the Mtb-Induced Inhibition of Phagosome Maturation
Mononuclear phagocyte cells have developed an array of strategies to control Mtb infection. MPCs recognize and phagocytose Mtb through PRRs such as C-type lectins, Dectin-1, Dectin-2, Mincle, macrophage C-type lectin, DC-SIGN, MR, scavenger receptors-A, and macrophage receptor with collagenous structure (MARCO) (scavenger receptors) (137). Consequently, activation through these receptors triggers various downstream signaling pathways mainly through Rac1–2, Cdc42, and most importantly GTPases. Arp2/3 is a key activator of actin polymerization. It is a primary step in instigating the process of phagocytosis and is triggered by the interaction of Wiskott–Aldrich syndrome protein with Rac1–2 and Cdc42 (43). Phagocytosis of Mtb triggers the formation and maturation of phagosome. During this process, a sequence of events occurs involving several molecules (25). Fc-gamma receptor along with MR is involved in the antigen trafficking to early phagosome. Early phagosome formation occurs upon interaction of MR with Mtb lipids such as PIMs and manLAM (138). The phosphorylation of immunoreceptor tyrosine-based activation motif by kinase of Src family, followed by downstream phosphorylation of Src homology region 2 domain-containing phosphatase-1 (SHP-1) and ras-related C3 botulinum toxin substrate (RAC) are critical for phagosome maturation (139). Membrane molecules exchange and deliver cargo either by “touch and run” or complete fusion with phagosome undergoing maturation. Motor proteins such as dynein and dynactin are key players to bring vesicles in an appropriate orientation for vesicular fusion, which is important for phagosome maturation. Many SNARE proteins such as vesicle-associated membrane proteins-7 and VAMP-8 are also involved in this event. During phagosome maturation, early endosome carrying Mtb undergo closure forming phagocytic cup by various coat proteins such as coronin or tryptophane aspartate-containing coat protein (140). Recruitment of proteins such as PX or FYVE motif proteins such as early endosome antigen 1 (EEA1) to early endosome for phagosome maturation is done through phosphotidylinositol-3-phosphate (PI(3)P) (141, 142). Endosome fusion to phagosome leads to oxidative and hydrolytic environment, which ultimately causes cargo degradation (143). Recruitment of lysosome-associated membrane proteins (LAMPs) 1 and 2 is a characteristic feature of late endosomal stage. Acidic pH of around 5 is an important marker of the late endosomal stage to control Mtb growth. This acidification process is controlled by Abl tyrosine kinase that functions as a negative regulator of phagosome maturation. Inhibition of this kinase by certain drugs such as imatinib results in controlling Mtb growth (144). The lipid body formation in the cell is induced by various bacterial infections such as S. aureus, M. leprae, and Mtb. The fusion of lipid compartments of the cells has been shown to be important for the maturation of phagosomes containing Mtb (145). However, Mtb resist this process of eradication by interfering in the maturation of phagosomes and subsequent fusion with lysosome (44). Mtb bearing phagosomes show reduced acidification due to halt H+-ATPase (146). The lipids produced by Mtb inside the macrophages mimic the host lipids such as phosphatidylinositol (PI3P) giving rise to inhibition of PI3P trafficking (147). EEA1 is an important molecule that inhibits Rab5 and acts as a key player in membrane fusion (148). There is a reduced recruitment of EEA1 in Mtb-infected macrophages, which inhibits the maturation of phagosome (149). Ca2+ is an important molecule involved in the phagosome maturation. However, Mtb inhibits sphingosine kinase dependent Ca2+ increase, leading to reduced phagosome maturation (150).
Phagosome containing Mtb express abnormal early endosomal markers for instance small GTPase Rab5, transferrin along with its receptor and absence of late endosomal markers viz small GTPase Rab7 along with vacuolar proton transporter v-ATPase (146). Furthermore, there is a reduced level of PI3P, EEA1 along with hepatocyte growth factor–regulated tyrosine kinase substrate (HRS) onto the Mtb phagosome membrane. These molecules are implicated in the protein sorting and fusion of the phagosome with late endosome followed by lysosome (151). Recently, it has been demonstrated that protein tyrosine phosphatase A (PtpA) binds to one of the subunits of v-ATPase leading to dephosphorylation of vacuolar-sorting protein 33B (152). Furthermore, Mtb glycolipids such as LAM as well as TDM inhibit the phagolysosome fusion (153). Mtb employs its type VII secretion system by exporting effector proteins EsxH and EsxG to destroy endosomal sorting complex required for transport and thereby impairing the maturation of phagosome (154). Overall, this signifies that the modulation of phagosome maturation of MPS can ultimately lead to the eradication of Mtb. In future, it can be used as an important therapeutic platform to control TB (Figure 2).
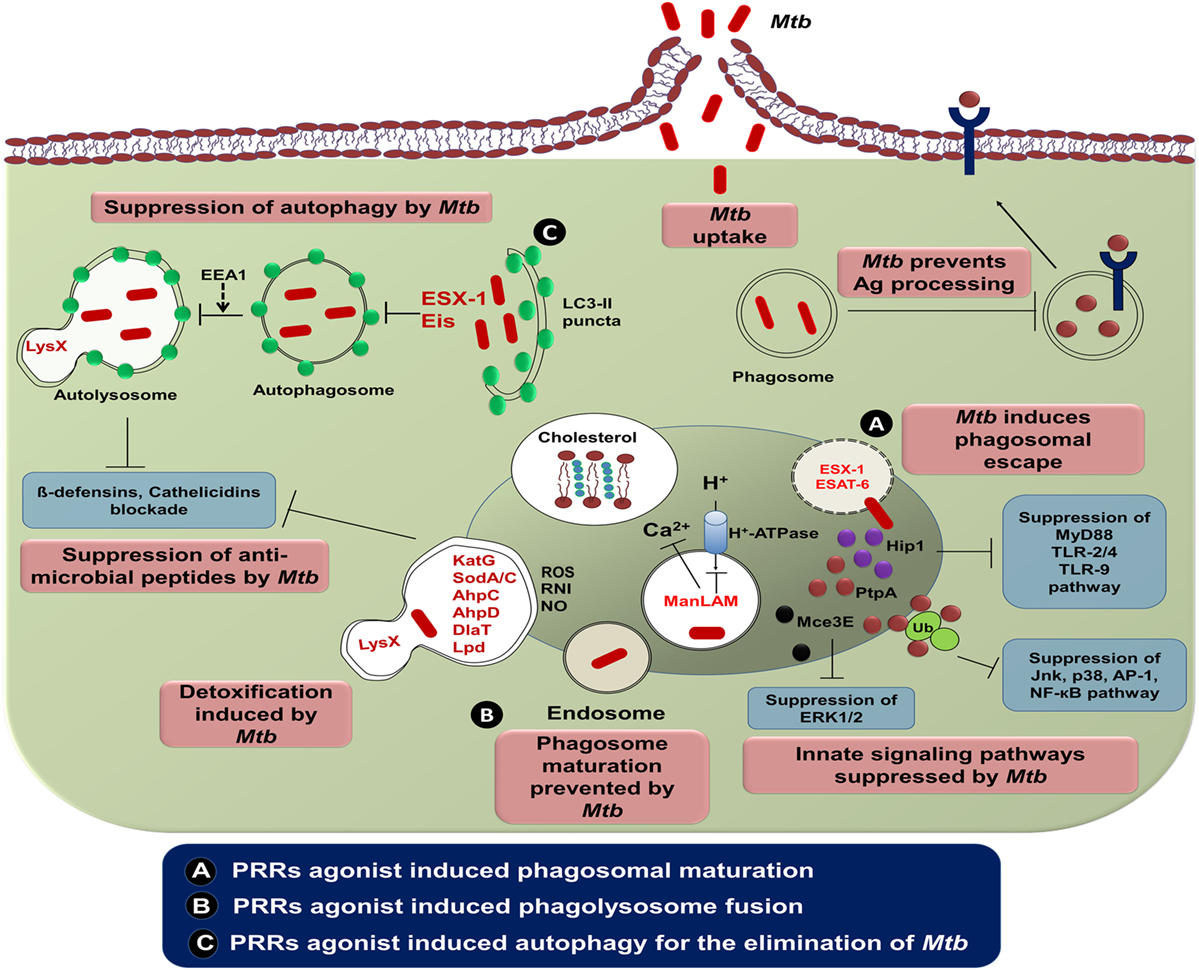
Figure 2. The intracellular evasion strategies adopted by Mycobacterium tuberculosis (Mtb) and its counteraction through cellular defense mechanism. Phagocytosis of Mtb is promoted by diverse cell-surface receptors and cholesterol present in the mononuclear phagocytic cells. Mtb utilizes the host cholesterol for its survival and impedes antigen processing and presentation by its lipoproteins. Consequently, ESAT-6 and ESX-1 of Mtb alter phagosome maturation process. The potential virulence factors, namely, PtpA and Mce3E of Mtb ultimately restrain various signaling cascades of innate immunity by binding with host ubiquitin. Another virulent factor of Mtb, ManLAM arrests phagosomal maturation via interrupting the transport of host H+-ATPase to phagosomes and blockading cytosolic Ca2+ release. Mtb enzymes such as KatG, SodA/C, NADH-dependent peroxidase, superoxide dismutases, and DlaT are involved in detoxification of ROI and RNI. Neutralization of antimicrobial peptides is accomplished through mycobacterial protein LysX. Suppression of autophagy in mononuclear cells is rendered by the Mtb encoded gene “enhanced intracellular survival (Eis).” (A) Several PRRs agonist such as TLRs (TLR-2, -4, and -9), NLRs (NOD-1 and NOD-2), and CLRs (Mincle, Dectin-1, and Dectin-2) induce phagosomal maturation and inhibit Mtb growth by membrane cholesterol reduction. (B,C) Involvement of these agonists triggers the phagolysosome fusion and subsequent process of autophagy. To monitor the effect of targeting various PRRs, a comprehensive investigation is required, before selecting the best combination of agonists to control Mtb infection. Abbreviations: Hip1, huntingtin-interacting protein 1; PtpA, protein tyrosine phosphatase A; Mce3E, mammalian cell entry operon 3E; ManLAM, mannose lipoarabinomannan; EEA1, early endosome antigen 1; ESAT-6, early secreted antigenic target of 6 kDa; ESX-1, ESTAT6 secretion system l; LysX, lysylphosphatidylglycerol biosynthesis bifunctional protein; KatG, catalase-peroxidase; SodA/C, superoxide dismutase A/C; AhpC/D, alkyl hydroperoxide reductase subunit C/D; DlaT, dihydrolipoamide acyltransferase; Lpd, lipoamide dehydrogenase; Ag, antigen; Ub, ubiquitin; MyD88, myeloid differentiation primary response gene 88; TLR, toll-like receptor; Jnk, c-Jun N-terminal kinase; AP-1, activator protein 1; NF-κB, nuclear factor-κB; ERK1/2, extracellular signal-regulated protein kinases 1 and 2; LC3, microtubule-associated protein 1A/1B-light chain 3; ROS, reactive oxygen species; RNI, reactive nitrogen intermediates; NO, nitric oxide; CLR, C-type lectin receptor; NOD, nucleotide-binding oligomerization domain; Mincle, macrophage-inducible C-type lectin.
Regulatory Role of MPCs in Inducing Autophagy Against Mtb
Autophagy is an intracellular degradation phenomenon that is developed during the stress response. It allows cells to alter their biomass and turn over components during starvation. Autophagy specifically targets the cytoplasmic components, which include organelles, macromolecules, and cells undergoing unintended cell death to lysosomes for their degradation. It ultimately leads to a periodical cleaning of the cell interiors. Similarly, autophagy has an essential role in numerous diseases, which includes cancer, degenerative diseases, as well as aging. In addition, autophagy augments the ability of cells to engulf and eliminate microbes and thereby protects the host. Treatment with rapamycin or IFN-γ or starvation can initiate and enhance autophagy. Furthermore, signaling through PRRs has been reported to have direct association with the induction of autophagy. It has been well established that triggering through various agonists of TLR-3, TLR-4, and TLR-7 can promote autophagy (155).
Autophagy boosts bactericidal mechanism by sequestering of the process in “double membrane envelope” structure called autophagosome. These processes follow the fusion of autophagosome with lysosomes by forming autolysosome for the subsequent elimination of the mycobacterium. Autophagy can be initiated within 30 min, as shown through the conversion of LC3-I to LC3-II, which is a fundamental indicator of this process. Autophagy helps in the clearance of Mycobacterium bovis BCG as well as Mtb by transporting them to the lysosome for their successive degradation (156). It has been reported that triggering infected macrophages through TLR-7 stimulates autophagy and can curb the intracellular growth of Mtb (155). Autophagy not only transports Mtb to lysosomes but also delivers bactericidal components to the Mtb degradation compartment. This observation was further confirmed by blocking the autophagy through knockdown of Atg5 and Beclin 1. Both these molecules are considered to be essential for autophagy. It may be an exciting line of study to identify the mechanism that triggers the autophagy-mediated clearance of Mtb. One of the possible mechanisms is an ubiquitination process, where the arrangement of “poly-ubiquitinylated protein aggregates” and contributes as an autophagy substrate. These protein aggregates are broken down into bactericidal peptides, which contribute in destroying Mtb (157).
Mycobacterium tuberculosis impedes MPCs bactericidal mechanism by deactivating the acidification of phagosome, lysosome and subsequently inhibiting the phagosome–lysosome fusion. Interestingly, autophagy directs the innate immunity to obstruct the evasion strategies adopted by Mtb by targeting the bacterium inhabiting inside, as well as outside the phagosome (156, 158). Similarly, BCG also hampers fusion of the phagosome with the lysosome, consequently resulting in the interference of antigen processing, presentation and therefore impairment in T cell response. This is considered as one of the possible reasons for BCG failure to safeguard people living in TB-endemic regions.
By contrast, autophagy in MPCs promotes both Mtb and BCG antigens processing and presentation. Mice adoptively transferred BCG infected APCs that were incubated with rapamycin, elicited Th1 cells that protected against Mtb. Rapamycin-induced autophagy with subsequent antigen processing and presentation was suppressed by treatment of cells with the autophagy inhibitors 3-MA or small interfering RNA against Beclin 1 (159). Designing of a novel approach with an appropriate adjuvant to induce autophagy may be an alternative and effective strategy to make BCG an effective vaccine for people living in TB-endemic zones. To achieve a full pharmacological evidence of the importance of the phagocytotic process in TB, it will be of interest to ascertain whether several PRR agonists or certain autophagy inducers are capable of stimulating the formation of autophagolysosome in MPCs (Figure 3).
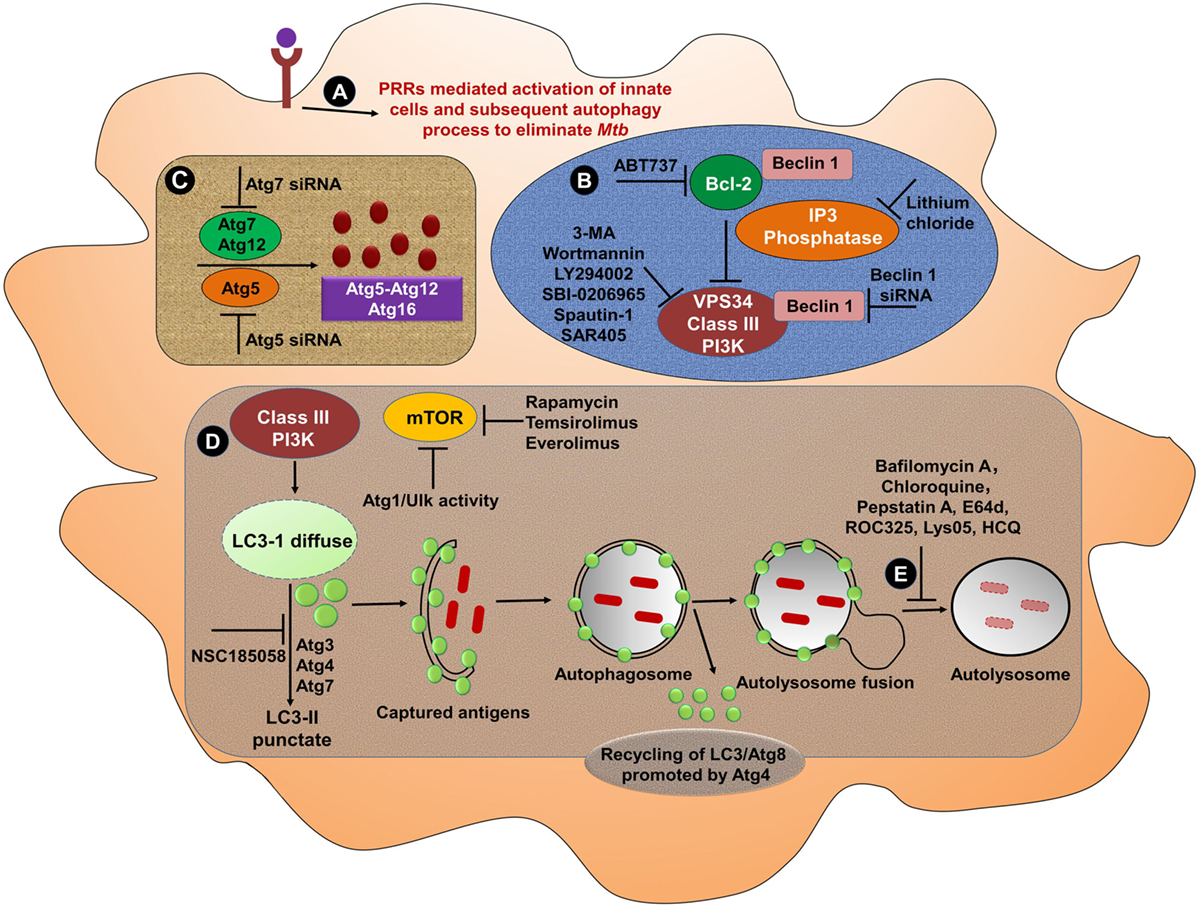
Figure 3. Bolstering the functionality of mononuclear phagocytic cells through PRRs and induction of autophagy. (A) Several PRRs-mediated approaches can be used to activate MPS. Activated cells then undergo autophagy to eliminate Mtb. Various strategies can be employed to induce or block different steps of autophagy from phagosome maturation to lysosomal fusion. (B) At the initiation stage of autophagy, the phagophore formation or nucleation processes occur. One of the most frequently used genetic approaches to inhibit autophagy is knockdown or knockout of Beclin 1 gene to demonstrate the specificity. The activation of Beclin 1 gene induces autophagy. The instigation of autophagy can be originated through the activation of PI3 kinase pathway. Treatment of cells with 3-MA, wortmannin, LY294002, SBI-0206965, spautin-1, and SAR405 inhibits the activity of class III PI3K for instance Vps34, which abrogates autophagy process. Autophagy can be artificially induced using lithium chloride, which inhibits inositol phosphatase, leading to augmented intracellular PI3P levels. Other targeted peptides, such as ABT737 that obstruct the interaction of Beclin 1 with Bcl-2 have also been validated to promote autophagy (160). (C) Similarly, the knockdown of Beclin 1 can effectively inhibit the autophagy, leading to the knockdown of Atg5 (161). Therefore, the knockdown of Beclin 1 may be the preferred approach to inhibit the autophagy. (D) The treatment with rapamycin induces the autophagy through its capacity to obstruct the inhibitory activity of mTOR. Subsequently, the conversion of LC3-I to LC3-II, capturing antigens and phagosome–lysosome fusion can effectively clear pathogens. (E) Several inhibitors such as bafilomycin A1 that inhibit the lysosomal Na+H+ ATPase are frequently used to reduce lysosomal turnover of autophagosomes. Other agents such as chloroquine, HCQ, Lys05, and ROC325 increase pH, lead to the prevention of the lysosomal acid proteases, as well as cause autophagosomes to accumulate in the lysosome (162, 163). The specific inhibitors of lysosomal proteases, for instance, pepstatin A or E64d abrogates the autophagy (164). Abbreviations: PRRs, pattern recognition receptors; Atg, autophagy-related protein; siRNA, small interfering RNA; Ulk, Unc-51-like kinase 1; ROC325, inhibitor of lysosomal-mediated autophagy; Lys05, dimeric chloroquine (lysosomal autophagy inhibitor); 3-MA, 3-methyl adenine; LC3, microtubule-associated protein 1A/1B-light chain 3; PIK3C3/Vps34, class III phosphatidylinositol-3-kinase; SAR405, selective ATP-competitive inhibitor of Vps34; HCQ, hydroxychloroquine; E64d, ethyl-ester of E64c; Bcl-2, B cell lymphoma-2; ABT-737, BH3 mimetic inhibitor of Bcl-2; NSC185058, inhibitor of Atg4B; mTOR, mammalian target of rapamycin; ATP, adenosine-5′-triphosphate; MPS, mononuclear phagocyte system; Mtb, Mycobacterium tuberculosis.
Development of the Possible Immunotherapeutic Strategies to Enhance Anti-TB Immunity
After the discovery of anti-TB drugs, it was assumed that the disease can be easily eliminated. Unfortunately, this could not be achieved due to the lengthy regimen, narrow therapeutic index and emergence of drug-resistant strains of Mtb (165). Currently, novel therapies are being explored for the treatment of numerous ailments such as cardiac diseases, cancer, and autoimmunity. In recent times, better information of host–pathogen interplay has given rise to a paradigm shift in remedial measures such as host-directed therapies, signaling pathway blockade, stem cells, signaling via receptors, adoptive transfer of antigen-loaded DCs to protect against cancers, treatment with immunomodulators and humanized Abs, probiotics as well as herbal remedies. In addition, Food and Drug Administration has permitted anti-cytotoxic T lymphocyte-associated antigen-4, CD80 as well as CD52 antibodies for treating cancer. Besides this, interferon-β is endorsed for the treatment of multiple sclerosis (166, 167). In spite of promising immunotherapies in diverse diseases, no thoughtful effort has been attempted in case of TB. Furthermore, immunotherapies with agonists of PRRs in conjunction with drugs have shown to improve the clinical outcome of the disease (168, 169). It will be of great interest to monitor the impact of drugs on Mtb in association with the immunomodulatory activity driven through PRRs. Such stratagem has dual advantage over the treatment with drugs alone. The drug will kill the bacterium residing in the MPCs, whereas immunomodulators will stimulate the host immunity to eliminate the pathogen, which had escaped the killing by the drug. Furthermore, this approach may not only reduce the dose as well as duration of the anti-TB drug regimen but can also curb the development of drug resistance in Mtb. Therefore, in future, immunotherapies may be the best choice to treat TB and its drug-resistant form.
The second most effective strategy to control and eliminate any disease is vaccine (3, 4). Presently, BCG is the only existing vaccine to treat TB. Ironically, BCG is the most controversial vaccine because of its highly variable efficacy worldwide. Moreover, it protects only children but not adults, as very categorically evident by 15 years follow-up study in India (170). The urgent necessity and challenge for the scientific communal is to improve the current drug regimen or develop alternative and innovative stratagems against TB. In essence, reinforcing the immunity against Mtb by triggering through the receptors of innate immunity might be a prudent idea to treat TB patients.
Conclusion and Future Prospects
There is no iota of doubt that for several years TB is treated with potent drugs. Unfortunately, the disease is neither controlled nor eradicated by these drugs; rather the regime has contributed in gifting resistant strains of the Mtb. Consequently, it is an important to devise and discover innovative and alternative therapies to control TB. In this connection, understanding the struggle between the bacterium and the host cells such as MPS may have an important impact on the disease outcome. The safe survival heaven for Mtb is MPCs. Targeting molecules such as PRRs that can optimally elicit MPCs to eliminate Mtb will be an interesting strategy to employ as an adjunct therapy along with the anti-TB drugs. There may be a distinct possibility of such therapeutic measurement in controlling TB by decreasing the dose and duration of drugs and also curbing the emergence of mono and MDR strains of the bacterium. In this article, we have mentioned about a possible combination of various PRRs such as TLRs (TLR-2, TLR-4, and TLR-9), NLRs (NOD-1 and NOD-2), and CLRs (Mincle, Dectin-1, and Dectin-2) as suggested by several studies, to control Mtb infection. However, studies are still required to select the best possible combination of PRRs to achieve complete elimination of Mtb. Exploration of this strategy may be an exciting line of future study to control TB.
Author Contributions
In this manuscript, the concept and theme were generated by JA and SP. The writing of manuscript and figures were done by SP, GK, SN, MA, DD, HB, SS, MN, JK, and JA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work is funded by the Council of Scientific and Industrial Research (CSIR), New Delhi, India. SP, GK, HB, and JK are the Council of Scientific and Industrial Research fellowship recipient; SN, DD, and MN of Department of Biotechnology (DBT); MA of Department of Science and Technology (DST), and SS of Indian Council of Medical Research (ICMR).
References
2. Gandhi NR, Nunn P, Dheda K, Schaaf HS, Zignol M, van Soolingen D, et al. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. Lancet (2010) 375(9728):1830–43. doi:10.1016/S0140-6736(10)60410-2
3. Francis DP. Successes and failures: worldwide vaccine development and application. Biologicals (2010) 38(5):523–8. doi:10.1016/j.biologicals.2010.06.003
4. Smith R, Renaud RC. Vaccines of the future. Nat Rev Drug Discov (2003) 2(10):767–8. doi:10.1038/nrd1204
5. Gowthaman U, Rai PK, Khan N, Jackson DC, Agrewala JN. Lipidated promiscuous peptides vaccine for tuberculosis-endemic regions. Trends Mol Med (2012) 18(10):607–14. doi:10.1016/j.molmed.2012.07.008
6. Andersen P, Doherty TM. The success and failure of BCG – implications for a novel tuberculosis vaccine. Nat Rev Microbiol (2005) 3(8):656–62. doi:10.1038/nrmicro1211
7. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22(2):240–73. doi:10.1128/CMR.00046-08
8. Hoebe K, Janssen E, Beutler B. The interface between innate and adaptive immunity. Nat Immunol (2004) 5(10):971–4. doi:10.1038/ni1004-971
9. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol (2014) 14(8):571–8. doi:10.1038/nri3712
10. Hume DA, Ross IL, Himes SR, Sasmono RT, Wells CA, Ravasi T. The mononuclear phagocyte system revisited. J Leukoc Biol (2002) 72(4):621–7.
11. Jenkins SJ, Hume DA. Homeostasis in the mononuclear phagocyte system. Trends Immunol (2014) 35(8):358–67. doi:10.1016/j.it.2014.06.006
12. Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol (2001) 2(8):675–80. doi:10.1038/9060990609
13. Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol (2001) 2(10):947–50. doi:10.1038/ni712
14. Steinman RM. Linking innate to adaptive immunity through dendritic cells. Novartis Found Symp (2006) 279:101–9; discussion 109–13, 216–9.
15. Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol (2006) 311:17–58.
16. Pahari S, Kaur G, Aqdas M, Negi S, Chatterjee D, Bashir H, et al. Bolstering immunity through pattern recognition receptors: a unique approach to control tuberculosis. Front Immunol (2017) 8:906. doi:10.3389/fimmu.2017.00906
17. McClean CM, Tobin DM. Macrophage form, function, and phenotype in mycobacterial infection: lessons from tuberculosis and other diseases. Pathog Dis (2016) 74(7). doi:10.1093/femspd/ftw068
18. van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ (1972) 46(6):845–52.
19. Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol (2007) 179(4):2509–19. doi:10.4049/jimmunol.179.4.2509
20. Marino S, Pawar S, Fuller CL, Reinhart TA, Flynn JL, Kirschner DE. Dendritic cell trafficking and antigen presentation in the human immune response to Mycobacterium tuberculosis. J Immunol (2004) 173(1):494–506. doi:10.4049/jimmunol.173.1.494
21. Fortsch D, Rollinghoff M, Stenger S. IL-10 converts human dendritic cells into macrophage-like cells with increased antibacterial activity against virulent Mycobacterium tuberculosis. J Immunol (2000) 165(2):978–87. doi:10.4049/jimmunol.165.2.978
22. Srivastava S, Ernst JD, Desvignes L. Beyond macrophages: the diversity of mononuclear cells in tuberculosis. Immunol Rev (2014) 262(1):179–92. doi:10.1111/imr.12217
23. Hanekom WA, Mendillo M, Manca C, Haslett PA, Siddiqui MR, Barry C III, et al. Mycobacterium tuberculosis inhibits maturation of human monocyte-derived dendritic cells in vitro. J Infect Dis (2003) 188(2):257–66. doi:10.1086/376451
24. Saraav I, Singh S, Sharma S. Outcome of Mycobacterium tuberculosis and Toll-like receptor interaction: immune response or immune evasion? Immunol Cell Biol (2014) 92(9):741–6. doi:10.1038/icb.2014.52
25. Flannagan RS, Cosio G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol (2009) 7(5):355–66. doi:10.1038/nrmicro2128
26. Goldberg MF, Saini NK, Porcelli SA. Evasion of innate and adaptive immunity by Mycobacterium tuberculosis. Microbiol Spectr (2014) 2(5). doi:10.1128/microbiolspec.MGM2-0005-2013
27. Poirier V, Av-Gay Y. Mycobacterium tuberculosis modulators of the macrophage’s cellular events. Microbes Infect (2012) 14(13):1211–9. doi:10.1016/j.micinf.2012.07.001
28. Humphreys IR, Stewart GR, Turner DJ, Patel J, Karamanou D, Snelgrove RJ, et al. A role for dendritic cells in the dissemination of mycobacterial infection. Microbes Infect (2006) 8(5):1339–46. doi:10.1016/j.micinf.2005.12.023
29. Thi EP, Hong CJ, Sanghera G, Reiner NE. Identification of the Mycobacterium tuberculosis protein PE-PGRS62 as a novel effector that functions to block phagosome maturation and inhibit iNOS expression. Cell Microbiol (2013) 15(5):795–808. doi:10.1111/cmi.12073
30. Deghmane AE, Soualhine H, Bach H, Sendide K, Itoh S, Tam A, et al. Lipoamide dehydrogenase mediates retention of coronin-1 on BCG vacuoles, leading to arrest in phagosome maturation. J Cell Sci (2007) 120(Pt 16):2796–806. doi:10.1242/jcs.006221
31. Bach H, Papavinasasundaram KG, Wong D, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe (2008) 3(5):316–22. doi:10.1016/j.chom.2008.03.008
32. Vergne I, Chua J, Lee HH, Lucas M, Belisle J, Deretic V. Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc Natl Acad Sci U S A (2005) 102(11):4033–8. doi:10.1073/pnas.0409716102
33. Khan N, Gowthaman U, Pahari S, Agrewala JN. Manipulation of costimulatory molecules by intracellular pathogens: veni, vidi, vici!! PLoS Pathog (2012) 8(6):e1002676. doi:10.1371/journal.ppat.1002676
34. Jozefowski S, Sobota A, Kwiatkowska K. How Mycobacterium tuberculosis subverts host immune responses. Bioessays (2008) 30(10):943–54. doi:10.1002/bies.20815
35. Khan N, Vidyarthi A, Amir M, Mushtaq K, Agrewala JN. T-cell exhaustion in tuberculosis: pitfalls and prospects. Crit Rev Microbiol (2017) 43(2):133–41. doi:10.1080/1040841X.2016.1185603
36. Luo M, Fadeev EA, Groves JT. Mycobactin-mediated iron acquisition within macrophages. Nat Chem Biol (2005) 1(3):149–53. doi:10.1038/nchembio717
37. Munoz-Elias EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med (2005) 11(6):638–44. doi:10.1038/nm1252
38. Marrero J, Rhee KY, Schnappinger D, Pethe K, Ehrt S. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc Natl Acad Sci U S A (2010) 107(21):9819–24. doi:10.1073/pnas.1000715107
39. McKinney JD, Honer zu Bentrup K, Munoz-Elias EJ, Miczak A, Chen B, Chan WT, et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature (2000) 406(6797):735–8. doi:10.1038/35021074
40. Arkhipov SA, Shkurupy VA, Solomatina MV, Akhramenko ES, Iljine DA. Study of macrophages in BCG granulomas in different compartments of the mononuclear phagocyte system. Bull Exp Biol Med (2013) 154(4):467–70. doi:10.1007/s10517-013-1979-6
41. Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol (2009) 27:451–83. doi:10.1146/annurev.immunol.021908.132532
42. Mege JL, Mehraj V, Capo C. Macrophage polarization and bacterial infections. Curr Opin Infect Dis (2011) 24(3):230–4. doi:10.1097/QCO.0b013e328344b73e
43. Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature (2014) 505(7482):218–22. doi:10.1038/nature12799
44. Lugo-Villarino G, Neyrolles O. Manipulation of the mononuclear phagocyte system by Mycobacterium tuberculosis. Cold Spring Harb Perspect Med (2014) 4(11):a018549. doi:10.1101/cshperspect.a018549
45. He XY, Xiao L, Chen HB, Hao J, Li J, Wang YJ, et al. T regulatory cells and Th1/Th2 cytokines in peripheral blood from tuberculosis patients. Eur J Clin Microbiol Infect Dis (2010) 29(6):643–50. doi:10.1007/s10096-010-0908-0
46. Lyadova IV, Panteleev AV. Th1 and Th17 cells in tuberculosis: protection, pathology, and biomarkers. Mediators Inflamm (2015) 2015:854507. doi:10.1155/2015/854507
47. Diebold J. [Mononuclear phagocyte system. Morphology and function of the principal constituting cells]. Ann Pathol (1986) 6(1):3–12.
48. Sareila O, Kelkka T, Pizzolla A, Hultqvist M, Holmdahl R. NOX2 complex-derived ROS as immune regulators. Antioxid Redox Signal (2011) 15(8):2197–208. doi:10.1089/ars.2010.3635
49. Koopman WJ, Nijtmans LG, Dieteren CE, Roestenberg P, Valsecchi F, Smeitink JA, et al. Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid Redox Signal (2010) 12(12):1431–70. doi:10.1089/ars.2009.2743
50. Arnoult D, Soares F, Tattoli I, Castanier C, Philpott DJ, Girardin SE. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J Cell Sci (2009) 122(Pt 17):3161–8. doi:10.1242/jcs.051193
51. Miner MD, Chang JC, Pandey AK, Sassetti CM, Sherman DR. Role of cholesterol in Mycobacterium tuberculosis infection. Indian J Exp Biol (2009) 47(6):407–11.
52. Trautmann A. Extracellular ATP in the immune system: more than just a “danger signal”. Sci Signal (2009) 2(56):e6. doi:10.1126/scisignal.256pe6
53. Hanley PJ, Musset B, Renigunta V, Limberg SH, Dalpke AH, Sus R, et al. Extracellular ATP induces oscillations of intracellular Ca2+ and membrane potential and promotes transcription of IL-6 in macrophages. Proc Natl Acad Sci U S A (2004) 101(25):9479–84. doi:10.1073/pnas.0400733101
54. Grahames CB, Michel AD, Chessell IP, Humphrey PP. Pharmacological characterization of ATP- and LPS-induced IL-1beta release in human monocytes. Br J Pharmacol (1999) 127(8):1915–21. doi:10.1038/sj.bjp.0702732
55. Qu Y, Ramachandra L, Mohr S, Franchi L, Harding CV, Nunez G, et al. P2X7 receptor-stimulated secretion of MHC class II-containing exosomes requires the ASC/NLRP3 inflammasome but is independent of caspase-1. J Immunol (2009) 182(8):5052–62. doi:10.4049/jimmunol.0802968
56. Ramachandra L, Qu Y, Wang Y, Lewis CJ, Cobb BA, Takatsu K, et al. Mycobacterium tuberculosis synergizes with ATP to induce release of microvesicles and exosomes containing major histocompatibility complex class II molecules capable of antigen presentation. Infect Immun (2010) 78(12):5116–25. doi:10.1128/IAI.01089-09
57. Kusner DJ, Adams J. ATP-induced killing of virulent Mycobacterium tuberculosis within human macrophages requires phospholipase D. J Immunol (2000) 164(1). doi:10.4049/jimmunol.164.1.379
58. Lammas DA, Stober C, Harvey CJ, Kendrick N, Panchalingam S, Kumararatne DS. ATP-induced killing of mycobacteria by human macrophages is mediated by purinergic P2Z(P2X7) receptors. Immunity (1997) 7(3):433–44. doi:10.1016/S1074-7613(00)80364-7
59. Biswas D, Qureshi OS, Lee WY, Croudace JE, Mura M, Lammas DA. ATP-induced autophagy is associated with rapid killing of intracellular mycobacteria within human monocytes/macrophages. BMC Immunol (2008) 9:35. doi:10.1186/1471-2172-9-35
60. Fairbairn IP, Stober CB, Kumararatne DS, Lammas DA. ATP-mediated killing of intracellular mycobacteria by macrophages is a P2X(7)-dependent process inducing bacterial death by phagosome-lysosome fusion. J Immunol (2001) 167(6):3300–7. doi:10.4049/jimmunol.167.6.3300
61. Cotton M, Claing A. G protein-coupled receptors stimulation and the control of cell migration. Cell Signal (2009) 21(7):1045–53. doi:10.1016/j.cellsig.2009.02.008
62. Manivannan S, Rao NV, Ramanathan VD. Role of complement activation and antibody in the interaction between Mycobacterium tuberculosis and human macrophages. Indian J Exp Biol (2012) 50(8):542–50.
63. Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev (2015) 264(1):182–203. doi:10.1111/imr.12266
64. Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor-beta1 promotes in vivo Th17 cell differentiation. Immunity (2011) 34(3):396–408. doi:10.1016/j.immuni.2011.03.005
65. Hidaka M, Wakabayashi I, Takeda Y, Fukuzawa K. Vitamin D(3) derivatives increase soluble CD14 release through ERK1/2 activation and decrease IL-8 production in intestinal epithelial cells. Eur J Pharmacol (2013) 721(1–3):305–12. doi:10.1016/j.ejphar.2013.09.014
66. Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science (2006) 311(5768):1770–3. doi:10.1126/science.1123933
67. Mortaz E, Adcock IM, Tabarsi P, Masjedi MR, Mansouri D, Velayati AA, et al. Interaction of pattern recognition receptors with Mycobacterium tuberculosis. J Clin Immunol (2015) 35(1):1–10. doi:10.1007/s10875-014-0103-7
68. Khan N, Pahari S, Vidyarthi A, Aqdas M, Agrewala JN. Stimulation through CD40 and TLR-4 is an effective host directed therapy against Mycobacterium tuberculosis. Front Immunol (2016) 7:386. doi:10.3389/fimmu.2016.00386
69. Khan N, Aqdas M, Vidyarthi A, Negi S, Pahari S, Agnihotri T, et al. Triggering through NOD-2 differentiates bone marrow precursors to dendritic cells with potent bactericidal activity. Sci Rep (2016) 6:27263. doi:10.1038/srep27263
70. Khan N, Vidyarthi A, Pahari S, Negi S, Aqdas M, Nadeem S, et al. Signaling through NOD-2 and TLR-4 bolsters the T cell priming capability of dendritic cells by inducing autophagy. Sci Rep (2016) 6:19084. doi:10.1038/srep19084
71. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol (2013) 13(10):722–37. doi:10.1038/nri3532
72. Moraco AH, Kornfeld H. Cell death and autophagy in tuberculosis. Semin Immunol (2014) 26(6):497–511. doi:10.1016/j.smim.2014.10.001
73. Pahari S, Khan N, Aqdas M, Negi S, Kaur J, Agrewala JN. Infergen stimulated macrophages restrict Mycobacterium tuberculosis growth by autophagy and release of nitric oxide. Sci Rep (2016) 6:39492. doi:10.1038/srep39492
74. Cunha LD, Zamboni DS. Subversion of inflammasome activation and pyroptosis by pathogenic bacteria. Front Cell Infect Microbiol (2013) 3:76. doi:10.3389/fcimb.2013.00076
75. Drennan MB, Nicolle D, Quesniaux VJ, Jacobs M, Allie N, Mpagi J, et al. Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am J Pathol (2004) 164(1):49–57. doi:10.1016/S0002-9440(10)63095-7
76. Shin DM, Yang CS, Lee JY, Lee SJ, Choi HH, Lee HM, et al. Mycobacterium tuberculosis lipoprotein-induced association of TLR2 with protein kinase C zeta in lipid rafts contributes to reactive oxygen species-dependent inflammatory signalling in macrophages. Cell Microbiol (2008) 10(9):1893–905. doi:10.1111/j.1462-5822.2008.01179.x
77. Lopez M, Sly LM, Luu Y, Young D, Cooper H, Reiner NE. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J Immunol (2003) 170(5):2409–16. doi:10.4049/jimmunol.170.5.2409
78. Reiling N, Holscher C, Fehrenbach A, Kroger S, Kirschning CJ, Goyert S, et al. Cutting edge: toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol (2002) 169(7):3480–4. doi:10.4049/jimmunol.169.7.3480
79. Amir M, Aqdas M, Nadeem S, Siddiqui KF, Khan N, Sheikh JA, et al. Diametric role of the latency-associated protein acr1 of Mycobacterium tuberculosis in modulating the functionality of pre- and post-maturational stages of dendritic cells. Front Immunol (2017) 8:624. doi:10.3389/fimmu.2017.00624
80. Siddiqui KF, Amir M, Gurram RK, Khan N, Arora A, Rajagopal K, et al. Latency-associated protein Acr1 impairs dendritic cell maturation and functionality: a possible mechanism of immune evasion by Mycobacterium tuberculosis. J Infect Dis (2014) 209(9):1436–45. doi:10.1093/infdis/jit595
81. Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med (2005) 202(12):1715–24. doi:10.1084/jem.20051782
82. Ferwerda G, Girardin SE, Kullberg BJ, Le Bourhis L, de Jong DJ, Langenberg DM, et al. NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog (2005) 1(3):279–85. doi:10.1371/journal.ppat.0010034
83. Juarez E, Carranza C, Hernandez-Sanchez F, Leon-Contreras JC, Hernandez-Pando R, Escobedo D, et al. NOD2 enhances the innate response of alveolar macrophages to Mycobacterium tuberculosis in humans. Eur J Immunol (2012) 42(4):880–9. doi:10.1002/eji.201142105
84. Rothfuchs AG, Bafica A, Feng CG, Egen JG, Williams DL, Brown GD, et al. Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J Immunol (2007) 179(6):3463–71. doi:10.4049/jimmunol.179.6.3463
85. Schoenen H, Bodendorfer B, Hitchens K, Manzanero S, Werninghaus K, Nimmerjahn F, et al. Cutting edge: mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J Immunol (2010) 184(6):2756–60. doi:10.4049/jimmunol.0904013
86. Wu T, Guo S, Wang J, Li L, Xu L, Liu P, et al. Interaction between mannosylated lipoarabinomannan and dendritic cell-specific intercellular adhesion molecule-3 grabbing nonintegrin influences dendritic cells maturation and T cell immunity. Cell Immunol (2011) 272(1):94–101. doi:10.1016/j.cellimm.2011.09.001
87. Sugawara I, Yamada H, Hua S, Mizuno S. Role of interleukin (IL)-1 type 1 receptor in mycobacterial infection. Microbiol Immunol (2001) 45(11):743–50. doi:10.1111/j.1348-0421.2001.tb01310.x
88. Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, et al. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol (2010) 12(8):1046–63. doi:10.1111/j.1462-5822.2010.01450.x
89. Kurenuma T, Kawamura I, Hara H, Uchiyama R, Daim S, Dewamitta SR, et al. The RD1 locus in the Mycobacterium tuberculosis genome contributes to activation of caspase-1 via induction of potassium ion efflux in infected macrophages. Infect Immun (2009) 77(9):3992–4001. doi:10.1128/IAI.00015-09
90. Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infect Immun (2002) 70(8):4501–9. doi:10.1128/IAI.70.8.4501-4509.2002
91. Santarlasci V, Cosmi L, Maggi L, Liotta F, Annunziato F. IL-1 and T helper immune responses. Front Immunol (2013) 4:182. doi:10.3389/fimmu.2013.00182
92. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature (2010) 467(7318):967–71. doi:10.1038/nature09447
93. Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol (2007) 8(4):369–77. doi:10.1038/ni1449
94. Kursar M, Koch M, Mittrucker HW, Nouailles G, Bonhagen K, Kamradt T, et al. Cutting edge: regulatory T cells prevent efficient clearance of Mycobacterium tuberculosis. J Immunol (2007) 178(5):2661–5. doi:10.4049/jimmunol.178.5.2661
95. Shafiani S, Tucker-Heard G, Kariyone A, Takatsu K, Urdahl KB. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J Exp Med (2010) 207(7):1409–20. doi:10.1084/jem.20091885
96. Domingo-Gonzalez R, Prince O, Cooper A, Khader SA. Cytokines and chemokines in Mycobacterium tuberculosis infection. Microbiol Spectr (2016) 4(5). doi:10.1128/microbiolspec.TBTB2-0018-2016
97. Flynn JL, Goldstein MM, Triebold KJ, Koller B, Bloom BR. Major histocompatibility complex class I-restricted T cells are required for resistance to Mycobacterium tuberculosis infection. Proc Natl Acad Sci U S A (1992) 89(24):12013–7. doi:10.1073/pnas.89.24.12013
98. Woodworth JS, Wu Y, Behar SM. Mycobacterium tuberculosis-specific CD8+ T cells require perforin to kill target cells and provide protection in vivo. J Immunol (2008) 181(12):8595–603. doi:10.4049/jimmunol.181.12.8595
99. Stenger S, Hanson DA, Teitelbaum R, Dewan P, Niazi KR, Froelich CJ, et al. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science (1998) 282(5386):121–5. doi:10.1126/science.282.5386.121
100. Serbina NV, Liu CC, Scanga CA, Flynn JL. CD8+ CTL from lungs of Mycobacterium tuberculosis-infected mice express perforin in vivo and lyse infected macrophages. J Immunol (2000) 165(1):353–63. doi:10.4049/jimmunol.165.1.353
101. Silva BDS, Trentini MM, da Costa AC, Kipnis A, Junqueira-Kipnis AP. Different phenotypes of CD8+ T cells associated with bacterial load in active tuberculosis. Immunol Lett (2014) 160(1):23–32. doi:10.1016/j.imlet.2014.03.009
102. D’Souza CD, Cooper AM, Frank AA, Mazzaccaro RJ, Bloom BR, Orme IM. An anti-inflammatory role for gamma delta T lymphocytes in acquired immunity to Mycobacterium tuberculosis. J Immunol (1997) 158(3):1217–21.
103. Tanaka Y, Sano S, Nieves E, De Libero G, Rosa D, Modlin RL, et al. Nonpeptide ligands for human gamma delta T cells. Proc Natl Acad Sci U S A (1994) 91(17):8175–9. doi:10.1073/pnas.91.17.8175
104. Boom WH, Chervenak KA, Mincek MA, Ellner JJ. Role of the mononuclear phagocyte as an antigen-presenting cell for human gamma delta T cells activated by live Mycobacterium tuberculosis. Infect Immun (1992) 60(9):3480–8.
105. Casetti R, Martino A. The plasticity of gamma delta T cells: innate immunity, antigen presentation and new immunotherapy. Cell Mol Immunol (2008) 5(3):161–70. doi:10.1038/cmi.2008.20
106. Yang CS, Yuk JM, Jo EK. The role of nitric oxide in mycobacterial infections. Immune Netw (2009) 9(2):46–52. doi:10.4110/in.2009.9.2.46
107. MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci U S A (1997) 94(10):5243–8. doi:10.1073/pnas.94.10.5243
108. Flesch IE, Kaufmann SH. Mechanisms involved in mycobacterial growth inhibition by gamma interferon-activated bone marrow macrophages: role of reactive nitrogen intermediates. Infect Immun (1991) 59(9):3213–8.
109. Akaki T, Sato K, Shimizu T, Sano C, Kajitani H, Dekio S, et al. Effector molecules in expression of the antimicrobial activity of macrophages against Mycobacterium avium complex: roles of reactive nitrogen intermediates, reactive oxygen intermediates, and free fatty acids. J Leukoc Biol (1997) 62(6):795–804. doi:10.1002/jlb.62.6.795
110. Nicholson S, Bonecini-Almeida Mda G, Lapa e Silva JR, Nathan C, Xie QW, Mumford R, et al. Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J Exp Med (1996) 183(5):2293–302. doi:10.1084/jem.183.5.2293
111. Sharma S, Sharma M, Roy S, Kumar P, Bose M. Mycobacterium tuberculosis induces high production of nitric oxide in coordination with production of tumour necrosis factor-alpha in patients with fresh active tuberculosis but not in MDR tuberculosis. Immunol Cell Biol (2004) 82(4):377–82. doi:10.1111/j.0818-9641.2004.01245.x
112. Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A (2000) 97(16):8841–8. doi:10.1073/pnas.97.16.8841
113. Ciccone R, Mariani F, Cavone A, Persichini T, Venturini G, Ongini E, et al. Inhibitory effect of NO-releasing ciprofloxacin (NCX 976) on Mycobacterium tuberculosis survival. Antimicrob Agents Chemother (2003) 47(7):2299–302. doi:10.1128/AAC.47.7.2299-2302.2003
114. Kuo HP, Wang CH, Huang KS, Lin HC, Yu CT, Liu CY, et al. Nitric oxide modulates interleukin-1beta and tumor necrosis factor-alpha synthesis by alveolar macrophages in pulmonary tuberculosis. Am J Respir Crit Care Med (2000) 161(1):192–9. doi:10.1164/ajrccm.161.1.9902113
115. Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol (2001) 19:93–129. doi:10.1146/annurev.immunol.19.1.93
116. Piddington DL, Fang FC, Laessig T, Cooper AM, Orme IM, Buchmeier NA. Cu,Zn superoxide dismutase of Mycobacterium tuberculosis contributes to survival in activated macrophages that are generating an oxidative burst. Infect Immun (2001) 69(8):4980–7. doi:10.1128/IAI.69.8.4980-4987.2001
117. Bryk R, Lima CD, Erdjument-Bromage H, Tempst P, Nathan C. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science (2002) 295(5557):1073–7. doi:10.1126/science.1067798
118. Colangeli R, Haq A, Arcus VL, Summers E, Magliozzo RS, McBride A, et al. The multifunctional histone-like protein Lsr2 protects mycobacteria against reactive oxygen intermediates. Proc Natl Acad Sci U S A (2009) 106(11):4414–8. doi:10.1073/pnas.0810126106
119. Pathania R, Navani NK, Gardner AM, Gardner PR, Dikshit KL. Nitric oxide scavenging and detoxification by the Mycobacterium tuberculosis haemoglobin, HbN in Escherichia coli. Mol Microbiol (2002) 45(5):1303–14. doi:10.1046/j.1365-2958.2002.03095.x
120. Hartman ML, Kornfeld H. Interactions between naive and infected macrophages reduce Mycobacterium tuberculosis viability. PLoS One (2011) 6(11):e27972. doi:10.1371/journal.pone.0027972
121. Chen M, Gan H, Remold HG. A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J Immunol (2006) 176(6):3707–16. doi:10.4049/jimmunol.176.6.3707
122. Lerner TR, Borel S, Greenwood DJ, Repnik U, Russell MR, Herbst S, et al. Mycobacterium tuberculosis replicates within necrotic human macrophages. J Cell Biol (2017) 216(3):583–94. doi:10.1083/jcb.201603040
123. Keane J, Remold HG, Kornfeld H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol (2000) 164(4):2016–20. doi:10.4049/jimmunol.164.4.2016
124. Balcewicz-Sablinska MK, Keane J, Kornfeld H, Remold HG. Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-alpha. J Immunol (1998) 161(5):2636–41.
125. Riendeau CJ, Kornfeld H. THP-1 cell apoptosis in response to Mycobacterial infection. Infect Immun (2003) 71(1):254–9. doi:10.1128/IAI.71.1.254-259.2003
126. Andersson H, Andersson B, Eklund D, Ngoh E, Persson A, Svensson K, et al. Apoptotic neutrophils augment the inflammatory response to Mycobacterium tuberculosis infection in human macrophages. PLoS One (2014) 9(7):e101514. doi:10.1371/journal.pone.0101514
127. Behar SM, Martin CJ, Booty MG, Nishimura T, Zhao X, Gan HX, et al. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol (2011) 4(3):279–87. doi:10.1038/mi.2011.3
128. Perskvist N, Long M, Stendahl O, Zheng L. Mycobacterium tuberculosis promotes apoptosis in human neutrophils by activating caspase-3 and altering expression of Bax/Bcl-xL via an oxygen-dependent pathway. J Immunol (2002) 168(12):6358–65. doi:10.4049/jimmunol.168.12.6358
129. Lim YJ, Choi JA, Choi HH, Cho SN, Kim HJ, Jo EK, et al. Endoplasmic reticulum stress pathway-mediated apoptosis in macrophages contributes to the survival of Mycobacterium tuberculosis. PLoS One (2011) 6(12):e28531. doi:10.1371/journal.pone.0028531
130. Choi JA, Lim YJ, Cho SN, Lee JH, Jeong JA, Kim EJ, et al. Mycobacterial HBHA induces endoplasmic reticulum stress-mediated apoptosis through the generation of reactive oxygen species and cytosolic Ca2+ in murine macrophage RAW 264.7 cells. Cell Death Dis (2013) 4:e957. doi:10.1038/cddis.2013.489
131. Pan H, Yan BS, Rojas M, Shebzukhov YV, Zhou H, Kobzik L, et al. Ipr1 gene mediates innate immunity to tuberculosis. Nature (2005) 434(7034):767–72. doi:10.1038/nature03419
132. Lee J, Hartman M, Kornfeld H. Macrophage apoptosis in tuberculosis. Yonsei Med J (2009) 50(1):1–11. doi:10.3349/ymj.2009.50.1.1
133. Saquib NM, Jamwal S, Midha MK, Verma HN, Manivel V. Quantitative proteomics and lipidomics analysis of endoplasmic reticulum of macrophage infected with Mycobacterium tuberculosis. Int J Proteomics (2015) 2015:270438. doi:10.1155/2015/270438
134. Lim YJ, Yi MH, Choi JA, Lee J, Han JY, Jo SH, et al. Roles of endoplasmic reticulum stress-mediated apoptosis in M1-polarized macrophages during mycobacterial infections. Sci Rep (2016) 6:37211. doi:10.1038/srep37211
135. Seimon TA, Kim MJ, Blumenthal A, Koo J, Ehrt S, Wainwright H, et al. Induction of ER stress in macrophages of tuberculosis granulomas. PLoS One (2010) 5(9):e12772. doi:10.1371/journal.pone.0012772
136. Lam A, Prabhu R, Gross CM, Riesenberg LA, Singh V, Aggarwal S. Role of apoptosis and autophagy in tuberculosis. Am J Physiol Lung Cell Mol Physiol (2017) 313(2):L218–29. doi:10.1152/ajplung.00162.2017
137. Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell (2012) 150(4):803–15. doi:10.1016/j.cell.2012.06.040
138. Kang PB, Azad AK, Torrelles JB, Kaufman TM, Beharka A, Tibesar E, et al. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J Exp Med (2005) 202(7):987–99. doi:10.1084/jem.20051239
139. Kant AM, De P, Peng X, Yi T, Rawlings DJ, Kim JS, et al. SHP-1 regulates Fcgamma receptor-mediated phagocytosis and the activation of RAC. Blood (2002) 100(5):1852–9.
140. Schuller S, Neefjes J, Ottenhoff T, Thole J, Young D. Coronin is involved in uptake of Mycobacterium bovis BCG in human macrophages but not in phagosome maintenance. Cell Microbiol (2001) 3(12):785–93. doi:10.1046/j.1462-5822.2001.00155.x
141. Gaullier JM, Simonsen A, D’Arrigo A, Bremnes B, Stenmark H, Aasland R. FYVE fingers bind PtdIns(3)P. Nature (1998) 394(6692):432–3. doi:10.1038/28767
142. Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, et al. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol (2001) 3(7):675–8. doi:10.1038/35083070
143. Johansson M, Rocha N, Zwart W, Jordens I, Janssen L, Kuijl C, et al. Activation of endosomal dynein motors by stepwise assembly of Rab7-RILP-p150Glued, ORP1L, and the receptor betalll spectrin. J Cell Biol (2007) 176(4):459–71. doi:10.1083/jcb.200606077
144. Bruns H, Stegelmann F, Fabri M, Dohner K, van Zandbergen G, Wagner M, et al. Abelson tyrosine kinase controls phagosomal acidification required for killing of Mycobacterium tuberculosis in human macrophages. J Immunol (2012) 189(8):4069–78. doi:10.4049/jimmunol.1201538
145. Melo RC, Dvorak AM. Lipid body-phagosome interaction in macrophages during infectious diseases: host defense or pathogen survival strategy? PLoS Pathog (2012) 8(7):e1002729. doi:10.1371/journal.ppat.1002729
146. Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, Haddix PL, Collins HL, Fok AK, et al. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science (1994) 263(5147):678–81. doi:10.1126/science.8303277
147. Vergne I, Chua J, Singh SB, Deretic V. Cell biology of Mycobacterium tuberculosis phagosome. Annu Rev Cell Dev Biol (2004) 20:367–94. doi:10.1146/annurev.cellbio.20.010403.114015
148. Simonsen A, Lippe R, Christoforidis S, Gaullier JM, Brech A, Callaghan J, et al. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature (1998) 394(6692):494–8. doi:10.1038/28879
149. Fratti RA, Chua J, Vergne I, Deretic V. Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci U S A (2003) 100(9):5437–42. doi:10.1073/pnas.0737613100
150. Malik ZA, Thompson CR, Hashimi S, Porter B, Iyer SS, Kusner DJ. Cutting edge: Mycobacterium tuberculosis blocks Ca2+ signaling and phagosome maturation in human macrophages via specific inhibition of sphingosine kinase. J Immunol (2003) 170(6):2811–5. doi:10.4049/jimmunol.170.6.2811
151. Philips JA. Mycobacterial manipulation of vacuolar sorting. Cell Microbiol (2008) 10(12):2408–15. doi:10.1111/j.1462-5822.2008.01239.x
152. Wong D, Bach H, Sun J, Hmama Z, Av-Gay Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc Natl Acad Sci U S A (2011) 108(48):19371–6. doi:10.1073/pnas.1109201108
153. Russell DG. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol Rev (2011) 240(1):252–68. doi:10.1111/j.1600-065X.2010.00984.x
154. Mehra A, Zahra A, Thompson V, Sirisaengtaksin N, Wells A, Porto M, et al. Mycobacterium tuberculosis type VII secreted effector EsxH targets host ESCRT to impair trafficking. PLoS Pathog (2013) 9(10):e1003734. doi:10.1371/journal.ppat.1003734
155. Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J (2008) 27(7):1110–21. doi:10.1038/emboj.2008.31
156. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell (2004) 119(6):753–66. doi:10.1016/j.cell.2004.11.038
157. Alonso S, Pethe K, Russell DG, Purdy GE. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc Natl Acad Sci U S A (2007) 104(14):6031–6. doi:10.1073/pnas.0700036104
158. Bento CF, Empadinhas N, Mendes V. Autophagy in the fight against tuberculosis. DNA Cell Biol (2015) 34(4):228–42. doi:10.1089/dna.2014.2745
159. Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL Jr, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med (2009) 15(3):267–76. doi:10.1038/nm.1928
160. Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J (2007) 26(10):2527–39. doi:10.1038/sj.emboj.7601689
161. Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell (2005) 120(2):237–48. doi:10.1016/j.cell.2004.11.046
162. Amaravadi RK, Winkler JD. Lys05: a new lysosomal autophagy inhibitor. Autophagy (2012) 8(9):1383–4. doi:10.4161/auto.20958
163. Chude CI, Amaravadi RK. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int J Mol Sci (2017) 18(6). doi:10.3390/ijms18061279
164. Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol (2010) 221(2):117–24. doi:10.1002/path.2694
165. Forget EJ, Menzies D. Adverse reactions to first-line antituberculosis drugs. Expert Opin Drug Saf (2006) 5(2):231–49. doi:10.1517/14740338.5.2.231
166. Rech AJ, Mick R, Martin S, Recio A, Aqui NA, Powell DJ Jr, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med (2012) 4(134):134ra62. doi:10.1126/scitranslmed.3003330
167. Weinstock-Guttman B, Ramanathan M, Zivadinov R. Interferon-beta treatment for relapsing multiple sclerosis. Expert Opin Biol Ther (2008) 8(9):1435–47. doi:10.1517/14712598.8.9.1435
168. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer (2012) 12(4):237–51. doi:10.1038/nrc3237
169. Khan N, Pahari S, Vidyarthi A, Aqdas M, Agrewala JN. NOD-2 and TLR-4 signaling reinforces the efficacy of dendritic cells and reduces the dose of TB drugs against Mycobacterium tuberculosis. J Innate Immun (2016) 8(3):228–42. doi:10.1159/000439591
Keywords: mononuclear phagocyte system, tuberculosis, monocyte, macrophage, dendritic cell, pattern recognition receptors, infection, immunotherapy
Citation: Pahari S, Kaur G, Negi S, Aqdas M, Das DK, Bashir H, Singh S, Nagare M, Khan J and Agrewala JN (2018) Reinforcing the Functionality of Mononuclear Phagocyte System to Control Tuberculosis. Front. Immunol. 9:193. doi: 10.3389/fimmu.2018.00193
Received: 15 September 2017; Accepted: 23 January 2018;
Published: 09 February 2018
Edited by:
Geanncarlo Lugo-Villarino, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceReviewed by:
Niyaz Ahmed, International Centre for Diarrhoeal Disease Research, BangladeshSaurabh Aggarwal, University of Alabama at Birmingham, United States
Giampietro Sgaragli, University of Siena, Italy
Copyright: © 2018 Pahari, Kaur, Negi, Aqdas, Das, Bashir, Singh, Nagare, Khan and Agrewala. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Javed N. Agrewala, javed@imtech.res.in